

Abstract
α-Synuclein (α-Syn) is enriched in nerve terminals. Two mutations in the α-Syn gene (Ala53→ Thr and Ala30→ Pro) occur in autosomal dominant familial Parkinson's disease. Mice overexpressing the human A53T mutant α-Syn develop a severe movement disorder, paralysis, and synucleinopathy, but the mechanisms are not understood. We examined whether transgenic mice expressing human wild-type or familial Parkinson's disease-linked A53T or A30P mutant α-syn develop neuronal degeneration and cell death. Mutant mice were examined at early- to mid-stage disease and at near end-stage disease. Age-matched nontransgenic littermates were controls. In A53T mice, neurons in brainstem and spinal cord exhibited large axonal swellings, somal chromatolytic changes, and nuclear condensation. Spheroid eosinophilic Lewy body-like inclusions were present in the cytoplasm of cortical neurons and spinal motor neurons. These inclusions contained human α-syn and nitrated synuclein. Motor neurons were depleted (∼75%) in A53T mice but were affected less in A30P mice. Axonal degeneration was present in many regions. Electron microscopy confirmed the cell and axonal degeneration and revealed cytoplasmic inclusions in dendrites and axons. Some inclusions were degenerating mitochondria and were positive for humanα-syn. Mitochondrial complex IV and V proteins were at control levels, but complex IV activity was reduced significantly in spinal cord. Subsets of neurons in neocortex, brainstem, and spinal cord ventral horn were positive for terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling, cleaved caspase-3, and p53. Mitochondria in neurons had terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-positive matrices and p53 at the outer membrane. Thus, A53T mutant mice develop intraneuronal inclusions, mitochondrial DNA damage and degeneration, and apoptotic-like death of neocortical, brainstem, and motor neurons.
Keywords: axonopathy, DNA damage, motor neuron disease, neuronal apoptosis, nitrative stress, peroxynitrite, synucleinopathy
Introduction
Parkinson's disease (PD) is a chronically progressive, age-related, fatal neurological disease in humans that affects at least 1% of the population over 55 years of age (Olanow and Tatton, 1999). Resting tremor, rigidity, bradykinesia, gait disturbance, and postural instability characterize PD clinically. The neuropathological hallmarks of PD are degeneration and elimination of dopamine neurons in substantia nigra and other brainstem regions as well as the formation of proteinaceous intraneuronal or intraglial inclusions, known as Lewy bodies (LBs), composed of a dense core of filamentous material enshrouded by filaments 10–20 nm in diameter (Goedert, 2001). The molecular pathogenesis of PD is still not understood. Two forms of PD exist: idiopathic and heritable (familial). Most PD cases are idiopathic, with no known genetic component. The cause of idiopathic PD is unknown. Epidemiological studies reveal several risk factors for developing idiopathic PD in addition to aging, including exposure to pesticides, herbicides, and some industrial chemicals (Olanow and Tatton, 1999). Approximately 5–10% of PD patients have familial patterns of inheritance. Gene mutations with autosomal dominant or autosomal recessive inheritance patterns have been identified in familial forms of PD. PD-linked mutations occur in the genes encoding α-synuclein (α-syn), parkin, ubiquitin C-terminal hydrolyase-L1, PTEN (phosphatase and tensin homolog)-induced kinase-1, DJ-1, and leucine-rich repeat kinase-2 (Shen, 2004). Two mutations in the α-syn gene (Ala-53→ Thr and Ala-30→ Pro) have been found in rare autosomal dominant inherited forms of PD (Polymeropoulos et al., 1997; Krüger et al., 1998). Duplication and triplication in the α-syn gene are also causes of PD (Singleton et al., 2003).
α-Syn is a relatively small (140 amino acids), abundant protein found in cells throughout the nervous system and is particularly enriched in presynaptic nerve terminals (Maroteaux et al., 1988; Murphy et al., 2000; Lesuisse and Martin, 2002). Although the functions of α-Syn are not well known, it has roles at the presynaptic terminal (Chandra et al., 2004). α-Syn is a major structural component of LBs, but in most neurodegenerative diseases, LBs are associated with accumulation of wild-type (wt), not mutant, α-Syn (Goedert, 2001). In vitro, overexpression of wt or mutant α-Syn elevates the generation of intracellular reactive oxygen species (Junn and Mouradian, 2002), and aggregation of wt and mutated α-Syn is associated with enhanced cell death (Giasson et al., 2002). Several transgenic (Tg) mouse lines have been generated with various different promoters to drive overexpression of human wt or mutant α-Syn (Kahle et al., 2000; Masliah et al., 2000; van der Putten et al., 2000; Matsuoka et al., 2001; Giasson et al., 2002; Lee et al., 2002; Richfield et al., 2002; Gispert et al., 2003; Gomez-Isla et al., 2003; Yazawa et al., 2005). Tg mice overexpressing A53T mutant α-Syn develop a severe movement disorder and synucleinopathy (van der Putten et al., 2000; Giasson et al., 2002; Lee et al., 2002); however, it is still uncertain whether Tg α-Syn mice develop cell death and neuronal loss. In this study, we determined whether neuronal degeneration and cell death occur in Tg mice expressing human wt or mutant α-Syn.
Materials and Methods
Tg mice. The generation of Tg mice expressing high levels of human wt or mutant (A53T and A30P) α-Syn under the control of the mouse prion protein (PrP) promoter has been described (Lee et al., 2002). Mice expressing A53T α-Syn (lines G2-3 and H5), but not mice expressing wt (line I2-2) or A30P (O2) α-Syn, develop adult-onset progressive motor deficits, including reduced spontaneous activity with bradykinesia, mild ataxia, and dystonia, at ∼10–15 months of age followed by rapidly progressive paralysis and death (Lee et al., 2002). For histological studies, we evaluated A53T mice (line G2-3) at early to mid stages of disease (n = 6), defined by the presence of bradykinesia and ataxia, and at near end-stage disease (n = 15), defined by paralysis. Age-matched non-Tg littermates served as controls. Tg mice harboring the A30P mutant (n = 6; mean age, ∼20 months) and wt α-Syn (n = 6; mean age, 26 months) were also evaluated.
Tissue harvesting and processing for histology. Mice were anesthetized with an overdose of sodium pentobarbital and perfused through the heart with ice-cold PBS (100 mm), pH 7.4, followed by ice-cold 4% paraformaldehyde or 4% paraformaldehyde with 0.1% glutaraldehyde. After perfusion–fixation, the brain, spinal cord, and skeletal muscle were removed and then were processed in one of three ways. Samples of brain and skeletal muscle were processed for paraffin histology. Brain and muscle samples were cut as serial sections (12 μm) on a rotary microtome in the sagittal plane, with every 10th section stained with hematoxylin and eosin (H&E) and every 11th section stained with the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) method (Portera-Cailliau et al., 1997; Martin and Liu, 2002b), as an assay for cell death based on the detection of DNA double-strand breaks. Alternatively, brains, spinal cords, and hindlimb skeletal muscles were placed in 20% glycerol for cryoprotection before frozen sectioning for cresyl violet staining, immunohistochemistry, or enzyme histochemistry or in phosphate buffer for vibratome sectioning for conventional electron microscopy (EM) and immunogold EM. Transverse serial symmetrical sections (40 μm) through brain and spinal cord were cut with a sliding microtome or vibratome and stored individually in 96-well plates.
Silver staining. Silver staining was used to visualize degenerating neuronal elements in brain and spinal cord sections of α-Syn mice and non-Tg age-matched control mice. Sections were processed with the FD NeuroSilver kit (FD Neurotechnologies, Baltimore, MD). For quantification of degeneration in spinal cord, entire transverse sections were digitized with an image analysis system. Inquiry software (Loats Associates, Westminster, MD) was used to measure the optical density of silver staining within the entire section (six to eight sections per mouse). The validation of this method has been described (Northington et al., 2001). Comparisons among groups were analyzed with a one-way ANOVA and a Newman–Keuls post hoc test.
Immunohistochemistry and enzyme histochemistry. The expression and localization patterns of human α-Syn, nitrated-Syn, cleaved caspase-3, p53, and phosphorylated neurofilament were examined at the light microscopic level in CNS or skeletal muscle by means of an immunoperoxidase method with diaminobenzidine as chromogen. Human α-Syn was detected with the Syn211 mouse monoclonal antibody (Sigma, St. Louis, MO) diluted at 1:1000, and human nitrated Syn was detected with the Syn12 mouse monoclonal antibody (Covance, Princeton, NJ) diluted at 1:500. The antibodies used to detect cleaved caspase-3 (Chemicon, Temecula, CA) and p53 (Cell Signaling, Beverly, MA) have been characterized previously and were used as described (Lok and Martin, 2002; Martin and Liu, 2002a; Martin, 2003). Phosphorylated neurofilament, an axonal marker, was detected with mouse monoclonal antibody SMI-31 (Sternberger Monoclonal, Lutherville, MD) diluted at 1:10,000.
Skeletal muscle was also examined for neurogenic atrophy and inflammatory changes. ATPase activity, pH 9.4, was visualized in sections of gastrocnemius and quadriceps with a histochemical protocol (Chayen et al., 1973). Cells positive for Mac-1 or CD68 were visualized in A53T mouse skeletal muscle with rat monoclonal antibody to mouse CD11b (Biosource, Camarillo, CA) or CD68 (Serotec, Indianapolis, IN) and immunoperoxidase labeling with diaminobenzidene as chromogen.
Cell counting. Profile counting was used to estimate the numbers of chomatolytic neurons in H&E sections, dying cells in TUNEL preparations, and cleaved caspase-3+ cells in immunolabeled sections. The regions analyzed were the retrorubral field and pontine reticular formation. In sagittal sections that were matched for level, the number of chromatolytic, TUNEL+, and caspase-3+ profiles were counted in six nonoverlapping microscopic fields at 1000× magnification. Motor neuron counts were made with the stereological optical dissector method as described (Calhoun et al., 1996; Martin and Liu, 2002a). Spinal cord sections were selected with a random start and then systematically sampled (every ninth section) to generate subsamples of sections from each mouse lumbar spinal cord that were mounted on glass slides and stained with cresyl violet for evaluation. Motor neurons were counted at 1000× magnification by using strict morphological criteria. These criteria included a round, open, pale nucleus (not condensed and darkly stained), globular Nissl staining of the cytoplasm, and a diameter of ∼30–40 μm. With these criteria, astrocytes, oligodendrocytes, and microglia were excluded from the counts. Group means and variances were evaluated statistically by one-way ANOVA and a Newman–Keuls post hoc test.
EM. Tg A53T mice and age-matched littermate control mice were evaluated for ultrastructural pathology in brainstem and spinal cord with conventional EM and immunogold EM. For conventional EM, brainstem and spinal cord samples were microdissected from vibratome sections, rinsed in phosphate buffer, placed in 2% osmium tetroxide for 2 h, dehydrated, and embedded in plastic. Semithin sections (1 μm) stained with 1% toluidine blue were screened for areas of interest, and then thin sections (gold interference color) were cut on an ultramicrotome (Sorvall, Norwalk, CT), contrasted with lead citrate and uranyl acetate, and viewed with a JEOL (Peabody, MA) 100S electron microscope.
DNA damage, human α-Syn, the mitochondrial marker cytochrome c oxidase subunit I (Cox-I), and p53 were examined in A53T and control mice by immunogold EM. Vibratome sections of brain and spinal cord were processed with an immunogold-silver intensification detection system and a flat sample–Epon embedding technique as described (Furuta et al., 1997; Martin and Liu, 2002a). For TUNEL–EM, vibratome sections were subjected to free-thaw and in situ DNA end-labeled as described (Portera-Cailliau et al., 1997), but with visualization of biotin-dUTP with goat anti-biotin conjugated with colloidal gold. For human α-Syn, Cox-I, and p53 localizations, sections were subjected to free-thaw, blocked in gelatin-bovine serum albumin, and then incubated (at 4°C) for 3–5 d with monoclonal antibody to human α-Syn (Sigma) or Cox-I (Invitrogen, Eugene, OR) or polyclonal antibody to p53 (Cell Signaling). After primary antibody incubation, sections were rinsed and incubated (at room temperature) with goat anti-mouse IgG or goat anti-rabbit IgG conjugated to 1 or 5 nm colloidal gold (Amersham Biosciences, Arlington Heights, IL) diluted 1:50 for 4 h, followed by silver enhancement solution for 10–20 min (Amersham Biosciences) or with no silver enhancement. Immunoreacted sections were examined microscopically, and areas of interest were microdissected. Samples were osmicated, dehydrated by graded concentrations of ethanol, and embedded in resin. Plastic blocks were thin-sectioned with ultramicrotomes (Sorvall). Ultrathin sections without contrasting were viewed and photographed with a JEOL electron microscope. Electron micrographs were made by an observer unaware of sample history.
Immunoblotting. The levels of mitochondrial complex IV protein, Cox-I, and the complex V protein, ATP synthase (β subunit), were evaluated in A53T and age-matched control mice by immunoblotting. Mice (n = 3 per genotype; 11–12 months old) were deeply anesthetized and decapitated, and the spinal cord and brain were removed quickly and frozen on dry ice. Spinal cord and frontal cortex samples were homogenized with a Brinkmann (Westbury, NY) Polytron in ice-cold 20 mm Tris-HCl, pH 7.4, containing 10% (w/v) sucrose, 200 mm mannitol, Complete Protease Inhibitor Cocktail (Roche Diagnostics, Basel, Switzerland), 0.1 mm phenylmethylsulfonyl fluoride, 10 mm benzamidine, 1 mm EDTA, and 5 mm EGTA. Crude homogenates were sonicated for 15 s and then centrifuged at 1000 × gav for 10 min (4°C). The supernatant was centrifuged at 54,000 × gav for 20 min (4°C) to yield soluble (S2) and mitochondria-enriched pellet (P2) fractions. The pellet fraction was washed (twice) by trituration in homogenization buffer followed by centrifugation and then finally resuspended in homogenization buffer (without sucrose) supplemented with 20% (w/v) glycerol. Protein concentrations were measured by a Bio-Rad (Hercules, CA) protein assay with bovine serum albumin as a standard. This subcellular fractionation protocol has been verified (Martin et al., 2003). Proteins from spinal cord and frontal cortex samples were subjected to 16% SDS-PAGE and transferred to nitrocellulose membrane by electroelution as described (Martin et al., 2003). The reliability of sample loading and electroblotting in each experiment was evaluated by staining nitrocellulose membranes with Ponceau S before immunoblotting. If transfer was not uniform, blots were discarded and gels were run again. Blots were blocked with 2.5% nonfat dry milk with 0.1% Tween 20 in 50 mm Tris-buffered saline, pH 7.4, and then incubated overnight at 4°C with antibody to Cox-I or ATP synthase (both from Invitrogen). The antibodies were used at concentrations for visualizing protein immunoreactivity within the linear range (Lok and Martin, 2002). After the primary antibody incubation, blots were washed and incubated with horseradish peroxidase-conjugated secondary antibody (0.2 μg/ml), developed with enhanced chemiluminescence (Pierce, Rockford, IL), and exposed to x-ray film.
To quantify protein immunoreactivity, films were scanned and densitometry was performed as described (Martin et al., 2003). Protein levels were expressed as relative optical density measurements. Immunodensities were normalized to Ponceau S-stained proteins. Statistical analyses of group means and variances were performed with a one-way ANOVA followed by a post hoc Newman–Keuls test for individual comparisons.
Cytochrome c oxidase activity assay. A colorimetric assay (Wharton and Tzagoloff, 1967) was used for biochemical measurement of Cox activity (Martin et al., 2000) in the spinal cord and frontal cortex of A53T and age-matched control mice (n = 3 mice per group; 11–12 months old). Briefly, reduced cytochrome c (ferrocytochrome c) was prepared freshly for each experiment by mixing 1% cytochrome c (Sigma) with sodium hydrosulfite in 10 mm potassium phosphate buffer. Samples (20 μg of protein) of mitochondrial-enriched membrane fractions, prepared as described previously (Martin et al., 2003), were reacted in a cuvette with ferrocytochrome c in potassium phosphate buffer (10 mm), pH 7.0, at room temperature. The decrease in absorbance was measured spectrophotometrically at 550 nm every 15 s for a period of 60 s. The activity was defined in terms of the first-order velocity constant. Statistical analyses of group means and variances were performed with a one-way ANOVA followed by a post hoc Newman–Keuls test for individual comparisons.
Results
We analyzed multiple lines of adult Tg α-Syn mice and age-matched non-Tg littermate control mice for neuropathology. These mice express high levels of wt human α-Syn (line I2-2) or familial PD-linked A30P (line O2) or A53T (line G2-3) mutant α-Syn (Lee et al., 2002). The brains and spinal cords of these mouse lines were studied for abnormalities with H&E and Nissl staining as well as immunohistochemical staining for human α-synuclein. A conspicuous feature of mice harboring mutant α-Syn, but not wt α-Syn, was that neurons in brainstem and spinal cord manifested somal chromatolytic changes (Fig. 1A,B), defined by distension of the cell body, displacement of the nucleus toward the periphery of the soma, and dissolution the Nissl substance. These neurons also showed nuclear condensation (Fig. 1A). Subsets of large neurons had this chromatolytic signature. Many were found in A53T mice, whereas their occurrence was lower in A30P mice. The number of brainstem chromatolytic neurons increased with disease progression in A53T mice (Fig. 1C). Very few neurons with chromatolytic changes were seen in age-matched non-Tg control mice (Fig. 1C). Large axonal swellings were also present throughout brainstem and spinal cord in A53T mice (Fig. 1B). Neurons containing inclusions were seen in A53T mice. Round eosinophilic LB-like inclusions were found in the cytoplasm of spinal motor neurons (Fig. 1D) and cortical neurons (Fig. 1G). Intraneuronal cytoplasmic inclusions were positive for human α-Syn (Fig. 1E) and nitrated Syn (Fig. 1F).
Figure 1.
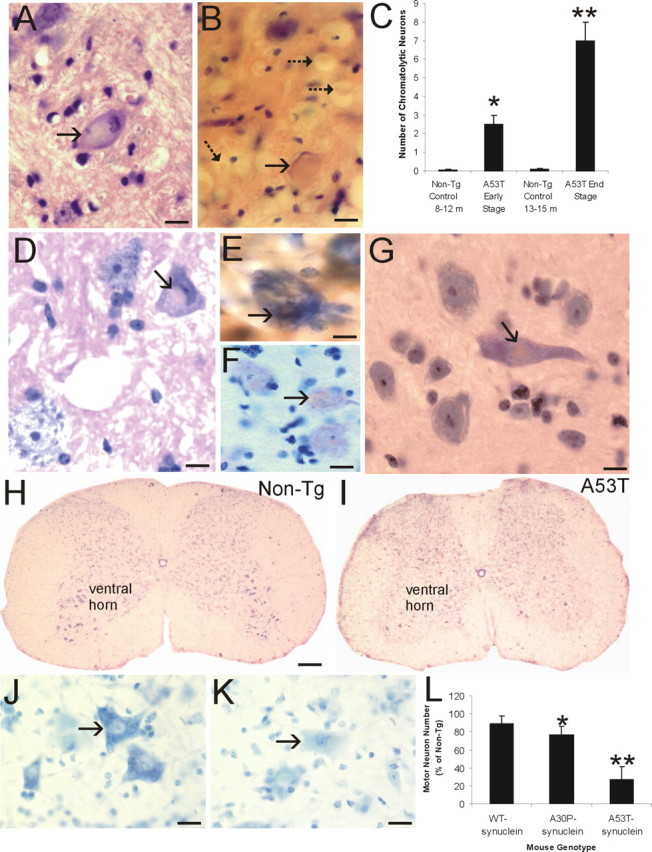
Axonal injury, inclusion formation, and motor neuron loss in A53T mice. A, H&E-stained section showing a large chromatolytic neuron (arrow) in brainstem with a ballooned cell body, dissolution of Nissl substance, nuclear displacement to the cell periphery, and chromatin condensation. B, H&E-stained section of upper cervical spinal cord showing a large chromatolytic neuron (solid arrow) with a ballooned cell body, homogenized cytoplasm, and a displaced nucleus that is compressed to a thin sliver. Also shown are several spheroids (dashed arrows). C, Graph showing the number of chromatolytic neurons in a brainstem area encompassing the retrorubal field and pedunculopontine tegmental nucleus in A53T mice killed at early and late stages of disease and in age-matched non-Tg littermate control mice. Values are mean ± SD.*p < 0.01 or **p < 0.001 (significant difference from control). D, H&E-stained section of lumbar spinal cord of A53T mouse showing an eosinophilic inclusion in a motor neuron (arrow). The inclusion is compressing the nucleus. The motor neuron in the bottom left appears normal. E, Intracytoplasmic inclusions in A53T mouse motor neurons (arrow) are immunopositive for human α-Syn (brown staining). Immunosections were counterstained with cresyl violet. F, Intracytoplasmic inclusions in A53T mouse motor neurons (arrow) are immunopositive for nitrated-Syn (brown staining). Immunosections were counterstained with cresyl violet. G, H&E-stained section of A53T mouse forebrain showing an eosinophilic, LB-like intracytoplasmic inclusion in a neocortical neuron (arrow). H, Cresyl violet-stained spinal cord section from a 14-month-old non-Tg mouse with a normal complement of motor neurons in ventral horn. I, Cresyl violet-stained spinal cord section from a 14-month-old paralyzed A53T mouse showing a marked depletion of motor neurons in ventral horn. J, High-magnification image of Nissl-stained spinal motor neurons (arrow) in non-Tg mouse characterized as large multipolar cells enriched in Nissl substance and with a large open nucleus. K, High-magnification image of Nissl-stained spinal motor neurons (arrow) in an A53T mouse showing the typical appearance of motor neurons before their loss. The cell body is devoid of Nissl substance and pale, and the nucleus is condensed. L, Graph showing the number of lumbar motor neurons in 19- to 25-month-old mice expressing wt α-Syn, 22-to 24-month-old A30P mutant α-Syn mice, and 9- to 15-month-old A53T mutant α-Syn mice. Values are mean ± SD. *p < 0.05 or **p < 0.001 (significant difference from non-Tg mice and human wt α-Syn Tg mice). Scale bars: A, B, 16 μm; D, 11 μm; E, 8 μm; F, 11 μm; G, 10 μm; H, I, 300 μm; J, K, 32 μm.
When Nissl-stained sections of spinal cord were examined, motor neurons appeared to be particularly lost at end-stage disease in mice harboring mutant α-Syn compared with age-matched littermate non-Tg mice (Fig. 1H,I). In the former mice, subsets of motor neurons had a pale cytoplasm and a condensed nucleus (Fig. 1J,K). Counts of lumbar motor neurons revealed a prominent (∼75%) depletion of motor neurons in A53T mice and a less dramatic loss in A30P mice (Fig. 1L). No significant loss of motor neurons was seen in Tg mice expressing wt human α-Syn at up to 22 months of age.
Prominent axonal degeneration was seen in silver-stained preparations of Tg mice with mutant α-Syn (Fig. 2). The most conspicuous degeneration was observed in the brainstem (Fig. 2A,B), spinal cord (Fig. 2C–H), and forebrain white matter (e.g., internal capsule; data not shown) of A53T mice. Axonal degeneration was seen less prominently in cerebral cortex and striatal white matter bundles (data not shown) of A53T mice. In spinal cord, axonal degeneration was selective and delineated specific tracts (Fig. 2D). The dorsal corticospinal tract in the dorsal column (Fig. 2D) and the rubrospinal tract in the dorsal lateral funiculus (Fig. 2D,E) had robust degeneration. Degenerating axons were also seen in the ventral root exit zones in the ventral funiculus (Fig. 2F). Densitometric quantification of silver staining revealed that spinal cord degeneration in A53T mice advanced with the progression of neurological deterioration (Fig. 2G). Moreover, subsets of motor neurons in A53T and A30P mice showed silver staining patterns indicative of apoptosis (Fig. 2 H). Axonal degeneration was sparse in Tg mice with wt human α-Syn (data not shown). Axonal degeneration was inconspicuous in age-matched littermate controls of mutant α-Syn mice (Fig. 2C,G).
Figure 2.
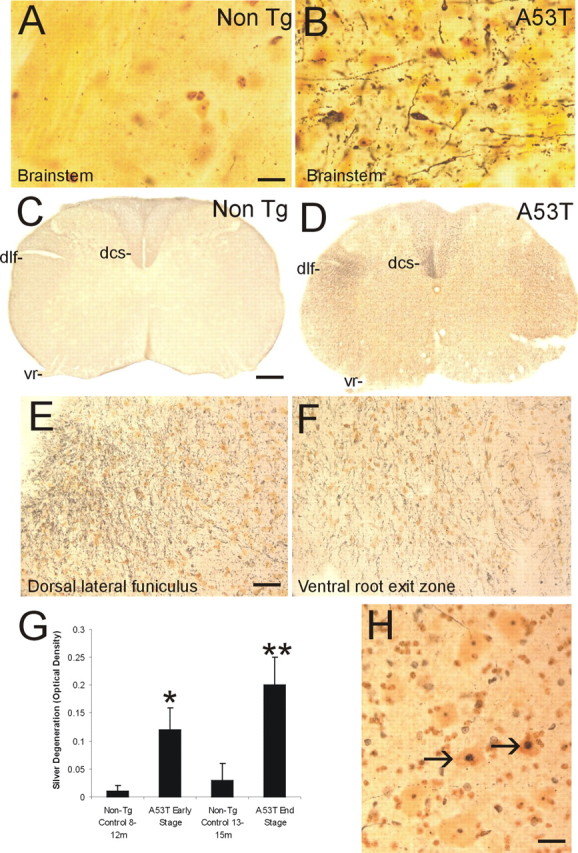
Axonal degeneration and motor neuron apoptosis in A53T mice. Silver staining was used to visualize axonal and neuronal cell body degeneration in brainstem and spinal cord of A53T mice. A, Axonal degeneration in the brainstem of age-matched non-Tg littermates is inconspicuous. The yellow-gold color is the typical background seen with FD NeuroSilver staining. B, The brainstem in A53T mice shows prominent axonal degeneration (black fibers), including axonal swellings (arrow). C, Axonal degeneration in the spinal cord of non-Tg littermates (age, 12–15 months) is minor. The dorsal corticospinal tract (dcs), dorsal lateral funiculus (dlf), and the ventral root (vr) are essentially free of degeneration. D, There is marked axonal degeneration in the dorsal corticospinal tract (dcs), dorsal lateral funiculus (dlf), ventral root (vr), and gray matter in A53T mouse spinal cord. E, Higher-magnification image of silver staining in the dorsal lateral funiculus. The fine black particles are degenerating axons seen in transverse profile. F, Degenerating axonal profiles (black fibers) are seen in the ventral root exit sites in A53T mice. G, Graph showing the quantification of degeneration in the spinal cord of A53T mice killed at early and late stages of disease and in age-matched non-Tg littermate control mice. Values are mean ± SD. *p < 0.01 or **p < 0.001 (significant difference from control). H, In silver-stained sections of spinal cord, subsets of motor neurons (arrows) undergo somal shrinkage and have a condensing nucleus with round, dark masses of chromatin similar to that seen in apoptosis. Other motor neurons (bottom left and top) appear normal. Scale bars: A, B, 20 μm; C, D, 150 μm; E, F, 32 μm; H, 25 μm.
EM was used to examine the subcellular localization of human α-Syn and the subcellular pathology in symptomatic A53T mice. Human α-Syn was detected in neuronal cell bodies, dendrites, and axons (Fig. 3F,G). It was localized within the cytosol and was associated with mitochondria and presynaptic active zones (Fig. 3F). Human α-Syn was seen occasionally at the nuclear membrane (data not shown). Immunogold labeling for human α-Syn was not seen in non-Tg mice, serving as a negative control. Pathologically, electron-dense cytoplasmic inclusions were observed in dendrites (Fig. 3A,B,D) and axons (Fig. 3C). Many of these inclusions had electron-dense perimeters with less dense cores. Other inclusions had long tubular shapes (Fig. 3D). Intradendritic inclusions were found near large, swollen mitochondria (Fig. 3B). Immunogold EM analysis confirmed that these structures contained human α-Syn (Fig. 3F,G). Some inclusions were degenerating mitochondria as identified by immunogold labeling for Cox-I (Fig. 3E). Cytoplasmic inclusions such as those shown in Figure 3 were not seen in age-matched non-Tg littermate controls.
Figure 3.
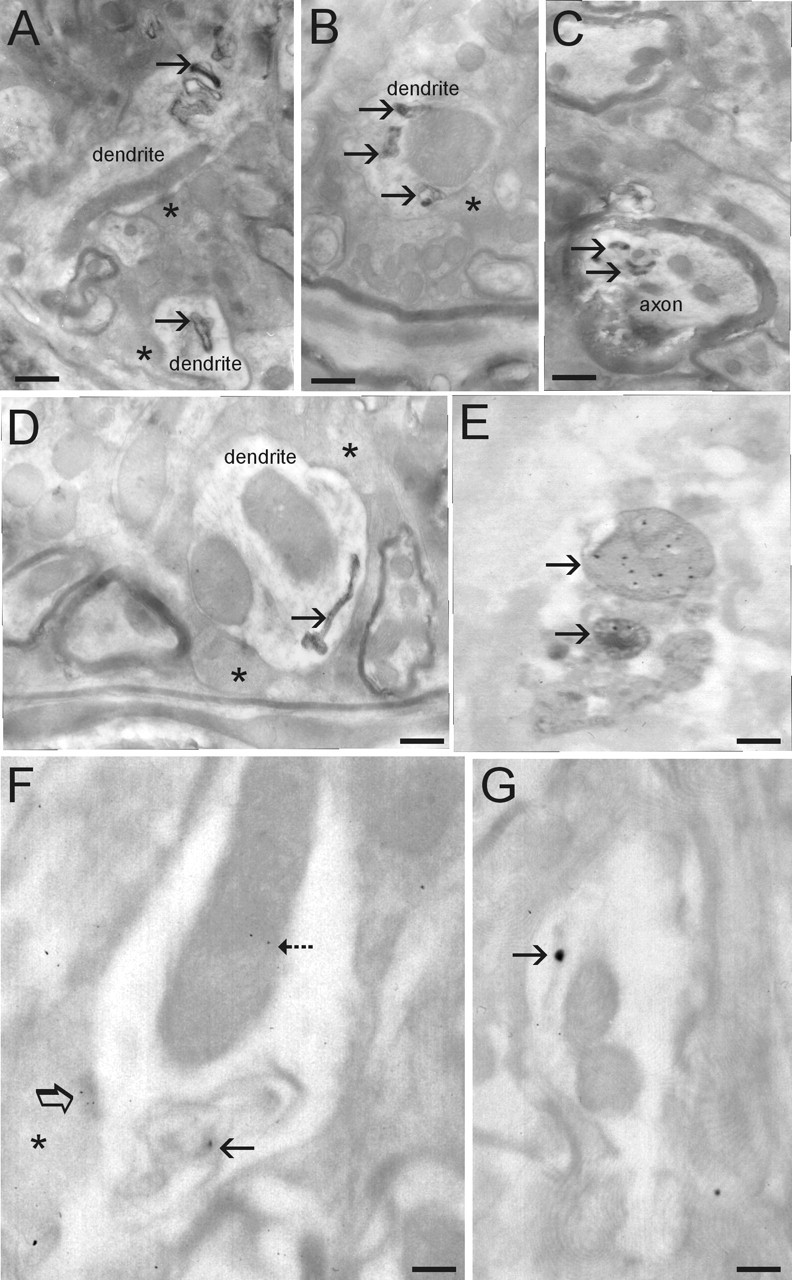
Ultrastructural evaluation of inclusions and mitochondrial degeneration in A53T mice. A, Dendrite profiles with inclusions (arrows). Asterisks identify presynaptic terminals on dendrites. B, Dendrite profile with large dilated mitochondrion (compare size with other mitochondria in the surrounding neuropil) that is associated with several electron-dense inclusions (arrows). C, Myelinated axonal profile containing several electron-dense inclusions (arrows). Axon profile at top of image appears normal. D, Dendrite profile containing anelectron-dense inclusion with a tubular shape and dilated at one end. E, Immunogold EM for Cox-I showing that some electron-dense inclusions are degenerating mitochondria. F, Immunogold EM for human α-syn showing its localization to mitochondria (dashed arrow) and abnormal inclusions (solid arrow) within a dendrite and to active zones of presynaptic terminals (asterisk, open arrow). G, Immunogold EM for human α-syn showing its localization to tubular inclusions (arrow; compare inclusion with one shown in D). E–G, Electron micrographs are fromt hins sections not contrasted to show more clearly the colloidal gold particles. Scale bars: A, 0.25 μm; B, 0.18 μm; C, 0.18 μm; D, 0.12μm; E, 0.1μm; F, 0.08μm; G, 0.09μm.
Because structural evidence for mitochondrial abnormalities was found in A53T mice, these mice were examined for additional independent measurements for mitochondrial changes (Fig. 4). Western blotting revealed no evidence for significant changes in the protein levels of Cox-1 and ATP synthase in spinal cord and frontal cortex of 11- to 12-month-old A53T mice (Fig. 4A,B). In contrast, the catalytic activity of Cox was significantly lower in the spinal cord of A53T mice compared with age-matched littermate non-Tg controls (Fig. 4C). This defect was regionally selective, because no difference in Cox activity was seen in the frontal cortex of A53T mice (Fig. 4C).
Figure 4.

Mitochondrial protein levels and functional activity in A53T mice. A, Western blot analysis of Cox-I and ATP synthase levels in mitochondrial-enriched membrane fractions of spinal cord and frontal cortex of 11- to 12-month-old A53T mice and wt age-matched littermate controls. Protein staining detected with Ponceau S was used as a loading control. B, Graph showing the quantitative densitometric analysis of Cox-I and ATP synthase protein levels in A53T mice and wt age-matched controls. The values are mean ± SD. No differences were detected. C, Biochemical assay for Cox-I enzyme activity in the spinal cord and frontal cortex of 11- to 12-month-old A53T mice and wt age-matched littermate controls. The values are mean ± SD. *p < 0.01; significant difference from wt control.
A53T mice at early and late stages of disease were evaluated for neuronal cell death. TUNEL was used to identify cells with double-stranded DNA breaks. Subsets of neurons in brainstem, neocortex, and spinal cord ventral horn were TUNEL positive (Fig. 5A,B). Some motor neurons had TUNEL-positive structures within the cytoplasm, having an appearance suggestive of mitochondria, but otherwise the neurons appeared normal (Fig. 5C). To determine whether the cytoplasmic TUNEL was caused by mitochondrial labeling, TUNEL-EM was done with colloidal gold. Subsets of mitochondria contained TUNEL gold (Fig. 5C, inset). EM also revealed evidence for apoptotic cell death in the spinal cord of A53T mice based on the nuclear morphology (Fig. 5J). To probe into the mechanisms of neuronal death, sections were immunolabeled for cleaved caspase-3 and p53. In A53T mice, brainstem and spinal motor neurons were immunopositive for cleaved caspase-3 (Fig. 5D,E). Age-matched non-Tg littermate controls were negative for cleaved caspase-3. The number of cleaved caspase-3-positive neurons in brainstem mirrored the number of TUNEL-positive cells as disease progressed (Fig. 5F,G). Spinal motor neurons in A53T mice accumulated p53, whereas spinal motor neurons in age-matched littermate controls were negative for p53 (Fig. 5H,I). In motor neurons of A53T mice, mitochondria had p53 associated with the outer mitochondrial membrane, whereas the mitochondria in motor neurons from age-matched non-Tg littermate control mice had sparse p53 immunolabeling (Fig. 5K,L).
Figure 5.

Neuronal cell death in A53T mice. A, B, Nuclear DNA fragmentation, identified by TUNEL, was detected in brainstem (A, arrow), sensorimotor cortex (B, arrows), and spinal motor neurons (data not shown) in A53T mice at early and end-stage disease. C, Subsets of spinal motor neurons had intracytoplasmic TUNEL seen as granules (arrow) that subsequent TUNEL–immunogold EM revealed to be mitochondria (inset). A–C, TUNEL preparations counterstained with cresyl violet. D, E, Subsets of large neurons in brainstem (D, arrow) and spinal motor neurons (E, arrow) displayed cleaved caspase-3 immunoreactivity indicative of apoptosis. F, Graph showing the quantification of dying cells (determined by TUNEL) in the brainstem retrorubal field and pontine reticular formation of A53T mice killed at early and late stages of disease and in non-Tg control mice. Values (cells per square millimeter) are mean ± SD. *p < 0.05 or **p < 0.01 (significant difference from control). G, Graph showing the quantification of the number of cleaved caspase-3 immunopositive cells in the brainstem retrorubal field and pontine reticular formation of A53T mice killed at early and late stages of disease and in non-Tg control mice. Values (cells per square millimeter)are mean ± SD. *p < 0.05 or **p < 0.01 (significant difference from control). H, p53 was not detected in spinal motor neurons in 12- to 15-month-old non-Tg mice. I, Subsets of spinal motor neurons in A53T mice accumulated p53 (arrows), but other motor neurons were not positive for p53 (bottom left). J, EM on A53T mouse spinal cord revealed the presence of degenerating cells with a nuclear morphology consistent with apoptosis (white asterisk). The nucleus is condensed into a uniformly dense round mass. K, L, Immunogold EM for p53 in spinal motor neurons showed that in age-matched non-Tg mice, p53 was at a low level in the cytoplasm (K, arrows) and was associated rarely with the mitochondria; in contrast, in A53T mice, p53 was associated with the outer mitochondrial membrane (L, arrows). Scale bars: A, 8 μm; B, 10 μm; C, 12 μm (inset, 0.18 μm); D, E, 22 μm; H, I, 22 μm; J, 2.5 μm; K, L, 6.5 μm.
A53T mice developed extensive peripheral nerve degeneration and skeletal muscle disease (Fig. 6). Axonal degeneration was conspicuous in intramuscular peripheral nerve (Fig. 6A). Subsets of axons in peripheral nerve were enlarged, swollen, distorted, and vacuolated (Fig. 6A). In all affected A53T mice, marked loss of motor neurons was accompanied by evidence of skeletal muscle denervation and extensive muscle atrophy. H&E staining of skeletal muscle sections revealed that the normal, tightly packed, plump, myofiber pattern was replaced in A53T mice by shrunken, angular, atrophic myofibers (Fig. 6B,C). Histochemical staining for ATPase, pH 4.6, to distinguish between fast type 1 fibers and slow type 2 fibers revealed a loss of the type 1 myofiber staining and grouped atrophy in A53T mice (Fig. 6D,E). The death of myofibers in A53T mice was revealed by TUNEL (Fig. 6F). These abnormalities were not evident in age-matched non-Tg littermate control mice. Severe muscle pathology was indicated further by the infiltration of many small cells and evidence of sarcolysis in individual myofibers (Fig. 6G). Skeletal muscle inflammatory changes in A53T mice were confirmed by the presence of infiltrated CD68+ cells (Fig. 6H) and CD11b+ cells (Fig. 6I) into the muscle. The presence of infiltrated CD68+ cells was associated with the rounding of myofiber nuclei and the condensation of nuclear chromatin (Fig. 6H). Inflammatory changes were not seen in the skeletal muscle of age-matched non-Tg littermate control mice (Fig. 6J).
Figure 6.
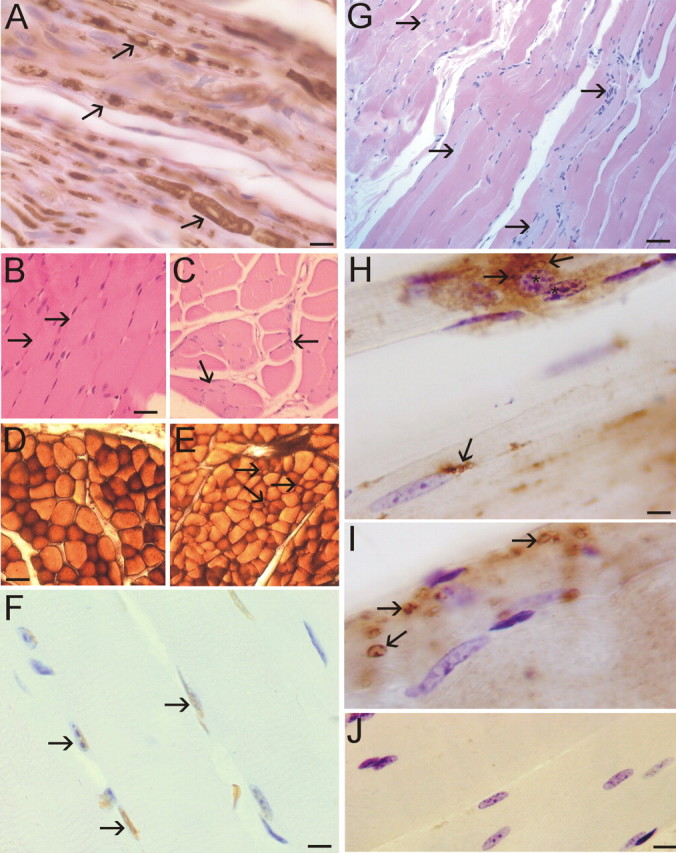
Peripheral nerve and skeletal muscle degeneration in A53T mice. A, Neurofilament staining of peripheral nerve in skeletal muscle of A53T mice revealed degenerating axons as indicated by swelling and vacuolization (arrows). B, H&E staining of skeletal muscle sections (quadriceps femoris) from non-Tg mice (12 months of age) revealed a normal histology with tightly packed plump myofibers (arrows). C, H&E staining of skeletal muscle sections (quadriceps femoris) from A53T mice (11 months of age) revealed the presence of myofiber atrophy, as indicated by the angular and shrunken myofibers and grouped atrophy (arrows). D, Enzyme histochemical staining for myofiber ATPase, pH 4.6, in non-Tg mice skeletal muscle revealed a pattern of darkly stained type 1 fibers and more lightly stained type 2 fibers. E, ATPase staining of A53T mouse skeletal muscle revealed a loss of type 1 fibers and the presence of grouped atrophy (arrows). F, The TUNEL assay showed that A53T skeletal muscle contained subsets of myofiber nuclei (arrows, brown staining) undergoing DNA fragmentation. Cresyl violet counterstaining showed that the TUNEL is specific for only a subset of nuclei (blue-stained nuclei are not labeled). Some TUNEL-positive nuclei (left arrow) also show evidence of nuclear condensation. G, In longitudinally cut skeletal muscle sections of A53T mice, H&E staining revealed atrophy of individual myofibers (arrows), indicated by the pale staining, and suggested tissue inflammation with the presence of many small nuclei. H, A53T mouse skeletal muscle showed evidence of tissue inflammation as indicated by the presence of CD68+ cells (arrows). Myofibers associated with intense CD68 immunolabeling showed evidence of chromatin condensation in the nucleus (asterisks). I, Additional evidence for inflammatory changes in A53T mouse skeletal muscle was revealed by infiltrated CD11b+ cells (arrow). J, Immunohistochemical evidence of skeletal muscle inflammation was not observed in age-matched non-Tg littermate control mice. Scale bars: A, 10 μm; B, C, 25 μm; D, E, 40 μm; F, 12 μm; G, 40 μm; H, I, 7 μm; J, 12 μm.
Discussion
This study provides new information about the development of neurological disease in α-Syn Tg mice. We found that mice harboring the A53T mutant variant of α-Syn have a robust neurodegeneration phenotype. These mice develop intraneuronal inclusions, mitochondrial degeneration, and cell death in neocortex, brainstem, and spinal cord. A53T mice formed LB-like inclusions in neocortical and spinal motor neurons and had profound loss of motor neurons that could account for their paralysis.
Various mouse lines expressing wt or mutant α-Syn have been described with varying pathological characteristics. Mice expressing wt α-Syn driven by a human platelet-derived growth factor-β promoter did not develop fatal disease, although they displayed modest impairments in motor function and developed amorphous nonfilamentous inclusions, most of which were intranuclear, and reduced striatal tyrosine hydroxylase (TH) immunoreactivity (Masliah et al., 2000). Mice expressing wt or mutant α-Syn driven by the TH promoter did not develop clinical or neuropathological phenotypes (Matsuoka et al., 2001). In contrast, mice expressing wt and mutant α-Syn under the control of the murine Thy-1 promotor developed pronounced motor deficits on the rotating rod task, intraneuronal α-Syn accumulation, axonal degeneration in spinal roots, and evidence of muscle denervation (van der Putten et al., 2000). With the mouse PrP promoter, Giasson et al. (2002) engineered mice expressing the A53T mutant and found that these mice become paralyzed and develop α-Syn inclusions, primarily in brainstem and spinal cord, and axonal degeneration in spinal roots. Similarly, Lee et al. (2002) generated Tg mice expressing human wt α-Syn as well as A53T and A30P mutant variants with the murine PrP promoter, with only the A53T mice developing profound adult-onset motor abnormalities that progressed to paralysis and death. These mice also showed neuronal accumulation of α-Syn and ubiquitin in brainstem and neocortex (Lee et al., 2002); however, no previous studies have reported evidence for neuronal cell death and loss in α-Syn Tg mice.
We show that the neurodegeneration in A53T mice is robust. Brainstem neurons and spinal motor neurons display a prominent chromatolysis reaction and axonal spheroids, typical of that seen with axonal injury (Lieberman, 1971). The presence of marked axonopathy was confirmed by silver staining, EM, and peripheral nerve neurofilament staining and was further indicated by neurogenic atrophy of skeletal muscle. Cell death, detected with the TUNEL method, was seen in neocortex, brainstem, and spinal cord. The considerable pathology in lower brainstem in A53T mice parallels the involvement of these regions in human PD (Braak et al., 2003). Early α-Syn lesions are confined chiefly to the lower brainstem in human PD, with involvement of the dorsal motor nucleus of the glossopharyngeal and vagal nerves (Braak et al., 2003). The substantia nigra was unremarkable in these mice, consistent other studies of α-Syn Tg mice failing to find evidence for substantial dopaminergic neuron degeneration and loss (Masliah et al., 2000; van der Putten et al., 2000; Giasson et al., 2002), even with expression of transgene driven by a TH promoter (Matsuoka et al., 2001).
We found that A53T mice develop mitochondrial pathology. Mitochondria in brainstem and spinal cord cells in A53T mice were dysmorphic as shown by EM. Subsets of mitochondria contained human α-Syn and were shrunken, swollen, or vacuolated. Some abnormal inclusions were degenerating mitochondria, as determined by Cox-I immunogold staining. A mitochondrial defect in A53T mice was further indicated by biochemical evidence revealing loss of Cox activity. Mitochondrial structure abnormalities and functional deficits have been seen in vitro in neurons overexpressing wt human α-Syn (Hsu et al., 2000). We observed a new type of mitochondrial pathology in A53T mice involving mitochondrial DNA damage as seen by TUNEL. Brainstem neurons and spinal motor neurons accumulated perinuclear TUNEL+ structures that were shown by TUNEL–immunogold EM to be mitochondria. Mitochondrial DNA damage was seen frequently in the absence of nuclear DNA damage in large brainstem neurons in A53T mice; thus, mitochondrial DNA damage could be an antecedent of neuronal cell death. The mechanisms for this mitochondrial DNA damage are possibly related to oxidative stress (Giasson et al., 2000). The presence of oxidative stress in mutant α-Syn Tg mice is shown by evidence that mitochondrial-associated metabolic proteins are oxidized in A30P mice (Poon et al., 2005). α-Syn can generate H2O2 (Turnbull et al., 2001). We found evidence for peroxynitrite-mediated oxidative–nitrative stress in A53T mouse motor neurons as indicated by the presence of nitrated synuclein. Nitrated synuclein formed inclusions in motor neurons consistent with in vitro data showing that peroxynitrite promotes the formation of stable α-Syn oligomers (Souza et al., 2000; Paxinou et al., 2001). Our data showing mitochondrial DNA damage is in line with the presence of peroxynitrite near mitochondria, because peroxynitrite is genotoxic by causing single- and double-strand breaks in DNA (Liu and Martin, 2001; Martin and Liu, 2002b). Moreover, our data showing loss of Cox-I enzyme activity without a change in protein level could be explained by oxidative inactivation.
The neuronal cell death seen in the brainstem and spinal cord of A53T mice appears to be a form of apoptosis. Motor neurons showed nuclear condensation as seen by silver and cresyl violet staining. EM showed that the nuclear morphology of degenerating cells in spinal cord is consistent with apoptosis (Martin, 2001). Moreover, subsets of affected neurons were positive for cleaved caspase-3 and p53. These changes seen in A53T mice are consistent with neuronal apoptosis induced by axotomy, particularly regarding the chromatolysis, chromatin condensation, and accumulation of DNA double-strand breaks detected with the TUNEL assay (Martin and Liu, 2002a). The mitochondria in motor neurons of A53T mice had p53 at the outer mitochondrial membrane. Recent studies have shown that p53 has a direct apoptogenic role at mitochondria (Mihara et al., 2003). p53 can mediate mitochondrial membrane permeabilization and apoptosis by activating Bax and Bak (Chipuk et al., 2004; Leu et al., 2004). Thus, the neuronal cell death seen in A53T mice appears to be apoptotic and involves p53 and caspase activation.
α-Syn has a role in cell death, but the precise pro-apoptotic versus anti-apoptotic roles in specific types of neurons remain to be delineated more clearly. Wild-type α-Syn can protect neurons from apoptosis by inhibition of caspase-3 (Alves da Costa et al., 2002). Mutations in α-Syn abolish its inhibitory modulation of caspase-3 (Alves da Costa et al., 2002). Aggregation of wt and PD-related mutated α-Syn is associated with enhanced cell death (Giasson et al., 2002). This process could be caused by the intrinsic pro-apoptotic properties of α-Syn or the loss of an anti-apoptotic function. The latter possibility is supported by evidence that α-Syn lowers the sensitivity of neurons to apoptosis driven by p53 (Alves da Costa et al., 2002) and activates MAPK (mitogen-activated protein kinase) survival pathways (Iwata et al., 2001). The functions of α-Syn might be context and neuron type dependent. For example, in vitro, α-Syn is pro-apoptotic in dopaminergic neurons, with toxicity requiring dopamine and the production of reactive oxygen species, but is neuroprotective in nondopaminergic cortical neurons (Xu et al., 2002); however, in vivo, TH promoter-driven expression of wt or mutant human α-Syn Tg mice is not toxic to mouse dopaminergic neurons (Matsuoka et al., 2001), but nigral dopamine neurons in rat and monkey are vulnerable to adeno-associated viral-mediated overexpression human wt and mutant α-Syn (Kirik et al., 2002, 2003).
The accountability for the high vulnerability of mouse motor neurons to A53T mutant human α-Syn is not clear. The forebrain, diencephalon, and midbrain of these mice express high levels of mRNA and protein for human α-Syn mRNA and protein (Lee et al., 2002), but these regions appear less vulnerable than spinal cord. Because A53T α-Syn causes axonopathy (van der Putten et al., 2000; Giasson et al., 2002), the high vulnerability of motor neurons to mutant α-Syn, including the formation of LB-like inclusions, could be related to their long myelinated axons and interactions with oligodendrocytes and Schwann cells for myelin support. Interestingly, the stability of α-Syn increases with aging (Li et al., 2004a), whereas axonal transport of α-Syn declines with aging (Li et al., 2004b), possibly contributing to axonal aggregation and inclusion formation. In human amyotrophic lateral sclerosis, spinal motor neurons can display LB-like filamentous inclusions (Kato et al., 1989; Murayama et al., 1989), most of which are ubiquitinated (Kato et al., 1989), and significant abnormal α-Syn staining of spheroids and corticospinal axons (Doherty et al., 2004).α-Syn is also upregulated in motor neurons in Tg mice expressing mutant human Cu/Zn superoxide dismutase (Chung et al., 2003); however, motor neuron degeneration in A53T mice might not be cell autonomous, because overexpression of wt α-Syn selectively in oligodendrocytes causes their degeneration as well as the degeneration of neurons, including motor axons in ventral roots (Yazawa et al., 2005). Moreover, distal axonopathy and muscle disease may have roles in the pathogenesis in A53T mice. A previous study and this work reveal skeletal muscle denervation in α-Syn Tg mice (van der Putten et al., 2000). We show that the mutant α-Syn A53T Tg mouse is an alternative model for studying mechanisms of motor neuron degeneration and could provide insight into the possible role of α-Syn in motor neuron disease.
Footnotes
This work was supported by grants from the United States Public Health Service, National Institutes of Health, National Institute of Neurological Disorders and Stroke (NS34100, NS52098), and National Institute on Aging (AG16282 to L.J.M. and NS38065, NS38377 to (M.K.L.)
Correspondence should be addressed to Dr. Lee J. Martin, Department of Pathology, Johns Hopkins University School of Medicine, 558 Ross Building, 720 Rutland Avenue, Baltimore, MD 21205-2196. E-mail: martinl@jhmi.edu.
DOI:10.1523/JNEUROSCI.4308-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260041-10$15.00/0
References
- Alves da Costa C, Paitel E, Vincent B, Checler F (2002) Alpha-synuclein lowers p53-dependent apoptotic of neuronal cell: abolishment by 6-hydroxydopamine and implication for Parkinson's disease. J Biol Chem 277: 50980–50984. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24: 197–211. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Jucker M, Martin LJ, Thinakaran G, Price DL, Mouton PR (1996) Comparative evaluation of synaptophysin-based methods for quantification of synapses. J Neurocytol 25: 821–828. [DOI] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon H-B, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Südhof TC (2004) Double-knockout mice for α- and β-synucleins: effects on synaptic functions. Proc Natl Acad Sci USA 101: 14966–14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chayen J, Bitensky L, Butcher R (1973) Practical histochemistry. New York: Wiley.
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303: 1010–1014. [DOI] [PubMed] [Google Scholar]
- Chung YH, Joo KM, Kim MJ, Cha CI (2003) Immunohistochemical study on the distribution of alpha-synuclein in the central nervous system of transgenic mice expressing a human Cu/Zn superoxide dismutase mutation. Neurosci Lett 342: 151–154. [DOI] [PubMed] [Google Scholar]
- Doherty MJ, Bird TD, Leverenz JB (2004) Alpha-synuclein in motor neuron disease: an immunohistologic study. Acta Neuropathol 107: 169–175. [DOI] [PubMed] [Google Scholar]
- Furuta A, Martin LJ, Lin CL, Dykes-Hoberg M, Rothstein JD (1997) Cellular and synaptic localization of the neuronal glutamate transporters, excitatory amino acid transporters 3 and 4. Neuroscience 81: 1031–1042. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM (2000) Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 290: 985–989. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM (2002) Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 34: 521–533. [DOI] [PubMed] [Google Scholar]
- Gispert S, Del Turco D, Garrett L, Chen A, Bernard DJ, Hamm-Clement J, Korf HW, Deller T, Braak H, Auburger G, Nussbaum RL (2003) Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol Cell Neurosci 24: 419–429. [DOI] [PubMed] [Google Scholar]
- Goedert M (2001) Alpha-synuclein and neurodegenerative diseases. Nat Rev 2: 492–501. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Irizarry MC, Mariash A, Cheung B, Soto O, Schrump S, Sondel J, Kotilinek L, Day J, Schwarzschild MA, Cha JH, Newell K, Miller DW, Ueda K, Young AB, Hyman BT, Ashe KH (2003) Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiol Aging 24: 245–258. [DOI] [PubMed] [Google Scholar]
- Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E (2000) Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol 157: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Maruyama M, Kanazawa I, Nukina N (2001) α-Synuclein affects the MAPK pathway and accelerates cell death. J Biol Chem 276: 45320–45329. [DOI] [PubMed] [Google Scholar]
- Junn E, Mouradian MM (2002) Human α-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci Lett 320: 146–150. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C (2000) Subcellular localization of wild-type and Parkinson's disease-associated mutant α-synuclein in human and transgenic mouse brain. J Neurosci 20: 6365–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Katagiri T, Hirano A, Kawanami T, Sasaki H (1989) Lewy body-like hyaline inclusions in sporadic motor neuron disease are ubiquitinated. Acta Neuropathol 77: 391–396. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C Burger C, Lindberg C, Johansen TE, Muzyczka N, Mandel RJ, Björklund A (2002) Parkinson-like neurodegeneration induced by targeted overexpression of α-synuclein in the nigrostriatal system. J Neurosci 22: 2780–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Björklund A (2003) Nigrostriatal α-synucleinopathy induced by viral vector-mediated overexpression of human α-synuclein: a new primate model of Parkinson's disease. Proc Natl Acad Sci USA 100: 2884–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Reiss O (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat Genet 18: 106–108. [DOI] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL (2002) Human α-synuclein-harboring familial Parkinson's disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA 99: 8968–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesuisse C, Martin LJ (2002) Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol 51: 9–23. [DOI] [PubMed] [Google Scholar]
- Leu JI-J, Dumont P, Hafey M, Murphy ME, George DL (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 6: 443–450. [DOI] [PubMed] [Google Scholar]
- Li W, Lesuisse C, Xu Y, Troncoso JC, Price DL, Lee MK (2004a) Stabilization of α-synuclein protein with aging and familial Parkinson's disease-linked A53T mutation. J Neurosci 24: 7400–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Hoffman PN, Stirling W, Price DL, Lee MK (2004b) Axonal transport of human alpha-synuclein slows with aging but is not affected by familial Parkinson's disease-linked mutations. J Neurochem 88: 401–410. [DOI] [PubMed] [Google Scholar]
- Lieberman AR (1971) The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int Rev Neurobiol 14: 49–124. [DOI] [PubMed] [Google Scholar]
- Liu Z, Martin LJ (2001) Motor neurons rapidly accumulate DNA single-strand breaks after in vitro exposure to nitric oxide and peroxynitrite and in vivo axotomy. J Comp Neurol 432: 35–60. [DOI] [PubMed] [Google Scholar]
- Lok J, Martin LJ (2002) Rapid subcellular redistribution of Bax precedes caspase-3 and endonuclease activation during excitotoxic neuronal apoptosis in rat brain. J Neurotrauma 19: 815–828. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH (1988) Synuclein: a neuronspecific protein localized to the nucleus and presynaptic nerve terminals. J Neurosci 8: 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ (2001) Neuronal cell death in nervous system development, disease, and injury. Int J Mol Med 7: 455–478. [PubMed] [Google Scholar]
- Martin LJ, Liu Z (2002a) Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and bax-dependent. J Neurobiol 5: 181–197. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Liu Z (2002b) DNA damage profiling in motor neurons: a single-cell analysis by comet assay. Neurochem Res 27: 1089–1100. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Price AC, Kaiser A, Agnew DM, Ichord RN, Traystman RJ (2000) Neuronal death in newborn striatum after hypoxiaischemia is necrosis and evolves with oxidative stress. Neurobiol Dis 7: 169–191. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Price AC, McClendon KB, Al-Abdulla NA, Subramaniam JR, Wong PC, Liu Z (2003) Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J Neurochem 85: 234–247. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000) Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 287: 1265–1269. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA (2001) Lack of nigral pathology in transgenic mice expressing alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis 8: 535–539. [DOI] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577–590. [DOI] [PubMed] [Google Scholar]
- Murayama S, Ookawa Y, Mori H, Nakano I, Ihara Y, Kuzuhara S, Tomonaga M (1989) Immunocytochemical and ultrastructural study of Lewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis. Acta Neuropathol 78: 143–152. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM (2000) Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 20: 3214–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ (2001) Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis 8: 207–219. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG (1999) Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci 22: 123–144. [DOI] [PubMed] [Google Scholar]
- Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, Ischiropoulos H (2001) Induction of α-synuclein aggregation by intracellular nitrative insult. J Neurosci 21: 8053–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276: 2045–2047. [DOI] [PubMed] [Google Scholar]
- Poon HF, Frasier M, Shreve N, Calabrese V, Wolozin B, Butterfield DA (2005) Mitochondrial associated metabolic proteins are selectively oxidized in A30P α-synuclein transgenic mice: a model of familial Parkinson's disease. Neurobiol Dis 18: 492–498. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ (1997) Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol 378: 70–87. [PubMed] [Google Scholar]
- Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuetzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ (2002) Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol 175: 35–48. [DOI] [PubMed] [Google Scholar]
- Shen J (2004) Protein kinases linked to the pathogenesis of Parkinson's disease. Neuron 44: 575–577. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. (2003) Alpha-synuclein locus triplication causes Parkinson's disease. Science 302: 841. [DOI] [PubMed] [Google Scholar]
- Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H (2000) Dityrosine cross-linking promotes formation of stable α-synuclein polymers. J Biol Chem 265: 18344–18349. [DOI] [PubMed] [Google Scholar]
- Turnbull S, Tabner BJ, El-Agnaf OM, Moore S, Davies Y, Allsop D (2001) Alpha-synuclein implicated in Parkinson's disease catalyses the formation of hydrogen peroxide in vitro. Free Radic Biol Med 30: 1163–1170. [DOI] [PubMed] [Google Scholar]
- van der Putten H, Wiederhold K-H, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WPJM, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G (2000) Neuropathology in mice expressing human α-synuclein. J Neurosci 20: 6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton DC, Tzagoloff A (1967) Cytochrome oxidase from beef heart mitochondria. Methods Enzymol 10: 245–250. [Google Scholar]
- Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA (2002) Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med 8: 600–606. [DOI] [PubMed] [Google Scholar]
- Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, Trojanowski JQ, Lee VM (2005) Mouse model of multiple system atrophy α-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 45: 847–859. [DOI] [PubMed] [Google Scholar]