

Supplemental Digital Content is available in the text.
Key Words: patient-derived tumor xenograft, colorectal cancer, preclinical models, chimeric antigen receptor
Abstract
Adoptive transfer of T cells engineered with a chimeric antigen receptor (CAR) is deemed as the silver bullet to overcome the barriers of solid tumor treatment; however, the therapeutic application against solid tumors faces major challenges largely owing to the complex heterogeneity and immunosuppressive microenvironment of solid tumors. Preclinical development of CAR-T-cell products necessitates an appropriate animal model for the evaluation and improvement of their therapeutic capacities. Patient-derived xenograft (PDX) resembles real patients in several ways, and may serve as an attractive alternative to generate and evaluate the efficacy of CAR-T-cell products. In this study, we established and characterized a PDX mouse model implanted with colorectal cancer (CRC) xenograft. Human epidermal growth factor receptor 2 (HER2) expression in CRC specimens was detected by immunohistochemistry. The fragments of patient tumors were subcutaneously implanted into immunodeficient NOD-NPG mice after surgery. Furthermore, HER2-specific CAR-T cells were engineered and tested in our model to show their effectiveness in tumor clearance. Adoptive transfer of HER2-specific CAR-T cells resulted in the regression or even elimination of CRC xenograft and protection of relapse from rechallenged colon cancer tissue in PDX model. Significant survival advantage was achieved in these mice as compared with those transplanted with green fluorescent protein-T cells. Thus, this study showed that CAR-T-cell treatment may be a promising approach for solid tumor clearance and that the PDX model may be useful to evaluate the effects of CAR-T cells.
Immunotherapy is an effective strategy for the treatment of tumors. Chimeric antigen receptor (CAR)-T-cell immune therapy has displayed good clinical response in the treatment of CD19+ leukemia.1,2 Recent studies have highlighted the potential of CAR-T cells in treating solid malignancies.3 However, further studies are warranted for the thorough application of CAR-T cells in solid tumor treatment. In comparison with hematological malignancies, solid tumors are more complex with special 3-dimensional (3D) structure, immunosuppressive microenvironment, and heterogenous surface marker expressions, posing a major challenge for T cells to completely eliminate tumor mass and maintain long-term remission.4 Therefore, good preclinical models that could authentically recreate the complex intratumor microenvironment are needed to evaluate the efficacies of different immunotherapy products. Most animal models used in cancer research are established by injecting tumor cell lines into graft-bearing hosts. However, the homogenous nature of these cells does not allow for the creation of the unique structure and heterogeneity observed in real tumors. Patient-derived xenograft (PDX) models are established by directly transplanting surgically dissected human tumor tissues into immune-deficient hosts. These models carry the patient-derived tumor cells and maintain the original tumor structure.5 The patient-derived tumor cells inside the xenograft recreate an immunosuppressive microenvironment that may hinder treatment, while the inherited organization of cancer cells provides a real test ground for tumor infiltration.6 Such PDX models have been widely used in drug discovery, but their applications in immunotherapy research have been rare.7,8
In this study, we reported the application of a PDX model in the preclinical development of immunotherapy products. The specific treatment included human epidermal growth factor receptor 2 (HER2)-targeted CAR-T-cell therapy. A variety of surface antigens enriched in solid tumors were developed as CAR-T-cell targets; these included HER2, mesothelin, and epidermal growth factor receptor (EGFR). Some of these targeted therapies have already entered clinical trials.7,8 HER2 is a member of the transmembrane EGFR family and is known to be overexpressed in breast, ovarian, gastric, colon, and lung cancers.9,10 Being the most studied tumor-associated antigen, HER2 was thought to be an effective target for CAR-T-cell–based immunotherapy. HER2 is overexpressed on ∼15% of colon cancer cells and HER2 antibody has been approved for routine therapeutic applications with good safety profile. The preliminary data of the clinical trial with HER2-targeted CAR-T cells indicated good safety profile but modest efficacy,10 demanding further preclinical development.
In this study, we successfully established and characterized a colon cancer PDX model using HER2+ patient-derived colon cancer tissues. Furthermore, we used this model to test the antitumor effect of the established HER2-specific CAR-T cells. These cells showed excellent tumor suppression capacity and protected the recipients from tumor rechallenge. The results of the present study highlight the effectiveness of the established PDX model as well as the good quality of the HER2-specific CAR-T cells.
MATERIALS AND METHODS
Establishment of PDX Model
Fresh tumor samples (F0) from surgery were immediately collected under sterile conditions in the operating room and placed in an antibiotic-containing Roswell Park Memorial Institute (RPMI)-1640 medium. After repeated washing with phosphate-buffered saline (PBS), tumor tissues were cut into pieces of ∼2 mm diameter with a scalpel. Tumor fragments were subcutaneously implanted in 6–10-week-old female severe combined immune deficiency (SCID)-NPG mice.11 Tumors (P0) were resected from mice upon reaching a size of ∼1000 mm3, cut into pieces, and implanted into the next generation of NPG mice, as previously described. The P1 xenograft models were then serially passaged to P2 and P3 generations. Tumor volume (V) was calculated by measuring 2 perpendicular diameters with calipers as follows: V=[length×(width)2]/2. All animal experiment protocols were authorized by the Biomedical Research Ethics Committee of Peking University and Peking University Health Science Center.
Hematoxylin and Eosin (H&E) and Immunohistochemistry (IHC) Staining
Tissue specimens were subjected to IHC staining by Service Bio. Patient tumor samples, every generation of xenografts, and xenograft tissues after CAR-T-cell treatment were analyzed as formalin-fixed, paraffin-embedded sections (5 mm thickness). H&E staining was performed on all specimens, and subsequent staining was performed in serial sections (5 mm thickness).
Cell Culture
Peripheral blood mononuclear cells were isolated from the peripheral blood of healthy adult donors under consent by density-gradient centrifugation using Ficoll-Paque Plus (Pharmacia Biotech, Piscataway, NJ). T cells were cultured in RPMI-1640 (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum and 100 U/mL of human interleukin-2 (hIL-2; PeproTech, Rocky Hill, CT). T cells were activated by CD3/CD28-specific magnetic beads at a concentration of one bead/cell (Invitrogen Life Technologies, Carlsbad, CA). 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum.
Generation of the HER2-specific CAR Lentivirus and T Cells
All kinds of lentiviruses were produced in 293T cells transfected with the pMD2.G plasmid, psPAX2 plasmid, and CAR-containing vector plasmid using calcium phosphate system. The transfected cells were incubated at 37°C for 12 hours in serum-containing DMEM medium. The transfected 293T cells were incubated for another 24–36 hours. Lentivirus-enriched supernatants were collected and filtered through a 45-μm filter (Millipore, Bedford, MA) to remove cellular debris and stored at −80°C. Two days after activation, T cells were added to culture flasks and loaded with the lentiviral HER2-specific CAR viral particles. The cells were transfected and cultured at 37°C and 5% CO2 for 24 hours in the presence of 10 mg/mL of polybrene (Sigma, St Louis, MO). The medium was changed every 2 days and the cells were collected at day 10.12
Flow Cytometry
We used a FACSCalibur instrument and Cellquest software for all flow cytometry analysis. Cells were washed with PBS before staining with antibodies. After 30–45 minute of incubation at 4°C in the dark, cells were washed twice with PBS (Gibco) and analyzed with FACSCalibur instrument. The samples were determined using BD FACSCalibur and Beckman Coulter Gallios. CD3, CD4, CD8, CD27, CD28, PD-1, Tim3, CCR7, and CD45RA antibodies (code no: 555333, 557922, 555367, 562297, 562613, 558694, 563422, 557734, and 563031, respectively) were obtained from BD Biosciences (San Jose, CA).
Isolation of Tumor Cells Stably Expressing Green Fluorescent Protein (GFP) and Luciferase
We designed a lentiviral vector that enabled reliable detection of the level of luciferase expression in cells. This lentiviral vector encoded a GFP-linked to firefly luciferase. Several types of tumor cell lines were stably transduced and purified by florescence-activated cell sorting (FACS) into different fractions based on increasing levels of GFP expression. Cell fractions were subjected to bioluminescence imaging to show that the level of GFP directly correlated with that of luciferase expression in tumor cells.
Cytotoxicity Assay and Cytokine Production
The ability of the HER2-specific CAR-T cells to lyse target tumor cells was analyzed using a bioluminescence assay, as previously described.12 HER2-specific CAR-T cells or GFP-T cells were coincubated with decreasing ratios of luciferase-expressing target cells [effector-to-target ratio (E:T)] in RMPI-1640 medium in 96-well U-bottom plates at 37°C for 16 hours. Percent lysis was measured as follows: % specific lysis=100×(spontaneous death relative light unit (RLU)−test RLU)/(spontaneous death RLU) where RLU represents RLU. GFP-T cells or HER2-specific CAR-T cells were cocultured with HER2+ human breast cancer cell line MCF7, HER2+ human ovarian cancer cell line SKOV3, and HER2− human lymphoma cell line Raji in 96-well culture plates in duplicates at effect T cell to target tumor cell ratios (E:T) of 0.3:1, 3:1, and 9:1 for 20 hours at 37°C. The targeted tumor cells were detected and the cell-free supernatant was collected and assayed for cytokine levels [IL-2, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, granzyme B] using different enzyme-linked immunosorbent assay (ELISA) kits (Biolegend, San Diego, CA).
In Vivo Studies of HER2-specific CAR-T Cells
The PDX model used in this experiment was at passage 2 and the successful establishment of the model was confirmed upon achieving a tumor volume of 50–100 mm3. A total of 2×106 GFP-T cells or HER2-specific CAR-T cells were intravenously injected into the tumor-bearing mice. Tumor diameter was monitored thrice a week and tumor volumes were calculated by measuring 2 perpendicular diameters with calipers. To measure T cells in mouse blood, a total of 100 μL of venous blood was obtained weekly from the orbital venous plexus and stained with anti-human CD3 antibody. Flow cytometry data were analyzed using FlowJo software. A total of 100 μL of venous blood was collected from all mice into K2EDTA anticoagulation centrifuge tubes (Beijing Emilion Science & Technology, Beijing, China) every week. The mice were anesthetized by an intraperitoneal injection of 10% chloral hydrate (0.05–0.07 mL). Before the puncture, iodophor was used to disinfect the skin around the eye. A capillary tube was slid flat along the nasal side of the eye and slowly to the eyelid below the eyeball, followed by rapid penetration into the plexus of the inner canthus. The blood was treated with BD FACS Lysing Solution (BD) and analyzed by flow cytometry.
The tumors treated with HER2-specific CAR-T cells disappeared; we reimplanted the PDX tissues (P1) into all the mice and monitored the tumor growth and T cells in the peripheral blood. Animals were killed when the mean tumor volume reached a dimension of ≥1200 mm3 and tumor tissues were collected for IHC assay.
Statistical Analyses
The differences in cytokine secretion and specific cytolysis are shown as means±SD. Other data are presented as means±SE of mean. The student t test was used to determine the statistical significance of differences between samples, and a value of P<0.05 was considered as statistically significant. All statistical analyses were performed using Prism software, version 5.0 (GraphPad, Inc., San Diego, CA).
RESULTS
Establishment of a Patient-derived Colon Carcinoma Xenograft Mouse Model
Clinical and pathologic characteristics of the original patient-derived colon cancer specimens are listed in Table 1. We established a PDX model for >10 patients, but only 1 patient-derived model expressed HER2 that was subsequently used for experiments. Fresh surgical samples (F0 tissue) from patients with colon cancer were cut into fragments (2×2 mm) and implanted into 8–10-week-old immunodeficient NPG mice. Tumor volume was measured by a caliper and the tumors were harvested upon reaching a volume of 500–1000 mm3. The excised tumors (P0) were dissected into small pieces and implanted into the next NPG mouse (P1). The succession rate of xenograft passage was nearly 100% in the first few generations. After 3 consecutive passages, tumors from each passage were harvested, cryopreserved, and embedded into paraffin for IHC analysis.
TABLE 1.
Clinical and Pathologic Characteristics of the Original Patient-derived Colon Cancer Specimen
Colon Cancer PDX Mouse Model Maintained the Heterogeneity of Human Tumor Tissue
To evaluate the level of intratumor heterogeneity and clinicopathologic characteristics of the tumor tissues, we analyzed the tumor tissues by H&E and IHC staining. Xenografts showed similar pathology patterns and maintained the morphology of the original patient-derived specimen (Fig. 1A). Furthermore, the tumor-associated marker HER2 was positively expressed in over 50% cancer cells of all the xenografts (Fig. 1B). Staining results showed that the immune percentage of fibroblast activation protein (FAP), a marker of tumor stromal cells (Fig. 1C), was comparable between the xenograft tissue collected from PDX models and original cancer tissues. As shown in Figure S1A (Supplemental Digital Content, http://links.lww.com/JIT/A518), the average optical density (calculated by Image Pro Plus) of HER2 and FAP was consistent from F0 to P3. The statistical results are shown in Figure S1B (Supplemental Digital Content, http://links.lww.com/JIT/A518). A slight increase in HER2 and FAP immune percentage from F0 to P3 (Figs. 1B, C) was observed. The immune percentage of HER2 was close to 40% at F0 and rose to nearly 60% at P3. The immune percentage of FAP similarly ranged from 60% to 70% at F0 but rose to 80% at P3 (Fig. 1D).
FIGURE 1.
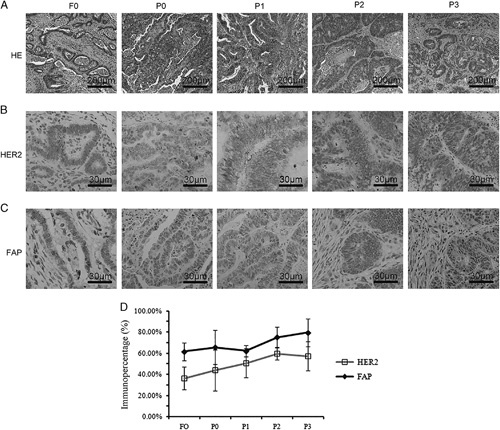
Comparison of the expression level of HER2 and FAP between patient-derived tumors and patient-derived xenograft models. Similar architecture (A), HER2 expression (B), and FAP (C) expression are shown. Some variations in HER2 expression were observed. D, Histologic analysis of HER2 and FAP expression in fresh tumor samples from P3 mice. At least 3 engrafts from each group were measured and the results are expressed as the mean and SE of mean. FAP indicates fibroblast activation protein; HER, human epidermal growth factor receptor.
To investigate the immune microenvironment of PDX tumors, we determined the expression level of programmed cell death ligand 1 (PD-L1), one of the most important immune checkpoints in tumor immunosuppression that is commonly expressed in solid tumors. The results showed that PD-L1 expression was detected in the original tissue sample and each generation of PDX (Fig. 2A). However, the immune percentage of tumor-infiltrating immunocytes, including CD3, and forkhead box P3 (foxp3)-positive cells immediately decreased during the passage of the xenograft (Figs. 2B–D). The average optical density of CD3 PD-L1 and foxp3 was consistent from F0 to P3 (Fig. S1A, Supplemental Digital Content, http://links.lww.com/JIT/A518). Statistical results are shown in Figure S1B (Supplemental Digital Content, http://links.lww.com/JIT/A518).
FIGURE 2.
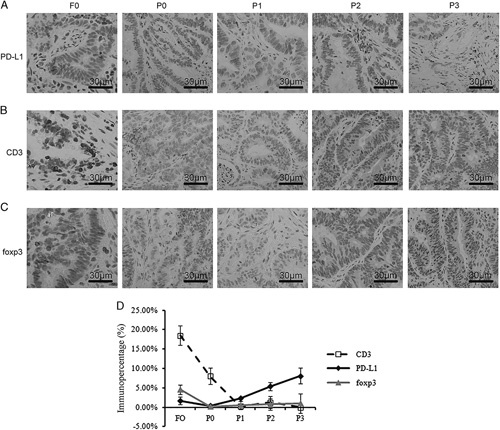
Disconcordance between primary tumors and patient-derived xenograft models. Tumors showed remarkable differences in the expressions of CD3 (A), PD-L1 (B), and foxp3 (C). D, Histologic analysis of CD3, PD-L1, and foxp3 expressions from fresh tumor samples obtained from P3-engrafted tumor tissues. At least 3 engrafts from each case were measured and the results are expressed as the mean and SE of mean.
HER2-specific CAR-T Cells Have Specific Cytotoxic Activities
In this study, the single-chain variable fragment was linked to the CD8α chain hinge and transmembrane region with 4-1BB and the CD3ζ intracellular signaling domains. This cassette was inserted into the pRRLSIN.cPPT vector, wherein the expression was driven from the EF-1α promotor (Fig. 3A).
FIGURE 3.
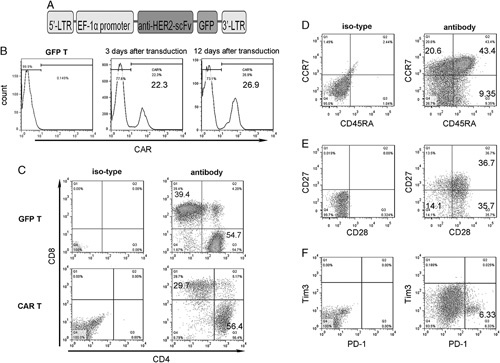
HER2-specific CAR is expressed on the surface of the activated human T lymphocytes. A, Schematic of the HER2-specific CAR cassette. B, Expression of CAR was examined both after 3 and 12 days of CAR transduction against GFP-T cells. Flow cytometry analysis of CD4 and CD8 (C), CD45RA and CCR7 (D), CD27 and CD28 (E), PD-1 and Tim3 (F) expressions in HER2-specific CAR+ T lymphocytes generated from healthy donors and stained for 12 days after retroviral gene transfer. The plots are gated on CD3+ cells, which accounted for >95% of the total cells in the cultures. The numbers on the plots are the percentages of cells in each quadrant. Data are representative of 3 experiments. CAR-T indicates chimeric antigen receptor T cell; HER, human epidermal growth factor receptor.
After the activation with anti-CD3/CD28 beads for 2 days, T lymphocytes isolated from healthy donor were transduced with lentiviral vectors encoding HER2-specific CAR. We first determined the percentage of T lymphocytes showing surface expression of HER2-specific CAR. Flow cytometry results indicated that HER2-specific CAR was expressed in 20%–40% of all transfected T lymphocytes after 3 days of transfection. HER2 expression was maintained in cells cultured for >12 days (Fig. 3B). Phenotypic analysis performed 12 days after gene transfer showed that both CAR-T and GFP-T cells were a mixture of CD4+ and CD8+ cells (Fig. 3C). Moreover, nearly 50% T cells simultaneously carried memory T-cell markers such as CCR7+, CD45RA−, CD27+, and CD28+, suggesting that these cells had a central memory T-cell phenotype (Figs. 3D, E). After transduction, HER2-specific CAR-T cells expanded well in vitro in the presence of IL-2. After prolonged culture, we evaluated the expression of the exhaustion marker on T cells and found that about 6% of cells were positive for PD-1, whereas very few cells were positive for Tim3 (Fig. 3F). CAR-T and GFP-T cells showed similar results (data not shown).
We used a standard luciferase-based bioluminescence assay to evaluate the cytotoxic activity of HER2-specific CAR-T cells. We used SKOV3 and HCT-116 cells that naturally expressed HER2 and HER2− Raji cells were used as the negative control. HER2+ cells incubated with CAR-T cells were efficiently eliminated, whereas the negative control cells were left intact (Fig. 4A). In contrast, GFP-T cells failed to kill both SKOV3 and HCT-116 cells (Figs. 4B, C). As >90% HER2+ cells were killed after coincubation with CAR-T cells, HER2-specific CAR-T cells could specifically kill the target cells. The lysis ability of HER2-specific CAR-T cells depended on the expression of HER2.
FIGURE 4.
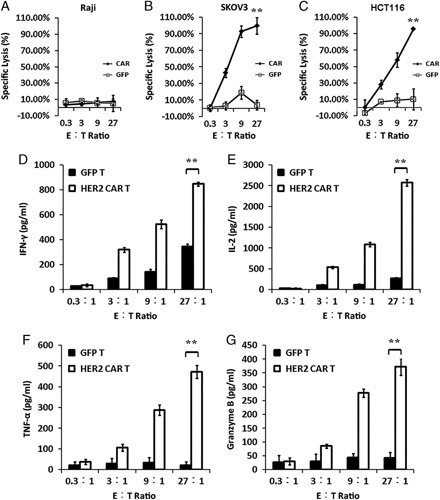
HER2-specific CAR-T cells secrete cytokines and kill target cells in the coculture. The cytotoxicity of CAR+ T cells against target antigen-negative Raji cells (A) as well as SKOV3 (B) and HCT-116 cells (C) that naturally express human HER2. The mean percentage of target cell lysis is shown in the results. HER2-specific CAR+ T cells as well as GFP-T cells were stimulated with HER2+ (SKOV3) cells. After 24 hours of stimulation, the expression levels of IFN-γ (D), IL-2 (E), TNF-α (F), and granzyme B (G) were determined. Error bars represent the SE of mean of 3 different experiments. **P<0.005. CAR-T indicates chimeric antigen receptor T cell; E:T, effector-to-target ratio; HER, human epidermal growth factor receptor; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
Next, we evaluated the capacity of HER2-specific CAR-T cells to secrete cytokines after the exposure to HER2+ cells. GFP-T or HER2-specific CAR-T cells were incubated with SKOV3 or Raji cells. After 24 hours of coincubation, the levels of IFN-γ, IL-2, TNF-α, and granzyme B were determined by ELISA. Only HER2-specific CAR-T cells produced high levels of these cytokines after the exposure to HER2+ targets, indicating that the cytokine production by CAR-T cells was associated with the presence of HER2+ antigen (Fig. 4D).
In Vivo Abilities of Persistence and Cytokine Secretions of HER2-specific CAR-T Cells in PDX Colon Cancer Mouse Model
For in vivo therapeutic efficacy studies, tumor xenograft-bearing NPG mice (tumor volumes of 50–100 mm3) were randomly assigned to HER2-specific CAR-T-cell treatment groups (6 animals per group) or GFP-T control group (Fig. 5A). Mice were intravenously injected with 2×106 HER2-specific CAR-T cells or GFP-T cells without exogenous cytokine injection.
FIGURE 5.
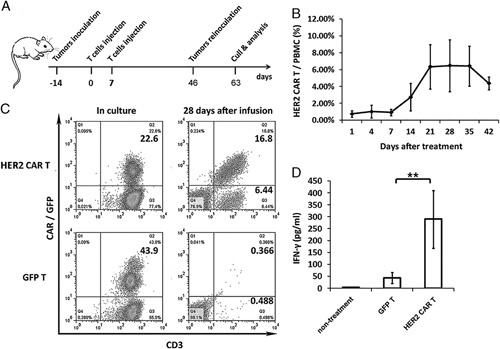
The expression and survival of engineered T cells in patient-derived xenograft model. A, Schematic diagram outlining the protocol of the experiment with the engraftment of the tumor tissue from patients to NPG mice. Mice were subcutaneously inoculated with dissected tumor masses from patients and infused with 5×106 T cells on days 7 and 14. The P1 xenograft tumor tissues were retransferred on day 46 and excised on day 63 for tumor analysis. B, In vivo persistence of HER2-specific CAR-T cells. Variable expression levels of HER2-specific CAR-T cells were detected in the blood samples of mice. CAR+ cells were detected by GFP fluorescence in the blood of all mice after the infusion. HER2-specific CAR-T cells were detected every week after infusion. Error bars represent the SEM of 6 mice. C, Both GFP-T and HER2-specific CAR-T cells were detected by florescence-activated cell sorting analysis before and after T-cell infusion. HER2-specific T cells showed significant proliferation in tumor-bearing mice. D, In vivo, HER2-specific CAR-T cells may produce IFN-γ. IFN-γ may be detected in mice peripheral blood 7 days after infusion. Error bars represent the SEM of 6 mice. **P<0.005. CAR-T indicates chimeric antigen receptor T cell; HER, human epidermal growth factor receptor; IFN, interferon; PBMC, peripheral blood mononuclear cell; SEM, SE of mean.
The expression level of HER2-specific CAR-T cells in the peripheral blood of recipients was weekly monitored by flow cytometry. CAR-T cells were detected in the peripheral blood of mice after 14 days of infusion. Continuous observation proved that HER2-specific CAR-T cells greatly expanded in the peripheral blood and showed good persistence for at least 11 weeks (Fig. 5B). GFP-T cells, on the other hand, disappeared before the first sampling. In mice injected with HER2-specific CAR-T cells, the peak blood concentration of these cells appeared after 28 days of infusion. Flow cytometry results showed that over 16% of peripheral blood mononuclear cells were positive for HER2-specific CAR (Fig. 5C).
The concentration of IFN-γ in the plasma was determined by ELISA at the first week after T-cell infusion. The production of IFN-γ was higher in HER2-specific CAR-T cells than in GFP-T control cells, as observed from the higher concentration of IFN-γ in the peripheral blood of CAR-T recipients (Fig. 5D).
HER2-specific CAR-T Cells Infiltrated Into the Tumor Xenograft, Repressed the Tumor Growth, and Protected the Recipients From the Colon Cancer Tumor Rechallenge
To investigate the therapeutic capacity of HER2-specific CAR-T cells, we sampled some tumor xenografts and determined the percentage of T-cell infiltration. IHC analysis of tumor xenografts showed T-cell infiltration in recipients treated with both control T cells and HER2-specific CAR-T cells. The percentage of infiltration of the tumor-reactive human T cells into the tumor highly correlated with tumor regression. We also performed staining for HER2 expression detection in HER2-specific CAR-T-cell–treated tissues and found that the number of HER2+ cells significantly reduced. Thus, HER2-specific CAR-T cells had the capacity to infiltrate into tumor tissues and selectively eliminated HER2+ cells (Fig. 6A).
FIGURE 6.
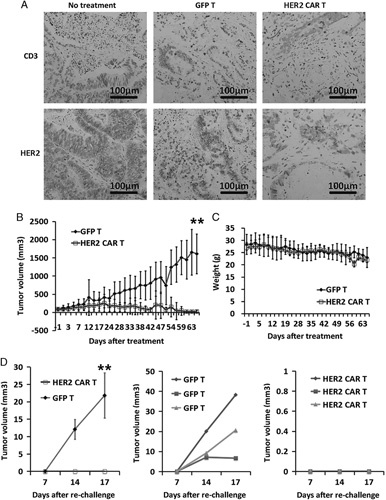
Observations following HER2-specific CAR-T-cell infusion. HER2-specific CAR-T cells controlled the tumor growth in patient-derived xenograft model. Mice bearing an established patient tumor were intravenously injected with 2×106 GFP group (n=6) or HER2-specific CAR-T cells (n=6). A, Both CAR-T and GFP-T cells were localized at tumor sites. CD3+ cells were observed and HER2+ tumor cells prominently vanished from the tumors after HER2-specific CAR-T-cell infusion, as detected by immunohistochemistry. B, Tumor growth was monitored in the form of tumor diameter every 3 days. Data represent the mean±SEM of 6 mice for each panel presented (**P<0.005 as compared with HER2-specific CAR+ T and GFP-T cells, respectively). C, Body weight was monitored after every 3 days. Data represent the means±SEM of 6 mice for each panel presented. D, Tumor growth after retransfer of P1 xenograft tumor tissues into mice treated with HER2-specific CAR+ T cells (n=3) and GFP-T cells (n=3) for 30 days. Data represent the mean±SEM of 3 mice for each panel presented (**P<0.005 as compared with HER2-specific CAR+ T and GFP-T lymphocytes, respectively). CAR-T indicates chimeric antigen receptor T cell; HER, human epidermal growth factor receptor; SEM, SE of mean.
To establish the growth curve of tumors, the subcutaneously xenografted tumors were measured thrice a week. Tumor volumes were calculated by measuring 2 perpendicular diameters with calipers. Size of tumor xenografts of GFP-T-treated mice consistently increased (n=6). The growth rate was similar to that of the xenograft of nontreated mice. In contrast, tumor xenograft size decreased in mice treated with HER2-specific T cells (Fig. 6B), and complete tumor elimination was achieved after 2 months of HER2-specific CAR-T-cell infusion. In addition, the growth parameters of mice infused with HER2-specific CAR-T cells were not significantly different from those of untreated control (Fig. 6C). These results show the excellent therapeutic effect of HER2-specific CAR-T cells.
To assess the long-term potent systemic antineoplastic effect of CAR-T cells, some of the surviving animals (n=3) were rechallenged in the hind limbs with viable P1 tumor tissues (Fig. 5A). All participating animals were monitored for additional 4 weeks; all 3 mice from GFP-T-cell treatment group developed tumors at the site of rechallenge. In contrast, none of the mice from HER2-specific CAR-T-cell group developed tumors at the site of rechallenge (Fig. 6D). These results indicate that HER2-specific CAR-T cells showed long-term persistence in vivo and effectively eliminated the freshly transplanted tumor tissues.
DISCUSSION
In this study, we established a patient-derived colon cancer mouse model and observed the therapeutic effect of HER2-specific CAR-T cells on the engrafted tumor. The results confirmed that HER2-specific CAR-T cells could play a therapeutic role in the treatment of colon cancer and show great potential in the treatment of solid tumors. This study also highlighted the ability of our PDX model to assess the therapeutic effect of various CAR-T-cell immunotherapy products.
Treatment with CAR-T cells has great potentials but toxic side effects, including cytokine release syndrome and macrophage activation syndrome, on-target off-tumor toxicity, neurotoxicity, and tumor lysis syndrome, may be observed.
As most targets of CAR-T cells are also expressed in normal cells, CAR-T cells may attack normal cells. Anti-HER2-targeting CAR-T cells caused fatal acute respiratory failure syndrome in patients with advanced colon cancer because HER2 expression is also detected in normal lung epithelial cells. Thus, the lung epithelial cells were also targeted by the circulating CAR-T cells. The first report of the lethal side effects of the treatment of a patient with colon cancer using CAR-T cells targeting HER2 was documented in 2010. This patient developed severe respiratory distress syndrome and died of multiple organ failure after 5 days of treatment.13
Clinical research is full of uncertainty. However, we should avoid as many risks as possible. The toxic side effects of CAR-T cells may be improved in the following ways. First, clinical trials require strict dose climbing and it is advisable to start with a relatively low dose. Second, the local confinement of CAR-T cells in the tumor may reduce the off-target effects. Smart structural designs can also improve the safety of CAR-T cells.14
At present, there is no effective animal model available for the preclinical development of cellular immunotherapy products. PDX models have been widely used for the screening of potential cancer therapeutic drugs. However, these models have been rarely used to evaluate cellular immunotherapy products. PDX models from various tumors, including gastric cancer,15 melanoma,16 ovarian cancer, glioblastoma,17 cervical cancer,18 and breast cancer19 have been developed. Most of these models were derived from surgical specimens.20–23 PDX models established by the direct surgical transplantation of human tumor tissues into immune-deficient mice, especially ones bearing early passage xenografts, offer several advantages and maintain the original tumor surface marker expression. These models serve better in the prediction of clinical tumor response.24–26 In the tumor model we established, the adenocarcinoma cells could be easily identified, and HER2+ tumor cells had distribution similar to that observed in the original specimen. Some cells were arranged in the form of flakes and cable, with a polygonal cavity structure. Tumor texture was hard and no festering or bleeding was observed. The fibrous tissue surrounding the tumor cells was clearly observed and formed a rigid 3D structure. The results confirmed that the established colorectal cancer (CRC) PDX model retained the stroma structure. It is possible that such a structure mimicking the real tumor microenvironment in patients provided gaps for the entry of T cells.
In the present study, the injected GFP-T cells and HER2-specific CAR-T cells were detected in the tumor tissue, but only HER2-specific CAR-T cells showed tumor-specific cytotoxicity. Histochemical staining showed that the percentage of HER2+ cells significantly reduced in the tumor. GFP-T cells in the control group also infiltrated into the tumor tissue but failed to kill the tumor cells or reduce the tumor volume. This observation clearly shows the effectiveness of CAR-T cells in treating CRC. Moreover, we found that HER2-specific CAR-T cells completely suppressed the formation and growth of rechallenged tumors, which was consistent with the existence of memory T cells in the peripheral blood of mice treated with HER2-specific CAR-T cells. Therefore, the PDX model could be used as an evaluation tool for future treatments against recurrence and metastasis of solid tumors.
Patient-derived tumor tissues contain immune cells, which formed a part of the immunosuppressive tumor microenvironment. In PDX models, foreign engraftments gradually lose human-sourced immune cells during the process of continuous passage.24,27 In this study, a few human immunocytes were observed in the PDX tumors; this observation was markedly different from that in the original cancer tissue. Although PDX model preserved the heterogeneity and structure of the original patient-derived tumor tissue, the lack of suppressive immune microenvironment made it an imperfect representation of the real patient tissue. Future studies with further improvements in PDX models are warranted. Humanized PDX model may be the answer to this challenge and may represent a PDX mouse model bearing humanized immune system. This system may not only recreate the structure and composition of the original tumor tissue but also have an authentic response to various immunotherapies. Although difficult to create, this system offers several benefits for the preclinical development of immunotherapy products.
In conclusion, we demonstrate the successful establishment of a PDX model of CRC and its application for the evaluation of the antitumor effects of the developed HER2-specific CAR-T cells. Histologic analysis of tumor xenografts showed that the structures and cellular compositions in this model were similar to those of the original tumor tissues. Furthermore, the infused HER2-specific CAR-T cells selectively killed HER2+ tumor cells and eliminated the tumors. The persistent or memory CAR-T cells provided protection from the tumor rechallenge. These observations show the usefulness of this established PDX model in the preclinical development of immunotherapies. The successful elimination of HER2+ tumor xenograft reveals the effectiveness of HER2-specific CAR-T cells. The concurrent development of a better representative model and more effective immunotherapy treatment in the future may help resolve the challenges posed by colorectal tumor.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.
CONFLICTS OF INTEREST/FINANCIAL DISCLOSURES
Supported by the National Natural Science Foundation of China (31521004, 31171417), the National Science and Technology Support Project (2014BAI02B01), the Guangdong Innovative and Entrepreneurial Research Team Program (2014ZT05S216), the Science and Technology Planning Project of Guangdong Province (2014B020226001), the Science and Technology Program of Guangzhou (2016B030232001). This work was also supported in part by a grant from BeiHao Stem Cell and Regenerative Medicine Translational Research Institute.
All authors have declared that there are no financial conflicts of interest with regard to this work.
Footnotes
R.D.T. and J.J.Z. contributed equally.
J.Z., J.W., and H.D.: conceptualization. R.T., J.Z., S.Z., J.G., H.L., and Z.L.: methodology. R.T.: formal analysis. J.Z.: investigation. M.Y., W.Y., and R.T.: resources. R.T.: data curation. R.T., J.W., and J.Z.: writing of the original draft. Y.Z. and J.W.: writing of the review and editing. J.W.: supervision and project administration. J.W. and H.D: funding acquisition.
REFERENCES
- 1.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed N, Brawley VS, Hegde M, et al. Human epidermal growth factor receptor 2 (HER2)—specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33:1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang R, Xu J, Juliette L, et al. Three-dimensional co-culture models to study prostate cancer growth, progression, and metastasis to bone. Semin Cancer Biol. 2005;15:353–364. [DOI] [PubMed] [Google Scholar]
- 6.Kopetz S, Lemos R, Powis G. The promise of patient-derived xenografts: the best laid plans of mice and men. Clin Cancer Res. 2012;18:5160–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei X, Lai Y, Li J, et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology. 2017;6:e1284722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang Z, Jiang X, Chen S, et al. Anti-GPC3-CAR T Cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front Immunol. 2016;7:690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Konig AM, Reeh M, Dancau AM, et al. Concordance of HER2 status in primary tumour and lymph node metastases in patients with esophageal carcinoma. Anticancer Res. 2013;33:4975–4982. [PubMed] [Google Scholar]
- 10.Bartley AN, Washington MK, Ventura CB, et al. HER2 testing and clinical decision making in gastroesophageal adenocarcinoma: guideline from the College of American Pathologists, American Society for Clinical Pathology, and American Society of Clinical Oncology. Am J Clin Pathol. 2016;146:647–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaufman DS, Hanson ET, Lewis RL, et al. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc Natl Acad Sci U S A. 2001;98:10716–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai Y, Kan S, Zhou S, et al. Enhancement of the in vivo persistence and antitumor efficacy of CD19 chimeric antigen receptor T cells through the delivery of modified TERT mRNA. Cell Discov. 2015;1:15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grupp SA. Advances in T-cell therapy for ALL. Best Pract Res Clin Haematol. 2014;27:222–228. [DOI] [PubMed] [Google Scholar]
- 15.Choi YY, Lee JE, Kim H, et al. Establishment and characterisation of patient-derived xenografts as paraclinical models for gastric cancer. Sci Rep. 2016;6:22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hidalgo M, Amant F, Biankin AV, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teng J, Carla DHC, Kantar RS, et al. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro Oncol. 2017;19:820–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murakami T, Murata T, Kawaguchi K, et al. Cervical cancer patient-derived orthotopic xenograft (PDOX) is sensitive to cisplatinum and resistant to nab-paclitaxel. Anticancer Res. 2017;37:61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vareslija D, Cocchiglia S, Byrne C, et al. Patient-derived xenografts of breast cancer. Methods Mol Biol. 2017;1501:327–336. [DOI] [PubMed] [Google Scholar]
- 20.Yoshiyuki T, Shimizu Y, Onda M, et al. Immunohistochemical demonstration of epidermal growth factor in human gastric cancer xenografts of nude mice. Cancer. 1990;65:953–957. [DOI] [PubMed] [Google Scholar]
- 21.Kubota T, Yamaguchi H, Watanabe M, et al. Growth of human tumor xenografts in nude mice and mice with severe combined immunodeficiency (SCID). Surg Today. 1993;23:375–377. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Yang J, Cai J, et al. A subset of gastric cancers with EGFR amplification and overexpression respond to cetuximab therapy. Sci Rep. 2013;3:2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin K, Teng L, Shen Y, et al. Patient-derived human tumour tissue xenografts in immunodeficient mice: a systematic review. Clin Transl Oncol. 2010;12:473–480. [DOI] [PubMed] [Google Scholar]
- 24.Brown KM, Xue A, Mittal A, et al. Patient-derived xenograft models of colorectal cancer in pre-clinical research: a systematic review. Oncotarget. 2016;7:66212–66225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta PB, Onder TT, Jiang G, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shorthouse AJ, Smyth JF, Steel GG, et al. The human tumour xenograft—a valid model in experimental chemotherapy? Br J Surg. 1980;67:715–722. [DOI] [PubMed] [Google Scholar]
- 27.Willey CD, Gilbert AN, Anderson JC, et al. Patient-derived xenografts as a model system for radiation research. Semin Radiat Oncol. 2015;25:273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.