

Abstract
Epilepsy is a brain disorder characterized by a predisposition to suffer epileptic seizures. Acquired epilepsy might be the result of brain insults like head trauma, stroke, brain infection, or status epilepticus (SE) when one of these triggering injuries starts a transformative process known as epileptogenesis. There is some data to suggest that, during epileptogenesis, seizures themselves damage the brain but there is no conclusive evidence to demonstrate that spontaneous recurrent seizures themselves injure the brain. Our recent evidence indicates calpain overactivation might be relevant for epileptogenesis. Here, we investigated if spontaneous recurrent seizures that occur during an early period of epileptogenesis show any correlation with the levels of calpain activation and/or expression. In addition, we also investigated a possible association between the occurrence of spontaneous seizures and increased levels of cell death, gliosis and inflammation (typical markers associated with epileptogenesis). We found that the number of spontaneous seizures detected prior to sample collection was correlated with altered calpain activity and expression. Moreover, the levels of hippocampal neurodegeneration were also correlated with seizure occurrence. Our findings suggest that, at least during early epileptogenesis, there is a correlation between seizure occurrence, calpain activity and neurodegeneration. Thus, this study opens the possibility that aberrant calpain reactivation by spontaneous seizures might contribute to the manifestation of future spontaneous seizures.
Keywords: Spontaneous Seizures, Calpain, Cell Death, gliosis, inflammation, epileptogenesis
INTRODUCTION
Epilepsy is a neuronal disorder characterized by increased predisposition to suffer epileptic seizures (Falconer et al., 1964; Chang and Lowenstein, 2003; Devinsky et al., 2018). In many instances, a brain damaging insult such as head trauma, stroke, brain infection or status epilepticus (SE) triggers a cascade of events leading to the manifestation of spontaneous seizures and to the diagnosis of epilepsy (Sharma et al., 2007; Curia et al., 2008; Devinsky et al., 2018). There is some evidence implying that epilepsy might be a progressive disorder and that recurrent seizures might contribute to its progression (Devinsky et al., 2018; Smith et al., 2018). Some data from animal models hint that seizures themselves damage the brain and result in self-propagation (Pitkanen and Sutula, 2002; Sutula et al., 2003) but the evidence provided by these studies do not conclusively demonstrate that seizure activity per se is sufficient to injure the brain (Rossini et al., 2018).
The existence of seizure-dependent damage during epileptogenesis has been difficult to prove because it is difficult to separate the effect of seizures themselves from the effects of other underlying factors driving epilepsy progression (Rossini et al., 2018). Thus, it is unclear which of the vast pathological abnormalities detected during epileptogenesis are indeed epileptogenic. It has been proposed that if a specific pathology is directly linked to epileptogenic events, then its severity might be correlated with the frequency/occurrence of spontaneous seizures (Hester and Danzer, 2013; Buckmaster et al., 2017). A recent study in rats demonstrated a positive correlation between seizure frequency and the percentage of ectopic granule cells, the levels of mossy fiber sprouting and mossy cell death (Hester and Danzer, 2013). Seizure frequency also correlated with the loss of GABAergic neurons in the dentate gyrus of epileptic mice (Buckmaster et al., 2017). However, the progressive changes impacting the brain at the molecular, cellular and network levels during epileptogenesis still remain poorly understood.
The calpains (ca/cium-dependent proteases with papain-like activity) represent a family of cysteine proteases activated by calcium (Campbell and Davies, 2012; Ono and Sorimachi, 2012). Sustained calpain dysregulation contributes to acute and chronic neurodegeneration in numerous pathologic conditions (Vanderklish and Bahr, 2000; Bevers and Neumar, 2008; Vosler et al., 2008; Saatman et al., 2010). In the brain, calpain-1 and calpain-2 are the most ubiquitously expressed isoforms (Liu et al., 2008; Saatman et al., 2010; Baudry and Bi, 2016). Brain tissue resected from epileptic patients shows increased expression of calpain-1 and calpain-2 (Feng et al., 2011; Das et al., 2012) and increased calpain expression is associated with clinicopathological changes key to epileptogenesis including neurodegeneration, astrogliosis and inflammation (Das et al., 2012). In particular, calpain dysregulation occurs after SE (Bi et al., 1996; Araujo et al., 2008; Wang et al., 2008b), its pharmacological inhibition provides neuroprotection (Araujo et al., 2008; Wang et al., 2008b) and ameliorates seizure burden (Howe et al., 2016; Lam et al., 2017).
Despite this knowledge, there is a lack of evidence showing a possible relationship between seizure occurrence and calpain activation. Here, we investigated if seizure occurrence during an early phase of epileptogenesis correlates with changes in calpain activity/expression. In addition, we analyzed if seizure frequency shows any correlation with typical markers associated with epileptogenesis such as cell death, gliosis and inflammation.
MATERIALS AND METHODS
Animal Subjects.
Adult male Sprague Dawley rats (Charles River, Wilmington, MA) were housed in a temperature-controlled facility with food and water ad libitum. Animal procedures were performed in accordance with Institutional Animal Care and Use Committee regulations and approved protocols by the University of Colorado Anschutz Medical Campus.
Electrode Implantation.
Animals were implanted with intracranial EEG electrodes as follows. Rats were anesthetized using a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg) and then placed in a stereotaxic apparatus. Anesthesia was maintained using isoflurane (1–4%) throughout the surgery. The area of incision was cleaned and anesthetized with 1% lidocaine. A midsagittal incision was made and the skin was retracted to allow the drilling of small burr holes onto the skull. Stainless steel screws were placed in the holes and used as electrodes (recording, ground and reference). Two screws were placed bilaterally at 4 mm caudal to Bregma and 2.5 mm lateral from midline over the temporolimbic cortices. Reference and ground electrodes were placed bilaterally behind lambda (i.e., over the cerebellum). Dental acrylic was used to secure the electrodes to a plastic connector (Plastics-One, Roanoke, VA) according to standard methods (Grabenstatter et al., 2014; Lam et al., 2017). Animals were allowed to recover from surgery for about a week before proceeding with any further experimentation.
Status Epilepticus.
Our standard protocol (Brooks-Kayal et al., 1998; Shumate et al., 1998; Gonzalez et al., 2013) was used to induce SE as follows: rats were intraperitoneally injected with scopolamine (1 mg/kg) 30 minutes before administration of pilocarpine (385 mg/kg). Animals that did not exhibit convulsive seizures and did not enter SE within one hour of the first pilocarpine injection were given up to two additional doses of pilocarpine (192.5 mg/kg) to produce an equivalent SE. As criteria for inclusion, all rats used had confirmed stage 5 behavioral seizures. Diazepam (6 mg/kg; Hospira, Lake Forest, IL) was administered one hour after the beginning of SE to slow seizure progression and reduce mortality. If necessary, additional doses of diazepam were administered every two hours (a maximum of two 3 mg/kg doses) until rats stopped exhibiting behaviors associated to seizures. Control rats were handled in a similar way but received a subconvulsive dose of pilocarpine (1/10 of the full dose, 38.5 mg/kg) and a reduced dose of diazepam (1/10 of the full dose, 0.6 mg/kg).
EEG Acquisition and Analysis.
Rats were placed in a recording chamber equipped with flexible cables attached to a commutator (i.e., electric swivel) to allow free animal movement. Animals were recorded 24 hours/day using an automatic Pinnacle digital video-EEG system. EEG signals were sampled at 2 kHz, amplified by 100×, and band-pass filtered between 0.3 Hz and 650 Hz. After seizure monitoring was completed, rats were sacrificed and half of the animals were used to collect samples for western blot (n=8) and the other half were perfused with 4% PFA to collect samples for histological analysis (n=8). A trained technician blinded to the experimental parameters performed the EEG analysis off-line. Electrographic recordings were examined to identify electrographic seizures and potential artifacts as follows: seizures differed from background noise by the presence of EEG signals with a progression of spike frequency; large-amplitude (at least three times baseline); and high-frequency activity (minimum of 5 Hz) that lasted at least 10 sec. Video recordings were then analyzed to corroborate that the abnormal EEG signals were not movement artifacts and to establish a behavioral correlate. The behavioral correlate was based on the Racine scale (Racine, 1972). Seizures scored as class 2 or below were considered non-convulsive and seizures scored as class 3 or above were categorized as convulsive. Animals were considered epileptic when one or more seizures were detected. All seizures detected (behavioral and non-behavioral) are reported. In addition, animals were arbitrarily labeled as enduring a “low” (~1–13), “medium” (~19–24) or “high” (~30–76) number of seizures.
Hippocampal Tissue Preparation.
Immediately after sacrifice, brains were removed and the hippocampus was isolated. Hippocampal slices (600 μm) were prepared using a McIlwan tissue chopper to facilitate microdissection of hippocampal regions as follows: first, the CA3 region was separated from CA1 and DG to then separate the CA1 from the DG (Silva et al., 2001). The pieces of tissue for each region taken from the same rat were pooled in one sample, quickly frozen on dry ice and stored at-80°C until use. Tissue lysates were prepared by briefly sonicating the pieces of tissue in RIPA buffer containing protease and phosphatase inhibitors. Lysates were cleared of cellular debris by centrifugation at 17,000 X g for 20 min. Protein concentration was determined using the Bio-Rad RC/DC reagent kit (Bio-Rad Laboratories, Hercules, CA, USA).
Western Blot.
Samples were separated using SDS-polyacrylamide gels and then transferred to nitrocellulose membranes. Blots were blocked with 5% non-fat dry milk in Tris-buffered saline (pH 7.4) plus 0.05% Tween-20 for one hour at room temperature. Blots were incubated overnight at 4°C with primary antibodies diluted in 1% non-fat dry milk. The AB38 antibody is a polyclonal rabbit antibody that recognizes spectrin fragments (~150 kDa) generated after calpain-cleavage that was previously produced and characterized (Roberts-Lewis et al., 1994) and was a generous gift from Dr. David R. Lynch (University of Pennsylvania, PA). The α-spectrin antibody (clone EPR3017), a rabbit monoclonal antibody (epitomics Cat. No. 2507–1, Abcam ID: ab75755) that detects the full-length and cleaved protein, was purchased from Epitomics (an Abcam company, Burlingame, CA). To assess calpain activation, samples were probed with antibodies than can detect a “pro-peptide domain” located at the amino-terminal domain of unprocessed calpain. These antibodies are specific for the “pro-peptide domain” of calpain-1 (Cat. No. RP1-Calpain-1) or calpain-2 (Cat. No. RP2-Calpain-2) and do not recognize amino-processed proteins and thus can be used to discriminate between latent and amino-processed (active) forms, as previously described (Stalker et al., 2005; Scalia et al., 2011). To detect total calpain expression, blots were probed with antibodies that detect both latent and aminoprocessed (activated) calpain-1 (Cat. No. RP3-Calpain-1) or calpain-2 (Cat. No. RP3-Calpain-2). All calpain antibodies were purchased from Triple Point Biologics (Forest Grove, OR). To estimate potential variability in protein content and loading, blots were probed with an anti-actin antibody (Cat. No. A2066) purchased from Sigma (St. Louis, MO). After washing the primary antibody, blots were incubated with secondary antibody for one hour at room temperature. Mouse anti-rabbit secondary antibodies conjugated to horseradish peroxidase were from GE Health Care (Piscataway, NJ) and goat anti-mouse secondary antibodies conjugated to horseradish peroxidase were from Jackson Immunoresearch laboratories (West Grove, PA). Immunoreactive bands were visualized using Super Signal West Dura chemiluminiscent substrate (Pierce, Rockford, IL, USA) and film. After scanning the films, immunoreactive bands of the appropriate size were quantified using Image J (NIH, Bethesda, MD, USA). Immunoreactivity for the bands of interest was normalized to actin immunoreactivity and compared to control values. Data is presented as the mean ± SEM.
Fixed Tissue Preparation.
Rats were deeply anesthetized and transcardially perfused, first with ice-cold PBS and then with ice-cold 4% PFA in 0.1 M phosphate buffer pH 7.4. Three controls and four SE animals were collected at four days post-SE and three controls and eight animals enduring SRS were collected at 15 days post-SE. Brains were removed from the skull and post-fixed overnight in 4% PFA solution. Fixed brains were cryoprotected in 30% sucrose solution and embedded in OCT compound (Tissue-Tek, Sakura Finetek, Torrance, CA). Whole brains were serially sectioned to obtain 15 μm coronal sections. For staining, three mounted sections were selected from a 1-in-10 series starting at approximately the same level of hippocampus (2.8 mm posterior to Bregma). For consistency and to minimize variability in the staining procedure, control and epileptic brains were processed and stained in parallel. Controls where the primary antibodies were omitted were run to confirm that the staining was dependent on the primary antibody. Images were obtained using a Nikon Eclipse TE2000-U fluorescence microscope. A low magnification (4X) image of the FJB staining has been included and the approximate location where 20X pictures were taken for the CA1, CA3 and Hilus of hippocampus is depicted as part of this image. For quantitation purposes, 20X images were obtained for the FJB, GFAP and Iba1 staining. Labeled cells found within the boundaries of the field of view in the 20X images were counted and included in the analysis. The number of stained cells was determined bilaterally on the three sections selected and the average of these counts represent the number of positive cells detected in each particular animal. Data is presented as the mean ± SEM. Cell counts were conducted blinded to seizure frequency.
Histological Analysis.
Degenerating neurons were detected in brain sections stained with the anionic fluorochrome Fluoro-Jade B (FJB, Cat. No. 1FJB, Histo-Chem Inc, Jefferson, AR) as previously described (Schmued and Hopkins, 2000; Lam et al., 2017). For this, mounted sections were dried at room temperature and rehydrated with 100% ethanol for 10 min, 70% ethanol for 2 min and rinsed in distilled water for 2 min. For staining, sections were immersed in 0.06% potassium permanganate for 10 min, rinsed with distilled water for 2 min and immersed in 0.0004% FJB solution for 10 min. Finally, sections were rinsed with distilled water, dried and immersed in CitriSolv (Fisher Scientific, Pittsburgh, PA). After staining, tissue sections were mounted using Permount (Fisher Scientific, Pittsburg, PA).
Astrogliosis was evaluated in brain sections stained with an antibody to detect GFAP (a marker for astrocytes). Tissue sections were first blocked 10% normal goat serum, 0.1% BSA, 0.01% glycine and 0.3% Triton X-100 in PBS. Then, sections were incubated overnight with a mouse monoclonal anti-GFAP antibody (Cat. No. G3893, Sigma, St. Louis, MO) diluted 1:1000 in blocking buffer. The next day, slices were washed and incubated with a highly cross-adsorbed Alexa Fluor 568 goat anti-mouse secondary.
Inflammation was estimated in brain slices stained with an antibody to detect Iba1 (a marker for microglia). Tissue sections were blocked with 10% normal goat serum and 0.3% Triton X-100 in PBS. Sections were then incubated overnight with a rabbit polyclonal anti-Iba1 antibody (Cat. No. 019–19741, Wako, Richmond, VA) diluted 1:500 in blocking buffer. Next day, slices were washed and incubated with a highly cross-adsorbed Alexa Fluor 568 goat anti-rabbit antibody. After staining, tissue sections were mounted on slides using Vectorshield (Vector Laboratories, Burlingame, CA, USA). The next day, slices were washed before incubation with a highly cross-adsorbed Alexa Fluor 568 goat anti-rabbit secondary antibody. After staining, tissue sections were mounted on slides using Vectorshield.
Nissl Stain was used to label cell bodies and reveal the overall morphology of the hippocampal structures to assess possible cells loss (Paul et al., 2008). Tissue sections were demyelinated, stain and destain by incubating the tissue sections in different solutions. After incubation of the tissue in different ethanol solutions to remove lipids and rinsing in distilled water, tissue sections were stained by immersion in a 0.1% solution of Cresyl Violet for 10 min. A clearing agent was then used to remove excess of stain solution before allowing them to air-dry. Finally, coverslips were mounted onto the sections using Permount (Fisher Scientific, Pittsburg, PA). Images were obtained using a Nikon Eclipse 6600 microscope. An image of the entire hippocampus after Nissl staining was obtained by tiling low magnification (10X) images. The approximate location where 40X images were taken is depicted in this image. For quantitation purposes, 40X images were obtained and labeled cells found within the boundaries of the field of view were counted and included in the analysis. The number of stained cells was determined bilaterally on three sections and the average of these counts represent the number of cells detected in each particular animal. Data is presented as the mean ± SEM. Cell counts were conducted blinded to seizure frequency.
Statistical analysis.
Statistical analyses were performed using GraphPad InStat (GraphPad Software, Inc., San Diego, CA, USA). For analyses, p values < 0.05 were considered significant. Differences between two groups were determined by unpaired t-test. Differences between three groups were determined by one-way analysis de variance (ANOVA) followed by a Bonferroni post hoc test.
RESULTS
There is an unresolved controversy about whether repeated spontaneous seizures can induce neuronal damage despite solid evidence of seizure-induced damage after experimental SE (Sutula et al., 2003). The neuronal loss observed following chemoconvulsant-induced SE has been clearly associated with calpain activation (Araujo et al., 2008; Wang et al., 2008b; Lopez-Meraz and Niquet, 2009), but the role that spontaneous recurrent seizures (SRS) might have on calpain (re)activation and neuronal loss during early epileptogenesis has not been investigated. Here, we addressed the effects that SRS might have on calpain activation and other pathologies associated with epileptogenesis. For this, rats were implanted, subjected to pilocarpine-induced SE and monitored to assess seizure burden. Continuous (24/7) video and EEG recordings were collected between three and fifteen days post-SE. Analysis of the EEG recordings showed that rats became epileptic and presented SRS after a seizure free period detected 4 to 6 days post-SE (Figure 1). In some animals, seizures were detected as early as three days post-SE but these early seizures might represent the tail-end of SE and not true SRS (Smith et al., 2018). The total number of seizures (behavioral and non-behavioral) detected during this period of monitoring was rater variable and ranged between 1 and 76 seizures. Animals were arbitrarily labeled as enduring a “low” (~1–16), “medium” (~19–24) or “high” (~30–76) number of seizures. Under these experimental conditions, rats endured an equivalent SE, presented a seizure free period at 4 to 6 days post-SE and endured differential levels of SRS on the days prior to sacrifice. Thus, the differential seizure burden observed before sample collection might be linked to possible changes in calpain activation and epileptogenesis.
Figure 1. Seizure Burden After Pilocarpine-Induced SE.
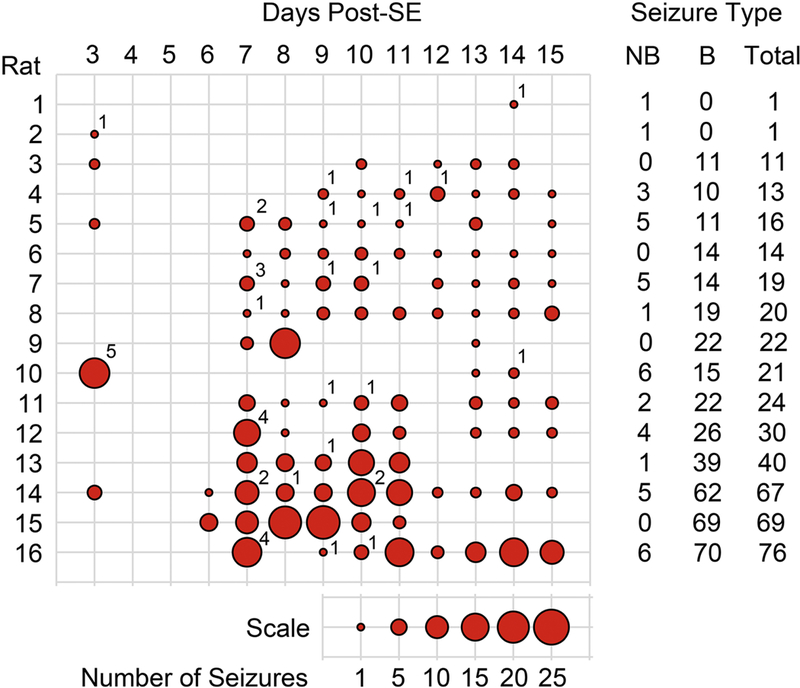
Implanted rats were recorded with an automatic video-EEG system to obtain continuous recordings (24 hours a day). A trained technician examined EEG and video recordings off-line to identify electrographic seizures. Behavioral characterization of seizures was based on the Racine scale. Summary of the number of seizures (behavioral and non-behavioral) detected on the 16 rats included in the current study presented as seizures per day. The size of the circles represents the number of seizures per day according to the scale provided. The number on the upper right of the circles depicts the number of non-behavioral seizures detected on that particular day. A summary of the number of non-behavioral (NB), behavioral (B) and total seizures for each animal is presented as columns on the right side of the figure. The number located at the upper right side of the circles represents the number of NB seizures detected on a particular day. After EEG and video recording, tissue of one group of 8 rats was collected for western blot analysis and another group of 8 rats was collected for histological analysis.
Having established the seizure burden, a western blot analysis was carried out in rats enduring SRS to evaluate calpain activation and to explore a possible correlation between seizure burden and calpain activity. Calpain activation was estimated indirectly by detecting the formation of α-spectrin breakdown products, a hallmark of calpain activity. For this analysis, two antibodies were used: (1) an anti-α-spectrin antibody, that detects both full-length spectrin and its break down products (BDPs); and (2), the AB38 antibody, that only detects spectrin BDPs generated after calpain cleavage. Analysis of lysates from the CA1 region of hippocampus showed a significant increase in BDPs detected with both spectrin and AB38 antibodies but a concomitant loss of full-length α-spectrin was not detected (Figure 2D). Analysis of CA3 lysates showed a significant increase in BDPs accumulation and, in this case, the increase in BDPs was concomitant with a significant loss of full-length α-spectrin (Figure 2E). Finally, analysis of lysates from DG showed a significant increase in BDPs (270 ± 24 of control, p<0.001) when the AB38 antibody was used but no loss of full-length α-spectrin was detected (data not shown).
Figure 2. Calpain Activation After SRS.
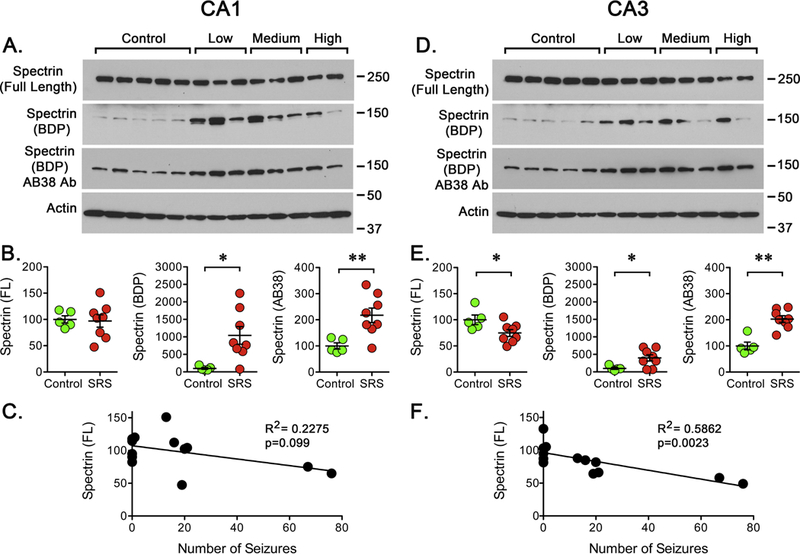
Hippocampal tissue was collected at 15 days post-SE. Western blot analysis was performed using antibodies to detect μ-spectrin and its BDPs (anti-μ-spectrin and AB38). The blots were also probed for actin immunoreactivity as control. (A, D) Representative western blots showing the detection of full-length μ-spectrin and BDPs in CA1 or CA3 of hippocampus. (B, E) Quantitation of the immunoreactivity detected for full-length μ-spectrin and BDPs in CA1 or CA3 regions of hippocampus in samples obtained from controls (n=5) and rats that experienced a “low”, “medium” or “high” number of SRS prior to sacrifice (n=8). Data is presented as the mean ± SEM. (C, F) Values were compared using an unpaired f-test, *p<0.05 or **p<0.01. Values obtained for full-length μ-spectrin were paired with the number of SRS detected during to days following the pilocarpine induced SE to perform a linear regression analysis. Plots display a significant correlation (R2=0.5862, p=0.0023) between the levels of full-length μ-spectrin and the number of SRS in CA3 but not in CA1.
Since a main difference detected between epileptic animals was the seizure burden endured prior to sample collection, a possible correlation between the number of SRS detected and the levels of full-length spectrin was investigated. For this, linear regressions were calculated between the number of SRS detected vs. full-length spectrin and SRS vs. BDPs but no significant correlation was found for CA1 or DG (Figure 2C and data not shown). However, when a correlation was calculated between SRS and full-length spectrin in the CA3 region, the loss of α-spectrin was significantly (p=0.0023) correlated (R2=0.5864) with the number of SRS detected before sample collection (Figure 2F). Despite the fact that all epileptic animals endured a similar SE, the impact of a delayed effect of SE on calpain activity cannot be completely ruled out. However, since the loss of full-length α-spectrin was more profound in the animals experiencing a “high” number of seizures and a correlation between seizure burden and spectrin loss was detected, the observations described above suggest that seizure occurrence is linkec to calpain (re)activation.
Calpain-1 and calpain-2 are the two isoforms more ubiquitously expressed in the brain (Liu et al., 2008; Saatman et al., 2010; Baudry and Bi, 2016). Thus, we decided to investigate the possible impact of SRS on its activation and expression. For this, western blot analysis was carried out using antibodies to detect a “pro-peptide domain” located at the amino-terminal domain of unprocessed calpain (inactive). In samples obtained from the CA1, the levels of unprocessed calpain-1 were significantly reduced while total levels of calpain-1 were significantly increased suggesting an increase in the levels of active calpain-1 (Figure 3). Despite a reduction in the levels of inactive calpain-1, no significant correlation (R2=0.08893, p=0.3224) was found between seizure number and the levels of inactive calpain-1. However, the highest lost of inactive calpain-1 was detected in rats enduring a “high” number of seizures. A significant correlation (R2=0.7021, p=0.0094) was observed between the number of SRS detected and total calpain-1 expression (Figure 3C). In the case of calpain-2, there was no change in inactive calpain despite a small but significant increase in the total expression of calpain-2 that is correlated with seizure occurrence (R2=0.3588, p=0.0305).
Figure 3. Calpain Activation/Expression After SRS.
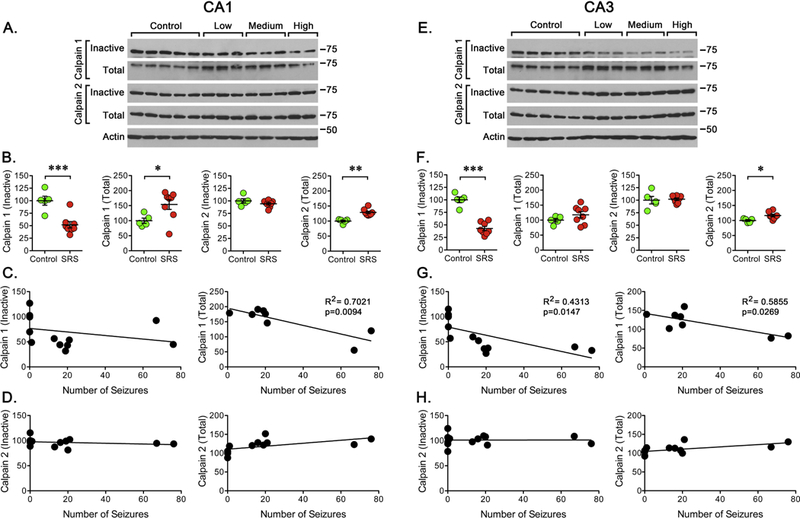
Western blot analysis was performed using antibodies to detect “inactive” and “total” calpain-1 or calpain-2. (A, E) Representative western blots for the detection of “inactive” and “total” calpain-1 or calpain-2 in CA1 or CA3 regions. (B, F) Quantitation of the immunoreactivity detected for “inactive” and “total” calpain-1 and calpain-2 in controls (n=5) and rats that experienced a “low”, “medium” or “high” number of SRS (n=8). Data is presented as the mean ± SEM of the values obtained from samples of CA1 or CA3 regions. Values obtained for controls and SRS animals were compared using an unpaired f-test, *p<0.05, **p<0.01, or ***p<0.001. (C, G) To further analyze the data, values obtained for “total” calpain-1 were paired with the number of SRS detected prior to sacrifice to perform a linear regression analysis. Plots displayed a significant correlation between total calpain-1 (R2=0.7021, p=0.0094) in CA1 and both “inactive” (R2=0.4313, p=0.0147) and “total” (R2=0.5855, p=0.0269) calpain-1 in CA3. (D, H) As for the inactive or total levels of calpain-2, no significant correlation was detected for the levels of “inactive” calpain but a significant correlation was found for “total” calpain-2 in CA1 (p=0.0366) and CA3 (p=0.0305).
In samples from the CA3 region, the levels of inactive calpain-1 were significantly reduced and a significant correlation (R2=0.4313, p=0.0147) was found between the number of seizures detected and the loss of inactive calpain-1 (Figure 3E, 3G) with the highest lost of calpain-1 detected in rats enduring a “high” number of seizures. A significant correlation (R2=0.5855, p=0.0269) between total calpain-1 and seizure occurrence was also observed in samples from CA3 (Figure 3G). The levels of inactive calpain-2 did not change but the total levels of calpain-2 show a small but significant change that is correlated (R2=0.3555, p=0.0315) with the number of seizures detected. Finally, in samples from DG, a significant reduction in the levels of inactive calpain-1 was detected (48 ± 4 percent of control, p<0.001) along with an increase in total calpain-1 (135 ± 4 of control, p<0.001) but there was no significant correlation with the number of SRS detected. In the case of the levels of inactive or total calpain-2 detected in DG, there was not change and no correlation with the number of seizures was detected.
Continuous or sporadic recurrent seizures can cause neuronal damage but the direct role of spontaneous seizures has been difficult to demonstrate (Pitkanen and Sutula, 2002). Here, we used FluoroJade B (FJB+) staining (Schmued and Hopkins, 2000) to assess if occurrence of SRS might correlate with the appearance of degenerating neurons during an early phase of epileptogenesis. In animals experiencing SRS, the bodies and dendrites of pyramidal neurons located within the CA1 and CA3 regions and hiliar neurons in DG were effectively labeled by FJB (Figure 4). The most prominent FJB labeling was observed in tissue obtained from rats enduring a “high” number of SRS and less evident staining was detected in animals enduring a “low” number of seizures. As depicted in Figure 4A, tissue obtained from the animal that presented the highest number of SRS shows a clear pattern of neurodegeneration that demarcates the layer of principal neurons along the CA1 and CA3 regions. In DG, the main site where positive cells were detected is the hilus. In contrast, no FJB+ cells were detected in tissue from control animals. Tissue sections obtained from rats enduring different levels of SRS showed a widespread and variable but significant increase in the number of FJB+ cells in CA1, CA3 and the hilus within the DG of hippocampus (Figure 4B, D). A further analysis showed that the number of FJB+ cells in CA1 (R2=0.5097, p=0.0136) and CA3 (R2=0.5144, p=0.0130) showed a significant correlation with the number of SRS detected during the two weeks prior to sacrifice (Figure 4D, 4G). In the case of DG, however, there was no significant correlation (R2=0.1073, p=0.3253).
Figure 4. Neuronal Degeneration After SRS.
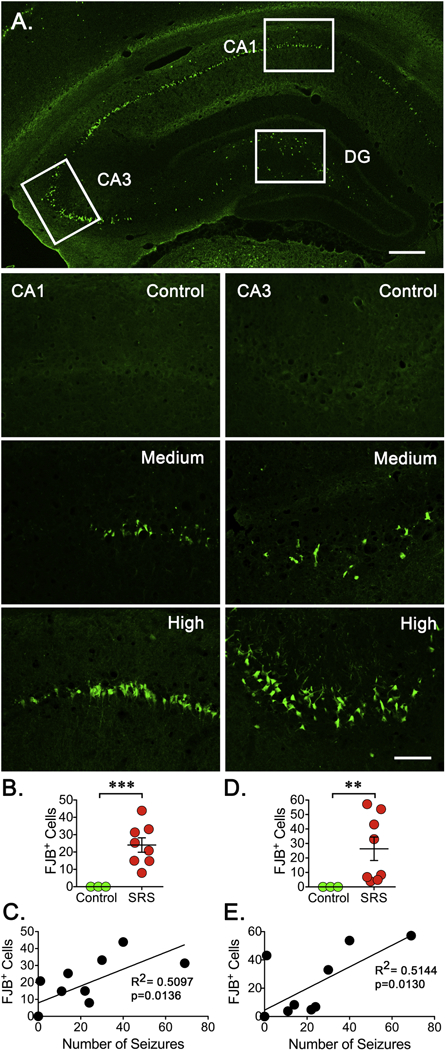
Three fixed coronal sections (15 μm) were selected from a 1 -in-10 series. A. Low magnification image (4X) from the hippocampus of the animal enduring the highest number of SRS was stained with the anionic fluorochrome Fluoro-Jade B (FJB) showing a clear pattern of neurodegeneration along the principal neurons within the CA1 and CA3 regions. In DG, the hilus is the main site where positive cells can be seen. The approximate location where 20X pictures for the CA1, CA3 and DG were taken is depicted. Scale bar represents 200 μm. (B, E) Representative sections (20X) stained with FJB show degenerating neurons in the CA1 and CA3 regions of hippocampus. Scale bar represents 100 μm. (C, F) The number of FJB positive (FJB+) cells was counted on the three sections selected and averaged to represent the number of FJB+ cells detected in a particular animal. Cell counts were conducted blinded to seizure frequency. Data is presented as the mean ± SEM of controls (n=3) and animals experiencing SRS (n=8). The number of FJB+ cells was compared using an unpaired f-test, **p<0.01 and ***p<0.001 represent a significant difference. (D, G) Values obtained for the number of FJB+ cells were paired with the total number of SRS detected to perform a linear regression analysis (n=8). Plots display a significant correlation between the levels of FJB+ cells and the number of SRS detected in the CA1 (R2=0.5097, p=0.0136) and CA3 (R2=0.5144, p=0.0130) regions of hippocampus.
Previous studies have shown that degenerating neurons, as detected by FJB staining, are detectable within hours of SE induction and reach peak levels during the first week post-SE to then gradually disappear (Poirier et al., 2000; Wang et al., 2008a). It is conceivable that subsequent waves of neurodegeneration might be triggered by SRS once rats become epileptic but this has nor been previously analyzed. To start evaluating if FJB+ cells detected after SRS might not only result from a delayed effect of SE but also result from the occurrence and frequency of spontaneous seizures, tissue samples were collected 4 days after SE, a time point previously described to be within the window of peak detection of FJB+ cells and small probability of seizure occurrence (Figure 5). Under these conditions, unsurprisingly since these cells more likely represent peak values of degenerating neurons directly resulting from the SE-induced injury, an increase in FJB+ cells was detected. In the CA1, the number of FJB+ detected after SE was not statistically different from the number of FJB+ cells detected in animals enduring SRS, but four animals enduring SRS showed higher levels of FJB+ cells than the average number of FJB+ cells detected at 4 days post-SE (Figure 5A). In the CA3 region, the average number of FJB+ cells detected after SE was not significantly different from the number of FJB+ cells detected in animals experiencing SRS. However, epileptic animals appear to segregate into two populations, one with a low and one with a high number of FJB+ cells. Animals with a low number of seizures presented a significantly lower number of FJB+ cells and animals with a high number of seizures presented a significantly higher number of FJB+ cells when compared to SE animals (Figure 5B). Finally, in DG, the number of FJB+ cells detected in rats enduring SRS was significantly higher than the number of cells detected after SE (Figure 5C). Since epileptic animals endured a similar degree of seizure activity during SE, the main differential factor between the seizure-free period and sample collection is the occurrence of SRS. The finding that neurodegeneration correlates with the number of SRS strongly suggest that SRS might exacerbate neuronal death and be a key factor involved in the continuous neurodegeneration observed during epileptogenesis. All together, these observations suggest that SRS might, at least in part, determine the trajectory of neurodegeneration during an early phase of epileptogenesis and that the number of degenerating neurons might be directly influenced by the seizure burden.
Figure 5. Neuronal Degeneration After SE or SRS.
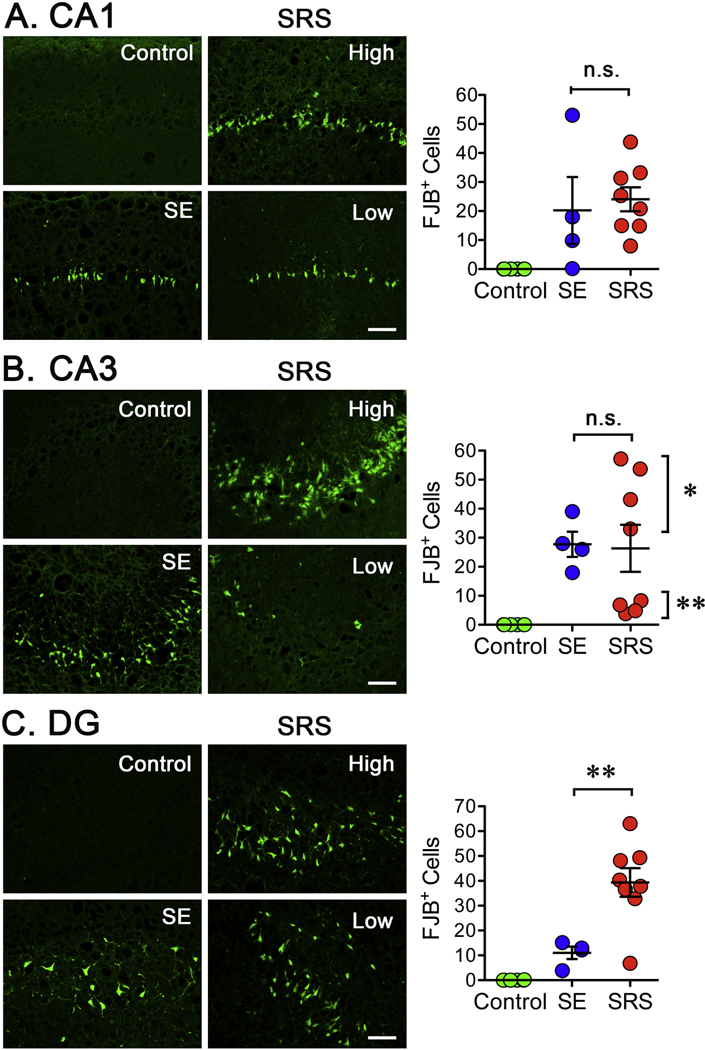
Brain tissue was collected at either 4 (SE) or 15 (SRS) days post-SE and stained with FJB. The images for the SRS are for animals presenting either a low or high number of seizures. A. Images from the CA1 region of control, SE or SRS samples. The number of FJB positive (FJB+) cells was counted on three sections and averaged to represent the number of FJB+ cells detected in a particular animal. On average, the number of FJB+ detected in tissue collected after SE or SRS was not significantly different. B. In the CA3 region, the number of FJB+ cells detected after SE or SRS was not significantly different but the number of FJB+ cells in SRS tissue appears to segregate into two distinct populations: low and high number of FJB+ cells. When tested independently, the number of FJB+ cells in the four animals with a low number of seizures is significantly lower that the number of FJB+ detected after SE. In addition, the number of FJB+ cells detected after SE is significantly different than those detected in animals with a higher number of SRS. Data is presented as the mean ± SEM of controls (n=6), SE (n=3) and SRS (n=8) rats. The number of FJB+ cells in the SE and the SRS groups was compared using ANOVA followed by a Bonferroni pos t test, *p<0.05 and **p<0.01 represent a significant difference.
Detection of degenerating neurons with the FJB staining suggested the continuous loss of neuronal cells during the early phase of epileptogenesis. A Nissl stain was performed in tissue from control and epileptic animals to assess the overall pattern of cell loss after SRS. An initial gross examination of the stained tissue revealed that the general distribution of principal neurons in the CA1, CA3 and DG layers was easily detectable in control and epileptic animals, but increased cell dispersion was observed in the different hippocampal regions (Figure 6). In addition, a reduction in number of cell bodies was apparent in the CA3 region of animals enduring a higher number of SRS. To more specifically assess a possible cell loss, cell bodies were counted in images obtained at a higher magnification. Quantitation of cells in the CA1 region revealed a downward trend in some animals but this change was not statistically significant (Figure 6B). In contrast, cell counts in the CA3 region showed a significant decrease but despite a downward trend, a linear regression analysis did not show a clear correlation between cell loss and the number of SRS (Figure 6E). In the case of DG, we focused on the hilus because this was the area were most FJB+ cells were detected, but no significant cell loss was detected (data not shown), perhaps because the loss of cells detected with the FJB staining did not amount to impact the total number of hiliar cells.
Figure 6. Neuronal Loss After SRS.
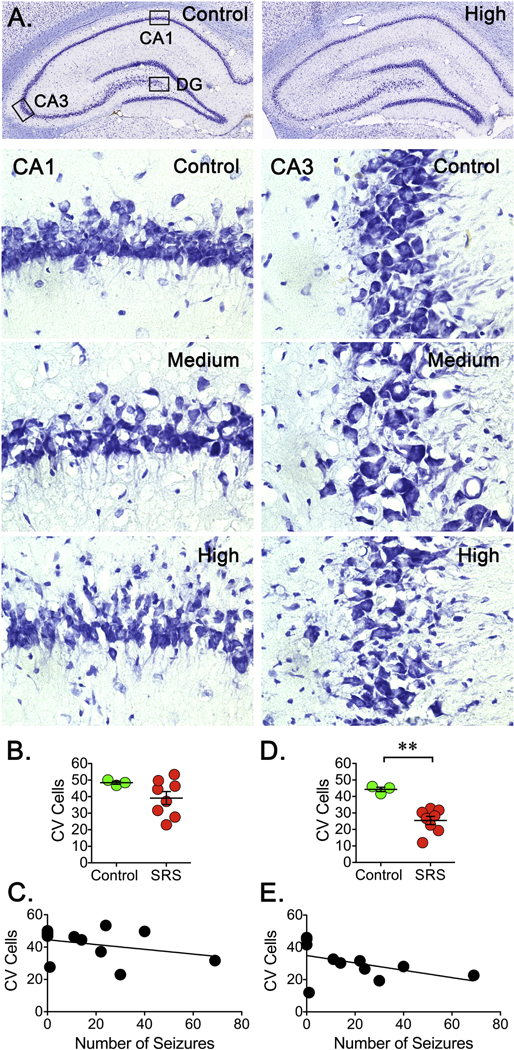
Three coronal sections (15 μm) from a 1-in-10 series were stained using Cresyl Violet. A. Low magnification images (10X) were obtained and assembled to provide a view of the entire hippocampus. Images from a control and the rat enduring the highest number of SRS are presented. Principal neurons along the CA1, CA3 and DG layers are easily detectable in both control and epileptic animals. Representative Nissl stained brain sections (40X) showing the overall pattern of neuronal bodies in CA1 and CA3 regions of hippocampus. A reduction in number of cell bodies is apparent in the CA3 region of animals experiencing a higher number of SRS. (B, D) Cell bodies were counted on three brain sections and averaged to represent the number of cells in a particular rat. Cell counts were conducted blinded to seizure frequency. Data is presented as the mean ± SEM of controls (n=3) and animals experiencing SRS (n=8). In the CA1 region, counts revealed a non-significant downward trend but counts in the CA3 region showed a significant decrease. The number of cells was compared using an unpaired t-test, **p<0.01 represents a significant difference. (C, E) Values obtained for the number of stained cells were paired with the total number of SRS detected in a linear regression analysis (n=8). Despite a trend, plots did not show a significant correlation between the levels of stained cells and the number of SRS detected in the CA3 region.
Neuroinflammation is another pathological finding commonly associated with epileptogenesis and this pathology can be detected in brain tissue as reactive gliosis. To investigate a possible correlation between SRS and inflammation, immunoreactivity for GFAP (astrogliosis) and Iba1 (microgliosis) was analyzed. The levels of GFAP+ cells detected in CA1, CA3 or DG of hippocampus showed a trend towards an increase in the number of GFAP+ cells but this increase did not reach significance (Figure 5B). An initial linear regression analysis did not show a clear correlation between the number of GFAP+ cells and the number of SRS detected. However, it was noticed that the one animal that endured a “high” number of SRS showed lower levels of GFAP+ cells when compared to animals that experienced a “medium” or “low” number of SRS. This suggested the possibility that animals experiencing a “low” or ‘‘medium” number of SRS follow a linear trend while animals experiencing a much higher number of seizures no longer follow a linear trend. To explore this possibility, the rat experiencing the highest number of seizures was excluded from the linear regression analysis (Figure 5C). In these conditions, a clear correlation emerged between the number of SRS and GFAP+ cells in CA1 (R2=0.6268, p=0.0063), CA3 (R2=0.5557, p=0.0133) and DG (R2=0.4578, p=0.0317).
In the case of the Iba1 staining, the number of Iba1+ cells was also widespread but a significant increase was detected in CA1, CA3 and DG (Figure 6). Similarly to the observations made for the GFAP staining, the animal with the highest number of SRS did not show the highest number of Iba1+ cells and no correlation was observed in an initial analysis (Figure 6C). If this specimen is not included in the analysis, a significant correlation between the number of SRS and the number Iba1+ cells can be detected in CA1 (R2=0.7814, p=0.0007), CA3 (R2=0.7645, p=0.0009) and DG (R2=0.6544, p=0.0046). Thus, a better fit was found between the number of SRS detected and the number of Iba1+ cells detected in animals that experienced a “low” and “medium” number of SRS. Together, these observations made using GFAP and Iba1 staining suggest that expression of these markers of inflammation might not be well correlated with seizure frequency and might not be a useful marker to predict seizure burden.
DISCUSSION
There is some evidence that seizures themselves damage the brain but there is no data to conclusively demonstrate the role of SRS in epileptogenesis. Here, we investigated if the number of SRS detected during early epileptogenesis is correlated with increased calpain activation and/or expression. In addition, a possible correlation between seizure burden and pathological changes typically associated to epileptogenesis was also investigated. Our findings demonstrate that the number of SRS detected during an early period of epileptogenesis is well correlated with the levels of calpain activity/expression and the levels of neuronal neurodegeneration detected in specific hippocampal regions. A general presumption is that epileptic animals endured a similar degree of seizure activity following the initial seizure induction of SE and that following the seizure free period the appearance of SRS might then drive further changes. Thus, the main differential factor among epileptic animals is the burden of SRS. The finding that seizure burden is well correlated with both calpain activation and neurodegeneration strongly suggest that occurrence of SRS might define the course of epileptogenesis and associated pathologies. In some instances, a linear effect was not detectable in animals enduring the highest seizure frequency, the reason for this effect is unclear but, as in many biological processes, the effects of seizure occurrence may saturate and reach a new steady state that no longer results in a linear effect. Thus, it is possible that analyzing tissue obtained during early epileptogenesis favored the detection of linear changes that might become undetectable during a more chronic period. All together, the results presented here imply that, at least during an early phase of epileptogenesis, there is a strong correlation between seizure occurrence with calpain activation and neuronal degeneration.
Seizures promote membrane depolarization and glutamate accumulation in the extracellular space that results in activation of voltage-gated calcium channels and NMDA receptors, respectively. Receptor activation then allows a rapid accumulation of intracellular calcium (Raimondo et al., 2017). Thus, seizure occurrence can promote a calcium influx in both neurons and astrocytes (Trevelyan et al., 2007). In hippocampal neurons, intracellular calcium levels are significantly elevated following SE and remain indefinitely elevated through the chronic period (Raza et al., 2004). Since the calpains are calcium-dependent proteases, seizure-dependent calcium influx represents a possible link between seizure occurrence and calpain activation during epileptogenesis. However, whether calcium dysregulation occurs first and it is followed by calpain activation or whether calpain activation promotes increased calcium dysregulation is unclear (O’Dell et al., 2012).
Calpain-dependent proteolysis represents one of the main mechanisms linked to cellular damage triggered by excitotoxic injuries like physical trauma, hypoxic/ischemic insult or chemical challenge (Liu et al., 2008; Curcio et al., 2016). The role of calpain in neuronal death in response to seizures and circuit dysregulation has been mostly demonstrated in samples taken acutely after SE (Araujo et al., 2008; Wang et al., 2008b; Lopez-Meraz and Niquet, 2009; Howe et al., 2016; Lam et al., 2017). Chemoconvulsant-induced SE triggers maximal calpain activation during the first week post-SE and acute administration of calpain inhibitors can partially reverse the appearance of spectrin BDPs and reduced neuronal death (Araujo et al., 2008; Wang et al., 2008b; Lam et al., 2017). In addition, calpain inhibition immediately after SE ameliorates seizure burden (Lam et al., 2017). It is unclear if calpain activity returns to control levels at any point after SE induction but it appears that the detection of spectrin BDPs trend towards control levels by 7–8 days post SE (Wang et al., 2008b; Lam et al., 2017). We decided to investigate if this downward trend could be affected by the spontaneous seizure activity that typically emerges during the second week post-SE (Figure 1). Thus, we decided to analyze if the levels of calpain activation could be affected by the repetitive bouts of SRS typically observed during early epileptogenesis. The results presented here suggest that increased occurrence of SRS is well correlated with augmented calpain expression and activity. In addition, these experiments show the existence of a good correlation between the number of seizures detected and higher calpain activation. Independently of possible differences during SE induction, the finding that animals with the higher seizure burden presented higher calpain activation supports the notion that calpain (re)activation is directly related to the occurrence of SRS since the main distinction detected between samples is the number of SRS endured prior to sacrifice. Moreover, increased levels of calpain activation were correlated with increased neuronal degeneration, astrogliosis and inflammation but the role of calpain activation on these markers of epileptogenesis remains to be fully investigated. Together, these observations suggest that SRS themselves can promote calpain activation and exacerbate cellular abnormalities typically found in epileptic tissue. Seizure-dependent activation of calpain during epileptogenesis might represent one of the underlying mechanism fueling the endless cycles of seizures characteristic of epilepsy, suggesting that delayed calpain inhibition might represent an alternative approach to prevent epilepsy progression.
Neurodegeneration is one of the best established hallmarks of temporal lobe epilepsy (O’Dell et al., 2012). Neuronal loss can be preferentially detected in hippocampus but extrahippocampal neuronal loss can also be detected in entorhinal cortex, piriform cortex and amygdala (de Lanerolle et al., 2003; Ben-Ari and Dudek, 2010). Seizure activity by itself can cause neuronal damage in calcium-dependent way (Walker, 2018); however, while some studies indicate progressive neuronal loss due to repetitive seizures (Ben-Ari and Dudek, 2010) other studies show no correlation between seizure frequency and cell damage (Gorter et al., 2001). The reason for this discrepancy is unclear but seizures may promote the loss of the most vulnerable neuronal populations first and once those susceptible cells are eliminated subsequent seizures might not promote further cell loss (Pitkanen et al., 2002). Thus, a clear demonstration of neuronal death caused by SRS has proven difficult. A key factor involved in these discrepancies might be the timing of tissue collection, since prolonged seizure occurrence might deplete the population of susceptible cells decreasing the probability of detecting further damage. Here, we were able to find a correlation between the number of SRS and the number of FJB+ cells at an early time point during epileptogenesis. Although it can be argued that these degenerating cells are the direct result of the initial SE, the tissue analyzed comes from animals that experienced an equivalent SE but different levels of chronically epileptic seizures. The finding of a significant correlation between the number of spontaneous seizures and the number of degenerating cells imply that the frequency of SRS can indeed drive differential levels of neurodegeneration. Assessment of the overall pattern of cell loss revealed that the main layers of principal neurons are easily detectable but increased cell dispersion was observed in epileptic animals. A significant reduction in number of cell bodies was apparent in the CA3 region of animals with a “high” number of SRS but, despite a trend, no correlation was found between cumulative cell loss and the number of SRS detected. However, although the levels of calpain activation and neuronal degeneration detected in this study might be linked, a direct correlation between cell death and calpain activation by SRS remains to be demonstrated.
At the moment, it is unclear if the pathological abnormalities detected in epileptic tissue are a cause or a result of seizure occurrence. An association between epilepsy and hippocampal sclerosis (characterized by neuronal loss and gliosis) has been clearly identified but whether seizures are a cause or effect of hippocampal sclerosis is still an unresolved controversy (Pitkanen and Sutula, 2002). In this sense, it has been postulated that if a particular pathology is epileptogenic, then its severity must correlate with the frequency of SRS (Hester and Danzer, 2013; Buckmaster et al., 2017). Recent evidence suggests that the number of ectopic granule cells (Hester and Danzer, 2013) and the loss of GABAergic interneurons (Hester and Danzer, 2013; Buckmaster et al., 2017) detected in the dentate gyrus of epileptic animals have a positive correlation with seizure frequency. However, despite multiple efforts, the molecular, cellular and network changes necessary for the manifestation of spontaneous seizures are only starting to emerge.
Gliosis is a standard finding in epileptic tissue obtained from human or experimental specimens (Pitkanen and Sutula, 2002; Sharma et al., 2007). In the epileptic foci, astrocytes undergo changes in morphology, molecular composition and proliferation (Devinsky et al., 2013). Reactive astrocytes, characterized by increased GFAP expression and hypertrophy are an important source of inflammatory molecules but are also the target of inflammatory molecules that amplify pro-epileptogenic inflammatory signaling and exacerbate glial scaring (Legido and Katsetos, 2014). Microglia are also integral for inflammatory processes in experimental models and human epilepsy. Activated microglia produce proinflammatory mediators and, in some types of drug resistant-epilepsy, the extent of microgial activation correlates with seizure frequency and disease duration (Devinsky et al., 2013). Due to the fact that cytokines and growth factors are upregulated during epileptogenesis it has been suggested that reactive astrocytes release tropic factors leading to axonal sprouting, synapse formation and hyperexcitability (Crespel et al., 2002). Here, the number of cells positive for GFAP and Iba1 show some correlation with the number of seizures detected. The correlation was mostly significant in animals that endured a moderated number of seizures.
The calpain activity detected here is more likely to result from activation of calpain-1 and calpain-2, the predominant calpain subtypes expressed in the brain. Recent evidence suggests that calpain-1 and calpain-2 might play different roles in neuroprotection and neurodegeneration (Seinfeld et al., 2016; Wang et al., 2016), calpain-1 activation appears to be necessary for neuroprotection while calpain-2 is directly be involved in neurodegeneration (Baudry and Bi, 2016). Unfortunately, the current study cannot determine the independent roles of calpain-1 or calpain-2 but future studies will be aimed to investigate the specific contribution of these isoforms to neurodegeneration and epileptogenesis.
Conclusion
This study found that the number of SRS occurring during early epileptogenesis was correlated with increased calpain activity and expression. Seizure occurrence was also correlated with the levels degenerating neurons detected in hippocampal tissue. Our findings demonstrate that there is a correlation between seizure occurrence, calpain activity and neurodegeneration during an early phase of epileptogenesis. Thus, this study raises the possibility that aberrant calpain activation triggered by SRS might contribute to the manifestation of future spontaneous seizures. In addition, our findings open the possibility that pharmacological inhibition of calpain during epileptogenesis might represent a novel therapeutic approach to reduce SRS.
Figure 7. Reactive Astrocytes After SRS.
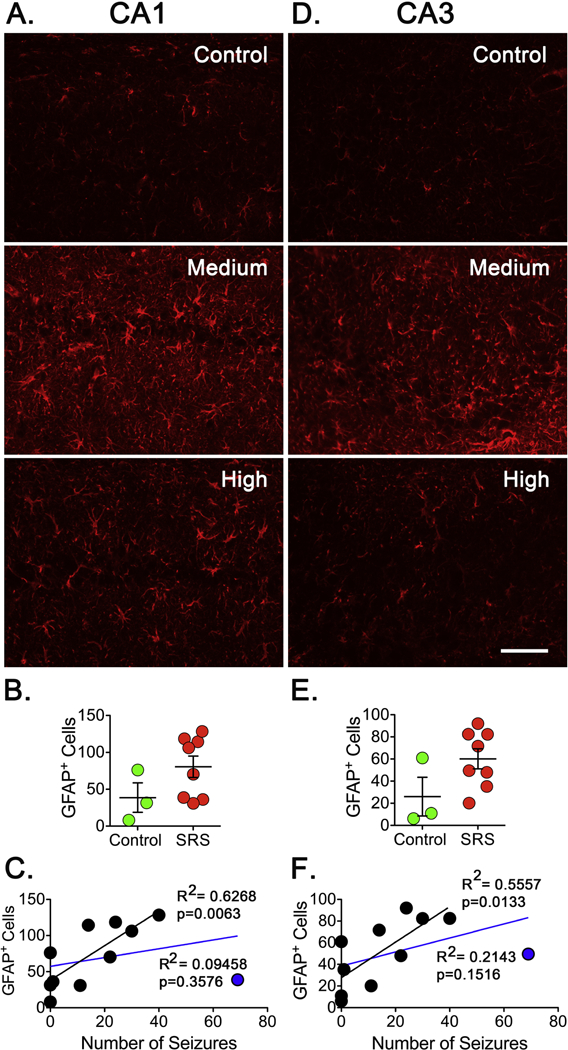
Coronal sections (15 μm) were selected from a 1-in-10 series started at approximately the same level of hippocampus. (A, D) Sections were stained with GFAP antibodies to detect reactive astrocytes in CA1 and CA3 regions of hippocampus. Scale bar represents 100 μm. (B, E). The number of GFAP+ cells was determined on the three sections and the average represents the number of GFAP+ cells in a particular animal. Cell counts were conducted blinded to seizure frequency. Data is presented as the mean ± SEM of controls (n=3) and animals that experienced SRS (n=8). No significant difference in the number of GFAP+ cells was detected using an unpaired t-test. (C, F) The number of GFAP+ cells was paired with the number of SRS detected in a linear regression analysis. Plots did not show a significant correlation in CA1 or CA3 regions of hippocampus when all animals were included (blue line). When an animal experiencing the highest number of seizures (blue dot) was not included in the analysis (black line), the plots displayed a significant correlation between the levels of GFAP+ cells and the number of SRS detected in CA1 (R2=0.6268, p=0.0063) and CA3 (R2=0.5557, p=0.0133).
Figure 8. Microglia Activation After SRS.
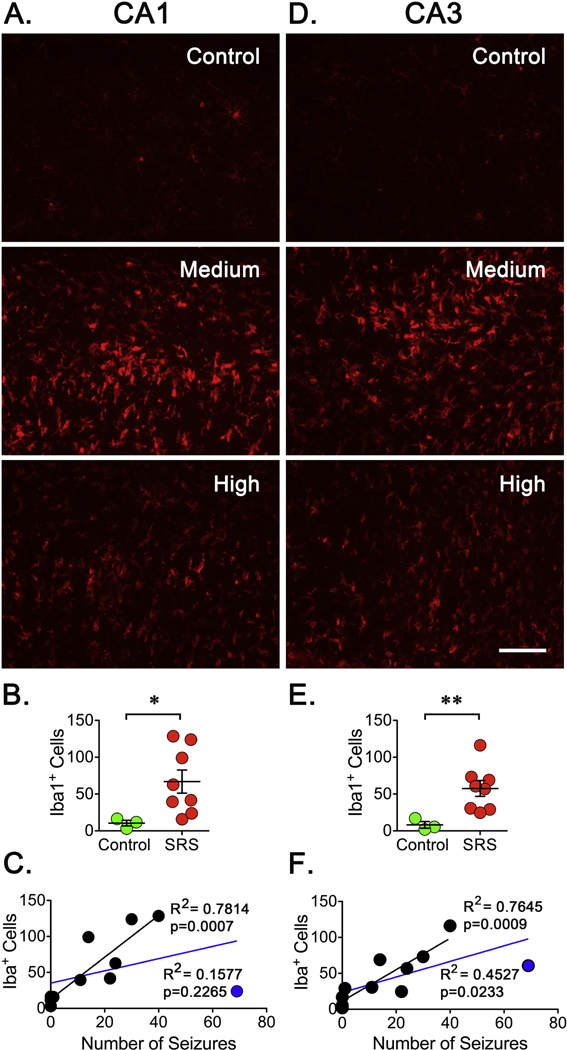
Brain sections (15 μm) from a 1-in-10 series were selected. (A, D) Sections were stained with Iba1 antibodies to detect reactive microglia in CA1 and CA3 regions of hippocampus. Scale bar represents 100 μm. (B, E). The levels of Iba1+ cells were obtained by counting the three sections and averaged to obtain a representative number of Iba1+ cells in a particular animal. Cell counts were conducted blinded to seizure frequency. Data is presented as the mean ± SEM of controls (n=3) and animals that experienced SRS (n=8). A significant difference in the number of Iba1+ cells in controls and animals experiencing SRS was detected using an unpaired t-test, *p<0.05 and **p<0.01 represent a significant difference. (C, F) A linear regression analysis between the number of Iba1+ cells and the number of SRS did not show a significant correlation when all animals are included (blue line). When an animal experiencing the highest number of seizures (blue dot) was not part of the analysis (black dots and line), the plots displayed a significant correlation in CA1 (R2=0.7814, p=0.0007) and CA3 (R2=0.7645, p=0.0009) regions.
HIGHLIGHTS.
Calpain activation correlates with the occurrence of spontaneous seizures.
Appearance of degenerating neurons is associated with the occurrence of spontaneous seizures.
Aberrant calpain reactivation might contribute to epileptogenesis.
Acknowledgements
We would like to thank the Rodent In Vivo Neurophysiology Core at the University of Colorado Anschutz Medical Campus for providing facilities to acquire and review the EEG/Video data. The AB38 antibody was a generous gift from Dr. David R. Lynch (University of Pennsylvania, PA). Grant R01-NS089698 from the National Institutes of Health to MIG supported this work.
Abbreviations used:
- CA1
Cornus Ammonis 1
- DG
Dentate Gyrus
- SE
status epilepticus
- SRS
spontaneous recurrent seizures
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Philip M. Lam, Department of Pediatrics, Division of Neurology and Translational Epilepsy Research Program, University of Colorado School of Medicine, Aurora, CO, 80045 USA,
Marco I. González, Department of Pediatrics, Division of Neurology and Translational Epilepsy Research Program, University of Colorado School of Medicine, Aurora, CO, 80045 USA,.
References
- Araujo IM, Gil JM, Carreira BP, Mohapel P, Petersen A, Pinheiro PS, Soulet D, Bahr BA, Brundin P, Carvalho CM (2008) Calpain activation is involved in early caspase-independent neurodegeneration in the hippocampus following status epilepticus. J Neurochem 105:666–676. [DOI] [PubMed] [Google Scholar]
- Baudry M, Bi X (2016) Calpain-1 and Calpain-2: The Yin and Yang of Synaptic Plasticity and Neurodegeneration. Trends Neurosci 39:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Dudek FE (2010) Primary and secondary mechanisms of epileptogenesis in the temporal lobe: there is a before and an after. Epilepsy Curr 10:118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevers MB, Neumar RW (2008) Mechanistic role of calpains in postischemic neurodegeneration. J Cereb Blood Flow Metab 28:655–673. [DOI] [PubMed] [Google Scholar]
- Bi X, Chang V, Siman R, Tocco G, Baudry M (1996) Regional distribution and time-course of calpain activation following kainate-induced seizure activity in adult rat brain. Brain Res 726:98–108. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA (1998) Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med 4:1166–1172. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Abrams E, Wen X (2017) Seizure frequency correlates with loss of dentate gyrus GABAergic neurons in a mouse model of temporal lobe epilepsy. J Comp Neurol 525:2592–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RL, Davies PL (2012) Structure-function relationships in calpains. Biochem J 447:335–351. [DOI] [PubMed] [Google Scholar]
- Chang BS, Lowenstein DH (2003) Epilepsy. N Engl J Med 349:1257–1266. [DOI] [PubMed] [Google Scholar]
- Crespel A, Coubes P, Rousset mC, Brana C, Rougier A, Rondouin G, Bockaert J, Baldy-Moulinier M, Lerner-Natoli M (2002) Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res 952:159–169. [DOI] [PubMed] [Google Scholar]
- Curcio M, Salazar IL, Mele M, Canzoniero LM, Duarte CB (2016) Calpains and neuronal damage in the ischemic brain: The swiss knife in synaptic injury. Prog Neurobiol 143:1–35. [DOI] [PubMed] [Google Scholar]
- Curia G, Longo D, Biagini G, Jones RS, Avoli M (2008) The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods 172:143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Wallace GCt Holmes C, McDowell ML, Smith JA, Marshall JD, Bonilha L, Edwards JC, Glazier SS, Ray SK, Banik NL (2012) Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 220:237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Williamson A, Spencer SS, Zaveri HP, Eid T, Spencer DD (2003) A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia 44:677–687. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA (2013) Glia and epilepsy: excitability and inflammation. Trends Neurosci 36:174–184. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, O’Brien TJ, Jette N, Scheffer IE, de Curtis M, Perucca P (2018) Epilepsy. Nat Rev Dis Primers 4:18024. [DOI] [PubMed] [Google Scholar]
- Falconer MA, Serafetinides EA, Corsellis JA (1964) Etiology and Pathogenesis of Temporal Lobe Epilepsy. Arch Neurol 10:233–248. [DOI] [PubMed] [Google Scholar]
- Feng ZH, Hao J, Ye L, Dayao C, Yan N, Yan Y, Chu L, Shi FD (2011) Overexpression of mu-calpain in the anterior temporal neocortex of patients with intractable epilepsy correlates with clinicopathological characteristics. Seizure 20:395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MI, Cruz Del Angel Y, Brooks-Kayal A (2013) Down-regulation of gephyrin and GABAA receptor subunits during epileptogenesis in the CA1 region of hippocampus. Epilepsia 54:616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Aronica E, Lopes da Silva FH (2001) Progression of spontaneous seizures after status epilepticus is associated with mossy fibre sprouting and extensive bilateral loss of hilar parvalbumin and somatostatin-immunoreactive neurons. Eur J Neurosci 13:657–669. [DOI] [PubMed] [Google Scholar]
- Grabenstatter HL, Del Angel YC, Carlsen J, Wempe MF, White AM, Cogswell M, Russek SJ, Brooks-Kayal AR (2014) The effect of STAT3 inhibition on status epilepticus and subsequent spontaneous seizures in the pilocarpine model of acquired epilepsy. Neurobiol Dis 62:73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hester MS, Danzer SC (2013) Accumulation of abnormal adult-generated hippocampal granule cells predicts seizure frequency and severity. J Neurosci 33:8926–8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe CL, LaFrance-Corey RG, Mirchia K, Sauer BM, McGovern RM, Reid JM, Buenz EJ (2016) Neuroprotection mediated by inhibition of calpain during acute viral encephalitis. Sci Rep 6:28699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam PM, Carlsen J, Gonzalez MI (2017) A calpain inhibitor ameliorates seizure burden in an experimental model of temporal lobe epilepsy. Neurobiol Dis 102:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legido A, Katsetos CD (2014) Experimental studies in epilepsy: immunologic and inflammatory mechanisms. Semin Pediatr Neurol 21:197–206. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu MC, Wang KK (2008) Calpain in the CNS: from synaptic function to neurotoxicity. Sci Signal 1:re1. [DOI] [PubMed] [Google Scholar]
- Lopez-Meraz ML, Niquet J (2009) Participation of mu-calpain in status epilepticus-induced hippocampal injury. Brain Res Bull 78:131. [DOI] [PubMed] [Google Scholar]
- O’Dell CM, Das A, Wallace Gt, Ray SK, Banik NL (2012) Understanding the basic mechanisms underlying seizures in mesial temporal lobe epilepsy and possible therapeutic targets: a review. J Neurosci Res 90:913–924. [DOI] [PubMed] [Google Scholar]
- Ono Y, Sorimachi H (2012) Calpains: an elaborate proteolytic system. Biochim Biophys Acta 1824:224–236. [DOI] [PubMed] [Google Scholar]
- Paul CA, Beltz B, Berger-Sweeney J (2008) The nissl stain: a stain for cell bodies in brain sections. CSH Protoc 2008:pdb prot4805. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Sutula TP (2002) Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol 1:173–181. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Nissinen J, Nairismagi J, Lukasiuk K, Grohn OH, Miettinen R, Kauppinen R (2002) Progression of neuronal damage after status epilepticus and during spontaneous seizures in a rat model of temporal lobe epilepsy. Prog Brain Res 135:67–83. [DOI] [PubMed] [Google Scholar]
- Poirier JL, Capek R, De Koninck Y (2000) Differential progression of Dark Neuron and Fluoro-Jade labelling in the rat hippocampus following pilocarpine-induced status epilepticus. Neuroscience 97:59–68. [DOI] [PubMed] [Google Scholar]
- Raimondo JV, Burman RJ, Katz AA, Akerman CJ (2017) Ion dynamics during seizures. Front Cell Neurosci 9:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza M, Blair RE, Sombati S, Carter DS, Deshpande LS, DeLorenzo RJ (2004) Evidence that injury-induced changes in hippocampal neuronal calcium dynamics during epileptogenesis cause acquired epilepsy. Proc Natl Acad Sci U S A 101:17522–17527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R (1994) Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci 14:3934–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossini L, Garbelli R, Gnatkovsky V, Didato G, Villani F, Spreafico R, Deleo F, Lo Russo G, Tringali G, Gozzo F, Tassi L, de Curtis M (2018) Seizure activity per se does not induce tissue damage markers in human neocortical focal epilepsy. Ann Neurol 82:331–341. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Creed J, Raghupathi R (2010) Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics 7:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalia R, Gong Y, Berzins B, Freund B, Feather D, Landesberg G, Mishra G (2011) A novel role for calpain in the endothelial dysfunction induced by activation of angiotensin II type 1 receptor signaling. Circ Res 108:1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued lC, Hopkins KJ (2000) Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874:123–130. [DOI] [PubMed] [Google Scholar]
- Seinfeld J, Baudry N, Xu X, Bi X, Baudry M (2016) Differential Activation of Calpain-1 and Calpain-2 following Kainate-Induced Seizure Activity in Rats and Mice. eNeuro 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AK, Reams RY, Jordan WH, Miller MA, Thacker HL, Snyder PW (2007) Mesial temporal lobe epilepsy: pathogenesis, induced rodent models and lesions. Toxicol Pathol 35:984–999. [DOI] [PubMed] [Google Scholar]
- Shumate MD, Lin DD, Gibbs JW 3rd, Holloway KL, Coulter DA (1998) GABA(A) receptor function in epileptic human dentate granule cells: comparison to epileptic and control rat. Epilepsy Res 32:114–128. [DOI] [PubMed] [Google Scholar]
- Silva AP, Carvalho AP, Carvalho CM, Malva JO (2001) Modulation of intracellular calcium changes and glutamate release by neuropeptide Y1 and Y2 receptors in the rat hippocampus: differential effects in CA1, CA3 and dentate gyrus. J Neurochem 79:286–296. [DOI] [PubMed] [Google Scholar]
- Smith ZZ, Benison AM, Bercum FM, Dudek FE, Barth DS (2018) Progression of convulsive and nonconvulsive seizures during epileptogenesis after pilocarpine-induced status epilepticus. J Neurophysiol 119:1818–1835. [DOI] [PubMed] [Google Scholar]
- Stalker TJ, Gong Y, Scalia R (2005) The calcium-dependent protease calpain causes endothelial dysfunction in type 2 diabetes. Diabetes 54:1132–1140. [DOI] [PubMed] [Google Scholar]
- Sutula TP, Hagen J, Pitkanen A (2003) Do epileptic seizures damage the brain? Curr Opin Neurol 16:189–195. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Yuste R (2007) Feedforward inhibition contributes to the control of epileptiform propagation speed. J Neurosci 27:3383–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderklish PW, Bahr BA (2000) The pathogenic activation of calpain: a marker and mediator of cellular toxicity and disease states. Int J Exp Pathol 81:323–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J (2008) Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol 38:78–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MC (2018) Pathophysiology of status epilepticus. Neurosci Lett 667:84–91. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu YH, Huang YG, Chen LW (2008a) Time-course of neuronal death in the mouse pilocarpine model of chronic epilepsy using Fluoro-Jade C staining. Brain Res 1241: 157–167. [DOI] [PubMed] [Google Scholar]
- Wang S, Shan P, Song Z, Dai T, Wang R, Chi Z (2008b) Mu-calpain mediates hippocampal neuron death in rats after lithium-pilocarpine-induced status epilepticus. Brain Res Bull 76:90–96. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lopez D, Davey PG, Cameron DJ, Nguyen K, Tran J, Marquez E, Liu Y, Bi X, Baudry M (2016) Calpain-1 and calpain-2 play opposite roles in retinal ganglion cell degeneration induced by retinal ischemia/reperfusion injury. Neurobiol Dis 93:121–128. [DOI] [PubMed] [Google Scholar]