

Abstract
The triggering receptor expressed on myeloid cells (TREM)-1 plays a crucial role during the onset of sepsis by amplifying the host immune response. The TREM-like transcript-1 (TLT-1) belongs to the TREM family, is selectively expressed on activated platelets, and is known to facilitate platelet aggregation through binding to fibrinogen. In this study, we show that a soluble form of TLT-1 is implicated in the regulation of inflammation during sepsis by dampening leukocyte activation and modulating platelet-neutrophil crosstalk. A 17-aa sequence of the TLT-1 extracellular domain (LR17) is responsible for this activity through competition with the TREM-1 ligand. Whereas early or late LR17 treatment of septic mice improves survival, treml-1−/− animals are highly susceptible to polymicrobial infection. The present findings identify platelet-derived soluble TLT-1 as a potent endogenous regulator of sepsisassociated inflammation and open new therapeutic perspectives. We anticipate soluble TLT-1 to be important in regulating leukocyte activation during other noninfectious inflammatory disorders.
Septic shock, a complex clinical syndrome resulting from a harmful and damaging host response to infection, is the leading cause of mortality in intensive care units. Sepsis develops when the initial appropriate host response to systemic infection becomes dysregulated and overamplified with an intimate crosstalk between inflammation and coagulation. Some of the potential candidates acting as amplifiers of the innate immune response belong to the triggering receptor expressed on myeloid cells (TREM) family (1–4). The human TREM gene cluster is located on chromosome 6p21.1 and encodes for six different proteins, TREM 1–5 and TREM-like transcript-1 (TLT-1) (5). Engagement of TREMs triggers a signaling pathway involving ZAP70 and SYK and the ensuing recruitment and tyrosine phosphorylation of adaptor molecules such as GRB2; the activation of PI3K, phospholipase C-γ, ERK-1,−2, and p38 MAPK (6–9); and the formation of a CARD9-BCL10-MALT1 complex (10). The triggering of these pathways ultimately leads to the activation of the transcription factor NF-κB.
Although crystallographic analyses (6, 7) can predict TREM-1 recognition by using Ab-equivalent CDR loops (such as TCRs, CD8, and CTLA-4), its natural ligand has yet to be determined.
TREM-1 is known to cooperate with several TLRs in a synergistic manner (8, 11, 12), whereas TREM-1 silencing downmodulates LPS-induced inflammatory gene activation in myeloid cells (13). Moreover, blocking experiments using a TREM-1 fusion protein or using a peptide designed for the CDR3 and the “F” β strand of the extracellular domain of TREM-1 have demonstrated a reduced inflammation resulting in improved survival in murine models of endotoxemia and polymicrobial sepsis (14–16). The protective effects of modulating TREM-1 signaling are also evident in other models of acute [ischemia-reperfusion (17), pancreatitis (18), hemorrhagic shock (19)] or chronic [inflammatory bowel diseases (20), inflammatory arthritis (21–23)] inflammation. All of these studies suggest a role of TREM-1 in amplifying infectious or sterile inflammation.
In addition to TREM-1, the TREM gene cluster includes TLT-1. TLT-1 is abundant, specific to the platelet and megakaryocyte lineage, and sequestered in platelet α granules. Upon platelet activation with thrombin or LPS, TLT-1 is translocated to the platelet surface (24). TLT-1 contains a v-set Ig-type extracellular domain, a transmembrane region, and a cytoplasmic tail that comprises an ITIM and a polyproline-rich domain. Unlike other TREM family members, TLT-1 does not couple to the DAP 12-activating chain, although it has been shown to enhance Ca2+ signaling in rat basophilic leukemia cells, suggesting that TLT-1 is a coactivating receptor (25). The specificity of TLT-1 expression suggests that it plays a unique role in hemostasis and/or thrombosis. The recent characterization of the TLT-1 extracellular domain may suggest a role of TLT-1 during the onset and progression of sepsis. First, similar to TREM-1, a soluble fragment of TLT-1 is identifiable in human serum and plasma, the level of which is highly correlated to disseminated intravascular coagulation scores during sepsis (26). Soluble TLT-1 (sTLT-1) binds to fibrinogen and augments platelet aggregation in vitro. Secondly, crystallographic studies reveal structural similarities between TLT-1 and TREM-1, especially in their exposed end-forming loops, therefore suggesting the existence of interactions between TLT-1 and TREM-1, and possibly revealing a new mechanism involved in platelet/myeloid cell crosstalk (27).
In this study, we show that TLT-1- and TLT-1-derived peptides exhibit anti-inflammatory properties by dampening TREM-1 signaling and thus behave as naturally occurring TREM-1 inhibitors. We further demonstrate that these same peptides also modulate in vivo the proinflammatory cascade triggered by infection, thus inhibiting hyperresponsiveness, organ damage, and death during sepsis in mice.
Materials and Methods
Peptides
Based on the TLT-1 and TREM-1 sequences in GenBank|European Molecular Biology Laboratory|DNA Data Base in Japan (www.ncbi.nlm.nih.gov; accession numbers AY078502, AF534822, AF241219, and AF287008), TLT-1 peptides were designed mimicking different portions of its extracellular domain: TLT-1-CDR2 (SAVDRRAPAGRR), TLT-1-CDR3 (CMVDGARGPQILHR), and a well-conserved sequence between TREM-1 and TLT-1: TLT-1-LR17 (LQEEDAGEYGCMVDGAR). These constructs were chemically synthesized (Pepscan Presto BV, Lelystad, The Netherlands) as a COOH terminally amidated peptide for in vitro and in vivo assays, and FITC labeled for flow cytometry experiments. The correct peptides were obtained with >99% yields and were homogeneous after preparative purification, as confirmed by mass spectrometry and analytic reversed-phase HPLC. These peptides were free of endotoxin. A scrambled peptide containing the same amino acids of TLT-1-LR17, but in a randomized sequence order, was synthesized and served as control peptide (TLT-1-LR17 scramble: EDGQIYVPCLQYSLPQV). Recombinant sTLT-1 was obtained, as previously described (26).
Isolation of human polymorphonuclear cells and monocytes
Human peripheral blood samples were collected on EDTA from healthy volunteer donors among the laboratory staff.
Polymorphonuclear cells (PMNs) were isolated by density gradient (polymorphprep; AbCys, Paris, France), and monocytes were isolated by negative cell sorting with Monocyte Isolation Kit II (Miltenyi Biotec SAS, Paris, France). Purity was assessed by flow cytometry (anti-CD45, anti-CD14, and anti-CD66b; all from Beckman Coulter, Villepinte, France).
Cell stimulation
Depending on the experiment, cells were stimulated in complete medium supplemented with 100 ng/ml Escherichia coli LPS (0111:B4; Sigma-Aldrich), 5 μg/ml anti-TREM-1 mAb (R&D Systems), and TLT-1 peptides. Supernatants were collected for cytokine measurements and cells subjected to flow cytometry or lysed for protein phosphorylation analyses.
Cytokine concentration measurements were done by ELISA (human and mouse Quantikine ELISA kits; R&D Systems) and cytokine panel assays (Proteome Profiler Human Cytokine Array Kit, Panel A and Proteome Profiler Mouse Cytokine Array Kit, Panel A; R&D Systems).
Quantitative RT-PCR
Total RNAs were extracted from isolated cells or tissues using RNeasy Plus Mini Kit (Qiagen, Courtaboeuf, France), quantified with NanoDrop (ThermoScientific) before being retrotranscripted using the iScript cDNA synthesis kit (Bio-Rad), and quantified by quantitative PCR using Qiagen available probes (Quantitect Primers) for human TREM-1, human IL-6, human IL-8, human TNF-α, human ActB, murine IL-6, murine TNF-α, and murine ActB. Alternatively, total RNAs were retrotranscripted with RT2 First Strand Kit (SABiosciences, Tebu-bio, Le Perray-en-Yvelines, France) for PCR arrays (Human/Mouse Innate and Adaptive Immune Responses RT2 Profiler PCR Arrays; SABiosciences). All PCRs were performed in a MyiQ Thermal Cycler and quantified by iQ5 software (Qiagen). PCR array results were analyzed using PCR Array Data Analysis Software (SABiosciences) and normalized with five housekeeping genes.
NF-κB activity measurement
A 150-μl aliquot of whole blood obtained from healthy donors was stimulated by 100 ng/ml LPS with or without LR17 peptide (20 μg/ml) at 37°C for the indicated time points. LR17 or unstimulated blood samples were used as controls. When indicated, cells were stained with anti-CD14-PC5 or anti-CD16-PE (Beckman Coulter).
RBCs were lysed by 10-min incubation in a 10-fold volume of Fix FACS lyse buffer (1:3:6 mix of FACS lysing solution [BD Biosciences]: formaldehyde 10% methanol free [Polysciences, Tebu-Bio]:distilled water). Cells were then permeabilized with Triton X-100 for intracellular staining with anti-NF-κB (Santa Cruz Biotechnology, Tebu-Bio) and corresponding secondary Ab-DyL649 (Jackson ImmunoResearch Laboratories, Beckman Coulter), fixed in 10% paraformaldehyde, and labeled with DAPI before analysis on the ImageStream (Amnis, Seattle, WA). Data were acquired with Inspire software and analyzed with Ideas software (Amnis).
Results are expressed in similarity score between NF-κB and DAPI staining, which represents the degree of NF-κB nuclear translocation (28, 29). Alternatively, cells were collected, nuclear extracts were obtained by nuclear extraction kit, and NF-κB activity was measured with human p50/ p65 combo transcription factor assay kit (Cayman Chemical, InterChim, Montluҫon, France), following the manufacturer’s instructions.
ROS production and phagocytosis assessment
The quantitative determination of neutrophil phagocytosis and oxidative burst was performed by flow cytometry using the Phagotest and the Bursttest (Orpegen Pharma, Heidelberg, Germany), following the manufacturer’s recommendations. FACS quantification of intracellular reactive oxygen species (ROS) production was assessed by the use of DCFDA, a fluorogenic substrate (Fisher Scientific, Illkirch, France), according to manufacturer’s instructions.
Protein phosphorylation analysis
Stimulated PMNs were lysed in PhosphoSafe Extraction Reagent (Novagen, Merck Biosciences, Nottingham, U.K.) and centrifuged for 5 min at 16,000 × g at 4°C to collect the supernatant. Protein concentration was determined according to Bradford’s method (BCA Protein Assay Kit; Pierce, Ther-moScientific, Brebiѐres, France). Lysates were then analyzed by Western blot (Criterion XT Bis-Tris Gel, 4–12%, Bio-Rad, Marnes-la-Coquette, France, and polyvinylidene difluoride membrane, Millipore, Saint-Quen-tin en Yvelines, France) and revealed with anti-phospho-p38 or antipERK1/2, and the corresponding secondary Ab was conjugated to HRP (Cell Signaling, Ozyme, Saint-Quentin en Yvelines, France) and Super-Signal West Femto Substrate (Pierce, ThermoScientific). Anti-p38 and anti-ERK1/2 (Cell Signaling) were used for normalization. Alternatively, PMNs were analyzed after 20 min of stimulation for a panel of multiple phosphorylated proteins by immunoblot (Human Phospho-Kinase Array; R&D Systems). Acquisition and quantitative signal density analyses were performed by a LAS-4000 imager (FSVT, Courbevoie, France) and Multi-Gauge software (LifeScience Fujifilm).
Immunoprecipitation
Cells were lysed with CytoBuster Protein Extraction Reagent (Novagen, Merck Biosciences, Nottingham, U.K.). Samples were normalized by total protein concentration, and lysates were precleared before performing immunoprecipitation. Precleared lysates were then incubated overnight at 4°C with rabbit anti-Bcl10 mAb (Cell Signaling) or rabbit anti-CARD9 (Antibodies Online, Aachen, Germany). Thereafter, anti-rabbit-Ig beads (eBio-science, CliniScience, Montrouge, France) were added for 1 h at room temperature. Beads were then washed three times, denatured in Laemmli buffer 5 min at 95°C, and centrifuged at 16,000 × g for 1 min. Supernatants containing retained proteins were analyzed by Western blotting and revealed with anti-Malt-1 and corresponding secondary Ab HRP conjugated (Cell Signaling).
Surface plasmon resonance
Detection of TREM-1 ligand on LPS-stimulated neutrophil supernatants was assessed by binding to a recombinant soluble form of TREM-1 (rsTREM-1) coated on a CM5 sensor chip with the BIAcore X instrument at 25°C with a flow rate of 5 μl/min. Binding specificity was verified through competition with the rsTREM-1. Inhibition of TREM-1 ligand binding by LR17 was assessed.
Platelet isolation
Platelets were obtained, as previously described (3). For activation, platelets were incubated for 30 min with 5 U/ml thrombin (Sigma-Aldrich) or 1 μg/ ml E. coli LPS (0111:B4; Sigma-Aldrich) at 37°C and subsequently fixed with 2% (w/v) paraformaldehyde. Residual paraformaldehyde was removed by two additional washing steps in Tyrode’s salt buffer. Activation was assessed by anti-CD62P (Beckman Coulter).
FACS analysis
Isolated cells (neutrophils, monocytes, or platelets) were blocked for aspecific binding with 10% human Ig (Sigma-Aldrich) for 1 h on ice. Cells were incubated with soluble rFITC-labeled TLT-1 peptides, FITC-labeled LP17 (or corresponding FITC-labeled scrambled peptides), PE-labeled anti-TREM-1 mAb (R&D Systems), CD62P-FITC, CD66b-PE, CD45-PE, and CD14-FITC (all from Beckman Coulter).
Preparation of Trem-1 knockdown monocytes
Trem-1 silencing was performed with the Human Monocyte Nucleofector Kit (Amaxa, Lonza, Verviers, Belgium) using small interfering RNA (siRNA) sequences obtained from Qiagen (HP GenomeWide siRNA; Qiagen, Court-aboeuf, France). Isolated monocytes were electroporated with Trem-1 siRNA and cultured for 24 h in Human Monocyte Nucleofector Medium (Amaxa) prior to stimulation. Monocytes electroporated without siRNA or cultured in presence of Trem-1 siRNA without electroporation served as negative controls, whereas monocytes transfected with a GFP-reporter plasmid served as positive controls. TREM-1 expression was subsequently assessed by quantitative RT-PCR and flow cytometry 24 h after transfection. Medium was changed, and monocytes were stimulated with LPS, anti-TREM-1 mAb, and TLT-1 peptides for 24 h, after which medium was collected for cytokine measurement.
LPS-induced endotoxemia in mice
Male BALB/c mice (4–6 wk) were randomly grouped and treated with LPS (LD50) i.p. in combination with LR17 (in 250 μl normal saline) or LR17 scrambled 1 h before or after LPS challenge. The viability of mice was examined every hour, and animals were sacrificed at regular intervals for blood sampling.
Caecal ligation and puncture polymicrobial sepsis model
Experiments were approved by our institutional Animal Care and Use Committee. Male BALB/c mice (4–6 wk) or male wild-type (WT) and treml-1−/−C57BL/6 mice (26) were anesthetized with isoflurane. Caecal ligation and puncture (CLP) was performed, as previously described (14). After surgery, all mice were injected s.c. with 0.5 μl 0.9% NaCl solution for fluid resuscitation. The animals were randomly grouped and treated with LR17 or LR17 scrambled as control in 250 μl 0.9% NaCl solution and administered i.p. and then monitored for survival. Five additional animals per group were sacrificed under anesthesia at 24 h after CLP for the determination of bacterial count, cytokine levels, platelet count, and organ samples. Bronchoalveolar lavage fluid and peritoneal lavage fluid were obtained, and blood was collected by cardiac puncture.
Thrombin-anti-thrombin complex quantification
Thrombin-anti-thrombin complex (TATc) concentration was measured from mouse plasma samples with the TAT ELISA Kit (USCN LifeScience, ICS, Munchen, Germany).
Patient population
Between August 2009 and September 2009, all consecutive patients admitted into a 16-bed medical intensive care unit of HÔpital Central (Nancy, France) were enrolled. Approval of the institutional review board and informed consent were obtained from patients or their relatives before inclusion. The diagnosis of sepsis was established on the basis of current definitions. Upon admission into the intensive care unit, age, sex, severity of medical condition stratified according to the simplified acute physiology score (30) and sepsis-related organ failure assessment (SOFA) score (31), routine blood test results, and microbial culture results were recorded. Plasma was obtained within 12 h after admission, and TREM-1 ligand concentration was determined by surface plasmon resonance. Outcome was assessed during a 28-d follow-up period.
Statistical analysis
All data, unless indicated, were normally distributed and then are presented as mean ± SD, and statistical significance of differences between two groups was analyzed using Student t test. Kaplan-Meier survival curves were analyzed using the log rank test. A p value <0.05 was deemed significant.
Results
sTLT-1 decreases LPS-mediated human neutrophil activation
Considering the structural similarities that exist between TLT-1 and TREM-1, we hypothesized that sTLT-1 may modulate the inflammatory response to LPS. We first determined whether sTLT-1 decreases LPS-induced cytokine production by human neutrophils. As seen in Fig. 1, TNF-α concentrations were reduced in a dose-dependent manner in the presence of sTLT-1 (Fig. 1A). Similar observations were also noted for IL-6 and IL-8.
FIGURE 1.
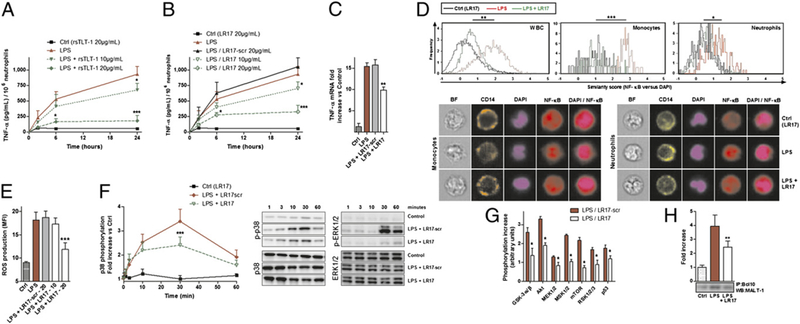
sTLT-1 decreases LPS-mediated neutrophil activation. (A and B) TNF-α concentrations. (C) TNF-α mRNA levels in neutrophils stimulated for 6 h by 100 ng/ml LPS and/or 20 μg/ml LR17. (D) ImageStream fluorescence quantification and imaging of 1-h LPS-induced NF-κB nuclear translocation in whole blood cells, CD14+ monocytes, and CD16+ neutrophils, with or without 20 μg/ml LR17. Brightfield (BF, white), CD14 (orange), CD16 (yellow), DAPI (violet), NF-κB (red), and DAPI/NF-κB composite images. (E) Neutrophil ROS production. (F and G) Protein phosphorylation of neutrophil lysates at indicated times (F) or after 20 min (G) 100 ng/ml LPS and/or 20 μg/ml LR17. (H) Immunoblot of MALT-1 after immunoprecipitation with anti-BCL10 mAb of lysates of neutrophils stimulated for 20 min. (A–H) Data are representative of at least five different experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus LPS.
To decipher which region of TLT-1 was responsible for this decrease in cytokine synthesis, several synthetic peptides were designed mimicking various portions of its extracellular domain, as follows: human TLT-1 (hTLT-1)-CDR2, hTLT-1-CDR3, hTLT-1-LR17, and hTLT-1-LR17 scramble. Only hTLT-1-LR17 (hereafter termed LR17) was able to decrease the production of TNF-α (Fig. 1B, 1C, and data not shown), IL-6, and IL-8 both at the gene and protein levels. Moreover, E. coli-induced neutrophil oxidative burst was also decreased by sTLT-1/LR17 (data not shown). In contrast, sTLT-1 and LR17 did not alter the phagocytic properties of neutrophils (data not shown).
The production of several other cytokines/chemokines was also modulated by LR17 (IL-1β, MCP-1, MIP-1β, RANTES). As observed above, among all the TLT-1-derived peptides tested, only LR17 showed an effect on cytokine production. These effects were also confirmed both in human monocytes and upon TLR2 (Pam3CSK4) stimulation (Supplemental Fig. 1).
TLR4 engagement upon LPS stimulation leads to NF-κB activation and ROS production by neutrophils. In this study, LR17 reduced LPS-induced NF-κB activation (Fig. 1D), intracellular ROS production (Fig. 1E), and p38 and ERK1/2 phosphorylation (Fig. 1F). The phosphorylation of other proteins involved in TREM-1 signaling was also reduced in the presence of LR17 (Fig. 1G). TREM-1 signals through its association with DAP12, an ITAM-containing adaptor protein. Because the formation of CARD9-BCL10-MALT1 has been proven essential in linking ITAM-coupled receptors to downstream NF-кB activation (10), we next examined the effect of LR17 on generation of this complex. LPS induced an increase in BCL10-MALT1 complex formation in neutrophils (Fig. 1H), an effect reversed by LR17. Similar effects were also observed for CARD9-BCL10.
The above data thus suggest that sTLT-1 is able to modulate LPS-induced neutrophil/monocyte activation through its residues 94–110 corresponding to LR17.
LR17 is a competitive inhibitor of the TREM-1 receptor
Although neutrophils do not express a membrane-bound TREM-1 ligand, these cells may release its soluble form. Using surface plasmon resonance, we observed that LPS-activated neutrophils time dependently secrete a TREM-1 ligand (Fig. 2A). LR17 was able to block the binding of the TREM-1 ligand to immobilized TREM-1 (Fig. 2B), suggesting that sTLT-1 could interfere with TREM-1/ TREM-1 ligand interactions. Human platelets are known to constitutively express TREM-1 ligand (3). To further prove that sTLT-1 binds to the TREM-1 ligand, experiments were performed to assess LR17 fixation on resting and activated platelets. FITC-labeled LR17 was found to bind to both resting and thrombin-activated platelets. This binding was decreased by coincubation of LP17, a TREM-1 derived peptide known to bind to the TREM-1 ligand (14). The opposite also held true: FITC-labeled LP17 binding to platelets was decreased by coincubation with sTLT-1 or LR17, but not by LR17 scrambled (Fig. 2C). As expected, LR17 did not bind to neutrophils or monocytes that do not express the TREM-1 ligand.
FIGURE 2.
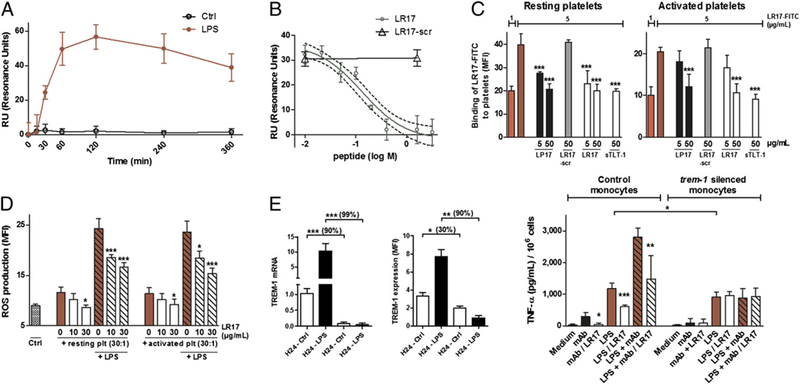
sTLT-1 blocks TREM-1/TREM-1 ligand interactions. (A) Surface plasmon resonance assessment of neutrophils’ supernatants during a time course stimulation with or without LPS for the release of the TREM-1 ligand. (B) Competition assay of TREM-1 ligand (derived from neutrophils stimulated with LPS for 30 min) binding to a rsTREM-1-coated sensorship with increasing concentrations of LR17 (green circles) or LR17 scrambled (black triangles). (C) Flow cytometry analysis of FITC-labeled LR17 (5 μg/ml) binding to resting (upper panel) or thrombin-activated (5 Ul/ml) human platelets (lower panel) is reversed by coincubation with unlabeled LR17, sTLT-1, or LP17, a TREM-1-derived peptide. (D) Neutrophil ROS production after a 2-h stimulation with platelets or platelets/LPS. (E) Trem-1 mRNA and TREM-1 expression in native and Trem-1-silenced monocytes at baseline and upon 24-h LPS stimulation (left), TNF-α concentrations produced by control (electroporated cells without siRNA), or Trem-1-silenced human monocytes stimulated by LPS or a TREM-1 agonist (mAb) (right). Data are representative of at least five different experiments. *p < 0.05, **p < 0.01, ***p < 0.001, LR17 versus LPS and/or anti-TREM-1 mAb.
TREM-1-specific signals are required for the platelet-induced augmentation of LPS-mediated leukocyte effector function, especially ROS production (3). Platelets have been shown to increase ROS production by neutrophils through TREM-1 engagement: this production was partly prevented in presence of LR17 (Fig. 2D).
To conclusively demonstrate that sTLT-1 modulates the LPS-induced inflammatory response via blockade of TREM-1, the effect of LR17 on monocytes treated with Trem-1 siRNA was investigated. Trem-1 silencing was achieved in monocytes with >90% efficiency from 24 to 96 h after transfection, as verified by quantitative RT-PCR. Gene silencing was associated with a concomitant ~30% decrease in TREM-1 baseline expression, as observed by flow cytometry. As expected, when native monocytes were stimulated with LPS, this yielded to a sharp increase of TREM-1 expression (mRNA and protein), whereas this upregulation was abolished in knocked down cells. Silenced monocytes were stimulated with LPS/anti-TREM-1 mAb: silenced monocytes’ response to LPS was decreased as compared with controls, whereas TREM-1 activation by anti-TREM-1 mAb was abrogated and LR17 did not show any effect on TNF-α synthesis (Fig. 2E).
To specifically induce TREM-1 engagement, we used an agonistic mAb (αTREM-1) (16). Myeloid cell activation through TREM-1 leads to p38 MAPK and ERK 1/2 phosphorylation (8) and CARD9-MALT1-BCL10 complex formation (10). We indeed observed in this study that neutrophil activation by LPS and αTREM-1 yielded to p-p38, p-ERK1/2, and CARD9-MALT1 -BCL10 appearance, with a synergy between these two stimuli. This effect was partly abrogated by LR17 (Fig. 3A-C). TREM-1 signaling pathway also leads to NF-κB activation. Again, LR17 decreased αTREM-1/LPS-induced NF-кB activity (Fig. 3D). Finally, we found that αTREM-1/LPS-induced cytokines, both at gene and protein levels, as well as ROS production were partly prevented by LR17 (Fig. 3E-G).
FIGURE 3.
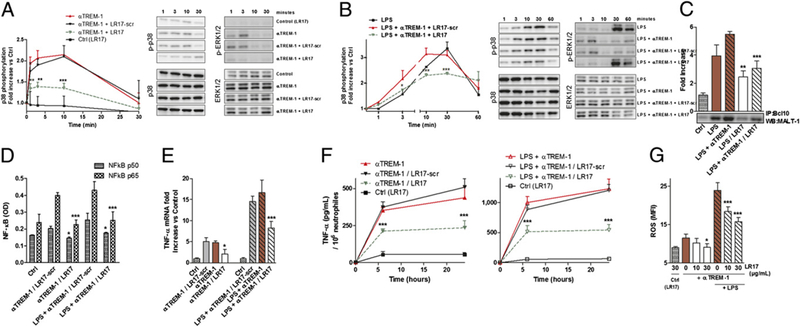
LR17 modulates the TREM-1 pathway. (A–E) Isolated human neutrophils were stimulated with 100 ng/ml LPS and/or 5 μg/ml αTREM-1, with 10–20 μg/ml LR17 or LR17 scrambled. (A and B) Western blot of lysates of neutrophils treated for 1, 3, 10, 30, and 60 min, analyzed with Ab to phospho (p)-p38, p38, p-ERK1/2, and ERK1/2. (C) Immunoblot of MALT-1 after immunoprecipitation with anti-BCL10 mAb of lysates of neutrophils stimulated for 20 min. (D) ELISA of nuclear p50 and p65 NF-κB subunits of neutrophils treated for 2 h. (E) TNF-α mRNA levels in neutrophils stimulated for 6 h. (F) ELISA quantification of TNF-α production after 6 and 24 h of stimulation. (G) FACS quantification of neutrophil ROS production. Data are representative of at least four different experiments. Results are mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, LR17 versus corresponding LPS and/or anti-TREM-1 mAb.
These results suggest that sTLT-1 regulates leukocyte activation through the modulation of TREM-1 activation.
LR17 protects endotoxemic mice from death
We next assessed whether sTLT-1 displays putative protective effects during sepsis. Adult male BALB/c mice were administered a single i.p. dose of mLR17 (LQEEDTGEYGCVVEGAA), mLR17 scrambled (EDGVGYVRCLQGEDAET), or 0.9% NaCl, 60 min prior to LPS administration (LD50, 25 mg/kg). Pretreatment as well as delayed administration of LR17 conferred significant protection (Supplemental Fig. 2A), with no late death occurring >10 d, indicating that LR17 did not merely delay the onset of LPS lethality, but also provided lasting protection. Control mice all developed lethargy, piloerection, and diarrhea before death. By contrast, LP17-treated mice remained well groomed and active, presented no diarrhea, and were lively. Compared with controls, both pre- as well as posttreatment by LR17 significantly reduced plasma concentrations of all four cytokines (Supplemental Fig. 2B-E).
LR17 protects mice against polymicrobial sepsis
To investigate the role of LR17 in a more relevant model of septic shock, CLP experiments were performed. First, plasma, bronchoalveolar, and peritoneal IL-6 and IL-10 concentrations were measured 24 h after surgery. Both cytokine concentrations were decreased in LR17-treated (100 μg i.p. 2 h after surgery) animals (Fig. 4A), as well as their respective gene mRNA levels in both lung and liver (Fig. 4B). Screening of plasma levels and mRNA tissue levels of various inflammatory mediators also revealed a decreased plasma concentration of several other important inflammatory cytokines/chemokines (C5a, IL-1ra, IL16, MCP-1, MIP-1α, MIP-2) as well as a downmodulation of various genes involved in TLR- and TREM-1 pathways in the lung and liver (Supplemental Table I).
FIGURE 4.
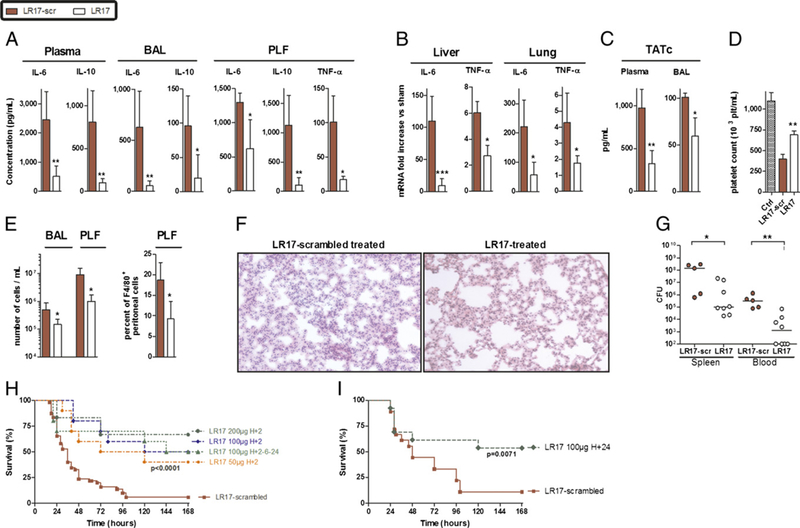
LR17 protects mice against polymicrobial sepsis. (A) TNF-α, IL-6, and IL-10 concentrations in plasma, peritoneal (PLF), and bron-choalveolar lavage (BAL) fluids. (B) Lung and liver IL-6 and TNF-α mRNA. (C) PLF and BALTATc concentrations. (D) Platelet count in whole blood. (A–D) Samples were obtained 24 h after CLP. (E) Total cell counts and F4/80+ cells in PLF and BAL 24 h after CLP. (F) Histopathological examination of the lungs 24 h after CLP. (G) Bacterial counts in spleen and blood obtained 24 h after CLP. (H and I) Survival curves (n = 25–40 per group) after CLP analyzed by log rank test. *p < 0.05, **p < 0.01 versus control animals.
Coagulation activation often occurs during sepsis as part of the inflammatory response. In this study, TATc plasma and alveolar concentrations were markedly elevated during peritonitis, along with a decrease of blood platelet count. Administration of LR17 prevented these coagulation abnormalities (Fig. 4C, 4D).
We next investigated whether LR17 affects local cell recruitment both at the site of infection (the peritoneum) as well as distally (alveolar space). Cell infiltration, especially macrophages, was reduced by LR17 treatment at both sites (Fig. 4E). Histological study of septic mice revealed severe lung injury, that is, intraalveolar hemorrhaging, protein precipitation, and leukocyte infiltration into the alveoli, as well as oedematous thickening of the perivascular space. These alterations were attenuated in LR17- treated animals (Fig. 4F). Hence, LR17 prevents the massive cellular infiltration and histological damage induced by peritonitis.
The effect of LR17 was also studied on bacterial clearance. As expected, very high bacterial counts were observed in the spleens of CLP mice 24 h after the onset of peritonitis. Moreover, all control animals were found to be bacteremic. By contrast, LR17 improved bacterial clearance and almost completely prevented septicemia (Fig. 4G). Of note, no antimicrobial activity was found for LR17 per se when tested according to Clinical and Laboratory Standards Institute recommendations. All of these protective effects translated into a dose-dependent survival rate improvement, even when LR17 was administered as late as 24 h after the onset of sepsis (Fig. 4H, 4I).
These results indicate that LR17 treatment is able to modulate sepsis-induced inflammatory response both locally and systemi- cally as well as improve bacterial clearance and survival rate.
The treml-1−/− mice are more susceptible to polymicrobial infection
All of the above experiments suggest that sTLT-1 plays a role in dampening inflammatory and organ damage-associated reaction to polymicrobial sepsis. To definitely validate this hypothesis, we performed CLP on mice lacking TLT-1 (treml-1−/− mice). Plasma and alveolar inflammatory cytokine concentrations were higher in treml-1−/− mice than in WT littermates both at the protein (Fig. 5A) and at the gene levels (Fig. 5B). Thrombocytopenia and lung damage were also more pronounced in treml-1−/− animals (Fig. 5C, 5D). This hyperinflammatory state translated into a higher mortality rate. By contrast, bacterial clearance did not differ between WT and treml-1−/−mice (Fig. 5E, 5F). When LR17 was administered to knockout mice, it still conferred significant protection, albeit less than in WT (Fig. 5G, Supplemental Table II).
FIGURE 5.
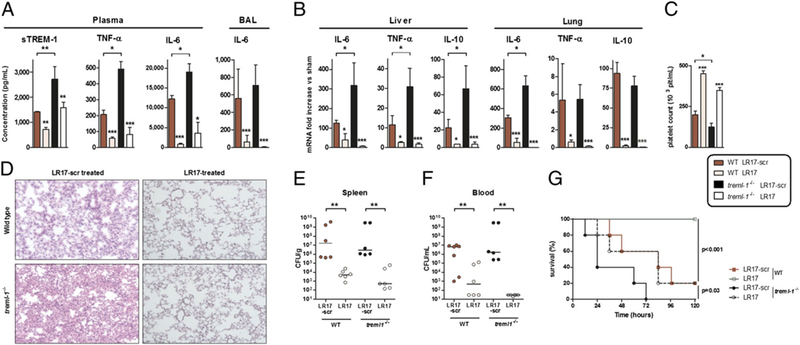
Knockout of treml-1 is deleterious, but reversible by LR17 administration during sepsis. (A) TNF-α, IL-6, and sTREM-1 concentrations in plasma and/or BAL. (B) Lung and liver IL-6, TNF-α, and IL-10 mRNA quantification. (C) Platelet count in whole blood. (D) Histopathological examination of the lungs 24 h after CLP. (E and F) Bacterial counts in spleen and blood obtained 24 h after CLP. (G) Survival curves after CLP analyzed by log rank test. *p < 0.05, **p < 0.01, ***p < 0.001 compared with control animals or between treml-1−/− and WT littermates. For survival analyses, p < 0.001 between LR17-scr and LR17 WT, and p = 0.03 between LR17-scr and LR17 treml-1−/− mice.
These data confirm a protective role of TLT-1 during polymicrobial sepsis.
A soluble form of the TREM-1 ligand is present in the plasma of infected patients and is associated with sepsis severity
Considering the potential implications of TLT-1/LR17 in interfering with the TREM-1/TREM-1 ligand binding, we investigated whether a soluble form of TREM-1 (sTREM-1) ligand exists in the plasma of critically ill patients. Fifty consecutive patients were included in this study. The main clinical and biological characteristics of this cohort are summarized in Supplemental Table II. Twenty-four patients were diagnosed with sepsis. Infection originated from the lung (community-acquired pneumonia, 72%), the urinary tract (pyelonephritis, 8%), the soft tissues (necrotizing fasciitis, 8%), or others (meningitis, peritonitis, 12%). Analysis of plasma TREM-1 ligand concentrations showed that infected patients presented much higher levels than noninfected ones (Fig. 6A). Indeed, TREM-1 ligand was undetectable in all but four nonseptic patients. We next sought to investigate the relationship between TREM-1 ligand and disease severity. We found a correlation between SOFA score, a usual severity score in critically ill patients, and the levels of plasma TREM-1 ligand (Fig. 6B). Moreover, nonsurviving patients presented with more increased TREM-1 ligand concentrations than survivors (Fig. 6C).
FIGURE 6.
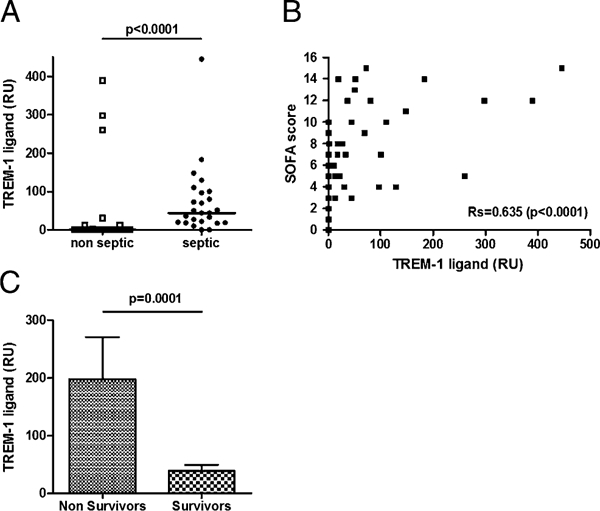
Elevated levels of plasma TREM-1 ligand in patients diagnosed with sepsis and correlation with severity. (A) Patients admitted into the intensive care unit were evaluated for the presence of plasma TREM-1 ligand by surface plasmon resonance according to the presence of a sepsis or not. Markers represent individual patients. Horizontal lines represent the median. The p values represent results from Mann-Whitney U test. Correlation between plasma TREM-1 ligand concentration and severity of the disease assessed by the SOFA score (B). Rs value was calculated via the Spearman test. (C) Plasma concentration of TREM-1 ligand according to the outcome.
Therefore, TREM-1 ligand can be found in the plasma of infected patients, the concentration of which correlates with severity. It may thus constitute a valuable target for the TLT-1/LR17 effect.
Discussion
In the current study, we report a crucial role for TLT-1 in regulating leukocyte activation and modulating sepsis-induced acute inflammatory response. A 17-aa residue of its extracellular domain mediates this effect. Whereas treml-1−/− mice are particularly susceptible to infection with the development of an intense inflammatory state that translated into organ damage and rapid mortality, LR17 administration confers a strong protection.
Until recently, the role of TLT-1 has remained unclear. TLT-1 is exclusively expressed in platelets, colocalized with CD62P in α-granules of resting platelets. Upon activation, TLT-1 is quickly exposed on the membrane (24) and subsequently cleaved, leading to the release of a soluble fragment. Structural analysis of the extracellular domain of TLT-1 suggests the existence of several distinct potential ligand binding sites (27). The first TLT-1 ligand was recently identified to be fibrinogen, positioning TLT-1 as a regulator of hemostasis by facilitating platelet aggregation: with membrane-bound TLT-1 linking fibrinogen and sTLT-1 crosslinking with fibrinogen, this receptor acts as a stabilizer of the fibrinogen network, and thus of platelet aggregates (26). Moreover, TLT-1 deficiency has been associated with prolonged bleeding times, increased cytokine production, and decreased survival in a mouse model of endotoxemia. Our results show that sTLT-1 binds to the TREM-1 ligand, thus acting as a decoy receptor. sTLT-1 inhibits TREM-1 receptor engagement, a phenomenon associated in vitro with limited neutrophil and monocyte TLR- induced activation.
To identify which portion of sTLT-1 was involved in this protective effect, we designed several TLT-1 peptides representative of various potential ligand-binding regions. Among these, a 17-aa sequence representative of residues 94–110, named in this work LR17, was shown to be directly responsible for the anti-inflammatory effect.
The role of TREM-1 as an amplifier of the inflammatory response has been confirmed in several models of septic shock in which blockade of TREM-1 signaling was shown to reduce mortality (14, 16). These studies suggest that during acute inflammation, activated neutrophils and monocytes/macrophages release a sTREM-1 that acts as a TREM-1 inhibitor by competing with its ligand. In this report, we show that sTLT-1 behaves in the same manner as sTREM-1 and specifically modulates TREM-1 activation, resuling in a decrease in the production of several cytokines as well as improved bacterial clearance. Organ failure was also partly prevented by administration of LR17 during experimental sepsis at the lung, liver, and kidney levels.
Supporting these data, we also found that treml-1−/− mice were particularly susceptible to infection with the development of an intense inflammatory state that translated into organ damage and rapid mortality. Although LR17 administration to treml-1−/− mice decreased inflammation, plasma IL-6 concentration elevation, thrombocytopenia, and lung damage still were more pronounced than in similarly treated WT animals. Protection conferred by LR17 was thus only partial, and mortality remained high in treml-1−/− mice.
The present work also has implications in the comprehension of how platelets and neutrophils cooperate during sepsis. Severe sepsis is characterized by a deregulated host immune response and disseminated coagulopathy, underlining the subtle crosstalk between platelets and immune cells in the regulation of inflammation and coagulation. Endothelial dysfunction caused by systemic inflammation leads to platelet activation, followed by their interaction with neutrophils and monocytes and their subsequent aggregation. Certain ligands and counter-receptors are implicated in the dialogue between platelets and neutrophils: P-selectin/P-selectin glycoprotein ligand 1 coupling initiates platelet binding to neutrophils, which is stabilized by Mac-1/GPIb-α interaction. This neutrophil activation is mediated by various membrane or soluble mediators. Among these, the interaction of the TREM-1 ligand (expressed in platelets) and TREM-1 (expressed in neutrophils) results in an enhancement of neutrophil activation (3). Results in this study reveal that this phenomenon is prevented by LR17. We thus suggest that sTLT-1 released in vivo during acute inflammation binds to platelet TREM-1 ligand, thereby inhibiting TREM-1 ligand/TREM-1 interaction, and ultimately preventing neutrophil activation. Moreover, in our experimental model of septic shock, LR17 administration partly protected from coagulation disorders as witnessed by the prevention of thrombocytopenia and the reduction of thrombin formation. In line with this finding, platelet count was more decreased in treml-1−/− animals following infection than in WT littermates.
We also showed in this study that sTREM-1 ligand is present in the plasma of critically ill patients, the concentration of which correlates with the severity of the disease. sTLT-1 may then also targets this sTREM-1 ligand, thus preventing overactivation of circulating immune cells. Interestingly, this TREM-1 ligand was almost only found in septic patients, and could therefore be a reliable marker of infection.
Overall, we show in this study that TLT-1 plays a pivotal role during sepsis by linking hemostasis and inflammation, and we propose that sTLT-1 is released by activated platelets to prevent a prolonged and sustained inflammation.
Supplementary Material
Acknowledgments
We are indebted to Chantal Montemont and Anne-Marie Carpentier for technical assistance. We also thank Dr. Frederic Cailotto for assistance with the siRNA experiments.
Abbreviations used in this article:
- CLP
caecal ligation and puncture
- hTLT-1
human triggering receptor expressed on myeloid cells-like transcript-1
- PMN
polymorphonuclear cell
- ROS
reactive oxygen species
- rsTREM
recombinant soluble form of triggering receptor expressed on myeloid cells
- siRNA
small interfering RNA
- SOFA
sepsis-related organ failure assessment
- sTLT-1
soluble triggering receptor expressed on myeloid cells-like transcript-1
- sTREM
soluble form of triggering receptor expressed on myeloid cells
- TATc
thrombin–anti-thrombin complex
- TLT-1
triggering receptor expressed on myeloid cells-like transcript-1
- TREM
triggering receptor expressed on myeloid cells
- WT
wild-type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Bouchon A, Dietrich J, and Colonna M. 2000. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 164: 4991–4995. [DOI] [PubMed] [Google Scholar]
- 2.Bleharski JR, Kiessler V, Buonsanti C, Sieling PA, Stenger S, Colonna M, and Modlin RL. 2003. A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J. Immunol. 170: 3812–3818. [DOI] [PubMed] [Google Scholar]
- 3.Haselmayer P, Grosse-Hovest L, von Landenberg P, Schild H, and Radsak MP. 2007. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood 110: 1029–1035. [DOI] [PubMed] [Google Scholar]
- 4.Gibot S,Massin F, Marcou M, Taylor V, Stidwill R, Wilson P, Singer M, and Bellingan G. 2007. TREM-1 promotes survival during septic shock in mice. Eur. J. Immunol. 37: 456–66. [DOI] [PubMed] [Google Scholar]
- 5.Allcock RJN, Barrow AD, Forbes S, Beck S, and Trowsdale J. 2003. The human TREM gene cluster at 6p21.1 encodes both activating and inhibitory single IgV domain receptors and includes NKp44. Eur. J. Immunol. 33: 567–577. [DOI] [PubMed] [Google Scholar]
- 6.Kelker MS, Debler EW, and Wilson IA. 2004. Crystal structure of mouse triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.76 A. J. Mol. Biol. 344: 1175–1181. [DOI] [PubMed] [Google Scholar]
- 7.Kelker MS, Foss TR, Peti W, Teyton L, Kelly JW, Wüthrich K, and Wilson IA. 2004. Crystal structure of human triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.47 A. J. Mol. Biol. 342: 1237–1248. [DOI] [PubMed] [Google Scholar]
- 8.Haselmayer P, Daniel M, Tertilt C, Salih HR, Stassen M, Schild H, and Radsak MP. 2009. Signaling pathways of the TREM-1- and TLR4-mediated neutrophil oxidative burst. J. Innate Immun. 1: 582–591. [DOI] [PubMed] [Google Scholar]
- 9.Gibot S 2005. Clinical review: role of triggering receptor expressed on myeloid cells-1 during sepsis. Crit. Care 9: 485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, et al. 2007. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol. 8: 619–629. [DOI] [PubMed] [Google Scholar]
- 11.Fortin CF, Lesur O, and Fulop T Jr. 2007. Effects of TREM-1 activation in human neutrophils: activation of signaling pathways, recruitment into lipid rafts and association with TLR4. Int. Immunol. 19: 41–50. [DOI] [PubMed] [Google Scholar]
- 12.Zheng H, Heiderscheidt CA, Joo M, Gao X, Knezevic N, Mehta D, and Sadikot RT. 2010. MYD88-dependent and -independent activation of TREM-1 via specific TLR ligands. Eur. J. Immunol. 40: 162–171. [DOI] [PubMed] [Google Scholar]
- 13.Ornatowska M, Azim AC, Wang X, Christman JW, Xiao L, Joo M, and Sadikot RT. 2007. Functional genomics of silencing TREM-1 on TLR4 signaling in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 293: L1377–L1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibot S, Kolopp-Sarda M-N, Béné M-C, Bollaert P-E, Lozniewski A, Mory F, Levy B, and Faure GC. 2004. A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis. J. Exp. Med. 200: 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibot S, Buonsanti C, Massin F, Romano M, Kolopp-Sarda M-N, Benigni F, Faure FC, Béné M-C, Panina-Bordignon P, Passini N, and Lévy B. 2006. Modulation of the triggering receptor expressed on the myeloid cell type 1 pathway in murine septic shock. Infect. Immun. 74: 2823–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouchon A, Facchetti F, Weigand MA, and Colonna M. 2001. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 410: 1103–1107. [DOI] [PubMed] [Google Scholar]
- 17.Gibot S, Massin F, Alauzet C, Montemont C, Lozniewski A, Bollaert P-E, and Levy B. 2008. Effects of the TREM-1 pathway modulation during mesenteric ischemia-reperfusion in rats. Crit. Care Med. 36: 504–510. [DOI] [PubMed] [Google Scholar]
- 18.Kamei K, Yasuda T, Ueda T, Qiang F, Takeyama Y, and Shiozaki H. 2009. Role of triggering receptor expressed on myeloid cells-1 in experimental severe acute pancreatitis. J. Hepatobiliary Pancreat. Surg. 17: 305–312. [DOI] [PubMed] [Google Scholar]
- 19.Gibot S, Massin F, Alauzet C, Derive M, Montemont C, Collin S, Fremont S, and Levy B. 2009. Effects of the TREM 1 pathway modulation during hemorrhagic shock in rats. Shock 32: 633–637. [DOI] [PubMed] [Google Scholar]
- 20.Schenk M, Bouchon A, Seibold F, and Mueller C. 2007. TREM-1-expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J. Clin. Invest. 117: 3097–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murakami Y, Akahoshi T, Aoki N, Toyomoto M, Miyasaka N, and Kohsaka F. 2009. Intervention of an inflammation amplifier, triggering receptor expressed on myeloid cells 1, for treatment of autoimmune arthritis. Arthritis Rheum. 60: 1615–1623. [DOI] [PubMed] [Google Scholar]
- 22.Kuai J, Gregory B, Hill A, Pittman DD, Feldman JL, Brown T, Carito B, O’Toole M, Ramsey R, Adolfsson O, et al. 2009. TREM-1 expression is increased in the synovium of rheumatoid arthritis patients and induces the expression of pro-inflammatory cytokines. Rheumatology 48: 1352–1358. [DOI] [PubMed] [Google Scholar]
- 23.Collins CE, La DT, Yang H-T, Massin F, Gibot S, Faure G, and Stohl W. 2009. Elevated synovial expression of triggering receptor expressed on myeloid cells 1 in patients with septic arthritis or rheumatoid arthritis. Ann. Rheum. Dis. 68: 1768–1774. [DOI] [PubMed] [Google Scholar]
- 24.Washington AV, Schubert RL, Quigley L, Disipio T, Feltz R, Cho EH, and McVicar DW. 2004. ATREM family member, TLT-1, is found exclusively in the alpha-granules of megakaryocytes and platelets. Blood 104: 1042–1047. [DOI] [PubMed] [Google Scholar]
- 25.Barrow AD, Astoul E, Floto A, Brooke G, Relou IAM, Jennings NS, Smith KGC, Ouwehand W, Farndale RW, Alexander DR, and Trowsdale J. 2004. Cutting edge: TREM-like transcript-1, a platelet immunoreceptor tyrosine-based inhibition motif encoding costimulatory immunoreceptor that enhances, rather than inhibits, calcium signaling via SHP-2. J. Immunol. 172: 5838–5842. [DOI] [PubMed] [Google Scholar]
- 26.Washington AV, Gibot S, Acevedo I, Gattis J, Quigley L, Feltz R, De La Mota A, Schubert RL, Gomez-Rodriguez J, Cheng J, et al. 2009. TREM-like transcript-1 protects against inflammation-associated hemorrhage by facilitating platelet aggregation in mice and humans. J. Clin. Invest. 119: 1489–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gattis JL, Washington AV, Chisholm MM, Quigley L, Szyk A, McVicar DW, and Lubkowski J. 2006. The structure of the extracellular domain of triggering receptor expressed on myeloid cells like transcript-1 and evidence for a naturally occurring soluble fragment. J. Biol. Chem. 281: 13396–13403. [DOI] [PubMed] [Google Scholar]
- 28.George TC, Fanning SL, Fitzgerald-Bocarsly P, Medeiros RB, Highfill S, Shimizu Y, Hall BE, Frost K, Basiji D, Ortyn WE, et al. 2006. Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J. Immunol. Methods 311: 117–129. [DOI] [PubMed] [Google Scholar]
- 29.Ouk C, Jayat-Vignoles C, Donnard M, and Feuillard J. 2011. Both CD62 and CD162 antibodies prevent formation of CD36-dependent platelets, rosettes, and artefactual pseudoexpression of platelet markers on white blood cells: a study with ImageStream. Cytometry A 79: 477–484. [DOI] [PubMed] [Google Scholar]
- 30.Le Gall JR, Lemeshow S, and Saulnier F. 1993. A new Simplified Acute Physiology Score (SAPS II) based on a European/North American multicenter study. JAMA 270: 2957–2963. [DOI] [PubMed] [Google Scholar]
- 31.Vincent JL, Moreno R, Takala J, Willatts S, De Mendonca A, Bruining H, Reinhart CK, Suter PM, and Thijs LG. 1996. The SOFA (Sepsis-Related Organ Failure Assessment) score to describe organ dysfunction/failure: on behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 22: 707–710. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.