

Abstract
Rates of degradation by the ubiquitin proteasome system depend not only on rates of ubiquitination but also on the level of proteasome activity, which can be regulated through phosphorylation of proteasome subunits. Many protein kinases have been proposed to influence proteasomal activity. However, for only two is there strong evidence that phosphorylation of a specific 26S subunit enhances the proteasome’s capacity to degrade ubiquitinated proteins and promotes protein breakdown in cells: 1) Protein Kinase A (PKA), which after a rise in cAMP phosphorylates the 19S subunit Rpn6, and 2) Dual Tyrosine Receptor Kinase 2 (DYRK2), which during S through M phases of the cell cycle phosphorylates the 19S ATPase subunit Rpt3. These findings were made possible by the development of methods to rapidly purify 26S proteasomes and to assay their multiple activities. The use of pulse-chase isotopic methods showed that PKA activation in cells enhances the degradation of short-lived cell proteins and misfolded proteins that cause neurodegenerative diseases, while DYRK2 enhances the breakdown of long-lived proteins and promotes progression through the cell cycle. The methods discussed here should be useful in clarifying the roles of other kinases and other post-translational modifications of proteasome subunits.
Keywords: Proteasome phosphorylation; protein degradation; protein kinase, proteasome activation, protein homeostasis; Protein Kinase A, DYRK2, Ubiquitin conjugate degradation
Introduction
Phosphorylation of 26S proteasome subunits is a newly appreciated mechanism for regulating protein degradation by the proteasome (1). Although more than 300 phosphorylation sites in proteasome subunits have been detected (2), for nearly all, the functional consequences of these modifications (if there are any) and the responsible kinases are unknown. While many kinases have been proposed to regulate proteasome activity, for only two are the sites of phosphorylation and their effects on proteasome function and on the degradation of cell proteins well established: Protein Kinase A (PKA) (3) and Dual Receptor Tyrosine Kinase 2 (DYRK2) (4). Pharmacological agents and physiological stimuli that raise cAMP and activate PKA enhance proteasome activity by phosphorylating the 19S subunit Rpn6 at Serine 14 (3). During S through M phases of the cell cycle, DYRK2 activates the proteasome by phosphorylating the 19S ATPase subunit Rpt3 at Threonine 25, which promotes cell proliferation (4). However, there is also evidence suggesting that phosphorylation of proteasome subunits occurs after a rise in cGMP (5, 6), following calcium influx following synaptic depolarization (7), and during apoptosis (8).
Our understanding of proteasome regulation by PKA and by DYRK2 originated through quite different studies. Several groups had suggested effects of cAMP on proteasome activity (9, 10), but the systematic studies of Lokireddy et al (3), using pharmacological agents affecting adenylate cyclase and phosphodiesterase 4 (PDE4), demonstrated that cAMP via PKA enhances multiple proteasome activities. This work and related studies in mice by Myeku et al. (11) demonstrated that these agents promote the selective degradation of mutant proteins that cause neurodegenerative diseases. Thus, the initial motivation for this work was to understand proteasome regulation and also mechanisms of proteotoxic diseases. The discovery of the role of DYRK2 came from an analysis by Guo et al. (4) of proteins phosphorylated during different stages of the cell cycle. Based on published findings that phosphorylation of Rpt3 on T25 increases during S through M phases, they screened a library of all human kinases and found that DYRK2 could catalyze this modification. This finding led to studies showing that phosphorylation by DYRK2 enhanced proteasome function and protein breakdown during these phases of the cell cycle (4).
These investigators used similar published approaches to analyze how subunit phosphorylation influenced proteasomal activities and intracellular protein degradation. First, 26S proteasomes were purified, and the particles’ multiple enzymatic activities were assayed. Then mass spectrometry could be performed on the proteasomes to identify the site of phosphorylation, and phosphosite-specific antibodies were generated to confirm modification of the site when the particles were activated. Finally, site-directed mutagenesis determined whether the identified phosphorylation site was in fact necessary and/or sufficient for activation of the proteasome. Measurement of overall rates of protein breakdown by pulse-chase labeling of cell proteins demonstrated that these 26S modifications have clear effects on protein half-lives.
Initial studies of proteasome activation by PKA and DYRK2 indicated that both stimulated the particles’ chymotrypsin-like activity in crude cell lysates (3, 4). For cAMP and PKA, this stimulation was studied in different cell types using pharmacological agents, (e.g. forskolin and the PDE4 inhibitor rolipram) and subsequently with neurotransmitters and hormones that raise cAMP (e.g. epinephrine and glucagon) (3, 25). Affinity-purification of 26S proteasomes by the UBL method of Besche and Goldberg (12) (See chapter by Kuo et al) and subsequent measurement of its multiple enzymatic activities (see chapter by Kim et al.) revealed an increase in the proteasome’s capacity to hydrolyze polyubiquitinated proteins, ATP, and short peptides (3). The UBL method (12) has proven particularly useful for such studies because it can be used in all cells and tissues without prior genetic manipulations.For example, this approach was used to show proteasome impairment in the nervous system of mouse models of Frontotemporal Dementia (FTD) (11) and Charcot Marie Tooth 1B peripheral neuropathy (13), and that treatment of the FTD mice with a PDE4 inhibitor could reduce the impairment of proteasome function and the accumulation of the mutant tau in the brain (11).
To test if phosphorylation was responsible for the PKA- or DYRK2-dependent increase in proteasomal activities, phosphorylation was prevented by inhibiting PKA in cells with the kinase inhibitor H89 (3), or by knocking down DYRK2 with shRNA (4). These treatments reduced proteasomal peptidase activity, presumably by preventing its modification by PKA or DYRK2. To further test if phosphorylation was responsible for the cAMP-mediated increase in proteasome activity, the purified 26S proteasomes were incubated with the Protein Phosphatase 1(PP1), which reduced the peptidase and ATPase activities toward control levels (3).
To identify the critical site of phosphorylation by PKA, 26S proteasomes were purified and analyzed by mass spectrometry. These studies suggested that Rpn6-Serine14 was phosphorylated when cAMP rose in cells (3). The PKA-mediated phosphorylation of Rpn6 was verified with Phos-tag SDS-PAGE (see below) (14). Of particular value was the subsequent production of an anti-phosphopeptide rabbit polyclonal antibody that recognizes specifically the phosphorylated Rpn6-S14 (Figure 1) (25). This antibody has proven very useful for the rapid and reliable demonstration of phosphorylated Rpn6-S14 in different cells and tissues in various physiological states (25). An antibody recognizing phosphorylated Rpt3-Thr25 was generated by Guo et al. (4) and was used in a screen of all overexpressed human kinases, which identified DYRK2 as capable of phosphorylating Rpt3 during S through M phases of the cell cycle (4).
Figure 1:
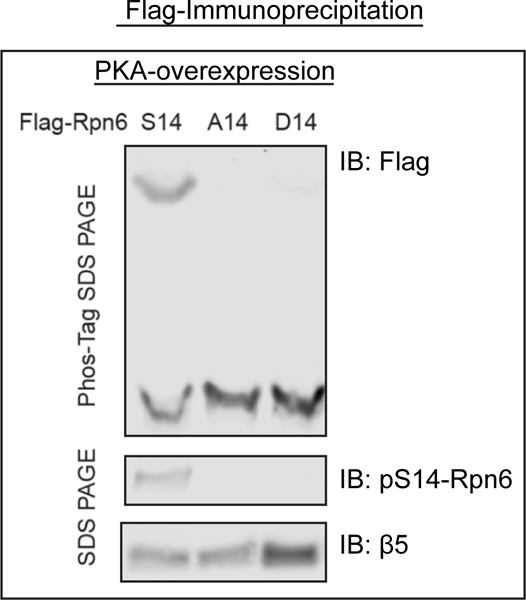
26S proteasomes are not phosphorylated by PKA on Rpn6 when ‘phosphomimetic’ or ‘phosphodead’ mutations at serine 14 on Rpn6 are expressed in HEK293 cells.
Identifying by mass spectrometry the phosphorylation site that regulates proteasome function has proven quite difficult for reasons that are not entirely clear. In fact, several proteasomal sites reported to be phosphorylated by kinases could not be confirmed in later studies (3, 8, 10, 15, 16) even though the changes in proteasome activity were reproduced. Detecting the critical phosphorylation site on the proteasome may be complicated because a kinase may modify hundreds of cellular targets, and these affinity purification methods could also isolate a number of contaminating phosphorylated proteins. Also, in addition to the more than 30 unique subunits that comprise the proteasome, several deubiquitinases, ubiquitin ligases, shuttling factors, and substrates associate in sub-stochiometric amounts with and co-purify with the proteasome (17). Consequently, proteasome preparations purified from HEK293 cells after kinase overexpression contain many p-Ser bands detected by western blot (Figure 2). Furthermore, phosphatases and kinases, including PKA, co-purify with the 26S proteasome (3) and may remove or add phosphate groups during particle isolation and analysis. Therefore, it is important in such studies to verify the site of phosphorylation by multiple approaches.
Figure 2:

26S proteasomes purified from HEK293 cells after kinase overexpression exhibit many phospho-serine positive bands by western analysis. The ovexpression of diffferent kinases results in distinct phospho-serine banding patterns on the 26S proteasomes.
To definitively demonstrate the biochemical importance of Rpn6-S14 phosphorylation, site-directed mutagenesis was used to generate a ‘phosphodead’ (serine to alanine) and ‘phosphomimetic’ (serine to aspartic acid) mutation at serine 14 of human Rpn6 (3). The DNA constructs encoding the Flag-tagged Rpn6 with these ‘phosphodead’, ‘phosphomimetic’, or no mutation were transiently expressed in HEK293 cells and proteasomes purified by the Flag-tag (see chapter by Kuo et al.). The phosphomimetic mutation stimulated the capacity of the 26S proteasomes to hydrolyze ATP and small peptides, and the ‘phosphodead’ mutation repressed these activities (3). Moreover, overexpression of the ‘phosphomimetic’ mutation in cells, like treatments that raise cAMP, stimulated the degradation of aggregation-prone, mutant proteins, and overexpressing the phosphodead mutation decreased it (3). Site directed mutagenesis was also used to test the importance of the modification of T25 of Rpt3 by DYRK2 except that CRISPR-Cas9 gene editing was used to generate the site-specific ‘phosphodead’ and ‘phosphomimetic’ mutants. Most importantly, the phospho-dead modification was found to reduce proteasome activity and to slow cell proliferation (4).
Another major challenge in such studies can be in determining whether a modification of a proteasome subunit that changes in its activity actually leads to detectable effects on intracellular protein breakdown. For PKA and DYRK2, a pulse-chase isotopic labelling of cell proteins was used to measure the overall rates of breakdown of long-lived and short-lived fractions of cell proteins (see chapter by Zhe et al) (18, 19). Both these 26S modifications had global effects in accelerating degradation of major classes of cell proteins, but surprisingly proteasome phosphorylation by DYRK2 or PKA, though seeming to activate proteasomes similarly, stimulated the degradation of a different class of proteins by the UPS. cAMP and PKA increased the degradation only of short-lived proteins, which are misfolded and regulatory proteins (3) while DYRK2 enhanced the breakdown of long-lived proteins (4) and thus affected cell proliferation.
An important goal of future studies will be to determine the specific proteins that are degraded faster in each case, and whether the mechanism responsible for this accelerated proteolysis is solely through proteasome activation or also involves some unknown concomitant effect of the kinase on ubiquitination of certain proteins (1, 20). These kinases were shown to promote the degradation of individual cell proteins using cycloheximide to block translation and western blot to follow disappearance of the protein. By this approach, cAMP was shown to stimulate the intracellular degradation of several mutant, aggregation-prone proteins that cause neurodegenerative diseases (3, 11) and DYRK2 to promote the degradation of certain cell cycle regulatory proteins (4). The use of cycloheximide in such studies serves another important function -- to ensure that the observed changes in the level of the plasmid-encoded protein were due to degradation and not to a different kinase-dependent mechanism; for example, PKA (and other kinases) can alter plasmid content and protein levels through their ability to stimulate gene transcription and translation.
This chapter will describe two methods to confirm phosphorylation by PKA on Serine 14 of Rpn6, but similar approaches can be applied to studies of any kinase and proteasome subunit. These approaches for establishing how phosphorylation can affect proteasome activities and protein degradation should also be useful in studying the many other reported post-translational modifications of the proteasome (e.g. O-GlcNAC (21), ADP-ribosylation (22), ubiquitination (23, 24)), whose biological significance remains unclear.
Materials
Affinity Purify Buffer (APB): 25 mM Hepes-KOH (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM ATP, 1 mM DTT
Sodium Fluoride (NaF) (Sigma): 1M stock solution in H20
β-glycerophosphate (Sigma): 1M stock solution in H20
Lambda phosphatase (NEB)
Proteasome Activity Buffer: 50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 1 mM ATP, 1 mM DTT, 0.05 mg/mL BSA
MnCl2: 1 M in H20
Mouse anti phosphorylated-serine (Santa Cruz, sc-81514)
4X SDS PAGE sample buffer (ThermoFisher Scientific) with 10% Beta-mercaptoethanol.
Precasted Phos Tag Zn2+ polyacrylamide gels (WAKO)
Precasted Bis Tris polyacrylamide gels (Thermofisher Scientific)
Transfer Buffer: 25 mM Tris, 192 mM glycine pH ~8.0, 10% methanol (v/v)
250 mM EDTA
PVDF (Millipore Immobilon FL)
Odyssey Blocking Buffer (LiCor)
Rabbit anti Rpn6 antibody (Cell Signaling, #14303)
1X TBS: 50 mM Tris, 150 mM NaCl
1X TBS/Tween: 50 mM Tris, 150 mM NaCl, 0.1% Tween
Methods
The procedures described below were useful in demonstrating proteasome activation by cAMP and PKA via subunit phosphorylation. For other kinases, some adaptations will probably be necessary.
Measuring PKA-mediated stimulation of 26S proteasome activities
To raise cAMP levels, treat cells for 6 hours with either 50 uM rolipram to raise cAMP by blocking its breakdown by phosphodiesterase 4 (PDE4) or 50 uM forskolin to raise cAMP by stimulating its synthesis by adenylyl cyclases (NOTE 1)(NOTE 2)(3).
To isolate 26S proteasomes from treated and control cells, lyse cells by sonication in APB including phosphatase inhibitors: 10 mM NaF and 25 mM β-glycerophosphate (see NOTE 3). Purify 26S proteasomes by the UBL method as described in the chapter by Kuo et al.
To characterize the enzymatic properties of the purified proteasomes, measure the hydrolysis of flurogenic peptides, ATP, and polyubiquitinated proteins as described in the chapter by Kim et al.
To determine whether the enhanced proteasome ATPase and peptidase activities are due to phosphorylation, incubate the 26S proteasomes for 90 minutes with lambda phosphatase in the proteasome assay buffer supplemented with 1 mM MnCl2 (NOTE 4). The phosphatase will hydrolyze phosphorylated serine and threonine residues. Comparison of these activities before and after dephosphorylation will indicate whether the stimulation is caused by kinase action. It is also advisable to verify the extent of dephosphorylation by performing western blot analysis with a phospho-serine specific antibody.
Phos-tag SDS PAGE to confirm phosphorylation of a specific subunit
Treat cells with agents to raise cAMP and purify proteasomes as described above.
Add 4X SDS PAGE sample buffer to the purified proteasomes (NOTE 5) to a final concentration of 1X and incubate at 95° C for 5 minutes.
Load 1–2 μg 26S proteasome preparation into pre-casted Zn2+ phos tag polyacrylamide gels (7%). Run SDS PAGE in MOPS running buffer at 155 V until the dye front runs off the bottom of the gel (approximately 2 hours). The same samples should also be run in a pre-casted bis tris polyacrylamide gel, to compare the amount of phosphorylated Rpn6 to the total amount of Rpn6.
Incubate the phos-tag gel with gentle shaking for 30 minutes in transfer buffer plus 1 mM EDTA to chelate the zinc ions from the gel. Replace the buffer every 5 minutes.
Incubate the phos-tag gel with gentle shaking for 5 minutes in transfer buffer without EDTA.
Transfer proteins in transfer buffer without EDTA to PVDF at 75 V for 1 hour, or 15 V overnight, at 4o C.
Incubate PVDF membranes in 5 % BSA/TBST or Odyssey Blocking Solution for 1 hour at room temperature, followed by an overnight incubation with a Rpn6 antibody diluted in 3% BSA/TBST (rabbit anti Rpn6, cell signaling technologies).
Wash PVDF membranes in TBST three times for 5 minutes each.
Incubate for 45 minutes at room temperature in secondary antibody (goat anti rabbit, 800) diluted 1:10,000 in 5% BSA/TBST or Odyssey blocking solution (NOTE 5).
Wash PVDF membranes in TBST three times for 5 minutes each.
Wash in TBS once for 5 minutes.
Image membranes on Odyssey (NOTE 6). To quantify the percent Rpn6 phosphorylated, normalize the amount of the up-shifted Rpn6 on the phos tag gel to the total amount of Rpn6 detected on the bis tris gel.
Notes
Note 1: Many pharmacological agents are known to raise cAMP and activate PKA and were useful as tools to investigate how raising cAMP stimulates proteasomal activities and intracellular degradation of some proteins. Other signaling systems may not offer this advantage.
Note 2: 6 hour treatments were used by Lokireddy et al. (3), but enhanced proteasomal activities can also be seen with shorter treatment times.
Note 3: NaF and β-glycerophosphate, which inhibit many Ser/Thr phosphatases, can be added to the reaction buffers used for measuring proteasome activities (see chapter by Kim et al.). The inclusion of these inhibitors during the assay does not reduce proteasome peptidase activities and can even result in a greater measured stimulation of peptidase activity. Calyculin A, an inhibitor of the PP1 and PP2A phosphatases, can also be added to lysis and reaction buffers (4).
Note 4: The PP1 phosphatase, another manganese-dependent protein phosphatase, can also be used (3).
Note 5: Phos-tag SDS PAGE analysis can also be performed on the cell lysates. In our experience, the phosphorylation-dependent shift is less detectable in lysates than with purified 26S proteasomes.
Note 6: We use an Odyssey CLX imager to visualize western blots, but chemiluminescent and film methods can also be used.
References
- 1.VerPlank JJS, Goldberg AL. Regulating protein breakdown through proteasome phosphorylation. The Biochemical journal. 2017;474(19):3355–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo X, Huang X, Chen MJ. Reversible phosphorylation of the 26S proteasome. Protein & cell. 2017;8(4):255–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lokireddy S, Kukushkin NV, Goldberg AL. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(52):E7176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo X, Wang X, Wang Z, Banerjee S, Yang J, Huang L, et al. Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nature cell biology. 2016;18(2):202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ranek MJ, Terpstra EJ, Li J, Kass DA, Wang X. Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation. 2013;128(4):365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranek MJ, Kost CK Jr., Hu C, Martin DS, Wang X. Muscarinic 2 receptors modulate cardiac proteasome function in a protein kinase G-dependent manner. Journal of molecular and cellular cardiology. 2014;69:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Djakovic SN, Schwarz LA, Barylko B, DeMartino GN, Patrick GN. Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. Journal of Biological Chemistry. 2009;284(39):26655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Um JW, Im E, Park J, Oh Y, Min B, Lee HJ, et al. ASK1 negatively regulates the 26 S proteasome. The Journal of biological chemistry. 2010;285(47):36434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asai M, Tsukamoto O, Minamino T, Asanuma H, Fujita M, Asano Y, et al. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. Journal of molecular and cellular cardiology. 2009;46(4):452–62. [DOI] [PubMed] [Google Scholar]
- 10.Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. The Journal of biological chemistry. 2007;282(31):22460–71. [DOI] [PubMed] [Google Scholar]
- 11.Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL, et al. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nature medicine. 2016;22(1):46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Besche HC, Goldberg AL. Affinity purification of mammalian 26S proteasomes using an ubiquitin-like domain. Methods in molecular biology (Clifton, NJ). 2012;832:423–32. [DOI] [PubMed] [Google Scholar]
- 13.VerPlank JJS, Lokireddy S, Feltri ML, Goldberg AL, Wrabetz L. Impairment of protein degradation and proteasome function in hereditary neuropathies. Glia. 2018;66(2):379–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinoshita E, Kinoshita-Kikuta E, Koike T. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nature protocols. 2009;4(10):1513–21. [DOI] [PubMed] [Google Scholar]
- 15.Lee SH, Park Y, Yoon SK, Yoon JB. Osmotic stress inhibits proteasome by p38 MAPK-dependent phosphorylation. The Journal of biological chemistry. 2010;285(53):41280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leestemaker Y, de Jong A, Witting KF, Penning R, Schuurman K, Rodenko B, et al. Proteasome Activation by Small Molecules. Cell chemical biology. 2017. [DOI] [PubMed] [Google Scholar]
- 17.Collins GA, Goldberg AL. The Logic of the 26S Proteasome. Cell. 2017;169(5):792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(52):15790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell metabolism. 2007;6(6):472–83. [DOI] [PubMed] [Google Scholar]
- 20.Filipcik P, Curry JR, Mace PD. When Worlds Collide-Mechanisms at the Interface between Phosphorylation and Ubiquitination. Journal of molecular biology. 2017;429(8):1097–113. [DOI] [PubMed] [Google Scholar]
- 21.Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, Kudlow JE. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell. 2003;115(6):715–25. [DOI] [PubMed] [Google Scholar]
- 22.Cho-Park PF, Steller H. Proteasome regulation by ADP-ribosylation. Cell. 2013;153(3):614–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Besche HC, Sha Z, Kukushkin NV, Peth A, Hock EM, Kim W, et al. Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. The EMBO journal. 2014;33(10):1159–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marshall RS, McLoughlin F, Vierstra RD. Autophagic Turnover of Inactive 26S Proteasomes in Yeast Is Directed by the Ubiquitin Receptor Cue5 and the Hsp42 Chaperone. Cell reports. 2016;16(6):1717–32. [DOI] [PubMed] [Google Scholar]
- 25.Lokireddy S, VerPlank JJS, Zhao J, Davogusotto G, Parker B, James D, Richter E, Taegetmeyer H, Goldberg AL. Hormones, Exercise, and Fasting Activate 26S Proteasome Function via cAMP-PKA pathway. Manuscript in revision . [Google Scholar]