

Abstract
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer death worldwide. Cancer stem cells (CSCs) have attracted attention as a novel therapeutic target for cancer because they play important roles in the development and aggravation of cancer. CD44 is expressed as a standard isoform (CD44s) and several variant isoforms. CD44v is a major isoform expressed on CSCs of a variety of tumors and has been extensively studied. However, HCC tissues dominantly express CD44s, whose function in CSCs remains unclear. In the present study, we investigated the roles of CD44s in CSCs of HCC. Knock‐out of the CD44 gene in HuH7 HCC cells on which only CD44s is expressed resulted in decreased spheroid formation and increased drug sensitivity. The expression of CSC marker genes, including CD133 and EpCAM, was significantly downregulated in the spheroids of CD44‐deficient cells compared with those in the spheroids of HuH7 cells. In addition, CD44 deficiency impaired antioxidant capacity, concomitant with downregulation of glutathione peroxidase 1 (GPX1) and thioredoxin. Because GPX1 uses the reduced form of glutathione (GSH) to regenerate oxidized cellular components, GSH levels were significantly increased in the CD44‐deficient cells. We also found that NOTCH3 and its target genes were downregulated in the spheroids of CD44‐deficient cells. NOTCH3 expression in HCC tissues was significantly increased compared with that in adjacent nontumor liver tissues and was correlated with CD44 expression. These results suggest that CD44s is involved in maintenance of CSCs in a HCC cell line, possibly through the NOTCH3 signaling pathway.
Keywords: cancer stem cell, CD44, hepatocellular carcinoma, NOTCH3, oxidative stress
1. INTRODUCTION
Primary liver cancer accounts for more than 30 000 deaths each year in Japan and is one of the leading causes of cancer death worldwide.1 Hepatocellular carcinoma (HCC) is the most prevalent subtype of primary liver cancer.1 Despite significant advances in HCC treatment, the recurrence rate is still high at 60%‐70%.2, 3 Thus, increasing attention is being focused on the mechanisms by which cancer cells acquire the malignant phenotype.
Accumulating evidence suggests that recurrence and metastasis in many cancers can be attributed to cancer stem cells (CSCs).4, 5 Because CSCs have the ability of self‐renewal and differentiation, CSCs are indispensable for initiating and maintaining tumor phenotypes while other cancer cells (non‐CSCs) do not have such properties.6 The existence of CSCs was first demonstrated in acute myeloid leukemia.7 Since then, using specific CSC markers, the existence of CSCs has been proven in various tumors such as brain, breast, lung, colon, and liver.8, 9, 10, 11, 12 Several studies reported that liver CSCs express several markers such as epithelial adhesion molecule (EpCAM), CD13, CD44, and/or CD133 and that the expression of these molecules on HCC cells is correlated with poor prognosis.12, 13, 14 Because CSCs display the features of tumorigenicity and resistance to conventional chemotherapy and radiotherapy, it is suggested that surviving CSCs eventually cause tumor recurrence and metastasis although conventional therapies could eliminate non‐CSCs.4, 5 Therefore, the removal of CSCs is important to cure cancer completely.
A transmembrane glycoprotein CD44 is the most frequently observed CSC marker.15 The gene encoding human CD44 consists of 20 exons, 10 of which are variant exons, and produces several isoforms.16 The CD44 variant isoform containing the variant exons 8 and 10 (CD44v) is a major CSC marker, and frequently upregulated on CSCs of many tumors such as gastric, colorectal, breast, and prostate cancers.17 Because this isoform promotes glutathione (GSH) synthesis by interacting with xCT, a glutamate‐cystine transporter, CD44v enhances antioxidant capacity in CSCs.18 In contrast, the CD44 standard isoform (CD44s) lacking variant exons is found in various cells such as mesenchymal stromal and hematopoietic cells.19 However, it has been reported that hepatic CSCs dominantly express CD44s and that CD44s is involved in epithelial mesenchymal transition (EMT).20, 21, 22 We have previously reported that CD44 expression in HCC tissues, which is assessed by an antibody against total CD44, was significantly associated with poor prognosis while no significant association was observed between the prognosis and CD44v expression, which is assessed by a CD44v‐specific antibody.23 These results prompted us to investigate the biological functions of CD44s in controlling antioxidant capacity and CSCs in HCC.
Genome editing technologies are widely used for investigating molecular functions of gene products.24 Clustered regularly interspaced short palindromic repeats/CRISPR‐associated proteins 9 (CRISPR/Cas9) system is one of the most established genome editing technologies derived from prokaryotes, where this system plays a role as an immune system against phages and plasmids.25, 26 Cas9 is an endonuclease and cleaves a specific DNA sequence indicated by a guide RNA at a position 3‐bp upstream of the trinucleotide (5′‐NGG‐3′) protospacer‐adjacent motif (PAM).24 Mutation(s) is thought to be induced during a DNA repair process known as nonhomologous end joining.24 The CRISPR/Cas9 system has proven its applicability for cancer research.27 This prompted us to study the function of CD44s in HCC cells.
In this study, we established CD44‐knocked out (CD44‐KO) cells from human HuH7 HCC cells, in which only CD44s is expressed, by means of the CRISPR/Cas9 system, and investigated the phenotypic changes in CSC properties of CD44‐KO cells.
2. MATERIALS AND METHODS
2.1. Knocking out the CD44 gene in HuH7 cells
Human hepatocellular carcinoma HuH7, HLE, HLF, HuH6, PLC/PRF/5, and HepG2 were purchased from the JCRB cell bank (Osaka, Japan) and was maintained in DMEM (Nissui Pharmaceutical; Tokyo, Japan) supplemented with 10% inactivated FBS (Sigma‐Aldrich; St. Louis, MO, USA).
Single‐guide RNA (sgRNA) targeting the CD44 gene was designed by GeneArt CRISPR Search and Design Tool (https://apps.thermofisher.com/apps/crispr/index.html), crispr grna design tool (https://www.atum.bio/eCommerce/cas9/input) and CRISPRdirect (https://crispr.dbcls.jp/). Off‐target sequence was searched by using GGGenome (https://gggenome.dbcls.jp/en/). The sequence CTACAGCATCTCTCGGACGGAGG (underline indicates PAM sequence) commonly provided by all three programs was used as the sgRNA. A double‐stranded oligo DNA harboring the sequence (Table S1) was ligated into pSpCas9(BB)‐2A‐GFP (PX458) (Addgene, #48138; Cambridge, MA, USA), which expresses both sgRNA and SpCas9.24 The plasmid DNA was transfected into HuH7 cells with Viofectin (Viogene, New Taipei City, Taiwan). It is of note that the cells used for the transfection was not sorted and thereby included both CD44‐positive and negative cells. Sequencing analysis of the genomic DNA of independent clones obtained by limited dilution was performed by BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific; Waltham, MA, USA). The HuH7 HCC cells in which the CD44 gene is knocked out were designated CD44‐KO cells.
2.2. Spheroid formation assay
HuH7 or CD44‐KO cells were plated in a ultra‐low attachment 24‐well plate (Corning; Corning, NY, USA) at 3000 cells/well and were cultured for 7 days in a spheroid culture medium, which was prepared by adding 20 ng/mL of recombinant human epidermal growth factor (PeproTech; Rocky Hill, NJ, USA), 20 ng/mL of recombinant human basic fibroblast growth factor (PeproTech), 1 × B27 (Thermo Fisher Scientific), and 0.52% methylcellulose (Sigma‐Aldrich), to Ham's F‐12 medium (Nacalai Tesque; Kyoto, Japan). The number of spheroids was counted by ImageJ software (Bethesda, MD, USA). NOTCH3‐specific siRNAs (siN3‐1 and siN3‐2) (Silencer Select siRNA s9640 and s532202, respectively, Thermo Fisher Scientific) and negative control siRNA (siNC) (Silencer Select Negative Control No. 2 siRNA, Thermo Fisher Scientific) were transfected with Lipofectamine RNAiMax (Thermo Fisher Scientific) into HuH7 cells one day before the start of spheroid culture.
2.3. Reverse transcription quantitative PCR (RT‐qPCR)
Cells in monolayer culture were prepared by culturing HuH7 or CD44‐KO cells at 6.5 × 104 cells/well in a 12‐well cell culture plate (Violamo; Osaka, Japan) for 4 days. Spheroids were prepared by plating cells at 5.0 × 104 cells/well in the ultra‐low attachment 24‐well plate, and culturing the cells in the spheroid culture medium for 7 days. Total RNA was recovered by TRIzol (Thermo Fisher Scientific). Following treatment with DNase (Nippon Gene; Tokyo, Japan), complementary DNA was synthesized by Super Script First‐Strand Synthesis for RT‐PCR Kit (Thermo Fisher Scientific). QPCR was performed by THUNDERBIRD SYBR qPCR Mix (Toyobo, Otsu, Japan) using ViiA 7 Real‐time PCR System (Thermo Fisher Scientific). Primers used in the present study were summarized in Table S1. Relative mRNA expression levels were calculated by using β‐actin as an internal control. For comparison of monolayer culture and spheroids, HPRT1 was used as an internal control instead of β‐actin, whose expression level was readily affected by the culture conditions.
2.4. Western blot analysis
Cells in monolayer culture were incubated in a 6‐cm dish (Violamo) at 5.0 × 105 cells/dish for 4 days. Spheroids were prepared by seeding cells to a Sumilon PrimeSurface 90‐mm dish (Sumitomo Bakelite; Tokyo, Japan) at 1.0 × 106 cells/dish and incubated in spheroid culture medium for 7 days. The cells were washed three times with phosphate‐buffered saline and were lysed in 150 µL RIPA buffer (50 mmol/L Tris‐HCl (pH 7.9), 150 mmol/L NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS)). After freezing the lysate at −80°C, the protein concentration was determined by the Bradford reagent (Bio‐Rad Laboratories; Hercules, CA, USA) and was adjusted with the RIPA buffer and 5 × sample buffer (60 mmol/L Tris‐HCl (pH 7.9), 14.4% β‐mercaptoethanol, 2% SDS, 25% glycerol). SDS‐polyacrylamide gel electrophoresis and immunoblotting were performed by standard procedures. Antibodies against actin (sc‐1616; Santa Cruz Biotechnology; Dallas, TX, USA), CD44 (#3570; Cell Signaling Technology; Danvers, MA, USA), glutathione peroxidase 1 (GPX1) (ab22604; Abcam; Cambridge, UK), glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) (sc‐365062; Santa Cruz Biotechnology), NOTCH1‐3 (#3608, #5732, and #5276, respectively; Cell Signaling Technology), superoxide dismutase 1 (SOD1) (sc‐11407; Santa Cruz Biotechnology), and thioredoxin (TXN) (M013‐3; MBL; Nagoya, Japan) were used.
2.5. Determination of cellular reactive oxygen species
Cellular reactive oxygen species were measured by using DCFDA‐Cellular Reactive Oxygen Species Detection Assay Kit (Abcam) with slight modifications. Cells were seeded to a dark 96‐well microplate (Thermo Fisher Scientific) at 1 × 104 cells/well and were allowed to attach overnight. The cells were washed twice with 1 × buffer solution and were incubated with 100 µL of DCFDA solution for 45 minutes at 37°C. After being washed twice with 1 × buffer solution, the cells were exposed to 0, 50, or 100 µmol/L of hydrogen peroxide (H2O2) (Nacalai Tesque) or tert‐butyl hydroperoxide (TBHP) (Abcam) for 5 hours at 37°C. Fluorescence intensity (excitation, 485 nm; emission, 535 nm) was measured by a plate reader (Tecan; Männedorf, Switzerland).
2.6. Determination of cellular GSH content
GSH and GSSG (oxidized glutathione) in cells at a confluency of approximately 80% on a 10‐cm dish were determined by GSH/GSSH Quantification Kit (Dojindo; Kumamoto, Japan) according to the manufacturer's protocol. Absorbance at 415 nm was measured by using the plate reader.
2.7. Cell viability assay
Cells were seeded to a 96‐well plate (Violamo) at 5.0 × 103 cells/well and were allowed to attach overnight. The cells were treated with 0‐20 µmol/L of sorafenib (Bayer; Leverkusen, Germany), or 0‐50 µg/mL of 5‐fluorouracil (5‐FU) (Nacalai Tesque) for 72 hours. The concentration of solvent (dimethyl sulfoxide) was kept at 0.1%. Cell counting kit‐8 (Dojindo) was used to determine cell viability according to the manufacturer's protocol. Absorbance at 450 and 600 nm was measured by using the plate reader.
2.8. Analysis of human samples
Liver specimens from 92 HCC patients (Table 1) were obtained at Tottori University Hospital between 2004 and 2013 and immediately stored in RNAlater (Qiagen; Valencia, CA, USA) at −80°C. RNA from frozen specimens was recovered and purified using TRIzol reagent and RNeasy Plus Kit (Qiagen), according to the manufacturer's instructions. CDNA synthesis and quantitative RT‐PCR were performed as described above. The clinical data of the patients were collected from medical records. Medical records were reviewed retrospectively after approval by the Institutional Review Board of our institution in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments (Institutional Review Board approval number: 18A071).
Table 1.
Clinical and demographic characteristics of hepatocellular carcinoma patients
| Number of patients | 92 |
| Age (y) | 67.2 ± 10.7a |
| Gender (male/female) | 79/13 |
| Etiology (HBV/HCV/nonB·nonC) | 40/23/29 |
| Child‐Pugh score (5/6/≥7) | 67/19/6 |
| Number of tumors (1/≥2) | 80/13 |
| Tumor size (cm) | 5.3 ± 4.1a |
| vp (0/1/2/3/NA) | 64/19/4/2/3 |
| TNM stage (IA/IB/II/IIIA/IIIB/IVA) | 8/30/38/7/7/2 |
| Fibrosis stage (HAI‐IVb) (≤3/4/NA) | 61/27/4 |
| Survival period (y) | 4.2 [2.8‐7.3]c |
Mean ± standard deviation.
Histological activity index IV (Ishak K, et al J Hepatol. 1995;22:696‐699).
Median [interquartile range].
2.9. Statistical analysis
Independent samples, of which numbers are over 3, were analyzed, and all experimental values were expressed as mean ±standard deviation (SD). The differences between the two groups were assessed by Student's t test. P value <0.05 was considered as statistically significant.
3. RESULTS
3.1. Knocking out the CD44 gene in HuH7 cells
Since HuH7 cells express only CD44s isoform,22 we knocked out the CD44 gene in HuH7 cells by employing the CRISPR/Cas9 system to clarify whether CD44s plays a role in HCC. Several clones were obtained by limited dilution of HuH7 cells transfected with plasmid DNA expressing SpCas9 and sgRNA targeting the CD44 exon 2, one of which showed 8‐bp deletion and 256‐bp insertion within each CD44 allele (Figure S1). This clone also showed remarkable decreases in expression of mRNA and protein of CD44 (Figure 1A,B). No mutation was found in the inhibitor of nuclear factor kappa B kinase subunit gamma (IKBKG) gene, which is the only one gene that was predicted to contain a potential off‐target site by searching with 12‐mer plus PAM sequence (Figure S2). We hereafter used this clone as CD44‐KO cells.
Figure 1.
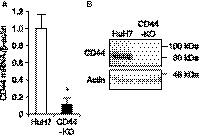
CD44 expression in HuH7 and CD44‐KO cells. A, relative mRNA expression levels of CD44 to β‐actin. *P < 0.05, vs HuH7 cells; Student's t test. B, CD44 protein expression. Actin is shown for loading control
3.2. Lowering of antioxidant capacity in CD44‐KO cells
CD44v was reported to increase cellular antioxidant capacity by activating the biosynthesis of GSH.18 High antioxidant capacity is a common feature of CSCs,18, 28, 29 prompting us to investigate the effect of CD44s deficiency on cellular antioxidant capacity. CD44‐KO cells treated with H2O2 or TBHP showed significantly higher oxidative stress than HuH7 cells at 0, 50, and 100 µmol/L of their concentrations (Figure 2A,B), suggesting that CD44s increases cellular antioxidant capacity in HuH7 cells.
Figure 2.
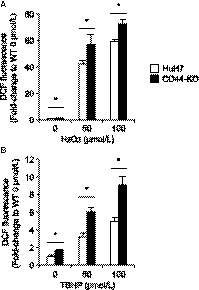
Cellular oxidative stress. A, oxidative stress in cells treated with 0‐100 µmol/L of H2O2 for 5 h. B, oxidative stress in cells treated with 0‐100 µmol/L of TBHP for 5 h. Open bars, HuH7 cells; filled bars, CD44‐KO cells. *P < 0.05; Student's t test
3.3. Decreased expression of antioxidant factors in CD44‐KO cells
The mRNA expression of antioxidant factors was determined by RT‐qPCR. In CD44‐KO cells, SOD1, TXN, GPX1, and GPX3 were significantly downregulated while SOD2 was significantly upregulated (Figure 3A). Because antioxidant capacity was lowered in CD44‐KO cells, we performed Western blotting for GPX1, TXN, and SOD1. The protein expression of GPX1 and TXN was markedly decreased while SOD1 protein expression was not changed (Figure 3B). GPX1 is an important antioxidant enzyme reducing oxidized cellular components by converting reduced GSH to the oxidized form (GSSG).30 The cellular GSH content of CD44‐KO cells was significantly increased, and the ratio of GSH to GSSG was also higher than that of HuH7 cells (Figure 3C,D), suggesting the suppression of GPX1 activity in CD44‐KO cells.
Figure 3.
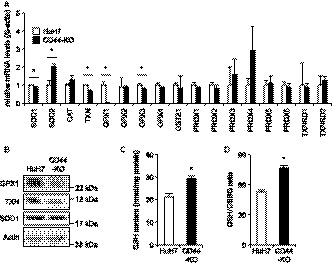
Expression of antioxidant factors. A, relative mRNA expression levels of antioxidant factors to β‐actin. B, protein expression levels of GPX1, TXN, and SOD1. Actin is shown for loading control. C, cellular GSH content. D, cellular GSH to GSSG ratio. Open bars, HuH7 cells; filled bars, CD44‐KO cells. *P < 0.05, vs HuH7 cells; Student's t test
3.4. Increased drug sensitivity of CD44‐KO cells
Drug resistance is another main feature of CSCs and is acquired by the high antioxidant capacity as well as by increased expression of drug transporters.6 As expected, CD44‐KO cells showed an increased sensitivity to sorafenib and 5‐FU at relatively high concentrations (Figure 4), suggesting that CD44s is an important factor for drug resistance in HuH7 cells.
Figure 4.
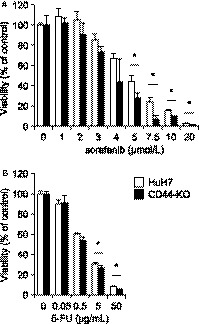
Drug sensitivity to sorafenib and 5‐FU. A, viability of cells exposed to the indicated concentrations of sorafenib for 72 h. B, viability of cells exposed to the indicated concentrations of 5‐FU for 72 h. Open bars, HuH7 cells; filled bars, CD44‐KO cells. *P < 0.05; Student's t test
3.5. Decreased cancer stemness of CD44‐KO cells
Because CSCs have the ability of anchorage‐independent growth like normal stem cells, the spheroid culture of cancer cells has been employed to assess cancer stemness and to enrich CSCs.31 This assay showed a significant decrease in cancer stemness of CD44‐KO cells (Figure 5A,B). CSC markers including CD44, CD133, and EPCAM were significantly upregulated in spheroids of HuH7 cells while such upregulation was not observed in CD44‐KO cells (Figure 5C). This observation suggests that CD44s is critical for maintaining cancer stemness in HuH7 cells.
Figure 5.
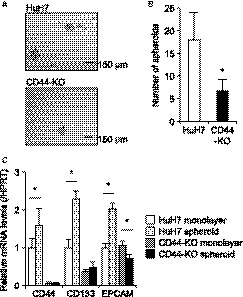
Cancer stemness. A, representative images of spheroids of HuH7 (upper panel) and CD44‐KO (lower) cells. scale bars, 150 µm. B, number of spheroids derived from HuH7 (open bar) and CD44‐KO (filled bar) cells. *P < 0.05, vs HuH7 cells; Student's t test. C, relative mRNA expression levels of CSC markers to HPRT White bars, HuH7 cells in monolayer culture; light gray bars, HuH7 spheroids; gray bars, CD44‐KO cells in monolayer culture; black bars, CD44‐KO spheroids. *P < 0.05, monolayer vs spheroids; Student's t test
3.6. Involvement of NOTCH3 in CD44‐mediated maintenance of cancer stemness
Hedgehog, WNT/β‐catenin, and NOTCH signaling pathways are well‐known and important regulatory mechanisms to maintain cancer stemness, and either of these pathways are activated in most types of CSCs.6 We determined the expression levels of target genes of hedgehog (GLI1), WNT/β‐catenin (HNF1α, CCND1, and c‐MYC), and NOTCH (HES1, HEY1, and HEYL) signaling pathways by RT‐qPCR. GLI1 was induced by the spheroid culture in HuH7 cells (Figure 6A). The expression of GLI1 was consistently high in CD44‐KO cells, but significantly decreased by the spheroid culture. Although HNF1α and CCND1 were not changed, c‐MYC was significantly upregulated by the spheroid culture of both cells (Figure 6A). The target genes of NOTCH signaling, such as HES1, HEY1, and HEYL, were significantly upregulated by the spheroid culture of HuH7 cells while their expression levels in CD44KO cells were not changed (Figure 6A).
Figure 6.
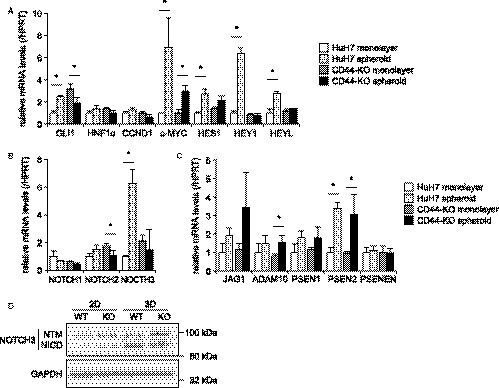
Relative mRNA expression of signaling factors to HPRT. A, hedgehog (GLI1), WNT/β‐catenin (HNF1α, CCND1, and c‐MYC), and NOTCH (HES1, HEY1, and HEYL) signaling pathways. B, NOTCH ligands. C, NOTCH signaling factors. White bars, HuH7 cells in monolayer culture; light gray bars, HuH7 spheroids; gray bars, CD44‐KO cells in monolayer culture; black bars, CD44‐KO spheroids. *P < 0.05, monolayer vs spheroids; Student's t test. D, Western blotting of cleaved transmembrane/intracellular fragment (NTM) and intracellular domain (NICD) of NOTCH3. GAPDH is shown for loading control
We next examined the expression of signaling molecules of the NOTCH pathway. Among NOTCH receptors, only NOTCH3 showed a significant change in its expression, similar to those of the target genes (Figure 6A,B). Note that NOTCH4 expression was not detected in HuH7 cells. Other components in the NOTCH pathway were not changed (Figure 6C). The expression of NOTCH intracellular domain (NICD), which is produced by the cleavage of the NOTCH receptors, is a hallmark of the activation of the NOTCH signaling.32 The expression of NOTCH3 NICD in HuH7 spheroids was higher than that in CD44‐KO spheroids (Figure 6D). NOTCH1 and NOTCH2 by the spheroid culture were unlikely activated both in HuH7 and CD44‐KO cells because cleaved bands corresponding to their NICDs were not observed (Figure S3A). In addition, siRNA‐mediated knockdown of NOTCH3 in HuH7 cells resulted in a decrease in spheroid formation (Figure S3B), suggesting that NOTCH3 is an important mediator of CD44‐mediated maintenance of cancer stemness in HuH7 cells.
3.7. Possible existence of a CD44‐NOTCH3 axis in human HCC tissue
Hepatocellular carcinoma tissues collected at Tottori University Hospital (Table 1) were used to determine the mRNA expression levels of CD44, NOTCH3, and GPX1. The expression of NOTCH3, but not GPX1, was significantly correlated with that of CD44 both in the HCC tissues (Figure 7).
Figure 7.
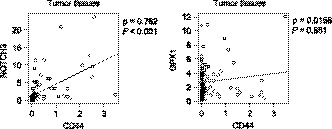
Correlation analysis of CD44 expression with NOTCH3 (right) and GPX1 (left) in HCC clinical samples. Spearman's coefficients (ρ) and P values are shown
4. DISCUSSION
Genome editing technologies including CRISPR/Cas9 have enabled us to knock‐out gene(s) in mammalian cells with high efficiency and low cost.24 It has been reported that NANOG‐deficient prostate cancer cells generated by the CRISPR/Cas9 system showed a significant decrease in tumorigenicity, compared with parental cells.27 Thus, we decided to employ this technology to study the pathological roles of CD44s in liver CSCs. However, although around 50 clones were analyzed for CD44 exon 2 sequence following the introduction of the CRISPR/Cas9 vector, we obtained only one clone that harbors the desired mutation in both alleles of the CD44 gene. Given that non‐CSCs are deficient to form a spheroid,31 the population of CSCs in HuH7 cells was assumed to be approximately 0.6% based on our spheroid formation assay (Figure 5B). This assumption suggests that the CD44 gene is expressed in only a small number of HuH7 cells. Because the targeting efficiency of transcriptionally inactive genes by CRISPR/Cas9 is low, additional treatment such as valproic acid may be necessary to enhance the knock‐out efficiency, in particular, of CSC marker genes.33
Off‐target effects are a disadvantage of the CRISPR/Cas9 system that needs to be considered, especially when knock‐out efficiency is low. Target specificity is determined mainly by a seed sequence within 12‐nt from the PAM since the introduction of a mutation into this region impaired specific cleavage by CRISPR nuclease.25, 26 Thus, we searched for potential off‐target genes by 12‐nt + PAM using an online program, by which only one gene, IKBKG was predicted (Figure S2A), possibly because the sgRNA sequence used in the present study was provided commonly by all of three online sgRNA design programs. Sequencing analysis demonstrated that no mutation was introduced into the gene, suggesting that CD44‐KO cells are valid to study the function of CD44s (Figure S2B).
Several CSC markers increase cellular antioxidant capacity.18, 28, 29 In colorectal carcinoma and neuroglioma cells, CD44s was reported to enhance NADPH production by activating the pentose‐phosphate pathway.34 This led to an increase in cellular GSH content because glutathione reductase reduces GSSG by using NADPH. CD44v reportedly also increased cellular GSH content by enhancing its biosynthesis in gastric cancer.18 Because CD44s lacks the domain interacting with xCT, which is required for the enhancement of GSH biosynthesis by CD44v, it has been unclear whether CD44s regulates redox status in HCC. Our data demonstrated that CD44s increases cellular antioxidant capacity by upregulating antioxidant enzymes, TXN and GPX1. The mechanism underlying how CD44s regulates GPX1 and TXN expression remains unknown. It was reported that a CD44s ligand, hyaluronic acid, upregulates and activates nuclear factor erythroid 2‐related factor 2 (NRF2) in bovine articular chondrocytes and then activated NRF2 further induces the expression of antioxidant enzymes including GPX1.35 The CD44s‐induced activation of NRF2 was also observed in a doxorubicin‐resistant breast cancer cell line, which possesses CSC‐like characteristics and dominantly expresses CD44s.36 It was demonstrated that NRF2 activation protects the breast cancer cells from oxidative stress and anticancer drugs.36
Among several isozymes of GPXs known in mammals, GPX1 is the most ubiquitously and abundantly expressed in most tissues, but has a limited role under a healthy condition because no obvious phenotype was observed in Gpx1 knock‐out mice.37 It was reported that docosahexaenoic acid (DHA) sensitized MDA‐MB‐231 breast cancer cell to doxorubicin by suppressing GPX1 activity.38 Of note, although oxidative stress was increased in the cells treated with doxorubicin and DHA, cellular GSH content was significantly increased.38 A similar observation was also made in Caco‐2 cell monolayer, where GSH content was increased by treating with mercaptosuccinate, a GPX inhibitor.30 In accordance with these reports, we also observed increased GSH content and GSH/GSSG ratio in CD44‐KO cells, suggesting that CD44s, unlike CD44v, enhances the consumption of GSH by activating GPX1. TXN is also another important antioxidant enzyme in normal and cancer cells.39 Increased oxidative stress due to the downregulation of GPX1 and TXN might contribute to sensitization of CD44‐KO cells to 5‐FU and sorafenib at least in part because both drugs increase oxidative stress in cells.40, 41
We found that GPX1 expression was significantly increased in HCC tissues (data not shown). However, CD44 makes little, if any, contribution to this upregulation at least in human HCC tissues, unlike in HuH7 cells, because no correlation between CD44 and GPX1 was observed (Figure 7). On the other hand, NOTCH3 expression was correlated with CD44 in the HCC tissues (Figure 7). In mammals, four NOTCH receptors (NOTCH1‐NOTCH4) are known, and reported to play roles in development, differentiation, and tumorigenesis.42 It has been demonstrated that NOTCH2 is involved in self‐renewal of CSCs in HCC.43, 44 NOTCH3 also reportedly regulates the stemness of CSCs of HCC.45, 46 Although no functional relationship between CD44s and NOTCH3 in HCC has been reported, Ma Y, et al observed that NOTCH3 was upregulated in nonsmall cell lung cancer tissues from chemoresistant patients and was positively associated with CD44.47 In addition, it was demonstrated that Her2‐negative breast tumors showed increased expression of NOTCH1 and NOTCH3.48 Knock‐down of NOTCH3 sensitized breast cancer cells to radiation more than did NOTCH1, and this effect was more prominent in CD44‐positive cells than in CD44‐negative cells.48 In the present study, it was observed that the upregulation of NOTCH3 mRNA was markedly suppressed in CD44‐KO cells while there was no significant change in the expression of NOTCH3 activators (Figure 6B,C). However, there was a discrepancy between mRNA and NICD protein expression levels of NOTCH3 in the spheroids of HuH7 and CD44‐KO cells (Figure 6B,D). These results imply that CD44 deficiency may impair the cleavage of NOTCH3 as well as its transcription. It is possible that other factors, such as ADAM17,49 might play a role in the activation of NOTCH3 in the downstream of CD44 in HCC cells. Although the detailed molecular mechanism remains to be clarified, our data suggest that NOTCH3 is an important mediator for the induction and maintenance of CSCs by CD44s in HCC as in other cancers.
The limitation of the present study is that we investigated the function of CD44s in one HCC cell line. However, although further studies should be carried out, the gene expression analysis of our cohort suggests that there is a close relationship between CD44 and NOTCH3 in HCC. Interestingly, although the target genes differed among cell lines, the upregulation of NOTCH3 as well as its target genes was observed in the spheroids of other HCC cell lines including HLE, HuH6, and PLC/PRF/5, whereas the NOTCH3 pathway was unlikely activated in those of HLF and HepG2 cells (Figure S3C). In addition, NOTCH3 was shown to be critical for the cancer stemness of HuH7 cells (Figure S3B). Considering these observations along with the significant upregulation of NOTCH3 in HCC tissues, NOTCH3 would be the therapeutic target of precision medicine for HCC.
In conclusion, we demonstrated that CD44s plays an important role in maintaining CSCs of HuH7 cells and regulates oxidative stress in HuH7 cells. CD44‐KO cells showed marked downregulation of NOTCH3 and GPX1, both of which are significantly increased in HCC tissues, compared with adjacent nontumor tissues. In addition, CD44 expression in HCC tissues was significantly correlated with NOTCH3 expression, suggesting that CD44 regulates CSC properties via NOTCH3. Because CD44 is expressed in normal cells as well, NOTCH3 may be a better therapeutic target for CSC‐directed therapy of CD44‐positive HCC. The clarification of molecular mechanism underlying CD44s‐induced NOTCH3 will deepen our understanding of CSCs in HCC and will provide new insights into the development and recurrence of HCC.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This work was supported in part by JSPS KAKENHI Grant Number JP16K09359 (HT).
Asai R, Tsuchiya H, Amisaki M, et al. CD44 standard isoform is involved in maintenance of cancer stem cells of a hepatocellular carcinoma cell line. Cancer Med. 2019;8:773–782. 10.1002/cam4.1968
REFERENCES
- 1. Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 2. Notake T, Kobayashi A, Shinkawa H, et al. Nomogram predicting long‐term survival after the diagnosis of intrahepatic recurrence of hepatocellular carcinoma following an initial liver resection. Int J Clin Oncol. 2017;22:715‐725. [DOI] [PubMed] [Google Scholar]
- 3. Shindoh J, Makuuchi M, Matsuyama Y, et al. Complete removal of the tumor‐bearing portal territory decreases local tumor recurrence and improves disease‐specific survival of patients with hepatocellular carcinoma. J Hepatol. 2016;64:594‐600. [DOI] [PubMed] [Google Scholar]
- 4. Vlashi E, Lagadec C, Vergnes L, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A. 2011;108:16062‐16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gao W, Chen L, Ma Z, et al. Isolation and phenotypic characterization of colorectal cancer stem cells with organ‐specific metastatic potential. Gastroenterology. 2013;145:636‐646. [DOI] [PubMed] [Google Scholar]
- 6. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. 2018;25:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730‐737. [DOI] [PubMed] [Google Scholar]
- 8. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396‐401. [DOI] [PubMed] [Google Scholar]
- 9. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983‐3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823‐835. [DOI] [PubMed] [Google Scholar]
- 11. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106‐110. [DOI] [PubMed] [Google Scholar]
- 12. Yamashita T, Ji J, Budhu A, Forgues M, et al. EpCAM‐positive hepatocellular carcinoma cells are tumor‐initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma S, Chan KW, Hu L, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542‐2556. [DOI] [PubMed] [Google Scholar]
- 14. Zhu Z, Hao X, Yan M, et al. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. 2010;126:2067‐2078. [DOI] [PubMed] [Google Scholar]
- 15. Zöller M. CD44: can a cancer‐initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11:254‐267. [DOI] [PubMed] [Google Scholar]
- 16. Prochazka L, Tesarik R, Turanek J. Regulation of alternative splicing of CD44 in cancer. Cell Signal. 2014;26:2234‐2239. [DOI] [PubMed] [Google Scholar]
- 17. Yan Y, Zuo X, Wei D. Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl Med. 2015;4:1033‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(‐) and thereby promotes tumor growth. Cancer Cell. 2011;19:387‐400. [DOI] [PubMed] [Google Scholar]
- 19. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signaling regulators. Nat Rev Mol Cell Biol. 2003;4:33‐45. [DOI] [PubMed] [Google Scholar]
- 20. Brown RL, Reinke LM, Damerow MS, et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial‐mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121:1064‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dang H, Steinway SN, Ding W, Rountree CB. Induction of tumor initiation is dependent on CD44s in c‐Met+ hepatocellular carcinoma. BMC Cancer. 2015;15:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okabe H, Ishimoto T, Mima K, et al. CD44s signals the acquisition of the mesenchymal phenotype required for anchorage‐independent cell survival in hepatocellular carcinoma. Br J Cancer. 2014;110:958‐966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sakabe T, Azumi J, Umekita Y, et al. Prognostic relevance of miR‐137 in patients with hepatocellular carcinoma. Liver Int. 2017;37:271‐279. [DOI] [PubMed] [Google Scholar]
- 24. Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc. 2013;8:2281‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim HM, Haraguchi N, Ishii H, et al. Increased CD13 expression reduces reactive oxygen species, promoting survival of liver cancer stem cells via an epithelial‐mesenchymal transition‐like phenomenon. Ann Surg Oncol. 2011;19:S539‐S548. [DOI] [PubMed] [Google Scholar]
- 26. Piao LS, Hur W, Kim TK, et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. 2012;315:129‐137. [DOI] [PubMed] [Google Scholar]
- 27. Rao RK, Li L, Baker RD, et al. Glutathione oxidation and PTPase inhibition by hydrogen peroxide in Caco‐2 cell monolayer. Am J Physiol Gastrointest Liver Physiol. 2000;279:332‐340. [DOI] [PubMed] [Google Scholar]
- 28. Cao L, Zhou Y, Zhai B, et al. Sphere‐forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol. 2011;11:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Giovannini C, Bolondi L, Gramantieri L. Targeting Notch3 in Hepatocellular Carcinoma: Molecular Mechanisms and Therapeutic Perspectives. Int J Mol Sci. 2016;18:E56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawamura N, Nimura K, Nagano H, et al. CRISPR/Cas9‐mediated gene knockout of NANOG and NANOGP8 decreases the malignant potential of prostate cancer cells. Oncotarget. 2015;6:22361‐22374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takayama K, Igai K, Hagihara Y, et al. Highly efficient biallelic genome editing of human ES/iPS cells using a CRISPR/Cas9 or TALEN system. Nucleic Acids Res. 2017;45:5198‐5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fu Y, Foden JA, Khayter C, et al. High‐frequency off‐target mutagenesis induced by CRISPR‐Cas nucleases in human cells. Nat Biotechnol. 2013;31:822‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pattanayak V, Lin S, Guilinger JP, et al. High‐throughput profiling of off‐target DNA cleavage reveals RNA‐programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tamada M, Nagano O, Tateyama S, et al. Modulation of Glucose Metabolism by CD44 Contributes to Antioxidant Status and Drug Resistance in Cancer Cells. Cancer Res. 2012;72:1438‐1448. [DOI] [PubMed] [Google Scholar]
- 35. Onodera Y, Teramura T, Takehara T, et al. Hyaluronic acid regulates a key redox control factor Nrf2 via phosphorylation of Akt in bovine articular chondrocytes. FEBS Open Bio. 2015;5:476‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ryoo IG, Choi BH, Ku SK, et al. High CD44 expression mediates p62‐associated NFE2L2/NRF2 activation in breast cancer stem cell‐like cells: Implications for cancer stem cell resistance. Redox Biol. 2018;17:246‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ho YS, Magnenat JL, Bronson RT, et al. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem. 1997;272:16644‐16651. [DOI] [PubMed] [Google Scholar]
- 38. Vibet S, Goupille C, Bougnoux P, et al. Sensitization by docosahexaenoic acid (DHA) of breast cancer cells to anthracyclines through loss of glutathione peroxidase (GPx1) response. Free Radic Biol Med. 2008;44:1483‐1491. [DOI] [PubMed] [Google Scholar]
- 39. Yoshihara E, Masaki S, Matsuo Y, et al. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol. 2014;4:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lamberti M, Porto S, Marra M, et al. 5‐Fluorouracil induces apoptosis in rat cardiocytes through intracellular oxidative stress. J Exp Clin Cancer Res. 2012;31:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coriat R, Nicco C, Chéreau C, et al. Sorafenib‐induced hepatocellular carcinoma cell death depends on reactive oxygen species production in vitro and in vivo. Mol Cancer Ther. 2012;11:2284‐2293. [DOI] [PubMed] [Google Scholar]
- 42. Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013;341:41‐45. [DOI] [PubMed] [Google Scholar]
- 43. Wu WR, Zhang R, Shi XD, et al. Notch2 is a crucial regulator of self‐renewal and tumorigenicity in human hepatocellular carcinoma cells. Oncol Rep. 2016;36:181‐188. [DOI] [PubMed] [Google Scholar]
- 44. Zhu P, Wang Y, Du Y, et al. C8orf4 negatively regulates self‐renewal of liver cancer stem cells via suppression of NOTCH2 signalling. Nat Commun. 2015;6:7122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Q, Lu C, Fang T, et al. Notch3 functions as a regulator of cell self‐renewal by interacting with the β‐catenin pathway in hepatocellular carcinoma. Oncotarget. 2015;6:3669‐3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu C, Liu L, Chen X, et al. LSD1 stimulates cancer‐associated fibroblasts to drive Notch3‐dependent self‐renewal of liver cancer stem‐like cells. Cancer Res. 2018;78:938‐949. [DOI] [PubMed] [Google Scholar]
- 47. Ma Y, Li M, Si J, et al. Blockade of Notch3 inhibits the stem‐like property and is associated with ALDH1A1 and CD44 via autophagy in non‐small lung cancer. Int J Oncol. 2016;48:2349‐2358. [DOI] [PubMed] [Google Scholar]
- 48. Hirose H, Ishii H, Mimori K, et al. Notch pathway as candidate therapeutic target in Her2/Neu/ErbB2 receptor‐negative breast tumors. Oncol Rep. 2010;23:35‐43. [PubMed] [Google Scholar]
- 49. Hong SW, Hur W, Choi JE, et al. Role of ADAM17 in invasion and migration of CD133‐expressing liver cancer stem cells after irradiation. Oncotarget. 2016;7:23482‐23497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ishak K, Baptista A, Bianchi L, et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22:696–699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials