

Abstract
This scientific commentary refers to ‘Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission’, by Shahni et al. (doi:10.1093/brain/awv182).
This scientific commentary refers to ‘Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission’, by Shahni et al. (doi:10.1093/brain/awv182).
Mitochondria exist in dynamic networks, which undergo rapid cycles of fission (division) and fusion (union). The adaptive roles of fission and fusion depend on the state of the cell and include: mitotic fission (mitochondrial division coordinated with mitosis supporting equitable distribution of mitochondria to daughter cells); mitophagic fission (mitochondrial division isolating dysfunctional portions of the organelle for elimination by mitophagy, thereby maintaining mitochondrial quality control); apoptotic fission (a prelude to apoptosis); and oxygen-sensitive fission [mitochondrial division occurring in response to a change in PO2 which modulates the production of reactive oxygen species (ROS)], as reviewed in Archer (2013). Mitochondrial fusion regulates mitochondrial calcium, mitochondrial metabolism and cell cycle progression (Archer, 2013). In this issue of Brain,Shahni et al. (2015) identify signal transducer and activator of transcription 2 (STAT2) as a novel activator of dynamin-related protein 1 (DRP1)-induced fission and report that a clinically asymptomatic STAT2 mutation in three patients predisposed them to develop severe, multiorgan dysfunction associated with impaired mitochondrial fission, subsequent to receiving the measles-mumps-rubella (MMR) vaccination. The patients included siblings aged 12 and 13 months and a previously reported, unrelated subject diagnosed at 18 months of age (Hambleton et al., 2013). All patients developed fever, and amongst the three of them showed other abnormalities including: conjunctivitis, lymphadenopathy, a sepsis-like syndrome, seizures, hepatitis and pneumonitis.
The major mediators of mitochondrial fusion are GTPases in the outer mitochondrial membrane [mitofusin 1 (MFN1) and mitofusin 2 (MFN2)], and the inner mitochondrial membrane protein optic atrophy 1 (OPA1). Fission is regulated by a single GTPase, DRP1. On activation, DRP1 is recruited from the cytosol to the mitochondrial outer membrane where it interacts with binding partner proteins, forming a ring-like multimeric structure that constricts and divides the mitochondrion (Zhu et al., 2004). DRP1 activity is differentially regulated at the post-translational level by phosphorylation at two key serines: serine 616 and serine 637. Phosphorylation at serine 616 activates DRP1; whereas phosphorylation at serine 637 inhibits DRP1.
Disorders of mitochondrial dynamics (Archer, 2013) can result from genetic abnormalities, such as mutations of parkin (encoded by PARK2), which encodes a ubiquitin E3 ligase that regulates mitochondrial quality control, and PINK1 (a serine–threonine kinase that regulates mitochondrial quality control and targeting of parkin). PINK1 and PARK2 mutations cause excessive DRP1 activation, mitochondrial fragmentation and neuronal death in autosomal recessive forms of familial parkinsonism. Disorders of mitochondrial dynamics can also occur by epigenetic mechanisms, such as the silencing of superoxide dismutase 2, which leads to fragmentation of mitochondria and a hyperproliferative diathesis in the vascular cells of patients with pulmonary arterial hypertension (Archer et al., 2010). More commonly, however, disorders of mitochondrial dynamics are acquired through changes in the activity of the kinases and phosphatases that regulate fissogenic and fusogenic enzymes. The many kinases that post-translationally regulate DRP1 (Table 1) respond to common pathogenic mediators, such as intracellular calcium, ROS, or cyclic adenosine monophosphate (cAMP) levels, thereby linking mitochondrial dynamics into the mechanisms of many common diseases, as reviewed in Archer (2013). Dysregulation of the balance between mitochondrial fission and fusion contributes to the pathogenesis of a wide spectrum of acquired diseases such as type 2 diabetes, Parkinson’s disease, Alzheimer’s disease, pulmonary arterial hypertension and lung cancer (Archer, 2013).
Table 1.
Kinases and phosphatases regulating DRP1 activity
| Kinases/phosphatases | Function | Reference |
|---|---|---|
| Cyclin-dependent kinase 1/cyclin B (CDK1/cyclin B) | Serine-threonine kinase | Archer, 2013 |
| Activates DRP1 by phosphorylating DRP1 serine 616 | ||
| Cyclin-dependent kinase 5 (CDK5) | Serine-threonine kinase | Cho et al., 2014 |
| Activates DRP1 by phosphorylating DRP1 serine 616 | ||
| Extracellular-signal regulated kinase 2 (Erk2) | Serine-threonine kinase | Kashatus et al., 2015 |
| Activates DRP1 by phosphorylating DRP1 serine 616 | ||
| Aurora-A kinase (AURKA) | Serine-threonine kinase | Kashatus et al., 2011 |
| Indirect phosphorylation of DRP1 serine 616 via CDK1/cyclin B | ||
| Calcium-calmodulin-dependent kinase (CamK) | Activates DRP1 (mechanism uncertain) | Archer, 2013 |
| Protein kinase A (PKA) | Serine-threonine kinase | Archer, 2013 |
| Deactivates DRP1 by phosphorylating DRP1 serine 637 | ||
| AMP-activated protein kinase (AMPK) | Serine-threonine kinase | Wikstrom et al., 2013 |
| Deactivates DRP1 by phosphorylating DRP1 serine 637 | ||
| Calcineurin | Calcium-dependent serine-threonine protein phosphatase | Archer, 2013 |
| Activates DRP1 by dephosphorylating DRP1 serine 637 | ||
| PGAM5 | Serine-threonine protein phosphatase | Wang et al., 2012 |
| Activates DRP1 by dephosphorylating DRP1 serine 637 |
Due to limitations on the number of references the citations in this table refer to review articles in some cases.
Shahni et al. (2015) provide strong evidence of dysfunctional mitochondrial dynamics in the skeletal muscle and skin fibroblast cells of three previously healthy children who became seriously ill shortly after receiving the MMR vaccination. Using whole exome sequencing they identified a homozygous STAT2 mutation in each subject that obliterated STAT2 expression. The STAT2 deficiency had not caused symptoms in these children until they were exposed to a viral challenge. Their clinical responses were heterogeneous (seizures in one child, sepsis-like and systemic illnesses in the other two); however, all three patients shared an interesting deep phenotype, characterized by elevated plasma and CSF lactate, mitochondrial elongation and reduced phosphorylation of DRP1 at serine 616. Although total DRP1 expression was normal, the authors focused on depressed DRP1 activity as a cause of the mitochondrial hyperfusion, because of the decreased expression of activated DRP1 (phosphorylated at serine 616) and increased expression of inactive DRP1 (phosphorylated at serine 637) in the patients’ fibroblasts. While the authors attributed the observed hyper-fused mitochondria to DRP1 inactivation, the expression of mitochondrial fusion proteins MFN1, MFN2, and OPA1 also appeared to be increased and may have contributed to the observed increased in fusion. Shahni et al. (2015) successfully reproduced the mitochondrial abnormality in SHSY5Y neuroblastoma cells by silencing STAT2. Furthermore, molecular rescue, by overexpression of wild-type STAT2, reversed the patients’ mitochondrial defect. A schematic representation of the authors’ proposed pathway is provided in Fig. 1.
Figure 1.
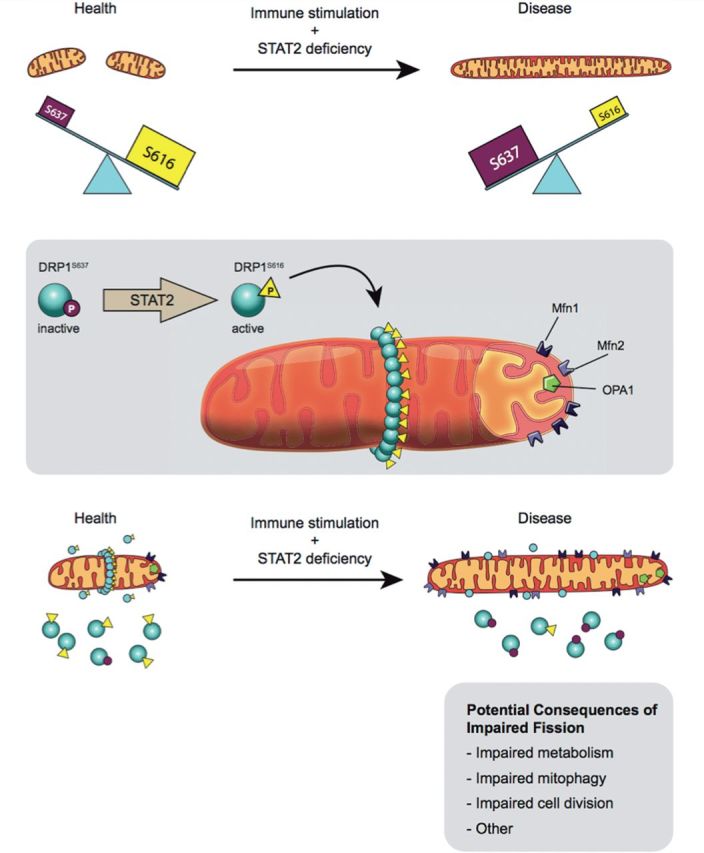
Proposed mechanism for induction of fusion as a consequence of STAT2 mutation. Top: Shahni et al. (2015) postulate that a non-canonical function of STAT2 is phosphorylation of DRP1 at serine 616, which causes a basal level of mitochondrial fission that is deemed to be beneficial. In the three children reported, a STAT2 mutation decreases STAT2 expression and eliminates DRP1 phosphorylation at serine 616, thereby creating elongated mitochondria. This pro-fusion environment caused by STAT2 mutation is compounded by an increase in the inhibitory form of DRP1, which is phosphorylated at serine 637. The details of the link to the immune system require further research, as does the mechanism by which mitochondrial hyperfusion leads to systemic illness or seizures. Middle: Normally, a basal level of fission is maintained by translocation of DRP1 phosphorylated at serine 616 to the outer mitochondrial membrane. Here, DRP1 multimerizes and, with its binding partners, forms the fission apparatus that divides the mitochondrion. The fusogenic GTPases, MFN1, MFN2 and OPA1 oppose fission. Bottom: DRP1 was present in the mitochondria of both patients and controls; however there was a deficiency of activated DRP1 serine 616 and a lack of fission in patient cells. The fusion mediators MFN1, MFN2 and OPA1 were upregulated in STAT2-deficient cells, thus it is likely that fusion was also disordered in these subjects. In addition, the patients showed increased levels of mitochondrial cytochrome c oxidase subunit II (MTCO2) and translocase of outer mitochondrial membrane 20 kDa (TOM20), suggestive of an increase in mitochondrial mass. Figure drawn by Julia Herr, M.Sc. Queen’s University.
Although Shahni et al. suggest that this is the first report of an association between STAT2, mitochondria and the immune system, there is prior evidence indicating the importance of this axis (Meier and Larner, 2014). While the canonical role of STATs is regulation of early response genes in the nucleus, numerous non-canonical, mitochondrial roles for STAT isoforms are emerging. Meier and Larner (2014) have reviewed the role of STATs in regulating diverse mitochondrial functions. They noted that STAT1, STAT2, STAT3, STAT5 and STAT6 are all expressed in mitochondria. STAT1 for example regulates mitochondrial biogenesis. STAT2 protects cells from viruses by an interferon-dependent mechanism, consistent with the adverse effects of STAT2 deficiency observed in Shahni and co-workers’ patients. In response to viral infection STAT2 translocates to the mitochondria, which may attenuate the cell’s anti-viral response (Goswami et al., 2013) and suppress innate immunity. Mitochondrial STAT2 also negatively regulates the transcription of certain mitochondrial RNAs (Meier and Larner, 2014); this would presumably increase mitochondrial encoded RNA transcripts in STAT2 deficiency, although the significance of any such changes for the patients in the current report is unknown. Mitochondrial STAT3 is perhaps best studied and is known to regulate mitochondrial respiration. The absence of STAT3 reduces ATP levels and impairs the function of the electron transport chain (Wegrzyn et al., 2009).
Shahni and co-workers endeavour to explain how STAT2 deficiency might promote the neurological disorders the children developed upon immunogenic challenge. Studies of the response of patients’ fibroblasts to interferon-α (IFNα) indicate that the STAT2 mutation impairs IFNα-induced apoptosis. However, there is no clear link between a failure of basal fission and the occurrence of a normal versus an abnormal immune response to viral exposure. Potential consequences of impaired fission are noted in the bottom panel of Fig. 1.
The report has several strengths, most notably highlighting the ability to apply new genetic methodologies, such as whole exome sequencing, to tease out basic mechanisms of disease in individual patients. In addition, it provides another example of the need for precision medicine. Perhaps individuals destined to have an adverse response to immune stimulation can be identified and the stimulus avoided or the consequences mitigated.
The study has limitations that might be considered in the design and implementation of future studies.
Optimizing mitochondrial imaging
Although the authors quantified mitochondrial fragmentation, the representative images suffer somewhat from limited resolution and substantial overlap of mitochondria, which compromises quantification of the mitochondrial network. Optimization of mitochondrial imaging can be achieved through careful attention to confocal imaging parameters and the choice of mitochondrial probes (Fig. 2). Tetramethylrhodamine (TMRM) is commonly used to image mitochondria and was used in this study. However, TMRM is somewhat suboptimal because it is sensitive to photobleaching and its uptake is decreased by mitochondrial depolarization, which commonly occurs in disease. Superior alternatives to TMRM (for morphology) include vectors that overexpress mitochondrial-targeted green fluorescent protein, as reviewed in Archer (2013) or fluorophores, such as MitoTracker Green®. Transfection of cells with mitochondrial-targeted photoactivated green fluorescent protein (mito-PA-GFP) and red fluorescent protein (mito-Ds-Red) allows quantification of fission and fusion (Archer, 2013). Recently, super-resolution confocal microscopy, as can be achieved using Stimulated Emission Depletion Microscopy (STED) technology, has allowed better definition of DRP1 localization and offers hope of directly imaging the composition of the fission complex. Using these techniques, future studies could better assess whether STAT2 is in the mitochondria and could better define STAT2’s binding partners.
Figure 2.

Representative images of mitochondrial network illustrating optimal network resolution. (A and B) Fibroblasts isolated from the right ventricle of normal rats and rats with monocrotaline-induced pulmonary arterial hypertension, respectively. Mitochondria were stained with MitoTracker Green®. Cells were imaged with Leica SP8 confocal laser scanning microscope using a 1.40 NA, ×63 oil immersion objective with 3×digital zoom. (C and D) Pulmonary artery smooth muscle cells isolated from normal subjects and patients with pulmonary arterial hypertension, respectively. Mitochondria were stained with TMRM. Cells were imaged as in A and B, but with a 2× digital zoom. Scale bars = 10 μm.
The relationship between STAT2 and DRP1 phosphorylation
The paper infers, but does not prove, that STAT2 directly phosphorylates DRP1 at serine 616. To strengthen this inference immunoprecipitation assays, using STAT2 and DRP1 antibodies, could be performed. Ideally, an in vitro kinase assay using STAT2 as a putative kinase and DRP1 as the putative substrate would allow definitive assessment of whether STAT2 can directly phosphorylate DRP1.
Use of immune cells
This study suggests a link between mitochondrial dynamics and innate immunity. However, experiments in this study were performed on skin fibroblasts and neuroblastoma cells. Ideally future studies of the STAT2-DRP1 pathway would be performed using immune cells (e.g. B and T cell lymphocytes).
Metabolic consequences of mitochondrial elongation
The potential bioenergetic consequences of STAT2 deficiency proposed by Shahni et al. (2015) should be directly tested using micropolarimetry.
Conclusion
Shahni et al. (2015) provide impressive data to support a linkage between innate immunity and mitochondrial dynamics. Their study suggests that disruption in the Janus kinase (JAK)-STAT signalling pathway may perturb mitochondrial dynamics in a manner that causes patients with asymptomatic STAT2 mutations to develop mitochondrial dysfunction and systemic illness in response to a minor viral infection. This rare adverse reaction is fascinating and instructive but should not be taken as a condemnation of the MMR vaccine, which has dramatically reduced death and disability by virtually eliminating measles in vaccinated populations.
Funding
Supported in part by U.S. National Institutes of Health (NIH) grants NIH 1R01HL113003-01A1 (S.L.A.) and NIH 2R01HL071115-08 (S.L.A.), Canada Foundation for Innovation (S.L.A.), the Canadian Institute for Health Research (CIHR), Tier 1 Canada Research Chair in Mitochondrial Dynamics and Translational Medicine (S.L.A.), the American Heart Association (S.L.A.), the William J. Henderson Foundation (S.L.A.), and Canadian Vascular Network Scholar Award (A.D. and L.T.).
References
- Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med 2013; 369: 2236–51. [DOI] [PubMed] [Google Scholar]
- Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 2010; 121: 2661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B, Cho HM, Kim HJ, Jeong J, Park SK, Hwang EM, et al. CDK5-dependent inhibitory phosphorylation of Drp1 during neuronal maturation. Exp Mol Med 2014; 46: e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami R, Majumdar T, Dhar J, Chattopadhyay S, Bandyopadhyay SK, Verbovetskaya V, et al. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res 2013; 23: 1025–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SM, Valappil M, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci USA 2013; 110: 3053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol 2011; 13: 1108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 2015; 57: 537–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JA, Larner AC. Toward a new STATe: the role of STATs in mitochondrial function. Semin Immunol 2014; 26: 20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahni R Cale CM Anderson G Osellame LD Hambleton S Jacques TS, et al. Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission. Brain 2015. Advance Access published on Jun 29, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012; 148: 228–43. [DOI] [PubMed] [Google Scholar]
- Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009; 323: 793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom JD, Israeli T, Bachar-Wikstrom E, Swisa A, Ariav Y, Waiss M, et al. AMPK regulates ER morphology and function in stressed pancreatic beta-cells via phosphorylation of DRP1. Mol Endocrinol 2013; 27: 1706–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem 2004; 279: 35967–74. [DOI] [PubMed] [Google Scholar]