

Summary:
Levels of the ribosome, the conserved molecular machine that mediates translation, are tightly linked to cellular growth rate. In humans, ribosomopathies are diseases associated with cell-type-specific pathologies and reduced ribosomal protein (RP) levels. Because gene expression defects resulting from ribosome deficiency have not yet been experimentally defined, we systematically probed mRNA, translation, and protein signatures that were either unlinked or linked to cellular growth rate in RP-deficient yeast cells. Ribosome deficiency was associated with altered translation of gene sub-classes, and profound general secondary effects of RP loss on the spectrum of cellular mRNAs were seen. Among these effects, growth-defective 60S mutants increased synthesis of proteins involved in proteasome-mediated degradation, whereas 40S mutants accumulated mature 60S subunits and increased translation of ribosome biogenesis genes. These distinct signatures of protein synthesis suggest intriguing and currently mysterious differences in the cellular consequences of deficiency for small and large ribosomal subunits.
Graphical Abstract
eTOC summary:
In this issue of Molecular Cell, Cheng et al. report specific and dose-dependent gene expression changes resulting from decreased ribosome levels, including distinct gene expression signatures resulting from loss of genes encoding members of either the large or small ribosomal subunit.
Introduction:
The universal importance of the cytosolic ribosome—a large protein complex containing ~80 proteins and four rRNAs in eukaryotes—is clear, based on its essential role in translating coding regions of mRNAs into protein. It has also been reported that deficiency in specific ribosomal proteins (RPs) can result in mRNA-specific translation defects. Determining the cause(s) of specific translational effects resulting from RP deficiency may be informative in guiding our understanding of an array of diseases called “ribosomopathies” in humans, which have been linked to mutation in any of a large set of RP genes (reviewed in (De Keersmaecker et al., 2015; McCann and Baserga, 2013; Mills and Green, 2017)).
The observation that ribosomopathies typically result in defects in only a subset of cell types in each case has prompted a surge in research on the potential of the ribosome itself to influence translation in specific ways, beyond homogenously converting mRNA sequences into protein. Multiple molecular models have been proposed to explain the specific phenotypes observed (reviewed in (Mills and Green, 2017)). First, it has been proposed that changes in cellular ribosome concentration may be a major driver of shifts in translatability of an mRNA pool (Lodish, 1974; Mills and Green, 2017). The central idea is that a change in ribosome relative to mRNA levels may cause changes in the translatability of different classes of messages based primarily on competition for ribosomes among the cellular mRNA complement. This model has been proposed to explain at least some cases of the ribosomopathy Diamond-Blackfan anemia (DBA), as several DBA-associated lesions have been seen to lower ribosome production during hematopoiesis, resulting in translation shifts in some mRNA classes (Khajuria et al., 2018).
A distinct (but not mutually exclusive) model, argues that specialized ribosomes, containing a different complement of RPs, or modifications to rRNAs or RPs, may be responsible (reviewed in (Dinman, 2016; Xue and Barna, 2012)). This model is based on the idea that a ribosome with an altered structure due to specialization in a certain condition or cell-type will interact with different affinities to a subset of mRNAs than a non-specialized or differently specialized ribosome, resulting in a shift in the population of mRNAs that are preferentially translated. A third model, which could co-exist with either of the other two but which has not been experimentally explored is that a change in ribosome quantity or functionality results in secondary compensatory effects on gene expression—for example, activation of a transcriptional response that changes the spectrum of mRNAs available for translation in the cell.
Identifying general and potentially specific effects of RP deficiency requires an approach to define the former. While a very small subset of RP genes show no general translation defect when absent, in most cases even partial RP deficiency is associated with slowed cellular growth and decreased bulk protein synthesis, which is likely to be due to defects in either ribosome assembly or stability of fully assembled ribosomes (Steffen et al., 2012). The degree of these defects varies greatly among RP gene mutants, but general defects due to RP deficiency do not preclude gene expression defects that might be directly or indirectly linked to specific to loss of a particular RP. Experimentally determining the potential relative contributions of RP-specific and -general phenotypes is challenging in complex eukaryotes. Budding yeast, however, offer a simple system in which to address this fundamental question because of the ease of modulating RP expression through mutation of paralogous RP-encoding genes in this organism. Towards this end, we measured mRNA, translation, and protein for a panel of RP mutants that showed a range of growth rate defects in rich media. In this system, cellular growth rate provides a robust proxy for total translation levels. This overall approach enabled growth rate matched comparisons between gene expression in mutants lacking different RPs, which to our knowledge has not been done before.
The resultant dataset revealed general signatures of protein synthesis that scale with total translation level, as predicted by the ribosome concentration hypothesis. Evidence for pervasive and strong secondary effects of general ribosome depletion on mRNA levels were also observed, which thus largely mirror translation changes measured by ribosome profiling. These results suggest the value of growth rate matched controls for studies investigating the effects of RP deficiency. To our surprise, these data also revealed dramatic differences in gene expression changes at the mRNA, translation, and protein levels in growth rate matched mutants depending on whether the mutation was in a gene encoding a component of the large 60S subunit (RPL) or the small 40S subunit (RPS). We report that rpl mutants show increased expression of a suite of genes involved in proteasome-mediated protein degradation with decreased growth rate, while rps mutants do not. rps mutants, in contrast, show a stronger upregulation in ribosome biogenesis than growth rate matched rpl mutants and show increased accumulation of mature 60S subunits with decreasing growth rate. These datasets provide a coherent framework to understand the interplay between specific and general, direct and indirect, and small and large subunit, in interpreting gene expression consequences of ribosome deficiency.
Results:
Growth-defective Rpl-deficient cells show specific signatures of protein synthesis
Based on the ribosome concentration hypothesis, one would expect to see a protein synthesis signature resulting from decreasing ribosome abundances. We reasoned that measuring translation globally in rp mutants with a range of different ribosome levels should provide the type of data necessary to define such an effect. To this end, ribosome profiling was performed for a panel of 14 rpl mutants that were constructed anew alongside two wild-type (WT) controls (Fig. 1A, left and right sides). Because, following the whole genome duplication in budding yeast, many RP genes have remained encoded by two paralagous loci, controlled depletion of total levels for a given RP was possible without resulting in cellular death, which is the outcome of full loss of the vast majority of RP genes (Steffen et al., 2012). De novo strain construction was necessary, as others have reported and we also observed that RP mutants have a tendency to become aneuploid (Steffen et al., 2012). Specifically, deletion of several individual RP genes resulted in cells gaining an extra copy of the chromosome carrying the paralog for that RP gene with high efficiency. For this reason, all experiments were performed using freshly thawed cell stocks, no more cell divisions than was necessary for the experiment were used, and every set of sequencing data were checked for evidence of increased dosage from any chromosome(s). In the case of non-sequencing experiments, tetrad dissection of the diploid strains was performed under the experimental conditions used to ensure that aneuploidy did not result. We did not continue to analyze data from any experiment that showed evidence of aneuploidy. All experiments were performed in diploid cells, as this also allowed inclusion of some heterozygous mutations, which was helpful in assembling a panel of mutants with a broad range of growth defects.
Figure 1:
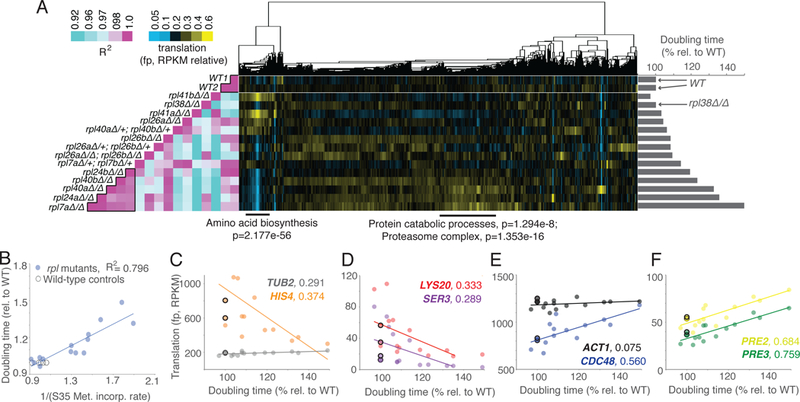
Growth rate-linked translation patterns can be seen among rpl mutant strains. See also Fig. S1, S2, S4, S5, File S1. A) A panel of mutants lacking genes encoding subunits of the large ribosomal subunit (60S) were subjected to growth rate analysis (bar graphs at right) and ribosome profiling (middle). Ribosome profiling data were clustered by similar expression patterns for genes (columns) across all mutants (rows). Columns are normalized to allow comparison. This analysis was highly reproducible (Fig. S5A), as two wild-type controls show a near perfect correlation (Pearson, left). Note that the rpl mutant strains that are most defective for growth showed the most highly correlated patterns of translation (Pearson, left, boxed in pink region at bottom). Below are GO Enrichment categories with Holm-Bonferroni (H-B) p-values for two discrete gene clusters. B) Growth rate analysis for a panel of rpl mutants shows that doubling time is inversely proportional to bulk translation rate, as determined by 35S-Methionine incorporation. C-F) Plots of protein synthesis rates, as assessed by ribosome footprints (RPKM), versus doubling times. WT are represented by open black circles. C) and D) Amino acid biosynthesis genes HIS4, LYS20, and SER3 show rates of protein synthesis that are negatively correlated with the degree of rpl growth rate defect. TUB2, a control gene, does not. TUB2 WT values are extremely similar and thus overlapping in panel C. E) and F) Genes involved in protein catabolism, CDC48, PRE2, and PRE3 show rates of protein synthesis that are positively correlated with the degree of rpl growth rate defect. ACT1, a control gene, does not. R2 -values are next to gene names, based on Pearson correlation.
Cellular doubling time is inversely proportional to growth rate and, in rich growth conditions, has also been reported to be inversely proportional to total translation rate, which is roughly proportional to ribosome number per cell (Marr, 1991; Vind et al., 1993; Warner, 1999). The panel of rpl mutants included doubling times varying from 97% to 149% of WT controls (Fig. 1A right, S1A, S1B). Recent studies have reported that under certain nutrient conditions, translation rate may not necessarily be reflective of ribosome number (Kafri et al., 2016; Metzl-Raz et al., 2017). This possibility was investigated in our conditions by performing 35S-Methionine incorporation experiments using all strains that were subjected to ribosome profiling. Cellular doubling time for this panel of mutants was strongly inversely proportional to the total level of translation, as expected if translation is limiting for cell growth in our experimental conditions (Fig. 1B, S1C). Growth rate is therefore used as a proxy for total translation levels for subsequent analyses.
WT replicates showed highly correlated patterns of translation and ordering translation data according to growth rate revealed that the mutants with the slowest growth rates also showed highly correlated translation patterns to each other (Fig. 1A, left; File S1). Analyses of specific clusters of similarly regulated genes revealed that several rpl mutants with no to mild growth defects exhibited translation increases in a group of genes that was strongly enriched for roles in amino acid biosynthesis (Fig. 1A, middle; 1C, 1D). Additionally, a group of rpl mutants with severe growth defects showed increases in translation of genes involved in protein catabolism and the proteasome (Fig. 1A, middle; 1E, 1F).
Growth-matched 40S and 60S mutants show distinct signatures of translation
If general translation defects in rpl mutants were due to overall translation rate, similar trends should exist in a panel of rps mutants with a similar spectrum of growth rates that were harvested, prepared, and sequenced in parallel to the rpl mutants. To address this, ribosome profiling data for 9 rps mutants was integrated together with the set of rpl mutants. Surprisingly, there were few clear trends in translation shared by rps and rpl mutants with matched growth rates (Fig. S1D, S1E; File S1). Clustering data after sequentially ordering rpl and rps mutants according to growth rate, however, revealed general trends that were shared among growth-defective large and small subunit mutants (Fig. 2A). For example, both growth defective rps and rpl mutants showed a mild upregulation in genes involved in transcription and regulation of metabolism and both showed decreases in translation of genes involved in amino acid biosynthesis compared to RP gene mutants without growth defects (Fig. 2A). The strongest trends observed in these data, however, differed based on whether the mutated RP gene encoded a member of the large or small ribosomal subunit. For example, the tendency for rp mutants with severe growth defects to upregulate proteasome-mediated protein catabolism was unique to rpl mutants (Fig. 2A, 2D, 2E). Genes involved in cytoplasmic translation were increased in translation only among rps mutants (Fig. 2A, 2B, 2C). Translation of genes involved in ribosome biogenesis was increased to some degree in both growth-defective rpl and rps mutants, but this effect was much stronger among growth-defective rps mutants (Fig. 2A).
Figure 2:
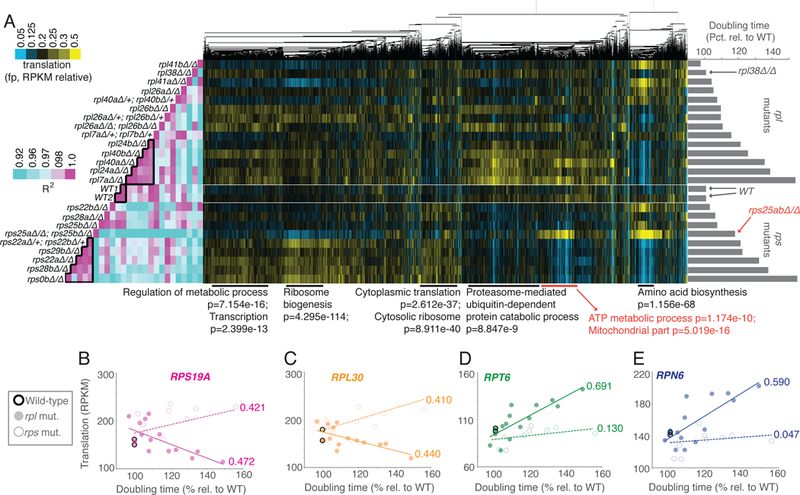
Growth rate-linked translation patterns differ between rps and rpl mutant strains. See also Fig. S1, S2, S4, S5, File S1. A panel of mutants lacking genes encoding components of the large ribosomal subunit (60S; rpl) or small ribosomal subunit (40S, rps) were subjected to growth rate analysis (bar graphs at right) and ribosome profiling (middle). Ribosome profiling data were clustered by similar expression patterns for genes across all mutants. Columns are normalized to allow comparison. Note that the rpl or rps mutant strains that are most defective for growth showed the most highly correlated patterns of translation to within each group but not between the two (Pearson correlation, left, two boxed in pink region in middle and at bottom). Note that the rpl data in this figure is the same as represented in Figure 1, analyzed in parallel with growth-matched rps mutants here. Below are GO enrichment categories and H-B p-values, with rps25-specific cluster information in red. B-E) Data for rpl (solid dot) and rps (open dot) mutants are plotted for representative RP genes B) RPS19A, C) RPL30 and representative proteasome genes D) RPT6 and E) RPN6. R2 -values based on Pearson correlation are included next to lines of best fit.
Isolation of rps from rpl mutant data also revealed a protein synthesis signature for cells lacking Rps25 (rps25a∆rps25b∆) that was distinct from other rps or rpl mutants with a similar growth defect as these cells (Fig. 2A). Translation patterns in rps25a∆rps25b∆ cells showed a generally poor correlation with all other RP deletes, suggesting a cellular effect due to loss of this specific RP that is distinct from lower overall ribosome number. In rps25a∆rps25b∆ cells, upregulation of a group of genes that are heavily enriched for mitochondrial roles, and ATP metabolism, in particular, was observed (Fig. 2A). Analyses of several of these genes reveal levels of expression in the rps25a∆rps25b∆ background that differs substantially from the trend for rps mutants, which instead show a slight downregulation of translation of such genes with increasing growth defect (Fig. 2A, 3A, 3B).
Figure 3:
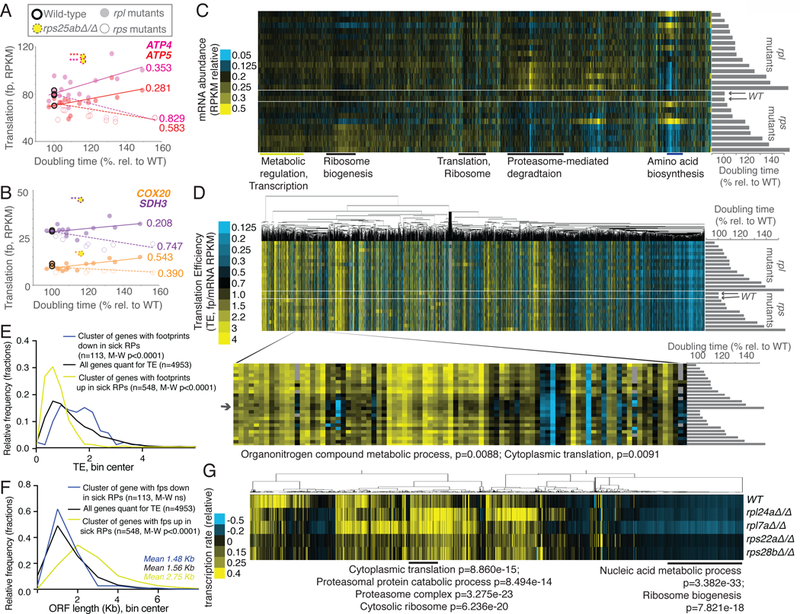
Analyses of genome-wide gene expression data suggest effects of ribosome concentration on translation and secondary effects of cellular growth rate on transcription. See also Fig. S1, S2, S4, S5, Files S1 and S2. A) and B) Data for rpl (solid dot) and rps (open dot) mutants are plotted for genes involved in ATP metabolism, ATP4, ATP5, COX20, and SDH3. Yellow dash-encircled dots represent rps25aΔrps25bΔ cells, which show a divergent trend from growth matched rps mutants. R2-values are included next to lines of best fit and are based on Pearson correlations, *values represent divergence from line of best fit for rps mutants, with *representing >2 standard deviations (SD) from expectation, **>4 SD, ***>7 SD. C) mRNA-seq data, resulting from total-RNA-seq (no polyA-selection) from matched samples collected in parallel for all rps and rpl mutants shown in Fig 2A are shown. Genes (columns) are ordered as in Fig 2A (top: rpl41bΔ/Δ, rpl38Δ/Δ, rpl41aΔ/Δ, rpl26aΔ/Δ, rpl40aΔ/+ rpl40bΔ/+, rpl26bΔ/Δ, rpl26aΔ/+ rpl26bΔ/+, rpl26aΔ/Δ rpl26bΔ/Δ, rpl7aΔ/+ rpl7bΔ/+, rpl24bΔ/Δ, rpl40bΔ/Δ, rpl40aΔ/Δ, rpl24aΔ/Δ, rpl7aΔ/Δ, WT1, WT2, rps22bΔ/Δ, rps28aΔ/Δ, rps25bΔ/Δ, rps25aΔ/Δ rps25bΔ/Δ, rps22aΔ/+ rps22bΔ/+, rps29bΔ/Δ, rps22aΔ/Δ, rps28bΔ/Δ, rps0bΔ/Δ: bottom). Columns are normalized to allow comparison. Note overall similarity to translation data (Fig. 2, S5D). D) TE (translation efficiency, footprint/mRNA) values are shown for all genes quantified for mRNA abundance in C) and translation in Fig. 2A. Order of genes is matched to C). Genes (columns) are clustered according to similar patterns over all mutants (rows). Inset shows a discrete cluster of genes that show modestly lower TE values in rpl mutants with severe growth defects. Enrichment by H-B p-value analysis is shown below. E) The average wild-type TE values for the cluster of genes that show decreased translation in both growth-defective rpl and rps mutants in Fig. 2A (n=113) and the cluster with increased translation in both growth-defective rpl and rps mutants in Fig. 2A (n=548) are plotted and compared to the average wild-type TE values for all genes quantified (n=4953). Two-tailed Mann-Whitney (M-W) tests show that the genes with decreased ribosome footprints in growth-defective rp mutants have WT TE values that are significantly higher than the overall TE distribution and genes with increased ribosome footprints in growth-defective rp mutants have WT TE values that are significantly lower than the overall TE distribution. F) The ORF length distributions for the cluster of genes that show decreased ribosome footprints in both growth-defective rpl and rps mutants in Fig. 2A (n=113) and the cluster with increased ribosome footprints in both growth-defective rpl and rps mutants in Fig. 2A (n=548) are plotted and compared to the average wild-type ORF lengths for all genes quantified (n=4953). Two-tailed M-W tests show that the genes with increased ribosome footprints in growth-defective rp mutants have ORF lengths that are significantly higher than the overall ORF length distribution. G) Metabolic labeling was used to assay new mRNA synthesis (Chan et al., 2018), with quantification of labeled mRNAs by mRNA-seq analysis. Total signal per row was normalized, as was total signal per gene. GO terms and H-B-based p-values enriched in discrete clusters are labeled below.
Evidence of secondary and specific translational effects of RP deficiency
mRNA-seq was performed on parallel samples from all strains analyzed and, surprisingly, the general patterns observed for translation largely reflected changes in mRNA abundance, suggesting homeostatic mechanisms in constitutive rp mutants cells that change the cell’s mRNA complement (Fig. 3C; File S1). When ribosome footprints were normalized to mRNA abundances to calculate translation efficiency (TE; File S1(Ingolia et al., 2009)), however, one cluster of genes showed a modest decrease in TE in rpl mutants with a severe growth defect (Fig. 3C, 3D). This group was enriched for genes involved in cytoplasmic translation, as well as organonitrogen compound metabolic processes. A set of shared mRNA features was not apparent in this grouping. The observation that strong TE changes were rarely observed, either for growth-defective rp mutants or for rps25a∆rps25b∆ mutants, led us to consider the possibility that secondary effects from altered translation efficiency might ultimately result in lowered steady-state mRNA abundances for affected transcripts. Such secondary effects have been observed following microRNA induction, for example. In vertebrate embryos and cell culture, decreased translation efficiencies for a set of transcripts targeted by microRNAs were shown to result in subsequent degradation of affected mRNAs (Bazzini et al., 2012; Djuranovic et al., 2012). The existence of such secondary effects is likely to make steady-state interpretation of TE values in constitutive translation-associated mutants challenging.
In support of specific TE changes among growth-defective rp mutants that may be masked by resultant mRNA abundance changes, among the genes that show a similar trend in ribosome footprint changes with growth rate in both rps and rpl mutants (Fig. 2A, 3C), a strong association with their WT TE emerged. Namely, the set of metabolic and transcription-enriched genes that increase in translation (as judged by ribosome footprints) in growth defective rps and rpl mutants have significantly lower TE values in WT cells (Fig. 3E) than the overall TE spectrum for genes in WT cells; and the set of genes that are heavily enriched for roles in amino acid biosynthesis (and that are seen to decrease in translation in growth-defective rpl and rps mutants) show a significantly higher TE in WT cells than the overall WT TE spectrum (Fig. 3E). This observation suggests, as the ribosome concentration hypothesis proposes, that shifts in the ribosome to mRNA concentration ratio within cells may result in shifts in translation of mRNAs that are associated with their TE. These results are consistent with a study that used human models of DBA, and observed translational downregulation of genes that were normally efficiently translated and shorter in ORF length than unaffected genes (Khajuria et al., 2018). Similarly, the genes for which translation was consistently lower in RP-deficient yeast cells were moderately but not significantly shorter than the overall spectrum of ORF lengths and the genes for which translation was consistently up in RP-deficient cells were dramatically and significantly longer than the overall spectrum of ORF lengths (Fig. 3F).
The mRNA complement in ribosome-deficient cells reflects homeostatic effects
When looking at the overall TE spectrum for WT controls compared to highly growth-defective rp mutants, a significant change in distribution was not seen (Fig. S1F). This fits with the mRNA measurements, and likely reflects the effects of homeostatic mechanisms on gene expression in constitutive mutants with strong cellular effects. To determine if evidence of such mechanisms could be detected, metabolic thio-Uracil-based labeling was performed to measure relative rates of transcription in WT cells compared to two growth-defective rpl mutants (rpl24a∆ and rpl7a∆) and two growth-defective rps mutants (rps22a∆ and rps28b∆; Fig. 3G, File S2;(Chan et al., 2018)). This analysis revealed that the share of transcription devoted to RP and proteasome genes was increased in rpl mutants specifically (Fig. 3G). This was interesting because it suggested that the increase in translation (as judged by ribosome profiling) seen for proteasome-related genes in sick rpl mutants could be explained by an increase in transcription. This also suggested that the mild decrease in TE seen for RP genes in rpl mutants relative to WT cells (Fig. 3D) is partially counterbalanced by an increase in their transcript production. These metabolic labeling data also revealed an increase in the share of ribosome biogenesis transcription in sick rps mutants, specifically (Fig. 3G). This was consistent with the increase in ribosome footprints seen for this class of genes in these mutants. Together, these data suggest that the shifts in which mRNAs are translated in rp mutants is multifactorial. Evidence is observed for effects from total ribosome concentration, modest rpl-specific translational effects, and pervasive secondary effects that shift which transcripts are present in growth-defective rp mutants. Together, these changes dramatically alter the spectrum of translation in a growth-defect-dependent and rpl/rps-dependent manner.
Ribosome composition of diverse rp mutant cells is comparable
While our data were consistent with a model in which large versus small subunit mutation status and total translation levels drive gene expression changes, we wondered if different rp mutations or reduced translation might also result in major shifts in RP composition of ribosomes that could contribute to observed gene expression effects. To test this, matched fractions were isolated from sucrose gradients corresponding to 80S ribosomes and monosomes from two WT controls, 8 rps strains, and 9 rpl strains. Quantitative mass spectrometry was performed on these fractions, revealing that RP proteins were greatly enriched, as expected [making up an average 87.2% of mass spec signal; File S3)]. When the relative RP abundance in 80S/monosomes from rp mutants compared to WT was investigated, few differences among the diverse set of mutants were observed (Fig. S2). Gratifyingly, the expected lack of the protein encoded by the deleted genes was seen, but even in cases in which this removed all genes encoding a given RP (rpl26ab∆ and rps25ab∆), the lack of these proteins did not generally affect the ability of other RPs to associate with the ribosome similarly as they did in WT cells (Fig. S2). These data suggested that gross ribosomal heterogeneity did not generally cause the gene expression changes observed in ribosome-deficient cells.
Rpl versus Rps deficiency result in distinct shifts in cellular protein content
For gene expression changes to result in cellular consequences, one would expect to observe effects on protein levels. This possibility was investigated by quantitative mass spectrometry analysis of matched extract from 18 of the 23 RP mutants that were analyzed for translation and mRNA (File S4, Fig. 4A). Clustering of these data revealed the expected effects based on other gene expression measurements. For example, a cluster of genes that were heavily enriched for roles in cytoplasmic translation were seen to be increased at the protein level specifically in growth-defective rps mutants, consistent with the increased ribosome footprint levels (Fig. 2A) seen for ribosome biogenesis genes. This is also consistent with the increased ribosome footprint levels seen for RP genes in growth-defective rps mutants, relative to WT and growth-defective rpl mutants (Fig. 4A). Similarly, upregulation of protein levels for a cluster of genes enriched for proteasome function was seen specifically in growth-defective rpl mutants (Fig. 4A), and mirrors the ribosome footprint trends that were observed for proteasome-related genes. A decrease in proteins involved in amino acid biosynthesis in both growth-defective rps and rpl mutants, that was expected based on ribosome footprint data, was also observed (Fig. 4A, 2A). The protein level measurements thus confirmed our expectations for the effects based on measurements of prior stages of gene expression (Fig. S5D) and show that significant proteome-composition changes can be seen that are dependent on the level of translation in these ribosome-deficient mutants and whether deficiency results from rpl or rps mutation.
Figure 4:
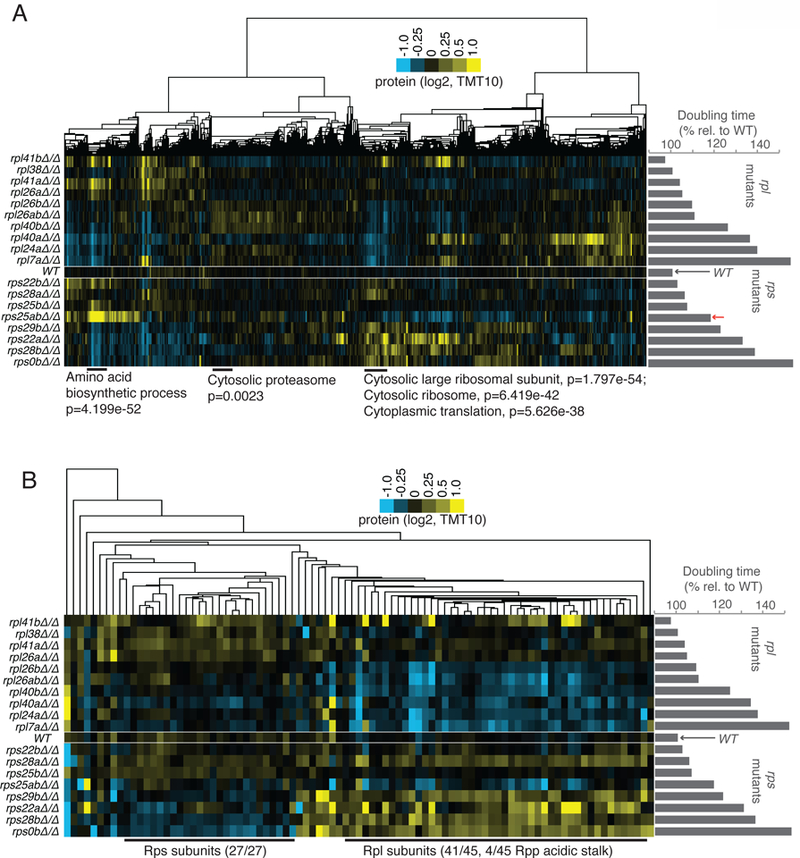
Protein levels in rpl and rps mutants generally match expectations based on translation patterns, but rpl mutants degrade all other RPs, while rps mutants only degrade other Rps subunits. See also Fig. S2, S4, S5, File S4. A) TMT-based mass spectrometry was performed on matched extract from experiment shown in Fig. 2A. Genes (columns) are clustered according to similar protein abundance patterns across all mutants. Red arrow indicates the position of rps25aΔrps25bΔ. GO terms and H-B-based p-values enriched in discrete clusters are labeled blow plot. Columns are normalized to allow comparison. Note similarity to translation data (Fig. 2, S5D). B) Data for RP-encoding genes only was isolated from dataset presented in A). Note that large cluster at the left contains only Rps subunits, the large cluster at right contains only Rpl and Rpp (60S acidic stalk) subunits.
60S mutants demonstrate general RP loss, 40S mutants retain 60S subunits
The proteomic data also offered an opportunity to directly test the degree to which loss of one RP affects the protein levels of all other RPs. It has been shown that reduced expression of even a single RP can result in degradation of others, but whether this is generally true and whether all other RPs are affected has been difficult to determine (Abovich et al., 1985). To investigate this, the mass spectrometry data for RPs only was isolated and clustered (Fig. 4B). A striking pattern emerged from this analysis, consisting of two major discrete clusters. The first contained 27 of the 37 Rps proteins that were measured by mass spectrometry and no other ribosomal proteins. This set was downregulated at the protein level in a manner dependent on degree of growth defect, but not dependent on whether the defect was due to loss of an Rps or Rpl (Fig. 4B). A larger cluster of 45 proteins contained 41 of the 47 Rpl proteins that were measured and 4 of the 6 Rpp proteins, which form the acidic stalk that is associated with the large 60S subunit (Fig. 4B). These proteins were downregulated at the protein level strongly and in a manner dependent on growth rate defect, but only in rpl mutants. All of these results were specific to protein level measurements and not observed in ribosome profiling data, suggesting that stability of the 40S subunit is dependent on individual 40S components and 60S components, while stability of individual 60S components appears to only depend on the presence of other 60S components.
An accumulation of 60S subunit components in growth-defective rps mutants suggests that an imbalance between 40S and 60S subunits is common to cells lacking Rps subunits, as previously suggested by analysis of individual rps cases in yeast and mammalian cells (Abovich et al., 1985; Fumagalli et al., 2009; O’Donohue et al., 2010; Volarevic et al., 2000). Polysome profiles for four rps and rpl strains were analyzed, with rps strains specifically showing a prominent peak at the point in the gradient corresponding to 60S subunits, proportional to their degree of growth defect. In the case of rps0b∆, this peak was so prominent that it exceeded the usually dominant monosome/80S peak (Fig. 5A). This was not observed in rpl mutants, as expected (Fig. 5B). Notably, in growth-defective rpl mutants, lower polysomes than WT were not observed (see rpl7a∆, in particular), despite strong evidence from 35S-Methionine incorporation data suggesting significantly lower translation in these mutants (Fig. 5B). This could be due to either slowed translation elongation in this case or it could simply reflect limitations in making quantitative conclusions from polysome gradient analysis. The build-up of 60S subunit components relative to 40S subunits in rps mutants could also be confirmed by quantification of peptides originating from Rps and Rpl mutants in whole cell extract (Fig. 5C). Similar ratios of the two in rpl mutants and WT cells were seen, but growth-defective rps mutants showed highly elevated Rpl peptide content. This was further reflected in measurements of the ratio of a 60S rRNA (25S) to the 40S rRNA (18S) content (Fig. 5D) in whole cell extract.
Figure 5:
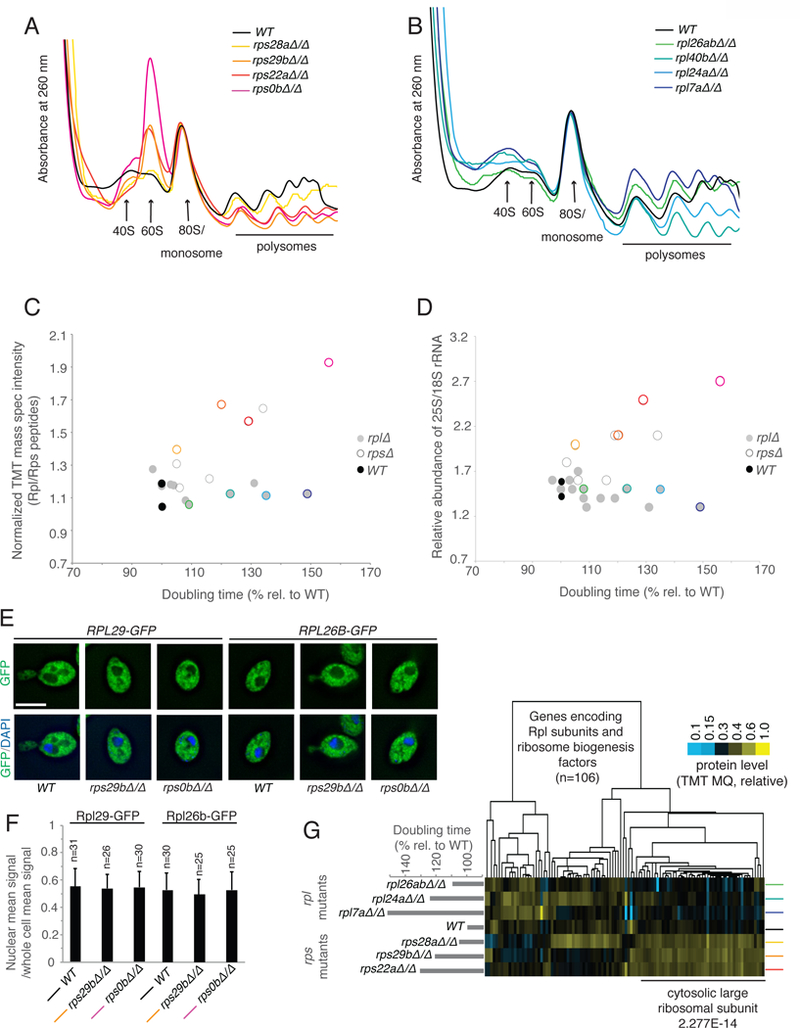
rps mutants accumulate mature 60S subunits. See also Fig. S3, File S5. A) and B) Polysome profiling by sucrose gradient-based centrifugation to assess composition of 40S, 60S, and 80S ribosomes. A wild-type control is compared to the four growth-defective rps mutants in A) and four growth defective rpl mutants in B). The positions of these mutants on the growth spectrum are noted by colored circles in C). C) The relative ratio of peptides from Rpl and Rps subunits yielded by our mass spectrometry approach is plotted on the y-axis compared for to rpl and rps mutants of varying growth rate as noted on the x-axis. The ratios are presented as relative to a WT control. D) The ratio of 25S and 18S rRNA populations by TapeStation analysis is shown, with this ratio plotted on the y-axis and compared to rpl and rps mutants of varying growth rate as noted on the x-axis. E). Either RPL29 or RPL26B was C-terminally tagged with GFP and its localization assessed by microscopy in wild-type and two growth-defective rps mutant strains, compared to DAPI signal. A representative image is shown in each case. F) Quantification of GFP intensity density in the nucleus compared to the whole cell is shown for the experiment represented in Figure 5E. Quantification was performed using ImageJ and z-section images. Error bar represents standard deviation. G) 60S fractions were collected from sucrose gradients for wild-type cells and rps and rpl mutants. Proteins from genes (columns) encoding RPL or ribosome biogenesis factors are shown and clustered according to similar pattern across mutants analyzed (rows). Enrichment as determined by H-B p-value analysis is below.
Accumulated 60S subunits in rps mutants are not RiBi intermediates
Accumulated 60S subunits in rps mutants might either result from a block in maturation of such subunits, or cellular tolerance for super-stoichiometric mature large versus small ribosomal subunits. To investigate which was the case, the localization of GFP-Rpl fusion proteins was determined. In WT cells during exponential growth, any nuclear GFP signal density represents ribosomes in the process of assembly and is approximately half of that in the cell as a whole (Fig. 5E, 5F). If rps mutants were blocked in maturation of 60S subunits, we should observe additional GFP-Rpl signal in the nucleus in these cells. This was not the case, and rather the distribution of Rpl protein between the nucleus and cytosol in these mutants was indistinguishable from WT cells (Fig. 5E, 5F). To investigate this further, a more sensitive approach to distinguish mature from maturing 60S subunits was employed. Quantitative mass spectrometry was performed on the fractions from WT, rpl, and rps polysome gradients that corresponded to the position of 60S subunits. This fraction contained a large peak in growth-defective rps cells and a much smaller peak in WT and rpl cells. Analysis of the relative abundance of proteins present in each case, as expected, showed an overall increase of Rpl proteins, specifically, in rps mutants (65.1% of total signal in average rps, 50.3% in average rpl, 51.2% in WT (Fig. 5G, Fig. S3A, File S5)). To determine the likelihood that these Rpl proteins were in mature 60S subunits, rather than maturing intermediates, data were isolated for Rpl proteins and proteins known to be involved in ribosome biogenesis (RiBi). We reasoned that if accumulated Rpl proteins represented maturing 60S subunits, we should observe a high level of specific RiBi proteins in the 60S fraction of rps mutants. This was not the case. A lower relative amount of RiBi proteins in this fraction was actually observed than in the equivalent fraction in WT or rpl mutants, suggesting that most of the excess 60S subunits in this peak in rps mutants were mature (Fig. 5G). Even proteins associated with ribosomes during late cytosolic 60S processing steps, such as Rei1, Reh1, Alb1 and Nmd3, were not seen to be enriched in the 60S fraction of growth-defective rps mutants (Fig. 5G; File S5;(Greber, 2016)). Consistently, we did not observe a build-up of large subunit-associated rRNA processing intermediates in these mutants (Fig. S3B–H). We concluded that in the absence of 40S subunits, mature 60S subunits remain stable and accumulate to high levels.
Discussion:
Ribosome deficiency results in strong and sometimes specific cellular effects. In humans, many such cases are thought to be responsible for diseases called ribosomopathies, which are characterized by tissue specific defects (reviewed in (McCann and Baserga, 2013)). We report here a systematic study of the link between RP deficiency and gene expression as a first step towards defining general signatures of ribosome depletion, and ultimately gaining a better understanding of the molecular underpinnings of ribosomopathies. Budding yeast offered several advantages for this study, including the ease of generating a panel of RP mutants with varying global levels of cellular translation, the presence of paralogs, which allow controlled depletion, and the lack of a p53-like checkpoint that responds to RP deficiency and introduces additional secondary gene expression effects (reviewed in (Bursac et al., 2014)). Our study uncovers robust gene expression signatures from RP loss that may result from primary translation defects due to a lack of ribosomes or from secondary effects of RP loss. Surprisingly, some of the strongest of these general signatures differ depending on whether ribosome deficiency results from loss of a 60S or 40S subunit component.
The reasons for the large differences in gene expression profiles in growth-matched rpl and rps mutants are unclear (Fig. 2, 3, 4). There have been cases in which 40S and 60S mutations have been seen to result in differing cellular phenotypes- for example, in a systematic screen for RP mutants that increase longevity, only a subset of Rpl-encoding genes, but no Rps-encoding genes, were found to show any effect when deleted (Steffen et al., 2008). In our datasets, we observe a dramatic and rpl-specific upregulation in proteasome-mediated degradation factors in a growth rate dependent manner. At least two models are attractive to explain this result. First, because rpl mutant cells degrade both super-stoichiometric Rpl and Rps subunits, it may be that cells require additional proteasome activity for this function (Abovich et al., 1985; Sung et al., 2016; Warner et al., 1985). Consistently, it has recently been shown that a proteasome-dependent process is indeed responsible for degradation of excess RP subunits (Sung et al., 2016). Alternatively, it is possible that mutant 60S subunits are more likely than mutant 40S subunits to result in production of poor quality nascent proteins, which subsequently increases the cellular demand for proteasome-mediated degradation. The major class of genes upregulated specifically in growth-defective rps mutants are involved in ribosome biogenesis. This, combined with the increased accumulation of mature 60S subunits seen in rps mutants of increasing growth defect was unexpected.
We cannot yet explain why rps mutants would continue to degrade 40S components, keep 60S components, and activate synthesis of more of both, although this is consistent with other studies of individual rps mutants in yeast and mammals (Abovich et al., 1985; Fumagalli et al., 2009; O’Donohue et al., 2010; Volarevic et al., 2000). Interestingly, deletion of some genes required for 60S biogenesis, NOP53 for example, results not only in deficiency in 60S maturation but also dramatic accumulation of 40S subunits (Sydorskyy et al., 2005). In contrast to this and the reciprocal situation in rps mutants, a loss of 40S proteins is seen in rpl mutants (Fig. 4B, 5C, 5D), which may suggest the existence of homeostatic mechanisms that prevent a high mature 40S to 60S ratio in the cytosol. It is notable that the late cytosolic maturation steps for 40S subunits require a non-productive “test drive” translation cycle through association with 60S subunits (Strunk et al., 2012). Thus perhaps the stockpiling of mature 60S subunits in Rps-deficient cells is a cellular adaptation to allow rapid maturation of the 40S subunits that remain. Alternatively, since translation initiation is limited by availability of 40S subunits and the small ribosomal subunit alone is the key hub for association of translation initiation factors, mRNAs, and the large subunit (Strunk et al., 2012), it may simply be that the presence of super-stoichiometric 60S subunits are tolerated by cells because they do not result in significant cellular cost, relative to excess 40S accumulation.
In addition to subunit-specific effects, we observe effects consistent with the concentration hypothesis (Fig. 3E;(Khajuria et al., 2018; Lodish, 1974; Mills and Green, 2017)). When considering growth rate-dependent gene expression effects that are shared by rps and rpl mutants, we find that the group of mRNAs that show upregulation of protein synthesis have a lower TE distribution than most genes in WT cells and those that show downregulation of protein synthesis have a higher TE distribution. These effects are difficult to discern by TE comparison in WT versus mutant cells, likely because the RP mutations are constitutive and translational and transcriptional changes result in secondary effects on the mRNA complement in these mutants relative to WT cells.
The ribosome concentration hypothesis consists of two parts. First, it was posited that ribosome levels could cause transcript-specific translation shifts. Second, and more specifically, it was hypothesized that mRNAs that are well translated in WT conditions should be able to outcompete poorly translated mRNAs when ribosomes are limiting (Lodish, 1974). Both parts of this model fit the observed patterns of translation in reticulocyte lysate for the two globin mRNAs originally studied. The idea that ribosome concentration alone can cause specific translation effects is also consistent with our data and with a recent human DBA study, the two studies thus far providing a direct experimental test of this hypothesis by manipulation of RP levels (Khajuria et al., 2018). In these two cases, the direction of the trend agrees with each other, but differs from the original globin–based prediction. The specific mechanisms underlying the selective translational shifts in RP-deficient cells may be based on mRNA features that are more complex than TE alone- for example, both our study and the DBA study also found an association with ORF length, which is complex to disentangle due to the known global association between ORF length with TE (Ingolia et al., 2009).
Ribosomal proteins were defined based on robust biochemical co-isolation. Subsequent structural studies have defined physical roles in the ribosome for most RPs, supporting a model in which ~80 small proteins and four RNA molecules work together as a machine to build self and other proteins from the information encoded in mRNAs (Warner, 1999). It has been proposed that the use of many small proteins in constructing a ribosome is important because it enables rapid autocatalytic production (Reuveni et al., 2017). RPs are among the most abundant proteins in cells and ribosome construction from many small pieces should be faster than if fewer, longer proteins were used. This structure provides a challenge to cells, however, which must keep many independent RPs in similar stoichiometry within cells. This seems to be largely achieved by cotranscriptonal regulation (Warner, 1999), as well as robust degradation of excess RP subunits, which appears to be highly effective for most RPs that we examined here.
We observe evidence of additional or distinct function for Rps25 in our system, beyond its general role as a core component of the ribosome, which is a valuable proof-of principle example suggesting that the approach used here should be useful in detecting cases in which specific RPs may serve additional or specialized roles. A specialized role of Rps25 has been shown in translation of viral and human mRNAs, with structural data supporting direct binding of Rps25 to IRES regions of mRNA (Hertz et al., 2013; Landry et al., 2009; Muhs et al., 2011; Nishiyama et al., 2007). Our data are consistent with a specialized role for this subunit in yeast, as well, but are also consistent with a non-ribosomal role for this protein that may produce a secondary effect on gene expression. Further work will be needed to clarify the molecular basis for this dramatic change in gene expression profile associated with yeast cells lacking Rps25. More generally, the question of whether to consider all RPs as cogs in a machine or if some of them serve distinct regulatory roles is a complex one. The fact that a handful of RPs do not result in a growth defect when deleted might be itself indicative of a specialized or condition-specific role (Fig. 1A; (Steffen et al., 2012)). This may, for example, be a temperature-specific role in the case of Rpl38 in yeast (Fig. S4A).
Overall, however, at least for the subset of RP mutants studied here and under the conditions used here, robust and specific gene expression changes were primarily the result of decreased overall cellular translation levels and whether the mutated RP gene encodes a member of the large or small ribosomal subunit. Our study argues that orthogonal growth-matched RP mutant controls are important in disentangling specific and general gene expression effects from mutations. The high degree of secondary gene expression changes that result from mutants that strongly affect cell growth—even with no evidence of secondary mutation—is an especially notable concern for RP mutant studies, based on its prevalence in our datasets. In severely growth-defective RP mutants, we find that the mRNA complement in cells differs dramatically from that in WT cells, making analysis of translation defects, specifically, difficult. While some of these secondary effects could be due to changes in the distribution of cell cycle stages (Fig. S4B, C; (Brauer et al., 2008)), the difference in effects that we observed between rpl and rps mutants with similar growth defects points to additional, yet-to-be-determined factors.
STAR Methods:
Contact for Reagent and Resource Sharing:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gloria Brar (gabrar@berkeley.edu)
Experimental Model and Subject Details:
Yeast material and growth conditions:
All experiments were performed using diploid Saccharomyces cerevisiae strains of the SK1 background. Strains were LEU URA TRP LYS HIS unless otherwise noted. Cells were grown in YEPD at 30°C and assayed in mid-log phase (OD6000.6) for all experiments.
| Strain number |
Genotype |
|---|---|
| 2374 | rpl26bΔ/Δ |
| 2435 | rpl7bΔ/RPL7B rpl7aΔ/RPL7A |
| 2700 | rpl40bΔ/Δ |
| 3654 | rpl40bΔ/RPL40B, rpl40aΔ/RPL40A |
| 3851 | rpl26aΔ/Δ |
| 3853 | rpl38Δ/Δ |
| 4472 | rps28aΔ/Δ |
| 4474 | rps28bΔ/Δ |
| 4480 | rps29bΔ/Δ |
| 4484 | WT |
| 4651 | rps0bΔ/Δ |
| 4654 | rpl26aΔ/RPL26A, rpl26bΔ/RPL26B |
| 4658 | rps25bΔ/Δ |
| 4659 | rps22aΔ/Δ |
| 4662 | rps22bΔ/Δ |
| 4665 | rps22bΔ/RPS22B, rps22aΔ/RPS22A |
| 5048 | rpl26bΔ/Δ, rpl26aΔ/Δ |
| 7461 | rpl41bΔ/Δ |
| 7463 | rps25bΔ/RPS25B, rps25aΔ/RPS25A |
| 7465 | rpl24bΔ/Δ |
| 7467 | rpl41aΔ/Δ |
| 7469 | rpl24aΔ/Δ |
| 7889 | rpl40bΔ/Δ |
| 7962 | rpl7aΔ/Δ |
| 13312 | rpl28bΔ/Δ HIS3 HIS4 |
| 13316 | rps22aΔ/Δ HIS3 HIS4 |
| 13318 | rpl24aΔ/Δ HIS3 HIS4 |
| 13323 | rpl7aΔ/Δ HIS3 HIS4 |
| 13390 | HIS3 HIS4 |
| 15346 |
lys2, ura3, leu2, his3 RPL26B/rpl26b::RPL26B-HA-TEV-AVI-GFP-KanMX |
| 15338 |
lys2, ura3, leu2, his3 RPL26B/rpl26b::RPL26B-HA-TEV-AVI-GFP-KanMX, rpl40bΔ/Δ |
| 15340 |
lys2, ura3, leu2, his3 RPL26B/rpl26b::RPL26B-HA-TEV-AVI-GFP-KanMX, rpl7aΔ/Δ |
| 15342 |
lys2, ura3, leu2, his3 RPL26B/rpl26b::RPL26B-HA-TEV-AVI-GFP-KanMX, rps29bΔ/Δ |
| 15345 |
lys2, ura3, leu2, his3 RPL26B/rpl26b::RPL26B-HA-TEV-AVI-GFP-KanMX, rps0bΔ/Δ |
| 15337 | lys2, ura3, leu2, his3 RPL29/rpl29::RPL29-HA-TEV-AVI-GFP-KanMX |
| 15347 |
lys2, ura3, leu2, his3 RPL29/rpl29::RPL29-HA-TEV-AVI-GFP-KanMX, rpl7aΔ/Δ |
| 15349 |
lys2, ura3, leu2, his3 RPL29/rpl29::RPL29-HA-TEV-AVI-GFP-KanMX, rpl40bΔ/Δ |
| 15335 |
lys2, ura3, leu2, his3 RPL29/rpl29::RPL29-HA-TEV-AVI-GFP-KanMX, rps0bΔ/Δ |
| 15333 |
lys2, ura3, leu2, his3 RPL29/rpl29::RPL29-HA-TEV-AVI-GFP-KanMX, rps29bΔ/Δ |
Yeast RP gene names compared to revised systematic nomenclature from (Ban et al., 2014)
| Yeast gene name (SGD) |
Revised systematic name |
|---|---|
| RPL7A | uL30 |
| RPL7B | uL30 |
| RPL24A | eL24 |
| RPL24B | eL24 |
| RPL26A | uL24 |
| RPL26B | uL24 |
| RPL38 | eL38 |
| RPL40A | eL40 |
| RPL40B | eL40 |
| RPL41A | eL41 |
| RPL41B | eL41 |
| RPS0B | uS2 |
| RPS22A | uS8 |
| RPS22B | uS8 |
| RPS25A | eS25 |
| RPS25B | eS25 |
| RPS28A | eS28 |
| RPS28B | eS28 |
| RPS29A | uS14 |
| RPS29B | uS14 |
Method Details:
Sample harvesting:
Vegetative exponential samples were collected by filtration after growth of 300 ml in YEPD to OD6000.6 from a dilution to OD6000.05. 1.5 mL flash frozen buffer was added to ribosome profiling aliquot (also to be used for mass spectrometry) of the standard ribosome profiling composition (20mM Tris pH8, 140mM KCl, 1.5mM MgCl2, 100ug/ml cycloheximide, 1% Trition X-100) supplemented with 2ug/ml Aprotinin, 10ug/ml Leupeptin, 1mM PMSF, 1:100 PIC2, 1:100 PIC3 (both Sigma inhibitor cocktails). Samples were lysed by Retsch mixermilling (6× 3 minute rounds at 15 Hz). Resulting powder was thawed, spun once at 4C for 5 min at 3000 RCF, sup was removed and spun at 20,000 RCF at 4C for 10 minutes. Extract was aliquoted in 200ul portions and flash frozen. Identical extract was used for ribosome profiling and mass spectrometry.
Ribosome Profiling:
Ribosome profiling was performed as described previously in (Brar et al., 2012). In short, samples were treated with RNase I (Ambion) at 15 U per A260 unit of extract for 1 hour at room temperature. Samples were then loaded onto sucrose gradients (10–50%) and centrifuged for 3 hrs. at 35,000 rpm at 4°C in a SW41Ti rotor (Beckman). 80S/monosome peaks were collected using a Gradient Station (BioComp). RNA was extracted using the hot acid phenol method, RNA was size selected from a polyacrylamide gel, dephosphorylated, polyA-tailed, subjected to rRNA subtraction, RT-PCR, circularization, and PCR. The enzymes used were PNK (NEB, lot 0951602), E.coli polyA polymerase (NEB, lot 0101309), Superscript III (Thermo, lot 1752971), Circ Ligase (Epicentre), Phusion polymerase (NEB). Oligos used were oCJ200-oligodT for Reverse transcription, oNTI231 and aatgatacggcgaccaccgagatcggaagagcacacgtctgaactccagtcac-barcode-cgacaggttcagagttc index primers, for PCR, all also PAGE purified from IDT, where the barcodes are six nucleotides in length. Sequencing was done for both reads with standard Illumina oligos. Results were highly reproducible, as shown in Fig. S5A.
mRNA sequencing:
The protocol followed was identical to above, except for the following: total RNA was isolated from frozen pellets, not subjected to mixermilling, by hot acid phenol extraction, total RNA was alkaline fragmented and size selected to 30–50 nt. Fragments were subjected to an identical library prep pipeline as the footprints, but no selective rRNA subtraction round was used. Results were highly reproducible, as shown in Fig. S5B.
Sequencing:
All samples were sequenced on an Illumina HiSeq 2500, 50SRR, with multiplexing, at the UC-Berkeley Vincent Coates QB3 Sequencing facility.
35S metabolic labeling:
Cells were inoculated into YEPD and grown overnight at 30°C with shaking. Cells were diluted to OD600 0.1 in 15 mL of YEPD and grown at 30°C with shaking. The 35S metabolic labeling was performed at log phase with OD600 of 0.5 to 0.7. The time to reach the proper cell density varied between strains due to difference in growth rate. 30 minutes before the labeling, 11 mL of cells were spun down at 3,000 rcf for 1.5 minutes, resuspended in 11 mL of pre-warmed fresh YEPD, and put back to 30°C with shaking. At the start of labeling, 1 mL of cells was transferred into a cuvette and 5 μL of EasyTag™ EXPRESS 35S protein labeling mix (PerkinElmer, Cat#NEG772002MC) were added into the rest of the cells. Cells were incubated with shaking at 30°C for 10 minutes. During the incubation, OD600 measurements were taken from the cells saved in the cuvette. When the incubation was complete, 900 μL of cells were immediately mixed with 100 mL of 100% trichloroacetic acid (TCA), incubated at 95°C for 15 minutes with 500 rpm shaking, and chilled on ice for 15 minutes. Samples were spun down at 20,000 rcf for 2 minutes at 4°C and the pellets were washed with 1 mL of ice-cold 10% TCA. Samples were pelleted again under the same condition and washed with 1 mL ice-cold 100% ethanol. Samples were pelleted again and resuspended in 5 mL of Econo-Safe™ scintillation fluid (RPI, Cat#111175). Scintillation was counted for 2 minutes and the 35S incorporation rates were derived from counts per minute (CPM) normalized to cell number. To ensure accurate and robust comparison, all 35S labeling experiments were performed using two ribosomal protein mutants in parallel with one wild-type control.
Growth rate assay:
Cells were inoculated into YEPD and grown overnight at 30°C with shaking. Cells were diluted in YEPD to OD600 0.2 and grown at 30°C with shaking to log phase with OD600 around 0.6. Cells were diluted again to OD600 0.2 and transferred into 96-well plates. Absorbance measurements were taken with a Tecan microplate reader at 15-minute interval for 8 hours. Each strain was measured in triplicate and a two-fold serial dilution of WT cells was included in each plate to generate the standard curve. Absorbance measurements of each strain were first transformed to relative cell concentrations using the standard curve, then plotted on a time-concentration graph with log-scale y axis. The slope of the linear region in this graph was used to calculate the doubling time.
Temperature sensitivity assay:
Cells were inoculated into YEPD and grown overnight at 30°C with shaking. Five-fold serial dilutions starting at OD600 0.25 were prepared from the overnight cell cultures and 3 μL of each dilution were transferred to two YEPD plates. One of the plates was incubated at 30°C and the other plate was incubated at 37°C. Images were taken after one day and two days.
DNA content analysis using flow cytometry:
Cells were inoculated into YEPD and grown overnight at 30°C with shaking. Cells were diluted to OD600 0.1 in YEPD and grown at at 30°C with shaking. At log phase with OD600 0.55 to 0.6, 1 mL of cells from each strain were pelleted by spinning at 6,000 rcf for 2 minutes. The cell pellets were resuspended in 1 mL of 70% ethanol and incubated overnight at 4°C. Cells were spun down at 12,000 rcf for 3 minutes and washed in 800 μL of 50 mM sodium citrate (pH 7.2). Cells were washed again and sonicated with ten 0.4-second pulses. The sonicated cells were pelleted at 12,000 rcf for 3 minutes, resuspended in 500 μL of 50 mM sodium citrate (pH 7.2) with 0.25 mg/mL RNase A (QIAGEN, Cat#19101) and 0.05% Triton X-100, and incubated overnight at 37°C with shaking at 500 rpm. After the incubation, 5 μL of 20 mg/mL proteinase K (Thermo Scientific, Cat#EO0491) were added and the samples were incubated at 50°C for 2 hours with shaking at 500 rpm. The samples were mixed with 500 μL of 1 nM SYTOX™ Green Nucleic Acid Stain (Invitrogen, Cat#S7020) solution in 50 mM sodium citrate (pH 7.2) and incubated for 5 minutes at room temperature. Single-cell green fluoresce measurements were taken on a Guava easyCyte flow cytometer. 20,000 cells were countered for each strain and the flow cytometry data were analyzed using FlowJo.
Measuring cellular localization of ribosomal proteins using microscopy:
Cells were inoculated into YEPD and grown overnight at 30°C with shaking. Cells were diluted to OD600 0.1 and grown at 30°C with shaking for 4.5 hours. Fixation was done by mixing 900 μL of cells with 100 μL of 37% formaldehyde and incubating at room temperature for 15 minutes. Cells were pelleted at 6,000 rcf for 2 minutes and washed in 100 μL of potassium phosphate/sorbitol buffer (1.2M sorbitol, 0.1M potassium phosphate pH 7.5). Cells were spun down again and resuspended in 100 μL of potassium phosphate/sorbitol buffer. After adhering cells to a polylysine-treated glass slide, permeabilization was performed by submerging the slide into 100% ethanol, taking it out after 1 second, and air-drying for 5 minutes. VECTASHIELD Mounting Medium with DAPI (Vector, Cat#H-1200) was added to the slide and fluorescent microscopy was done on a DeltaVision microscope with a 100X objective. The nuclear region was determined by the DAPI signal and the average green fluoresce intensity in the nucleus and in the whole cell was measured with ImageJ (Schneider et al., 2012).
ThioU labeling
mRNA synthesis was determined using non-invasive metabolic labeling of RNA exactly as described in (Chan et al., 2018). In short, cells were grown in synthetic dextrose media with half the standard concentration of uracil to exponential phase, treated with 1 mM 4-thiouracil (4TU) and then collected over a time series by filtration. total RNA was extracted, biotinylated, and mRNA was enriched. Labeled and unlabeled mRNAs were separated using strepavidin beads and the two pools were measured either using qPCR for transcript-specific measurements or by RNASeq for whole transcriptome stability profiling.
Polysome gradient analysis
Extract from mixermilling flash-frozen cells (as harvested per “Sample Harvesting” method above) was subjected to polysome gradient analysis as described in (Ingolia et al., 2009). In short, 200 ul extract was loaded on 10–50% sucrose gradients with or without prior RNAse I treatment, depending on if sample would be used for ribosome profiling or simple polysome analysis, respectively. Samples were centrifuged in a Beckman XL-70 Ultracentrigue, using a Sw-Ti41 rotor for 3 hours at 35,000 rpm at 4°C. Tube was loaded on a Bio-Comp Gradient Station and analyzed for absorbance at 260 nm. For mass spectrometry of 60S or monosome fractions, sucrose fraction was collected and flash frozen prior to precipitation and mass spectrometry.
Northern blotting for rRNA intermediates
Northern blotting was performed as in (Babiano and de la Cruz, 2010), except that the 5’ A0 and C1/C2 regions were detected by a mixture of three probes to increase signal strength. In short, 6ug of total RNA was loaded on 1.1% glyoxal agarose gel, and ran at 100V for 3 hours. The gel was transferred onto a nylon membrane (Hybond, GE), UV-crosslinked, and stained with methylene blue. The blot was preincubated for 2 hours at 42°C in ULTRAhyb™ Ultrasensitive Hybridization Buffer (Invitrogen, AM8670) with 5X denhardt's solution and 0.1mg/ml salmon sperm DNA. Oligonucleotide probes was end-labelled using [γ- 32 P]-ATP (PerkinElmer, NEG502A250UC) and T4 Polynucleotide Kinase (NEB, M0201S). Hybridization was performed at 37˚C overnight. The blot was washed twice with 2X SSC and twice with 2X SSC, 0.1% SDS, andvisualized using Typhoon phosphor-imaging.
Probes are listed below:
| probe 5’ A0 (1) | GGTCTCTCTGCTGCCGG |
| probe 5’ A0 (2) | GCTTTTACACTCTTGACCAGC |
| probe 5’ A0 (3) | CCATAGCACTCTTTGAGTTTCC |
| probe D/A2 | GACTCTCCATCTCTTGTCTTCTTG |
| probe A2/A3 | TGTTACCTCTGGGCCC |
| probe 5.8S | TTTCGCTGCGTTCTTCATC |
| probe C1/C2 (1) | GAACATTGTTCGCCTAGA |
| probe C1/C2 (2) | TCTTCTTATCGATAACGTTCC |
| probe C1/C2 (3) | AGATTAGCCGCAGTTGGTAA |
Mass spectrometry
Mass spectrometry based protein quantification of total cell extracts by TMT-labeling
Proteins were precipitated by adding −20°C cold acetone to the lysate (acetone to eluate ratio 10:1) and overnight incubation at −20°C. The proteins were pelleted by centrifugation at 20,000 g for 15 min at 4°C. The supernatant was discarded and the pellet was left to dry by evaporation. The protein pellet was reconstituted in 200 μl urea buffer (8 M Urea, 75 mM NaCl, 50 mM Tris/HCl pH 8.0, 1 mM EDTA). As we had more than 20 samples (not all are shown), which had to be distributed to three different TMT-10plex mixes for relative quantification, we also generated a Master-sample, that was an equal volume mix of all samples. This Master-sample was included as a single reference sample in each of the 3 TMT-10plex mixes in order to allow relative normalization that would improve comparison between the three different TMT-10plex, as it should correct for systematic biases in each TMT-10plex mix. Protein concentrations of all samples plus the Master-sample were determined by BCA assay (Pierce). 20 μg of total protein per sample were processed further. Disulfide bonds were reduced with 5 mM dithiothreitol and cysteines were subsequently alkylated with 10 mM iodoacetamide. Samples were diluted 1:4 with 50 mM Tris/HCl (pH 8.0) and sequencing grade modified trypsin (Promega) was added in an enzyme-to-substrate ratio of 1:50. After 16 h of digestion, samples were acidified with 1% formic acid (final concentration). Tryptic peptides were desalted on C18 StageTips according to (Rappsilber et al., 2007) and evaporated to dryness in a vacuum concentrator. Desalted peptides were labeled with the TMT10plex mass tag labeling reagent according to the manufacturer’s instructions (Thermo Scientific) with small modifications. Briefly, 0.5 units of TMT10plex reagent was used per 20 μg of sample. Peptides were dissolved in 50 μl of 50 mM Hepes pH 8.5 solution and the TMT10plex reagent was added in 20.5 μl of MeCN. After 1h incubation the reaction was stopped with 4 μl 5% Hydroxylamine for 15 min at 25°C. Differentially labeled peptides were mixed for each replicate (see mixing scheme below) and subsequently desalted on C18 StageTips (Rappsilber et al., 2007), evaporated to dryness in a vacuum concentrator and reconstituted in 200 μl of 3% acetonitrile and 0.1% formic acid.
| TMT mix | knocked out gene(s) | strain | TMT label |
|---|---|---|---|
|
| |||
| 1 | RPS25A and RPS25B | 7463 | 127N |
| 1 | RPL41B | 7461 | 128N |
| 1 | RPL40B | 2700 | 128C |
| 1 | Rpl41A | 7467 | 129N |
| 1 | RPL24A | 7469 | 129C |
| 1 | RPL26A and RPL26B | 5048 | 130N |
| 1 | Master-mix (mix of all 27 samples) |
N/A | 131N |
| 2 | RPL26A | 3851 | 126C |
| 2 | RPS22A | 4659 | 127N |
| 2 | RPS28B | 4474 | 127C |
| 2 | RPS0B | 4651 | 128N |
| 2 | RPL26B | 2374 | 128C |
| 2 | RPS25B | 4658 | 129N |
| 2 | RPS28A | 4472 | 130N |
| 2 | Master-mix (mix of all 27 samples) |
N/A | 131N |
| 3 | RPL38 | 3853 | 126C |
| 3 | RPS29B | 4480 | 127C |
| 3 | RPS22B | 4662 | 128N |
| 3 | RPL40A | 7889 | 129N |
| 3 | RPL7A | 7962 | 130N |
| 3 | WT-2 | 4484 | 130C |
| 3 | Master-mix (mix of all 27 samples) |
N/A | 131N |
| TMT mix | knocked out gene(s) | strain |
TMT label |
| 1 | RPS25A and RPS25B | 7463 | 127N |
| 1 | RPL41B | 7461 | 128N |
| 1 | RPL40B | 2700 | 128C |
| 1 | Rpl41A | 7467 | 129N |
| 1 | RPL24A | 7469 | 129C |
| 1 | RPL26A and RPL26B | 5048 | 130N |
| 1 | Master-mix (mix of all 27 samples) |
N/A | 131N |
LC-MS/MS analysis on a Q-Exactive HF was performed as previously described (Cheng et al., 2018; Keshishian et al., 2015). Briefly, around 1 μg of total peptides were analyzed on an EASY-nLC 1000 UHPLC system (Thermo Fisher Scientific) coupled via a 20 cm C18 column ID picofrit column (New Objective, Woburn, MA) packed in house with Reprosil-Pur C18 AQ 1.9 μm beads (Dr. Maisch, GmbH, Entringen, Germany) to a benchtop Orbitrap Q Exactive HF mass spectrometer (Thermo Fisher Scientific). Peptides were separated at a flow rate of 200 nL/min with a linear 206 min gradient from 2% to 25% solvent B (100% acetonitrile, 0.1% formic acid), followed by a linear 5 min gradient from 25 to 85% solvent B. Each sample was run for 270 min, including sample loading and column equilibration times. Data was acquired in data dependent mode using Xcalibur 2.8 software. MS1 Spectra were measured with a resolution of 60,000, an AGC target of 3e6 and a mass range from 375 to 2000 m/z. Up to 15 MS2 spectra per duty cycle were triggered at a resolution of 60,000, an AGC target of 2e5, an isolation window of 1.6 m/z and a normalized collision energy of 36. Results were highly reproducible, as shown in Fig. S5C.
Mass spectrometry based protein quantification of the 60S and the monosome (80S) peaks by TMT-labeling
Proteins were precipitated by adding −20°C cold acetone to the lysate (acetone to eluate ratio 10:1) and overnight incubation at −20°C. The proteins were pelleted by centrifugation at 20,000 g for 15 min at 4°C. The supernatant was discarded and the pellet was left to dry by evaporation. The protein pellet was reconstituted in 100 μl urea buffer (8 M Urea, 75 mM NaCl, 50 mM Tris/HCl pH 8.0, 1 mM EDTA) and protein concentrations were determined by BCA assay (Pierce). 10 μg of total protein per sample were processed further. Disulfide bonds were reduced with 5 mM dithiothreitol and cysteines were subsequently alkylated with 10 mM iodoacetamide. Samples were diluted 1:4 with 50 mM Tris/HCl (pH 8.0) and sequencing grade modified trypsin (Promega) was added in an enzyme-to-substrate ratio of 1:50. After 16 h of digestion, samples were acidified with 1% formic acid (final concentration). Tryptic peptides were desalted on C18 StageTips according to (Rappsilber et al., 2007) and evaporated to dryness in a vacuum concentrator. Desalted peptides were labeled with the TMT-11plex mass tag labeling reagent according to the manufacturer’s instructions (Thermo Scientific) with small modifications. Briefly, 0.2 units of TMT-11plex reagent was used per 10 μg of sample. Peptides were dissolved in 30 μl of 50 mM Hepes pH 8.5 solution and the TMT-11plex reagent was added in 12.3 μl of MeCN. After 1 h incubation the reaction was stopped with 2.5 μl 5% Hydroxylamine for 15 min at 25°C. Differentially labeled peptides were mixed for each replicate (see mixing scheme below) and subsequently desalted on C18 StageTips (Rappsilber et al., 2007), evaporated to dryness in a vacuum concentrator and reconstituted in 50 μl of 3% acetonitrile and 0.1% formic acid.
| TMT mix | knocked out gene(s) | strain | TMT label |
|---|---|---|---|
| 80S-1 | Wild-type | 4484 | 126C |
| 80S-1 | RPL41B | 7461 | 127N |
| 80S-1 | RPL26A | 3851 | 128N |
| 80S-1 | RPL26B | 2374 | 128C |
| 80S-1 | RPL26A and RPL26B | 5048 | 129N |
| 80S-1 | RPL40B | 2700 | 130N |
| 80S-1 | RPL40A | 7889 | 130C |
| 80S-1 | RPL24A | 7469 | 131N |
| 80S-1 | RPL7A | 7962 | 131C |
| 80S-2 | wild-type | 4484 | 126C |
| 80S-2 | RPL41A | 7467 | 127N |
| 80S-2 | wild-type | 4484 | 127C |
| 80S-2 | RPS22B | 4662 | 128N |
| 80S-2 | RPS28A | 4472 | 128C |
| 80S-2 | RPS25B | 4658 | 129N |
| 80S-2 | RPS25A and RPS25B | 7463 | 129C |
| 80S-2 | RPS22A het and RPS22B het | 4665 | 130N |
| 80S-2 | RPS29B | 4480 | 130C |
| 80S-2 | RPS22A | 4659 | 131N |
| 80S-2 | RPS28B | 4474 | 131C |
| 60S |
RPL26A and RPL26B (60S full region) |
5048 | 127N |
| 60S | RPL40B (60S full region) | 2700 | 127C |
| 60S | RPL7A (60S full region) | 7962 | 128N |
| 60S | WT (60S full region) | 4484 | 129N |
| 60S | RPS28A (60S full region) | 4472 | 129C |
| 60S | RPS29B (60S full region) | 4480 | 131N |
| 60S | RPS22A (60S full region) | 4659 | 131C |
| TMT mix | knocked out gene(s) | strain | TMT label |
| 80S-1 | Wild-type | 4484 | 126C |
| 80S-1 | RPL41B | 7461 | 127N |
| 80S-1 | Wild-type 2 | 1362 | 127C |
| 80S-1 | RPL26A | 3851 | 128N |
| 80S-1 | RPL26B | 2374 | 128C |
| 80S-1 | RPL26A and RPL26B | 5048 | 129N |
| 80S-1 | RPL24B | 7465 | 129C |
| 80S-1 | RPL40B | 2700 | 130N |
| 80S-1 | RPL40A | 7889 | 130C |
| 80S-1 | RPL24A | 7469 | 131N |
| 80S-1 | RPL7A | 7962 | 131C |
| 80S-2 | wild-type | 4484 | 126C |
| 80S-2 | RPL41A | 7467 | 127N |
| 80S-2 | wild-type | 4484 | 127C |
| 80S-2 | RPS22B | 4662 | 128N |
| 80S-2 | RPL28A | 4472 | 128C |
| 80S-2 | RPS25B | 4658 | 129N |
| 80S-2 | RPS25A and RPS25B | 7463 | 129C |
| 80S-2 | RPS22A het and RPS22B het | 4665 | 130N |
| 80S-2 | RPS29B | 4480 | 130C |
| 80S-2 | RPS22A | 4659 | 131N |
| 80S-2 | RPS28B | 4474 | 131C |
| 60S | RPL26A (60S full region) | 3851 | 126C |
| 60S |
RPL26A and RPL26B (60S full region) |
5048 | 127N |
| 60S | RPL40B (60S full region) | 2700 | 127C |
| 60S | RPL7A (60S full region) | 7962 | 128N |
| 60S | empty | empty | 128C |
| 60S | WT (60S full region) | 4484 | 129N |
| 60S | RPS28A (60S full region) | 4472 | 129C |
| 60S | RPS29A (60S full region) | 4479 | 130N |
| 60S | RPS29B (extra 60S alone) | 4480 | 130C |
| 60S | RPS29B (60S full region) | 4480 | 131N |
| 60S | RPS22A (60S full region) | 4659 | 131C |
The samples were afterwards analyzed by LC-MS/MS on a Q-Exactive HF was performed as previously described (Cheng et al., 2018; Keshishian et al., 2015). Around 1 μg of total peptides were analyzed on an Eksigent nanoLC-415 HPLC system (Sciex) coupled via a 25 cm C18 column (inner diameter of 100 μm, packed in-house with 2.4 μm ReproSil-Pur C18-AQ medium, Dr. Maisch GmbH) to a benchtop Orbitrap Q Exactive HF mass spectrometer (Thermo Fisher Scientific). Peptides were separated at a flow rate of 200 nL/min with a linear 106 min gradient from 2% to 25% solvent B (100% acetonitrile, 0.1% formic acid), followed by a linear 5 min gradient from 25 to 85% solvent B. Each sample was run for 170 min, including sample loading and column equilibration times. Data was acquired in data dependent mode using Xcalibur 2.8 software. MS1 Spectra were measured with a resolution of 60,000, an AGC target of 3e6 and a mass range from 375 to 2000 m/z. Up to 15 MS2 spectra per duty cycle were triggered at a resolution of 60,000, an AGC target of 2e5, an isolation window of 1.6 m/z and a normalized collision energy of 36.
Quantification and Statistical Analyses:
Mass spectrometry based protein quantification of total cell extracts by TMT-labeling
All raw data were analyzed with MaxQuant software version 1.6.0.16 (Cox and Mann, 2008) using a UniProt yeast database (release 2014_09, strain ATCC 204508 / S288c), and MS/MS searches were performed with the following parameters: TMT-10plex labeling on the MS2 level, oxidation of methionine and protein N-terminal acetylation as variable modifications; carbamidomethylation as fixed modification; Trypsin/P as the digestion enzyme; precursor ion mass tolerances of 20 p.p.m. for the first search (used for nonlinear mass re-calibration) and 4.5 p.p.m. for the main search, and a fragment ion mass tolerance of 20 p.p.m. For identification, we applied a maximum FDR of 1% separately on protein and peptide level. We required 1 or more unique/razor peptides for protein identification and at least two MS/MS spectra ratio counts for quantification for each TMT channel in each of the three TMT mixes. This gave us a total of 2132 quantified protein groups.
Next, we normalized the corrected TMT MS2 intensity such that at each condition (sample) these intensity values added up to exactly 1,000,000, therefore each protein group value can be regarded as a normalized microshare (we did this separately for each TMT channel for all proteins that made our filter cutoff in all the TMT channels). Finally, in order to correct for systematic biases in each TMT-10plex mix, we did relative normalization for each protein group in each sample by calculating the relative ratio of the microshare intensity values relative to the microshare intensity values of the “Master-sample” in its corresponding TMT 10plex mix. This step of relative normalization improves comparison between the three different TMT-10plex. These values are provided in File S4.
Mass spectrometry based protein quantification of the 60S and the monosome (80S) peaks by TMT-labeling
All raw data were analyzed with MaxQuant software version 1.6.0.16 (Cox and Mann, 2008) using a UniProt yeast database (release 2014_09, strain ATCC 204508 / S288c), and MS/MS searches were performed with the following parameters: TMT11plex labeling on the MS2 level, the “precursor ion fraction” (PIF) was set to 0.75 (as we were here mainly interested in quantifying the ribosomal proteins and that at best accurarcy), oxidation of methionine and protein N-terminal acetylation as variable modifications; carbamidomethylation as fixed modification; Trypsin/P as the digestion enzyme; precursor ion mass tolerances of 20 p.p.m. for the first search (used for nonlinear mass re-calibration) and 4.5 p.p.m. for the main search, and a fragment ion mass tolerance of 20 p.p.m. For identification, we applied a maximum FDR of 1% separately on protein and peptide level. We required 1 or more unique peptides for protein identification and a ratio count for each of the 11 TMT channels of the corresponding TMT-11plex mix. This gave us a total of 1384 quantified protein groups and among these 93 ribosomal proteins for the 60S peak samples. This gave us a total of 1238 quantified protein groups and among these 96 ribosomal proteins for the 80S-1 peak samples. This gave us a total of 1138 quantified protein groups and among these 96 ribosomal proteins for the 80S-1 peak samples. The combined number (union) of protein groups that passed the cut-off in either the 80S-1 and/or 80S-2 peak samples is 1526.
Finally, each protein group of a TMT labeled sample got its proportional fraction of the MS1 based iBAQ intensities based on its labeling channel specific TMT MS2 intensity relative to the sum of TMT MS2 intensities of all labeled channels for the corresponding protein group. Afterwards we normalized these fractional MS1 iBAQ intensities such that at each condition/time point these intensity values added up to exactly 1,000,000, therefore each protein group value can be regarded as a normalized microshare (we did this separately for each TMT channel for all proteins that made our filter cutoff in all the TMT channels of the corresponding TMT-11plex mix).
Ribosome footprint and mRNA-seq analyses:
Sequencing data were analyzed as in (Ingolia et al., 2009). In short, bowtie2-based alignment (Langmead and Salzberg, 2012) was used and only unique sequences were mapped. Gene expression quantification involved summing unique reads over annotated ORFs and adjustment for RPKM (reads per kilobase million) values.
Data Clustering and Visualization:
We used Cluster 3.0 (de Hoon et al., 2004) for our hierarchical clustering, using uncentered correlation clustering with the centered setting. We visualized the results using Java Treeview (Saldanha, 2004).
Statistics and correlations:
All correlation measurements used throughout this manuscript are Pearson correlations. Enrichment in clusters was determined using H-B analyses to generate p-values within YeastMine (yeastmine.yeastgenome.org). Two-tailed Mann-Whitney tests were used for Figures 3E and 3F.
Data and Software Availability:
Sequencing and mass spectrometry data included in this manuscript are publically available through NCBI GEO (accession number GSE121189) and MassIVE (accession number MSV000083033).
Supplementary Material
Highlights:
Decreased ribosome levels result in dose-dependent changes in gene expression
Ribosomal protein deficiency leads to strong secondary changes to the transcriptome
Mutation of genes for 60S proteins causes upregulation of protein degradation genes
RPs are posttranslationally regulated, with distinct outcomes from Rpl or Rps loss
Acknowledgements:
We thank Nick Ingolia and Nick Guydosh for critical reading of this manuscript and helpful suggestions. We thank Basil Greber, Jamie Cate, and Ray Deshaies for feedback on conceptual aspects of this project. This work has been funded by NIH grants (DP2-GM-119138; P50-GM-31535), investigator awards from the Alfred P. Sloan Foundation (FG-2016–6229) and Pew Charitable Trusts (00029624), and UC-Berkeley start-up funding, including from the Bowes Foundation, to GAB. KW and CFM are supported by a grant from the Swiss National Science Foundation (SNF 159731). AR is supported by NIHGRI CEGS (P50 HG006193), the Klarman Cell Observatory, and HHMI. MJ is supported by a Maximizing Investigators’ Research Award for Early Stage Investigators (R35GM128802). AK is supported as a Dean’s Fellow by Columbia University Graduate School of Arts and Sciences. LYC is funded by the Shurl and Kay Curci Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
The authors declare no competing interests.
References:
- Abovich N, Gritz L, Tung L, and Rosbash M (1985). Effect of RP51 gene dosage alterations on ribosome synthesis in Saccharomyces cerevisiae. Mol. Cell. Biol 5, 3429–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiano R, and de la Cruz J (2010). Ribosomal protein L35 is required for 27SB pre-rRNA processing in Saccharomyces cerevisiae. Nucleic Acids Res 38, 5177–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, Beckmann R, Cate JHD, Dinman JD, Dragon F, Ellis SR, Lafontaine DLJ, Lindahl L, Liljas A, Lipton JM, et al. (2014). A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol 24, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, and Giraldez AJ (2012). Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 336, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, and Weissman JS (2012). High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, and Botstein D (2008). Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol. Biol. Cell 19, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursac S, Brdovcak MC, Donati G, and Volarevic S (2014). Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta 1842, 817–830. [DOI] [PubMed] [Google Scholar]
- Chan LY, Mugler CF, Heinrich S, Pascal Vallotton, and Weis K (2018). Non-invasive measurement of mRNA decay reveals translation initiation as the major determinant of mRNA stability. ELife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Otto GM, Powers EN, Keskin A, Mertins P, Carr SA, Jovanovic M, and Brar GA (2018). Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. Cell 172, 910–923.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- De Keersmaecker K, Sulima SO, and Dinman JD (2015). Ribosomopathies and the paradox of cellular hypo- to hyperproliferation. Blood 125, 1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinman JD (2016). Pathways to Specialized Ribosomes: The Brussels Lecture. J. Mol. Biol 428, 2186–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, and Green R (2012). miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 336, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli S, Di Cara A, Neb-Gulati A, Natt F, Schwemberger S, Hall J, Babcock GF, Bernardi R, Pandolfi PP, and Thomas G (2009). Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat. Cell Biol 11, 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber BJ (2016). Mechanistic insight into eukaryotic 60S ribosomal subunit biogenesis by cryo-electron microscopy. RNA N. Y. N 22, 1643–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz MI, Landry DM, Willis AE, Luo G, and Thompson SR (2013). Ribosomal protein S25 dependency reveals a common mechanism for diverse internal ribosome entry sites and ribosome shunting. Mol. Cell. Biol 33, 1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJL, Imoto S, Nolan J, and Miyano S (2004). Open source clustering software. Bioinforma. Oxf. Engl 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafri M, Metzl-Raz E, Jona G, and Barkai N (2016). The Cost of Protein Production. Cell Rep 14, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshishian H, Burgess MW, Gillette MA, Mertins P, Clauser KR, Mani DR, Kuhn EW, Farrell LA, Gerszten RE, and Carr SA (2015). Multiplexed, Quantitative Workflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidates for Early Myocardial Injury. Mol. Cell. Proteomics MCP 14, 2375–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajuria RK, Munschauer M, Ulirsch JC, Fiorini C, Ludwig LS, McFarland SK, Abdulhay NJ, Specht H, Keshishian H, Mani DR, et al. (2018). Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 173, 90–103.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry DM, Hertz MI, and Thompson SR (2009). RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev 23, 2753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish HF (1974). Model for the regulation of mRNA translation applied to haemoglobin synthesis. Nature 251, 385–388. [DOI] [PubMed] [Google Scholar]
- Marr AG (1991). Growth rate of Escherichia coli. Microbiol. Rev 55, 316–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann KL, and Baserga SJ (2013). Genetics. Mysterious ribosomopathies. Science 341, 849–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzl-Raz E, Kafri M, Yaakov G, Soifer I, Gurvich Y, and Barkai N (2017). Principles of cellular resource allocation revealed by condition-dependent proteome profiling. ELife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EW, and Green R (2017). Ribosomopathies: There’s strength in numbers. Science 358. [DOI] [PubMed] [Google Scholar]
- Muhs M, Yamamoto H, Ismer J, Takaku H, Nashimoto M, Uchiumi T, Nakashima N, Mielke T, Hildebrand PW, Nierhaus KH, et al. (2011). Structural basis for the binding of IRES RNAs to the head of the ribosomal 40S subunit. Nucleic Acids Res 39, 5264–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama T, Yamamoto H, Uchiumi T, and Nakashima N (2007). Eukaryotic ribosomal protein RPS25 interacts with the conserved loop region in a dicistroviral intergenic internal ribosome entry site. Nucleic Acids Res 35, 1514–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donohue M-F, Choesmel V, Faubladier M, Fichant G, and Gleizes P-E (2010). Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J. Cell Biol 190, 853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Reuveni S, Ehrenberg M, and Paulsson J (2017). Ribosomes are optimized for autocatalytic production. Nature 547, 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview--extensible visualization of microarray data. Bioinforma. Oxf. Engl 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen KK, MacKay VL, Kerr EO, Tsuchiya M, Hu D, Fox LA, Dang N, Johnston ED, Oakes JA, Tchao BN, et al. (2008). Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell 133, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen KK, McCormick MA, Pham KM, MacKay VL, Delaney JR, Murakami CJ, Kaeberlein M, and Kennedy BK (2012). Ribosome deficiency protects against ER stress in Saccharomyces cerevisiae. Genetics 191, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunk BS, Novak MN, Young CL, and Karbstein K (2012). A translation-like cycle is a quality control checkpoint for maturing 40S ribosome subunits. Cell 150, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung M-K, Reitsma JM, Sweredoski MJ, Hess S, and Deshaies RJ (2016). Ribosomal proteins produced in excess are degraded by the ubiquitin-proteasome system. Mol. Biol. Cell 27, 2642–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydorskyy Y, Dilworth DJ, Halloran B, Yi EC, Makhnevych T, Wozniak RW, and Aitchison JD (2005). Nop53p is a novel nucleolar 60S ribosomal subunit biogenesis protein. Biochem. J 388, 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vind J, Sørensen MA, Rasmussen MD, and Pedersen S (1993). Synthesis of Proteins in Escherichia coli is Limited by the Concentration of Free Ribosomes. J. Mol. Biol 231, 678–688. [DOI] [PubMed] [Google Scholar]
- Volarevic S, Stewart MJ, Ledermann B, Zilberman F, Terracciano L, Montini E, Grompe M, Kozma SC, and Thomas G (2000). Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 288, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Warner JR (1999). The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci 24, 437–440. [DOI] [PubMed] [Google Scholar]
- Warner JR, Mitra G, Schwindinger WF, Studeny M, and Fried HM (1985). Saccharomyces cerevisiae coordinates accumulation of yeast ribosomal proteins by modulating mRNA splicing, translational initiation, and protein turnover. Mol. Cell. Biol 5, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, and Barna M (2012). Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol 13, 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.