

Abstract
Matrix assisted laser desorption/ionization (MALDI) imaging mass spectrometry (IMS) is a powerful technology used to investigate the spatial distributions of thousands of molecules throughout a tissue section from a single experiment. As proteins represent an important group of functional molecules in tissue and cells, the imaging of proteins has been an important point of focus in the development of IMS technologies and methods. Protein identification is crucial for the biological contextualization of molecular imaging data. However, gas-phase fragmentation efficiency of MALDI generated proteins presents significant challenges, making protein identification directly from tissue difficult. This review highlights methods and technologies specifically related to protein identification that have been developed to overcome these challenges in MALDI IMS experiments.
Graphical abstract
1). Introduction
Since the introduction of matrix assisted laser desorption/ionization imaging mass spectrometry (MALDI IMS) in the late 1990’s, the technology has seen tremendous growth in utility, being employed to analyze biological substrates ranging from plants and insects, to mammalian tissues specimens [1–4]. MALDI IMS allows for the label-free, multiplex analysis of thousands of analytes across a samples surface yielding 2-dimensional molecular maps that elucidate both the localization and relative abundance of endogenous species. The technology has been used to study a wide range of analyte classes, including metabolites, drugs, lipids, peptides, and proteins [5–8]. The imaging of proteins has garnered particular attention due to the role the proteins play in cellular processes [9], and because MALDI IMS allows for the visualization of a protein and its various proteoforms (i.e. varying post-translational modifications) in a single imaging experiment [10–12]. As highlighted in Figure 1, MALDI IMS is performed by first coating a tissue section with a MALDI matrix, which assists in desorption and ionization of endogenous biomolecules during laser irradiation. Individual mass spectra are then collected at discrete x,y coordinates allowing for signal intensity maps to be plotted across the sample area creating ion images. A single MALDI IMS experiment can produce thousands of ion images, providing molecular context to classical histological analysis. Fragmentation data is often collected in separate experiments either directly from tissue [13] or by LC-MS/MS following extraction [14].
Figure 1:
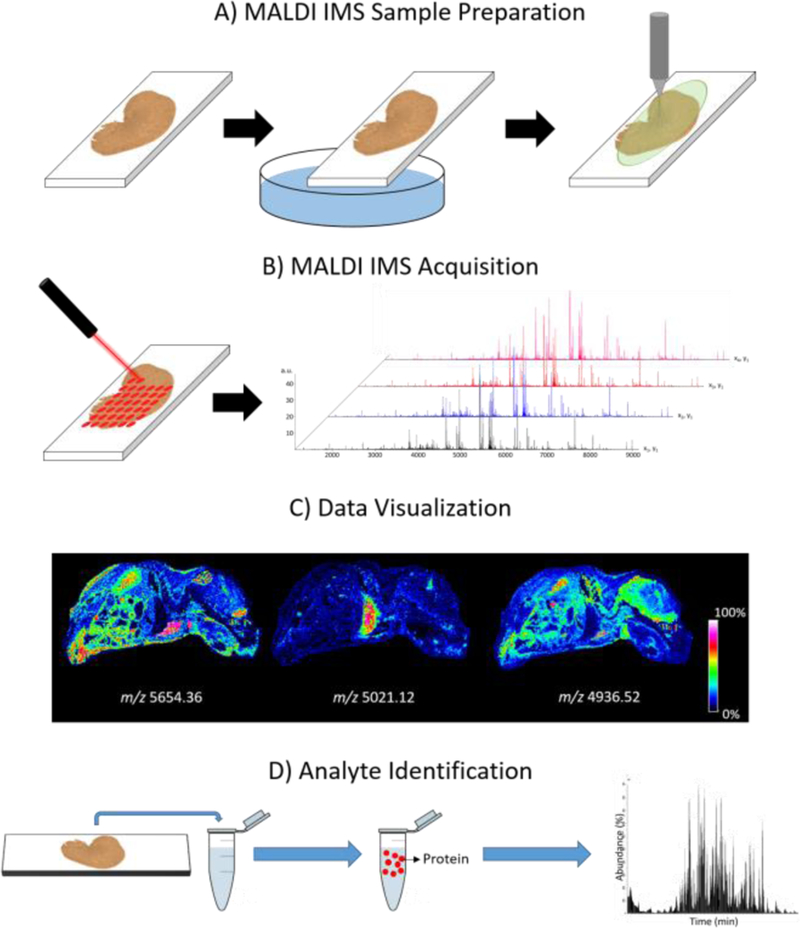
An overview of the MALDI IMS workflow for protein imaging. A) A tissue sample is sectioned, washed to remove interfering salts and lipids, and coated homogenously with a MALDI matrix. After sample preparation, the sample is loaded into the instrument and the section is irradiated by a laser, moving a defined lateral distance which dictates the spatial resolution of the image. B) A mass spectrum is generated at each pixel location. C) Ion intensities for a selected mass range are then plotted in a coordinate system within the sampled tissue area, creating an ion image. D) Following, or in parallel to IMS experiments, orthogonal experiments are completed in order to generate protein identifications that can be correlated to the imaging data.
Protein identification in MALDI IMS is crucial in helping to understand the physiological role of biomolecular and cellular systems. However, identifying proteins observed in many imaging experiments can be challenging because ALDI generated ions typically have low charge states (≤ 3), greatly reducing the gas-phase fragmentation efficiency resulting in limited sequence coverage [15,16]. In general, protein identification in mass spectrometry is performed using either bottom-up or top-down sequencing [17]. Bottom-up protein identification methods rely on enzymatic digestion to hydrolyze larger proteins into smaller peptides that are easier to fragment, resulting in higher sequence coverage [18]. In top-down methods, intact proteins are injected into the mass spectrometer and subjected to fragmentation without prior digestion [19]. Aside from better tracking of protein modifications, an advantage of top-down protein sequencing is that it complements imaging experiments by enabling mass measurement of the intact protein that relates more directly to the MALDI IMS generated signals.
For both bottom-up and top-down proteomics experiments, proteins and peptides are commonly fragmented using collision induced dissociation (CID) or electron transfer dissociation (ETD). In CID, ions are accelerated and collide with a neutral gas leading to increased internal energy of the ion. Should the deposited energy exceed the critical energy of a bond, fragmentation will occur [20]. The ‘mobile proton model’ is used to describe the dissociation of proteins and peptides in CID studies [21]. This model postulates that sequence fragments of highly charged proteins result from charge directed fragmentation after the mobilization of a proton to a carbonyl on the peptide backbone. However, MALDI primarily produces low charge state ions with few protons which tend to be sequestered on highly basic amino acid side chains (e.g. Lys and Arg). Thus, MALDI generated protein ions produce fragments with poor sequence coverage. In ETD, ions are bombarded with radical anions in an ion trap resulting in electron transfer and formation of radical cations. Once the radical cation is formed, rapid dissociation along the peptide backbone occurs resulting in informative sequence fragments [22]. ETD requires that multiple protons are present, and the fragmentation efficiency for a given protein or peptide is highly correlated to an increased charge state. For these reasons, the applicability of ETD to MALDI generated proteins is also limited.
To overcome the challenges associated with identification of MALDI generated proteins, technologies and methods have been developed to enable separate, complimentary proteomics experiments to be performed as part of IMS workflows. These orthogonal experiments are typically performed on the sample following LDI image analysis or using a serial tissue section. In general, these experiments involve the extraction of proteins from the tissue with subsequent analysis of the sample by electrospray ionization (ESI). ESI typically produces highly charged ions that are more amenable to CID and ETD fragmentation [23]. By completing these experiments offline, protein identifications can be made using more traditional proteomics workflows and the resulting identifications can be correlated to the imaging data through accurate mass matching [6,10,11,13,24,25].
2). Protein Identification Strategies in MALDI Imaging Mass Spectrometry Workflows
Analytical methods for generating protein identifications in MALDI IMS workflows must balance trade-offs between the effective spatial resolution and sensitivity with respect to the number of proteins identified. The goal is to provide complimentary data by mapping protein identifications to distinct tissue substructures or cell types in the sample. Each approach has its own unique performance characteristics including spatial resolution, throughput, intact protein/peptide compatibility, and fragmentation method compatibility. Figure 2 provides a visual description of the complimentary proteomic experiments discussed below.
Figure 2:
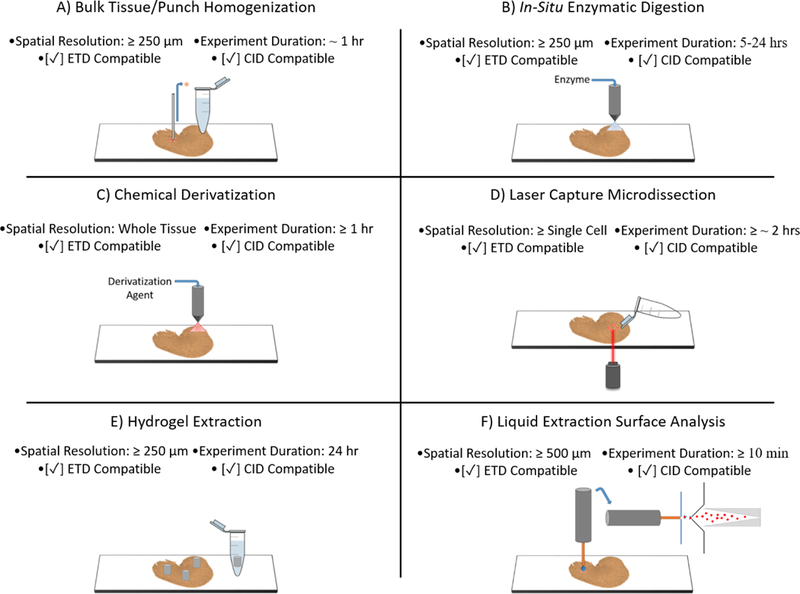
A visual representation of common orthogonal proteomics experiments used to generate protein identifications in MALDI IMS workflows. The achievable spatial resolution, experimental duration, and CID/ETD compatibility is also presented.
2A). Tissue homogenization:
Many common approaches for extracting and identifying proteins from tissue utilize bulk tissue homogenization [23,26]. A number of protocols have been developed to enhance the detection of specific types of proteins (e.g. membrane and phosphorylated proteins) [27–29], as well as quantitative workflows [30,31]. In general, the sample to be homogenized can be an unsectioned area of tissue, serial tissue section, or a punch biopsy if attempting to isolate analytes from discrete areas of tissue (> 250 µm). he sample is pulverized and undergoes a cell lysis step. Cell lysis is typically performed mechanically, [32,33] and is followed by various solvent washes to solubilize and extract the endogenous proteins. Once extracted, proteins are either analyzed directly (top-down analysis) or undergo enzymatic digestion prior to MS analysis (bottom-up). Purification procedures are typically employed to increase sensitivity (i.e. removing salts, detergents, etc.). In both bottom-up and top-down workflows the complex protein mixture is fractionated by gel electrophoresis or liquid chromatography. There are many examples of researchers employing this strategy in protein IMS workflows. For example, investigators have extracted and combined proteins from serial sections of rat brain tissue that were subsequently analyzed using top-down MS in order to link identifications to a MALDI Fourier transform ion cyclotron IMS data set using mass accuracy [10]. Recently, a tissue homogenization protocol was used in order to identify extracellular matrix proteins using collagenase type III and elastase enzymes enabling previously inaccessible collagens and elastin’s to be identified in IMS experiments [34]. Although tissue homogenization is the most utilized strategy for tissue proteomics workflows, spatial fidelity is lost in most cases, limiting its application for the analysis of discrete foci in tissue.
2B). In-Situ Enzymatic Digestions:
Ideally, proteins would be identified in an imaging experiment workflow from their host environment; that is, directly from tissue. This allows both mass accuracy and spatial information to be used to relate imaging data to protein identification experiments. As described above, MALDI generated proteins suffer from inefficient fragmentation in the gas phase. To overcome this challenge, on-tissue enzymatic digestions can be performed to generate peptides of larger proteins that are more amenable to MS/MS experiments directly from tissue. Modern approaches utilizing in-situ enzymatic digestions are often performed using robotic sprayers to apply a homogenous coating of enzyme across the tissue section or by robotically depositing small (> ~200 µm), discrete droplets of enzyme on the surface [35,36]. An incubation step is performed prior to MALDI MS analysis. Trypsin is the most commonly employed enzyme, however, other endoproteinases such as Glu-C and Asp-N have been utilized for IMS studies [37,38]. Once digested, proteins can be identified by spatially targeting and sequencing peptides using traditional MS/MS approaches directly from tissue. In an early example where trypsin was applied to lung tumor tissue micro-arrays using an automated spotter, researchers were able to differentiate specific cancer types (e.g. adenocarcinoma from squamous cell carcinoma biopsies) by peptide imaging [39]. In another approach, investigators combined MALDI IMS with ion mobility mass spectrometry and in-situ digestions from both fresh frozen and formalin fixed paraffin embedded tissue samples [13]. The use of ion mobility provided separation of isobaric species that would not normally be observed using conventional MALDI IMS, enabling higher peak capacity in the tissue-based bottom-up proteomics analysis. Enzymes other than trypsin are also being utilized for on-tissue digestion studies. Investigators profiled the distribution of multiple N-linked glycans using MALDI IMS by completing an in-situ digestion with the enzyme peptide-N-glycosidase F (PNGaseF). They went on to demonstrate the usefulness of N-glycan profiling in order to help distinguish tumor from non -tumor regions human liver tissue in a micro-array format [40] Although effective for identifying proteins, there is currently no software available to allow for automated peptide fragmentation from tissue, severely limiting throughput and data interpretation.
2C). Chemical Derivatization:
Chemical derivatization entails modification of analytes present on the surface of the tissue to enhance analytical characteristics such as sensitivity and fragmentation efficiency [41]. The majority of this work has been applied to the study of small molecule analytes to enhance sensitivity of species with low ionization efficiency. However, there are a number of studies that have used chemical derivatization of proteins to simplify the resulting fragmentation data generated from bottom-up MALDI data sets [42,43]. Protein IMS workflows have been developed that include N-terminal derivatization strategies that yields only y-type sequence ions during MALDI TOF/TOF experiments [44]. In this work a negative charge was added to the N-terminus of tryptic peptides using 3-sulfobenzoic acid succinimidyl ester (3-SBASE). This sulfonation agent was used to generate complete peptide y-fragment series from MALDI generated ions. Also, a sialic acid derivatization approach was developed for N-Glycan MALDI IMS from formalin fixed, paraffin-embedded tissues. In order to preserve the sialic acid group, which is often lost by in-source decay during ionization and ion transfer, they utilized a linkage-specific demethylation and amidation for stabilization [45]. Similar to in-situ enzymatic digestion, these methods allow for the identification of proteins directly from tissue, but are throughput-limited and are better suited for targeted studies.
2D). Laser Capture Microdissection:
Laser capture microdissection (LCM) facilitates the interrogation of tissue foci and is well suited for cellular analysis, having the capability to sample single cells from heterogeneous environments using microscopy combined with a laser targeting system. In LCM, a laser is used to perforate the tissue and effectively cut out the target region, separating it from adjacent tissue. The separated tissue is then collected using a non-contact approach such as laser induced forward transfer (LIFT). complete overview of LCM and its applications is outside the scope of this review and can be found elsewhere [46]. LCM instrumentation commonly rely on ultraviolet (UV) and/or infrared (IR) lasers for tissue perforation and collection. Once collected, the tissue sample can be subjected to homogenization protocols and bottom-up or top-down workflows. In recent studies, high resolving power FT-ICR MS with spatially targeted LCM was used to aid in identifying proteins from an IMS data set [6]. These two platforms were employed to study and identify proteins in a mouse model of glioblastoma and enabled proteins to be identified specifically from tumor and non-tumor regions of tissue [6]. In other work, a combined workflow for integrating LCM and MALDI IMS was reported using a parafilm-assisted microdissection (PAM) [47]. This approach enabled rapid and inexpensive LCM-based proteomic analyses in IMS workflows [47]. LCM allows for spatially targeted proteomic information to be gathered for specific cell types in tissue, but can suffer from poor sensitivity due to tissue loss, and requires long collection times.
2E). Hydrogel Protein Extraction:
Hydrogels are a superabsorbent polymer that are characterized by the ability to retain a large amount of liquid relative to volume and are inexpensive to fabricate. Hydrogels have found use in a diverse range of applications including microfluidics [48], wound care and drug delivery [49,50], and protein extractions [51,52]. An extensive review of hydrogel preparation has been previously described [53], but a concise overview of their utility is worthwhile discussing. Briefly, a hydrogel is cast into sheets through polymerization of water soluble monomeric units such as acrylamide. After casting, a punch biopsy is used to retrieve a gel of a specific diameter. The hydrogel can then be dehydrated and subsequently rehydrated with an enzyme solution, most commonly trypsin. The gel is then placed onto the surface of the tissue, leading to spatially targeted protein digestion and peptide extraction into the gel. Peptides are then washed from the gel and analyzed by LC-MS. Hydrogels were initially investigated with applications to IMS as a cost-effective approach to generate spatially targeted protein identifications from tissue [51]. This approach utilized laser printed molds creating an ionotropic hydrogel that were employed to extract and digest proteins from the cerebellum of a rat brain. Hydrogel fabrication was further optimized for tissue analysis using a polyacrylamide hydrogel [52]. With this strategy, investigators were able to fabricate hydrogels with diameters down to 260 µm while still identifying hundreds of protein species from rat liver and various substructures in rat brain (Figure 3) [54]. Although efficient at generating spatially targeted protein identifications, hydrogels are limited in throughput due to the lengthy preparation and incubation time required for peptide extraction, as well as difficulties when attempting to manipulate a small gel on a tissue surface.
Figure 3:
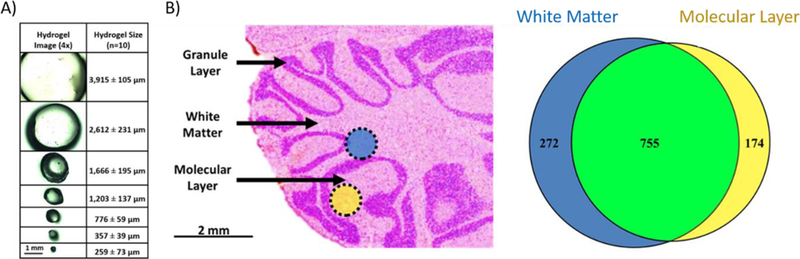
Spatial resolution and sensitivity of hydrogel-based proteomic workflows: A) Using dermal punch biopsy tools, hydrogels can be fabricated down to ~260 µm in diameter. B) Extracting peptides using a hydrogel with a diameter of 777 µm, the white matter and molecular layers of rat brain cerebellum were profiled and the proteomic profiles were compared. Figure 3 was adapted from reference [54].
2F). Liquid Extraction Surface Analysis:
Liquid micro-extractions, commonly referred to as a liquid extraction surface analysis (LESA), utilize a small volume (~0.5–3 µL) of extraction solvent that can be placed on a samples surface. present in the tissue diffuse into the solvent that is subsequently aspirated off the tissue and analyzed by traditional proteomic workflows. Analyte classes ranging from small molecule metabolites to intact proteins have been studied using LESA technology [55–57]. Initial development of the technology focused on using robotics to complete the extraction [58–60]. Modeled after work utilizing continuous-flow liquid microjunctions [61], new instrumentation allows for liquid extractions to be directly coupled with online LC-MS/MS. Several investigators have reported the utility of integrating a LESA LC-MS system into MALDI IMS workflows for the analysis of intact proteins [62,63]. Spatially targeted extractions followed by top-down LC-MS/MS enables protein identifications to be directly correlated with the species observed in the IMS data (Figure 4). A challenge to the LESA experiment is the size of the droplet diameter on tissue. Typically, LESA is limited to a spatial resolution of ~500 µm for routine use. Although robotic LESA platforms allow for robust, reproducible analysis, performing liquid micro-extractions using a hand pipette can also produce quality proteomics data. Using this approach, researchers detected upwards of 100 intact protein species from thin tissue sections by simply dispensing 1–2 µL of extraction solvent onto a tissue using a gel loading pipette tip, aspirating the solvent, and taking it offline for top-down LC-MS/MS [64]. Although limited in resolution, LESA provides a high-throughput approach to gather spatially targeted protein identifications from tissue.
Figure 4:
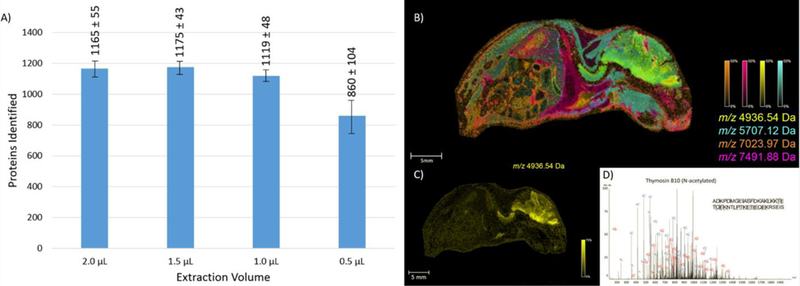
LESA sensitivity and applications: A) Extracting from enzymatically digested rat brain, the number of unique protein identifications was evaluated as a function of solvent extraction volume B) Intact protein extracts were gathered from select regions of whole sections of mouse pup. Top-down MS was utilized to generate protein identifications which could then be correlated to the IMS data through accurate mass matching. The ETD spectrum of thymosin B10 is presented along with its corresponding ion overlay. Figure 4 was adapted from citation [14].
3). Perspective:
Advancements in MALDI IMS instrumentation and sample preparation methods have enabled routine imaging of peptides and proteins from tissue. To fully elucidate the underlying biology that these images represent, it is important to employ methods for the identification of protein signals afforded from IMS experiments. Recent developments in orthogonal, spatially targeted proteomics technologies and methods have allowed for more robust identification of peptides and proteins, driving the continued use of protein IMS towards next-generation biological applications. There is a focus in the field towards combining technologies to maximize sensitivity and fragmentation efficiency at higher spatial resolutions. For example, instrument source modifications to allow for finer control over source pressure are improving sensitivity of intact protein analysis on high-performance imaging platforms [12]. Additionally, the implementation of charge state independent ion activation techniques, such as ultraviolet photodissociation, may hold the key to allowing spatially targeted fragmentation of intact proteins directly from tissue using MALDI [65,66] It is this synergistic combination of analytical technologies and methods that will enable a visualization of a greater fraction of the proteome and ultimately a deeper understanding of pathologically relevant processes. This will usher in a new era of systems biology by providing spatial contextualization of the dynamic molecular interactions and protein networks found in intact tissues, opening new possibilities for probing disease-perturbed systems and drug target discovery.
Acknowledgments
Funding: The authors acknowledge support by the NIH/NIGMS (5P41 GM103391-08).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Caprioli RM, Farmer TB, Gile J: Molecular imaging of biological samples: Localization of peptides and proteins using MALDI-TOF MS. Analytical Chemistry 1997, 69:4751–4760. [DOI] [PubMed] [Google Scholar]
- ••2.Kompauer M, Heiles S, Spengler B: Atmospheric pressure MALDI mass spectrometry imaging of tissues and cells at 1.4-mu m lateral resolution. Nature Methods 2017, 14:90–96.Using a modified MALDI source, the authors present an atmospheric pressure MALDI MSI setup capable of 1.4 µm lateral resolution and a mass resolution greater than 100,000. They achieve this by using a focusing objective with a numerical aperature of 0.9 at 337 nm and a free working distance of 18 mm in coaxial geometry, coupled to an orbitrap mass spectrometer.
- 3.Qin L, Zhang YW, Liu YQ, He HX, Han MM, Li YY, Zeng MM, Wang XD: Recent advances in matrix-assisted laser desorption/ionisation mass spectrometry imaging (MALDI-MSI) for in situ analysis of endogenous molecules in plants. Phytochemical Analysis 2018, 29:351–364. [DOI] [PubMed] [Google Scholar]
- 4.Pratavieira M, Menegasso ARD, Esteves FG, Sato KU, Malaspina O, Palma MS: MALDI Imaging Analysis of Neuropeptides in Africanized Honeybee (Apis mellifera) Brain: Effect of Aggressiveness. Journal of Proteome Research 2018, 17:2358–2369. [DOI] [PubMed] [Google Scholar]
- 5.Ly A, Buck A, Balluff B, Sun N, Gorzolka K, Feuchtinger A, Janssen KP, Kuppen PJK, van de Velde CJH, Weirich G, et al. : High-mass-resolution MALDI mass spectrometry imaging of metabolites from formalin-fixed paraffin-embedded tissue. Nature Protocols 2016, 11:1428–1443. [DOI] [PubMed] [Google Scholar]
- •6.Dilillo M, Ait-Belkacem R, Esteve C, Pellegrini D, Nicolardi S, Costa M, Vannini E, de Graaf EL, Caleo M, McDonnell LA: Ultra-High Mass Resolution MALDI Imaging Mass Spectrometry of Proteins and Metabolites in a Mouse Model of Glioblastoma. Scientific Reports 2017, 7.The authors utilized high mass accuracy, high mass resoltuion MALDI FTICR IMS to detect and observe tumor-specific proteins in a mouse model of high-grade glioma. The use of the FTICR allowed for overlapping and isobaric distributions to be resolved. Proteins detected via IMS were identified by completing regio-specific proteomics via LCM.
- •7.Duenas ME, Essner JJ, Lee YJ: 3D MALDI Mass Spectrometry Imaging of a Single Cell: Spatial Mapping of Lipids in the Embryonic Development of Zebrafish. Scientific Reports 2017, 7.The authors utilized high mass accuracy, high mass resoltuion MALDI FTICR IMS to detect and observe tumor-specific proteins in a mouse model of high-grade glioma. The use of the FTICR allowed for overlapping and isobaric distributions to be resolved. Proteins detected via IMS were identified by completing regio-specific proteomics via LCM.
- 8.Llombart V, rejo SA, Bronsoms S, Morancho A, Ma FF, Faura J, Garcia-Berrocoso T, Simats A, Rosell A, Canals F, et al. : rofiling and identification of new proteins involved in brain ischemia using MALDI-imaging-mass-spectrometry. Journal of Proteomics 2017, 152:243–253. [DOI] [PubMed] [Google Scholar]
- 9.Walther TC, Mann M: Mass spectrometry-based proteomics in cell biology. Journal of Cell Biology 2010, 190:491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spraggins JM, Rizzo DG, Moore JL, Rose KL, Hammer ND, Skaar EP, Caprioli RM: MALDI FTICR IMS of Intact Proteins: Using Mass Accuracy to Link Protein Images with Proteomics Data. Journal of the American Society for Mass Spectrometry 2015, 26:974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••11.Spraggins JM, Rizzo DG, Moore JL, Noto MJ, Skaar EP, Caprioli RM: Next-generation technologies for spatial proteomics: Integrating ultra-high speed MALDI-TOF and high mass resolution MALDI FTICR imaging mass spectrometry for protein analysis. Proteomics 2016, 16:1678–1689.The authors utilized both high speed MALDI TOF IMS and high mass accuracy, high mass resolution MALDI FTICR IMS for protein imaging mass spectrometry. Protein images were acquired at speeds >25 pixels/s and structures as small as 50 µm were resolved in cystic fibrosis lung tissue and rat brain tissue using a time-of-flight system. Alternatively, MALDI FITCR IMS was used to prduce data with high mass accuracy (< 5ppm) and high resolving power ( ca. 75,000) from human clear cell renal cell carcinoma tissue.
- 12.Prentice BM, Ryan DJ, Van de Plas R, Caprioli RM, Spraggins JM: Enhanced Ion Transmission Efficiency up to m/z 24 000 for MALDI Protein Imaging Mass Spectrometry. Edited by. Anal. Chem; 2018:5090–5099. vol 90.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stauber J, MacAleese L, Franck J, Claude E, Snel M, Kaletas BK, Wiel I, Wisztorski M, Fournier I, Heeren RMA: On-Tissue Protein Identification and Imaging by MALDI-Ion Mobility Mass Spectrometry. Journal of the American Society for Mass Spectrometry 2010, 21:338–347. [DOI] [PubMed] [Google Scholar]
- 14.Ryan DJ, Nei D, Prentice BM, Rose KL, Caprioli RM, Spraggins JM: Protein identification in imaging mass spectrometry through spatially targeted liquid micro-extractions. Rapid Communications in Mass Spectrometry 2018, 32:442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boersema PJ, Taouatas N, Altelaar AFM, Gouw JW, Ross PL, Pappin DJ, Heck AJR, Mohammed S: Straightforward and de Novo Peptide Sequencing by MALDI-MS/MS Using a Lys-N Metalloendopeptidase. Molecular & Cellular Proteomics 2009, 8:650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mekecha TT, Amunugama R, McLuckey SA: Ion trap collision-induced dissociation of human hemoglobin alpha-chain cations. Journal of the American Society for Mass Spectrometry 2006, 17:923–931. [DOI] [PubMed] [Google Scholar]
- 17.Reid GE, McLuckey SA: ‘Top down’ protein characterization via tandem mass spectrometry. Journal of Mass Spectrometry 2002, 37:663–675. [DOI] [PubMed] [Google Scholar]
- 18.Zhang YY, Fonslow BR, Shan B, Baek MC, Yates JR: Protein Analysis by Shotgun/Bottom-up Proteomics. Chemical Reviews 2013, 113:2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Catherman AD, Skinner OS, Kelleher NL: Top Down proteomics: Facts and perspectives. Biochemical and Biophysical Research Communications 2014, 445:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wells JM, McLuckey SA: Collision-induced dissociation (CID) of peptides and proteins. Biological Mass Spectrometry 2005, 402:148–185. [DOI] [PubMed] [Google Scholar]
- 21.Wysocki VH, Tsaprailis G, Smith LL, Breci LA: Special feature: Commentary - Mobile and localized protons: a framework for understanding peptide dissociation. Journal of Mass Spectrometry 2000, 35:1399–1406. [DOI] [PubMed] [Google Scholar]
- 22.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF: Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America 2004, 101:9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schey KL, Hachey AJ, Rose KL, Grey AC: MALDI imaging mass spectrometry of Pacific White Shrimp L. vannamei and identification of abdominal muscle proteins. Proteomics 2016, 16:1767–1774. [DOI] [PubMed] [Google Scholar]
- 24.Alberts D, Pottier C, Smargiasso N, Baiwir D, Mazzucchelli G, Delvenne P, Kriegsmann M, Kazdal D, Warth A, De auw E, et al. : MALDI Imaging-Guided Microproteomic Analyses of Heterogeneous Breast Tumors- Pilot Study. Proteomics Clinical Applications 2018, 12. [DOI] [PubMed] [Google Scholar]
- 25.Yajima Y, Hiratsuka T, Kakimoto Y, Ogawa S, Shima K, Yamazaki Y, Yoshikawa K, Tamaki K, Tsuruyama T: Region of Interest analysis using mass spectrometry imaging of mitochondrial and sarcomeric proteins in acute cardiac infarction tissue. Scientific Reports 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maccarrone G, Nischwitz S, Deininger SO, Hornung J, Konig FB, Stadelmann C, Turck CW, Weber F: MALDI imaging mass spectrometry analysis-A new approach for protein mapping in multiple sclerosis brain lesions. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences 2017, 1047:131–140. [DOI] [PubMed] [Google Scholar]
- 27.Svensson M, Boren M, Skold K, Falth M, Sjogren B, Andersson M, Svenningsson P, Andren PE: Heat Stabilization of the Tissue Proteome: A New Technology for Improved Proteomics. Journal of Proteome Research 2009, 8:974–981. [DOI] [PubMed] [Google Scholar]
- 28.Wu CC, MacCoss MJ, Howell KE, Yates JR: A method for the comprehensive proteomic analysis of membrane proteins. Nature Biotechnology 2003, 21:532–538. [DOI] [PubMed] [Google Scholar]
- 29.Whiteaker JR, Zhao L, Saul R, Kaczmarczyk JA, Schoenherr RM, Moore HD, Jones-Weinert C, Ivey RG, Lin CW, Hiltke T, et al. : A Multiplexed Mass Spectrometry-Based Assay for Robust Quantification of Phosphosignaling in Response to DNA Damage. Radiation Research 2018, 189:505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pelkonen L, Sato K, Reinisalo M, Kidron H, Tachikawa M, Watanabe M, Uchida Y, Urtti A, Terasaki T: LC-MS/MS Based Quantitation of ABC and SLC Transporter Proteins in Plasma Membranes of Cultured Primary Human Retinal Pigment Epithelium Cells and Immortalized ARPE19 Cell Line. Molecular Pharmaceutics 2017, 14:605–613. [DOI] [PubMed] [Google Scholar]
- 31.Overmyer KA, Tyanova S, Hebert AS, Westphall MS, Cox J, Coon JJ: Multiplexed proteome analysis with neutron-encoded stable isotope labeling in cells and mice. Nature Protocols 2018, 13:293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM: Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nature Biotechnology 2002, 20:301–305. [DOI] [PubMed] [Google Scholar]
- 33.Liu F, Rijkers DTS, Post H, Heck AJR: Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nature Methods 2015, 12:1179–+. [DOI] [PubMed] [Google Scholar]
- •34.Angel PM, Comte-Walters S, Ball LE, Talbot K, Mehta A, Brockbank KGM, Drakes RR: Mapping Extracellular Matrix Proteins in Formalin-Fixed, Paraffin-Embedded Tissues by MALDI Imaging Mass Spectrometry. Journal of Proteome Research 2018, 17:635–646.The authors investigated collagen and elastin networks in tissue using a newly developed workflow combining MALDI IMS with matrix metalloproteinase enzymes in order to observe the spatial distributions of such species in FFPE tissue. Using this new method, the authors were able to visualize the distribution of species that were previously unattainable.
- 35.Wisztorski M, Quanico J, Franck J, Fatou B, Salzet M, Fournier I: Droplet-Based Liquid Extraction for Spatially-Resolved Microproteomics Analysis of Tissue Sections. Imaging Mass Spectrometry: Methods and Protocols 2017, 1618:49–63. [DOI] [PubMed] [Google Scholar]
- 36.Lefcoski S, Kew K, Reece S, Torres MJ, Parks J, Bras LED, Virag JAI: Anatomical-Molecular Distribution of EphrinA1 in Infarcted ouse Heart Using MALDI Mass Spectrometry Imaging. Journal of the American Society for ass Spectrometry 2018, 29:527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grey AC, Schey KL: Age-Related Changes in the Spatial Distribution of Human Lens alpha-Crystallin Products by MALDI Imaging Mass Spectrometry. Investigative Ophthalmology & Visual Science 2009, 50:4319–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pote N, Alexandrov T, Le Faouder J, Laouirem S, Leger T, Mebarki M, Belghiti J, Camadro JM, Bedossa P, Paradis V: Imaging Mass Spectrometry Reveals Modified Forms of Histone H4 as New Biomarkers of Microvascular Invasion in Hepatocellular Carcinomas. Hepatology 2013, 58:983–994. [DOI] [PubMed] [Google Scholar]
- 39.Groseclose MR, Massion PR, Chaurand P, Caprioli RM: High-throughput proteomic analysis of formalin-fixed paraffin-embedded tissue microarrays using MALDI imaging mass spectrometry. Proteomics 2008, 8:3715–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powers TW, Neely BA, Shao Y, Tang HY, Troyer DA, Mehta AS, Haab BB, Drake RR: MALDI Imaging Mass Spectrometry Profiling of N-Glycans in Formalin-Fixed Paraffin Embedded Clinical Tissue Blocks and Tissue Microarrays. Plos One 2014, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toue S, Sugiura Y, Kubo A, Ohmura M, Karakawa S, Mizukoshi T, Yoneda J, Miyano H, Noguchi Y, Kobayashi T, et al. : Microscopic imaging mass spectrometry assisted by on- tissue chemical derivatization for visualizing multiple amino acids in human colon cancer xenografts. Proteomics 2014, 14:810–819. [DOI] [PubMed] [Google Scholar]
- 42.Keough T, Lacey MP, Youngquist RS: Derivatization procedures to facilitate de novo sequencing of lysine-terminated tryptic peptides using postsource decay matrix-assisted laser desorption/ionization mass spectrometry. Rapid Communications in Mass Spectrometry 2000, 14:2348–2356. [DOI] [PubMed] [Google Scholar]
- 43.Samyn B, Debyser G, Sergeant K, Devreese B, Van Beeumen J: A case study of de novo sequence analysis of N-sulfonated peptides by MALDI TOF/TOF mass spectrometry. Journal of the American Society for Mass Spectrometry 2004, 15:1838–1852. [DOI] [PubMed] [Google Scholar]
- 44.Franck J, El Ayed M, Wisztorski M, Salzet M, Fournier I: On-Tissue N-Terminal Peptide Derivatizations for Enhancing Protein Identification in MALDI Mass Spectrometric Imaging Strategies. Analytical Chemistry 2009, 81:8305–8317. [DOI] [PubMed] [Google Scholar]
- •45.Holst S, Heijs B, de Haan N, van Zeijl RJM, Briaire-de Bruijn IH, van Pelt GW, Mehta AS, Angel PM, Mesker WE, Tollenaar RA, et al. : Linkage-Specific in Situ Sialic Acid Derivatization for N-Glycan Mass Spectrometry Imaging of Formalin-Fixed Paraffin-Embedded Tissues. Analytical Chemistry 2016, 88:5904–5913.The authors developed a linkage-specific sialic acid derivatization using dimethylamidation, followed by amidation, transferred onto FF E tissue for the imaging of N-linked glycans. The use of a sialic acid derivatization not only increased the detection range, but also showed no trace of lateral diffusion during sample preparation.
- 46.Datta S, Malhotra L, Dickerson R, Chaffee S, Sen CK, Roy S: Laser capture microdissection: Big data from small samples. Histology and Histopathology 2015, 30:1255–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quanico J, Franck J, Wisztorski M, Salzet M, Fournier I: Combined MALDI Mass Spectrometry Imaging and Parafilm-Assisted Microdissection-Based LC-MS/MS Workflows in the Study of the Brain. Neuroproteomics: Methods and Protocols, 2nd Edition 2017, 1598:269–283. [DOI] [PubMed] [Google Scholar]
- 48.Liu EY, Jung S, Weitz DA, Yi HM, Choi CH: High-throughput double emulsion-based microfluidic production of hydrogel microspheres with tunable chemical functionalities toward biomolecular conjugation. Lab on a Chip 2018, 18:323–334. [DOI] [PubMed] [Google Scholar]
- 49.Li JY, Mooney DJ: Designing hydrogels for controlled drug delivery. Nature Reviews Materials 2016, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu H, Wang CY, Li C, Qin YG, Wang ZH, Yang F, Li ZH, Wang JC: functional chitosan-based hydrogel as a wound dressing and drug delivery system in the treatment of wound healing. Rsc Advances 2018, 8:7533–7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris GA, Nicklay JJ, Caprioli RM: Localized in Situ Hydrogel-Mediated Protein Digestion and Extraction Technique for on-Tissue Analysis. Analytical Chemistry 2013, 85:2717–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicklay JJ, Harris GA, Schey KL, Caprioli RM: MALDI Imaging and in Situ Identification of Integral Membrane Proteins from Rat Brain Tissue Sections. Analytical Chemistry 2013, 85:7191–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmed EM: Hydrogel: Preparation, characterization, and applications: A review. Journal of Advanced Research 2015, 6:105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •54.Rizzo DG, Prentice BM, Moore JL, Norris JL, Caprioli RM: Enhanced Spatially Resolved Proteomics Using On-Tissue Hydrogel Mediated Protein Digestion. Analytical Chemistry 2017, 89:2948–2955.In this work, a peptide extraction from tissue, via a hydrogel, was optimized and applied to study sub-regions of a rat brain. The authors were able to achieve upwards of 600 protein identifications from a gel with a diameter of 260 µm and subsequently analyzed both the white matter and molecular layer of the brain.
- •55.Wisztorski M, Desmons A, Quanico J, Fatou B, Gimeno JP, Franck J, Salzet M, Fournier I: Spatially-resolved protein surface microsampling from tissue sections using liquid extraction surface analysis. Proteomics 2016, 16:1622–1632.Using a liquid micro-junction with a detergent extraction sovlent, the athors were able to extract and identify upwards of 1,400 proteins from spot sizes of 1mm in diamter from the surface of a tissue. The were able to achieve reproducable extractions that allowed for both qualitative and quantitative measurements. They applied their method to study and differntiate different regions of rat brain.
- 56.Hall Z, Chu YJ, Griffin JL: Liquid Extraction Surface Analysis Mass Spectrometry Method for Identifying the Presence and Severity of Nonalcoholic Fatty Liver Disease. Analytical Chemistry 2017, 89:5161–5170. [DOI] [PubMed] [Google Scholar]
- 57.Griffiths RL, Randall EC, Race AM, Bunch J, Cooper HJ: Raster-Mode Continuous-Flow Liquid Microjunction Mass Spectrometry Imaging of Proteins in Thin Tissue Sections. Analytical Chemistry 2017, 89:5684–5688. [DOI] [PubMed] [Google Scholar]
- 58.Ford MJ, Van Berkel GJ: An improved thin-layer chromatography/mass spectrometry coupling using a surface sampling probe electrospray ion trap system. Rapid Communications in Mass Spectrometry 2004, 18:1303–1309. [DOI] [PubMed] [Google Scholar]
- 59.Wachs T, Henion J: A device for automated direct sampling and quantitation from solid-phase sorbent extraction cards by electrospray tandem mass spectrometry. Analytical Chemistry 2003, 75:1769–1775. [DOI] [PubMed] [Google Scholar]
- 60.Kertesz V, Van Berkel GJ: Fully automated liquid extraction-based surface sampling and ionization using a chip-based robotic nanoelectrospray platform. Journal of Mass Spectrometry 2010, 45:252–260. [DOI] [PubMed] [Google Scholar]
- 61.Van Berkel GJ, Kertesz V: Continuous-flow liquid microjunction surface sampling probe connected on-line with high-performance liquid chromatography/mass spectrometry for spatially resolved analysis of small molecules and proteins. Rapid Communications in Mass Spectrometry 2013, 27:1329–1334. [DOI] [PubMed] [Google Scholar]
- •62.Ryan DJ, Nei D, Prentice BM, Rose KL, Caprioli RM, Spraggins JM: Protein identification in imaging mass spectrometry through spatially targeted liquid micro‐extractions. Edited by: Rapid Commun Mass Spectrom; 2018:442–450. vol 32.The authors combine robotic liquid micro-extractions online with liquid chromtography to improve protein identifacation strategies in MADLI IMS. Initially optimizing the technology, the authors then imaged and detected proteins localized to regions of the brain and kidney of a mouse pup. Using a liquid micro-extraction, they generated identifications and correlated them with signals in the imaging data.
- 63.Lamont L, Baumert M, Potocnik NO, Allen M, Vreeken R, Heeren RMA, Porta T: Integration of Ion Mobility MSE after Fully Automated, Online, High-Resolution Liquid Extraction Surface Analysis Micro-Liquid Chromatography. Analytical Chemistry 2017, 89:11143–11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schey KL, Anderson DM, Rose KL: Spatially-Directed Protein Identification from Tissue Sections by Top-Down LC-MS/MS with Electron Transfer Dissociation. Analytical Chemistry 2013, 85:6767–6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klein DR, Brodbelt JS: Structural Characterization of Phosphatidylcholines Using 193 nm Ultraviolet Photodissociation Mass Spectrometry. Analytical Chemistry 2017, 89:1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parthasarathi R, He Y, Reilly JP, Raghavachari K: New Insights into the Vacuum UV Photodissociation of Peptides. Journal of the American Chemical Society 2010, 132:1606–1610. [DOI] [PubMed] [Google Scholar]
- 67.Shen M, Xiang P, Shi Y, Pu H, Yan H, Shen BH: Mass imaging of ketamine in a single scalp hair by MALDI-FTMS. Analytical and Bioanalytical Chemistry 2014, 406:4611–4616. [DOI] [PubMed] [Google Scholar]