

Abstract
During development, neurons require highly integrated metabolic machinery to meet the large energy demands of growth, differentiation, and synaptic activity within their complex cellular architecture. Dendrites/axons require anterograde trafficking of mitochondria for local ATP synthesis to support these processes. Acute energy depletion impairs mitochondrial dynamics, but how chronic energy insufficiency affects mitochondrial trafficking and quality control during neuronal development is unknown. Because iron deficiency impairs mitochondrial respiration/ATP production, we treated mixed-sex embryonic mouse hippocampal neuron cultures with the iron chelator deferoxamine (DFO) to model chronic energetic insufficiency and its effects on mitochondrial dynamics during neuronal development. At 11 days in vitro (DIV), DFO reduced average mitochondrial speed by increasing the pause frequency of individual dendritic mitochondria. Time spent in anterograde motion was reduced; retrograde motion was spared. The average size of moving mitochondria was reduced, and the expression of fusion and fission genes was altered, indicating impaired mitochondrial quality control. Mitochondrial density was not altered, suggesting that respiratory capacity and not location is the key factor for mitochondrial regulation of early dendritic growth/branching. At 18 DIV, the overall density of mitochondria within terminal dendritic branches was reduced in DFO-treated neurons, which may contribute to the long-term deficits in connectivity and synaptic function following early-life iron deficiency. The study provides new insights into the cross-regulation between energy production and dendritic mitochondrial dynamics during neuronal development and may be particularly relevant to neuropsychiatric and neurodegenerative diseases, many of which are characterized by impaired brain iron homeostasis, energy metabolism and mitochondrial trafficking.
SIGNIFICANCE STATEMENT This study uses a primary neuronal culture model of iron deficiency to address a gap in understanding of how dendritic mitochondrial dynamics are regulated when energy depletion occurs during a critical period of neuronal maturation. At the beginning of peak dendritic growth/branching, iron deficiency reduces mitochondrial speed through increased pause frequency, decreases mitochondrial size, and alters fusion/fission gene expression. At this stage, mitochondrial density in terminal dendrites is not altered, suggesting that total mitochondrial oxidative capacity and not trafficking is the main mechanism underlying dendritic complexity deficits in iron-deficient neurons. Our findings provide foundational support for future studies exploring the mechanistic role of developmental mitochondrial dysfunction in neurodevelopmental, psychiatric, and neurodegenerative disorders characterized by mitochondrial energy production and trafficking deficits.
Keywords: dendrite, energy metabolism, iron deficiency, mitochondria dynamics, mitochondria motility, mitochondria trafficking
Introduction
The newborn human brain accounts for 60% of total body oxygen consumption (Kuzawa, 1998). The dynamic functions of developing neurons require an integrated metabolic system to meet this high energetic demand throughout the cell. The mitochondrion is the cellular hub of energy metabolism. In developing neurons, mitochondria are involved in many critical functions, including mitochondrial respiration/ATP production, calcium buffering, apoptosis regulation, heme and Fe/S cluster biosynthesis, and ROS signaling/quenching (Mattson et al., 2008; Mishra and Chan, 2016). These functions are tightly regulated in time and space through three main processes: mitochondrial trafficking, fusion/fission, and recycling (mitophagy).
Mitochondrial trafficking to distal sites of high-energy demand is a critical component of normal neurophysiology and the pathophysiology of several neurodevelopmental and neurodegenerative disorders (Mattson et al., 2008; Sheng and Cai, 2012; Lin and Sheng, 2015). During neuronal development, mitochondria are recruited to sites of dendritic outgrowth/branching and spine formation. Disruption of this trafficking impairs dendritic arborization and synapse formation, resulting in structural abnormalities (Li et al., 2004; Dickey and Strack, 2011; Fukumitsu et al., 2015, 2016; López-Doménech et al., 2016; Das Banerjee et al., 2017). The signaling pathways and trafficking machinery that regulate dendritic and axonal mitochondrial trafficking have been extensively reviewed (Mattson et al., 2008; Sheng and Cai, 2012; Lin and Sheng, 2015). Mitochondria are mainly trafficked along microtubules by ATPase-dependent kinesin and dynein motor proteins and dendrite- and axon-specific adaptor proteins. In distal dendrites (the focus of this study) and the entire axon, the plus-end of microtubules is oriented toward the growth cone and kinesins mediate anterograde transport while dyneins mediate retrograde transport (Baas et al., 1988). In proximal dendrites, microtubule polarity is mixed and motor proteins are not selective for either anterograde or retrograde transport (Baas et al., 1988).
Fusion allows for exchange of mitochondrial components between damaged and healthy mitochondria to mitigate the effects of cellular stressors. The GTPases Optic atrophy 1 (Opa1) and Mitofusion 1 (Mfn1) and 2 control mitochondrial membrane fusion. Dynamin related protein 1 (Drp1) mediates mitochondrial fission after recruitment by fission adaptor proteins Mitochondrial fission factor (Mff), Mitochondrial fission 1 (Fis1), and Mitochondrial dynamics proteins of 49 and 51 kDa (Mid49 and Mid51). Fission, in concert with fusion, controls mitochondrial size to optimize mitochondrial ATP production and trafficking. Fission also promotes mitophagy by sequestering damaged mitochondrial components for degradation. Disruption of fusion, fission, or mitophagy genes alters axonal/dendritic mitochondrial trafficking and impairs neuronal maturation and synaptic function (Li et al., 2004; Dickey and Strack, 2011; Fukumitsu et al., 2016; Das Banerjee et al., 2017).
Significant crosstalk exists between mitochondrial quality control, dynamics and neuronal energy requirements. Iron is a critical component of the developing brain's energy machinery because it is required for the activity of multiple tricarboxylic acid cycle and electron transport chain enzymes (Dallman, 1986). We have previously shown that chronic iron deficiency (ID) impairs mitochondrial respiration and dendritic complexity without severely impairing overall neuronal health (Bastian et al., 2016), and thus provides a unique model to study the effects of ongoing energy insufficiency on mitochondrial dynamics during neuronal development. From a human health perspective, early-life ID affects ∼40%-50% of fetuses and children, resulting in long-term neurobehavioral dysfunction (Yip, 1994; McLean et al., 2009; Georgieff, 2011; Chen et al., 2013). Preclinical models of ID anemia and hippocampal neuronal-specific ID during development result in impaired brain energy metabolism, altered neuronal dendritic structure, reduced electrophysiologic capacity/synaptic efficacy, and long-lasting neurocognitive abnormalities (de Deungria et al., 2000; Jorgenson et al., 2003, 2005; Carlson et al., 2009; Brunette et al., 2010; Fretham et al., 2012; Pisansky et al., 2013; Bastian et al., 2016). Understanding the cellular mechanisms underlying these neurodevelopment-based abnormalities is imperative to improving long-term neurobehavioral function following early-life ID.
We hypothesized that metabolic insufficiency induced by chronic iron chelation in developing neurons would impair mitochondrial motility in distal growing dendrites, providing insight into interactions between energy metabolism, mitochondrial trafficking, and neuronal structural maturation.
Materials and Methods
Animals.
CD1 mice (Charles River Laboratories) were given free access to food and drinking water and were housed at constant temperature and humidity on a 12 h light/dark cycle. All animal procedures were conducted in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and in accordance with the principles and procedures outlined in the National Institutes of Health's Guide for the care and use of laboratory animals. The University of Minnesota Institutional Animal Care and Use Committee approved all animal procedures.
Cell culture.
Our primary hippocampal neuron culture model of developmental ID was previously characterized (Bastian et al., 2016). Briefly, primary hippocampal neuron cultures were prepared from mixed-sex embryonic day 16 (E16) embryos of CD1 mice at 170 cells/mm2 in 35 mm dishes containing five acid-washed, poly-D-lysine/laminin-coated glass coverslips (for immunohistochemistry and qPCR experiments). For live imaging experiments, special dishes were prepared in-house. Briefly, a 20 mm hole was drilled in the bottom center of a 35 mm dish, and a 25 mm acid-washed German glass coverslip was glued to the dish bottom with aquarium glue. Dishes were dried for several hours or overnight, then rinsed 3 times to remove excess glue, and coated with poly-D-lysine/laminin.
Beginning at 3 DIV, cultures were treated with 10 μm deferoxamine (DFO, Cayman Chemical #14595;), a high-affinity iron chelator (Liu and Hider, 2002), and 5-FU (67.5 μm 5-fluoro-2′-deoxyuridine (Sigma-Aldrich #F0503; Burlington, MA)/136 μm uridine (Sigma-Aldrich #U6381; Burlington, MA)) to inhibit glia proliferation as described previously (Bastian et al., 2016). Previous dose–response experiments (Bastian et al., 2016) demonstrated that this DFO dose creates a similar degree of functional neuronal ID as in the brains of neonatal iron-deficient rodents (Carlson et al., 2007, 2009) and human neonates (Petry et al., 1992).
Neuronal bioenergetics.
At 11 DIV, real-time oxygen consumption rate (OCR), a sensitive measure of cellular respiration, and extracellular acidification rate (ECAR), a sensitive measure of cellular glycolysis, were simultaneously measured using a Seahorse XFe24 Extracellular Flux Analyzer (Agilent Technologies) at baseline and following treatment with 1 μm oligomycin (ATP synthase inhibitor), 4 μm FCCP (uncouples oxygen consumption from ATP production), and 1 μm antimycin A/rotenone (electron transport chain complex III and I inhibitors, respectively) as described previously (Bastian et al., 2016). This enables calculation of key indicators of mitochondrial and glycolytic function, including basal respiration, maximal respiration, ATP-coupled respiration, spare respiratory capacity, coupling efficiency, cellular respiratory control ratio, glycolytic rate, glycolytic capacity, and glycolytic reserve, as described previously (Wu et al., 2007; Bastian et al., 2016).
Cellular ATP assay.
To measure intracellular ATP levels, 5000 hippocampal cells were grown on poly-D-lysine/laminin-coated 96-well white-wall tissue culture plates. At 11 DIV, to eliminate extracellular sources of ATP, the media was briefly removed, and replaced with 0.2% Triton X-100 in PBS containing Quant-iT PicoGreen Reagent (1:200; Thermo Fisher Scientific). The total DNA content of each well (as a surrogate for total cell number) was determined by measuring the dsDNA-specific fluorescence, according to the manufacturer's instructions, with a fluorescent microplate reader (Molecular Devices). Next, an equal volume of CellTiter-Glo 2.0 reagent (Promega) was added, the plate was mixed, and luminescence was measured with a microplate reader. To determine intracellular ATP concentrations, a standard curve, with ATP concentrations ranging from 0 to 400 ng/μl (diluted in 0.2% Triton X-100), was included on each plate. ATP concentrations were normalized to the relative DNA content for each well. DFO treatment does not alter cellular density at 11 DIV (Bastian et al., 2016; and data not shown), the main time point assessed in this study.
Plasmids and neuronal transfection.
To enable visualization of individual neurons, an hrGFP expressing plasmid (Bastian et al., 2016) or a plasmid expressing an eGFP-tagged intrabody specific for PSD95 (pCAG_PSD95.FingR-eGFP-CCR5TC was a gift from Don Arnold ((Gross et al., 2013); Addgene plasmid # 46295; Cambridge, MA)) was electroporated into hippocampal cells prior to plating using an Amaxa Nucleofector II system and the Lonza Mouse Neuron Nucleofector Kit (Lonza #VAPG-1001; Basel, Switzerland) as previously described (Bastian et al., 2016). To visualize mitochondria, cells were also electroporated with the mCherry-mito-7 plasmid (Addgene plasmid #55102), a gift from Michael Davidson (Olenych et al., 2007). Electroporated cells were then mixed 1:1 with nonelectroporated cells before plating.
Live imaging and mitochondrial motility analysis.
Two independent time-lapse live-cell imaging experiments were performed on 11 DIV neurons coexpressing mCherry-Mito-7 and hrGFP. Dishes were transferred to a microscope humidified stage incubator that maintains the cultures at 37°C and 5% CO2. Two-channel images of terminal dendrite segments were collected every 5 s for 3 min using a Carl Zeiss Axiovert 200M microscope with 63× plan-apo objective (NA = 1.4) and MicroManager version 1.4 software (Edelstein et al., 2014) or every 1 s for 3 min using a Carl Zeiss “Axio Observer” Z.1 with a 63× plan-apo objective (NA = 1.4).
Mitochondrial motility was analyzed in individual terminal dendrite segments using FIJI (Schindelin et al., 2012) and the semiautomated KymoAnalyzer version 1.01 suite of plugins (Neumann et al., 2017). KymoAnalyzer parameters were adjusted for mitochondria-specific motility features as suggested by the authors. The threshold for the detection of mobile versus stationary tracks and for defining a directional switch was increased from 350 nm to 2 μm. The specific motility outcomes that were analyzed are outlined and defined in Table 1.
Table 1.
Mitochondrial motility parameter definitions
| Parameter | Definition | Figure |
|---|---|---|
| Anterograde density | No. of mitochondria with only anterograde motion and/or pauses per micrometer | — |
| Retrograde density | No. of mitochondria with only retrograde motion and/or pauses per micrometer | — |
| Reversal density | No. of moving mitochondria with at least one reversal of direction per micrometer | — |
| Moving density | No. of moving mitochondria per micrometer (sum of anterograde, retrograde, and reversal densities) | — |
| Stationary density | No. of stationary mitochondria per micrometer (<2 μm movement from starting position) | — |
| Total density | Total number of mitochondria per micrometer (sum of moving and stationary densities) | — |
| Average speed | The average speed of individual moving mitochondria regardless of direction, including pause segments, and including reversal tracks with no net movement | 3A |
| Net velocity | Average net velocity of all moving mitochondria with net motion regardless of direction | 3A |
| Net anterograde velocity | Net velocity (net distance traveled/time) of mitochondria with net anterograde motion | 3A |
| Net retrograde velocity | Net velocity (net distance traveled/time) of mitochondria with net retrograde motion | 3A |
| Average segmental speed | The average combined segmental speed regardless of direction | 3B |
| Anterograde segmental velocity | Velocity of combined anterograde segments | 3B |
| Retrograde segmental velocity | Velocity of combined retrograde segments | 3B |
| Average run length | Average length of combined segmental runs regardless of direction | — |
| Anterograde run length | Length of anterograde combined segmental runs | — |
| Retrograde run length | Length of retrograde combined segmental runs | — |
| Percent time moving | Percentage of total time spent in either anterograde or retrograde motion for all moving mitochondria | 4A |
| Percent time anterograde motion | Percentage of total time spent in anterograde motion for all moving mitochondria | 4A |
| Percent time retrograde motion | Percentage of total time spent in retrograde motion for all moving mitochondria | 4A |
| Percent time pausing | Percentage of total time spent pausing for all moving mitochondria | 4A |
| Total pause frequency | Average number of pauses per moving mitochondria regardless of direction preceding or following the pause | 4B |
| Anterograde pause frequency | No. of pauses per moving mitochondria with anterograde motion preceding and following the pause | 4B |
| Retrograde pause frequency | No. of pauses per moving mitochondria with retrograde motion preceding and following the pause | 4B |
| Reversal pause frequency | No. of pauses per moving mitochondria with a change in direction following the pause | 4B |
| Total pause duration | Average duration of all pauses for a given moving mitochondria regardless of direction preceding or following the pause | — |
| Anterograde pause duration | Average duration of pauses with anterograde motion preceding and following the pause, per moving mitochondria | — |
| Retrograde pause duration | Average duration of pauses with retrograde motion preceding and following the pause, per moving mitochondria | — |
| Reversal pause duration | Average duration of pauses with a change in direction following the pause, per moving mitochondria | — |
Dendritic mitochondrial density analysis.
At 18 DIV, hippocampal cultures on 12 mm coverslips were fixed and MAP2 immunocytochemistry was performed as described with minor modifications (Bastian et al., 2016). In this study, the AMCA-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch Laboratories #715-155-151, RRID:AB_2340807) secondary antibody was used at 1:100 to visualize dendrites in the blue channel. Three-channel images of dendritic arbors for individual hrGFP/mCherry-expressing neurons were collected with a Carl Zeiss Axiovert 200M microscope with 100× plan-neofluor objective (NA = 1.3) and MicroManager version 1.4 software. Images of terminal dendrite branch segments were isolated and the GFP and mCherry signals thresholded using FIJI and the “adaptiveThr” plugin (Tseng, 2015). The “Analyze Particles” function (size >0.05 μm2) was used on the thresholded images to clear the remaining background. The resulting dendrite and mitochondria masks were selected and the area of each measured. The mitochondrial areal density was calculated as the percent of dendrite area taken up by mitochondria area.
mRNA expression analysis.
Total RNA was extracted from hippocampal cultures at 11 DIV and 18 DIV using a Quick-RNA MicroPrep kit (Zymo Research) or NucleoSpin RNA XS (Macherey-Nagel) according to the manufacturers' protocol as previously described (Bastian et al., 2016). cDNA was synthesized from 100 to 500 ng total RNA using a High Capacity RNA-to-cDNA Kit (Applied Biosystems). qRT-PCR was performed using a 2× FastStart Universal Probe Master (Rox) kit (Roche Applied Science) and Stratagene MX3000P or Applied Biosystems QuantStudio3 qPCR machines. PCRs were performed on cDNA equivalent to 3–10 ng of total RNA according to the manufacturer's protocol, except that a final volume of 10 μl was used. TaqMan qPCR probes for the genes assessed are described in Table 2. Relative mRNA levels were calculated as described previously (Bastian et al., 2016, 2017) and were normalized to a reference gene, TATA box binding protein, Tbp, which has been shown to be stable in our hippocampal neuron cultures with various conditions that deplete intracellular iron (Bastian et al., 2016, 2017).
Table 2.
TaqMan qPCR probes
| Gene name | GenBank mRNA accession no. | TaqMan probe assay ID | Functional category |
|---|---|---|---|
| Drp1 | NM_001025947.2, NM_001276340.1, NM_001276341.1, NM_152816.3, NR_075074.1 | Mm01342903_m1 | Mitochondrial/peroxisomal fission |
| Fis1 | NM_001163243.1, NM_025562.3 | Mm00481580_m1 | Mitochondrial fission |
| Mff | NM_029409.2 | Mm01273401_m1 | Mitochondrial/peroxisomal fission |
| Mfn1 | NM_024200.4 | Mm00612599_m1 | Mitochondrial fusion |
| Mfn2 | NM_001285920.1, NM_001285921.1, NM_001285922.1, NM_001285923.1, NM_133201.3 | Mm00500120_m1 | Mitochondrial fusion |
| Opa1 | NM_001199177.1, NM_133752.3 | Mm01349707_g1 | Mitochondrial fusion |
| Tbp | NM_013684.3 | Mm00446971_m1 | Basal transcription factor (reference gene) |
Experimental design and statistical analyses.
For each individual outcome measure, the data from two or three independent cultures were pooled, and statistical outliers were identified using the ROUT test with Q = 1%. Student's t test (α = 0.05) was used to determine differences between experimental groups for each parameter. When variances were unequal, as determined by F test with α = 0.01, Welch's correction was applied. When multiple null hypotheses were tested on a single dataset family, the false discovery rate (FDR) method (with Q = 5%) of Benjamini et al. (2006) was used to adjust for multiple comparisons and determine which p values could be considered significant “discoveries.” Discoveries are denoted with asterisks in each graph. All data are presented as mean ± SEM. Statistical analyses and data graphing were performed using Prism (GraphPad Software) software.
Results
Neuronal energy metabolism
We previously showed that our hippocampal neuron culture model of ID creates a similar degree of functional neuronal ID as in the brains of neonatal iron-deficient rodents (Carlson et al., 2007, 2009) and human neonates (Petry et al., 1992) and causes blunted hippocampal neuron mitochondrial respiration and glycolytic rates at 18 DIV (Bastian et al., 2016), during the period of peak dendritic arborization and synaptogenesis. Mitochondrial respiration, due to oxidative phosphorylation, is the main determinant of cellular OCR (Wu et al., 2007). ECAR is predominantly controlled by lactic acid formation and thus is a specific read-out of glycolysis (Wu et al., 2007). Therefore, to determine the effect of neuronal iron chelation on mitochondrial and glycolytic energy metabolism during the beginning stage of dendritic branching and synaptogenesis (i.e., 11 DIV), real-time OCR and ECAR were measured in untreated or DFO-treated neurons at 11 DIV (Fig. 1A). DFO treatment impaired overall mitochondrial respiration (Fig. 1A), reducing basal respiration, maximal respiration, ATP-coupled OCR, and spare respiratory capacity by 31%–62% (Fig. 1B), without altering ATP coupling efficiency (78.0 ± 2.7% vs 77.3 ± 5.0%; t(36) = 0.12, p = 0.91, unpaired t test). DFO-treated neurons had a significantly lower cellular respiratory control ratio compared with control neurons (2.25 ± 0.15 vs 2.84 ± 0.19, t(34) = 2.31, p = 0.027, unpaired t test). Glycolytic capacity (84% lower) and reserve were also significantly reduced following iron chelation (Fig. 1C), suggesting an overall impairment of energy metabolism in developing hippocampal neurons with reduced iron availability. As a result, total intracellular ATP levels were 21% lower in DFO-treated 11 DIV neuron cultures compared with controls (p < 0.0001).
Figure 1.
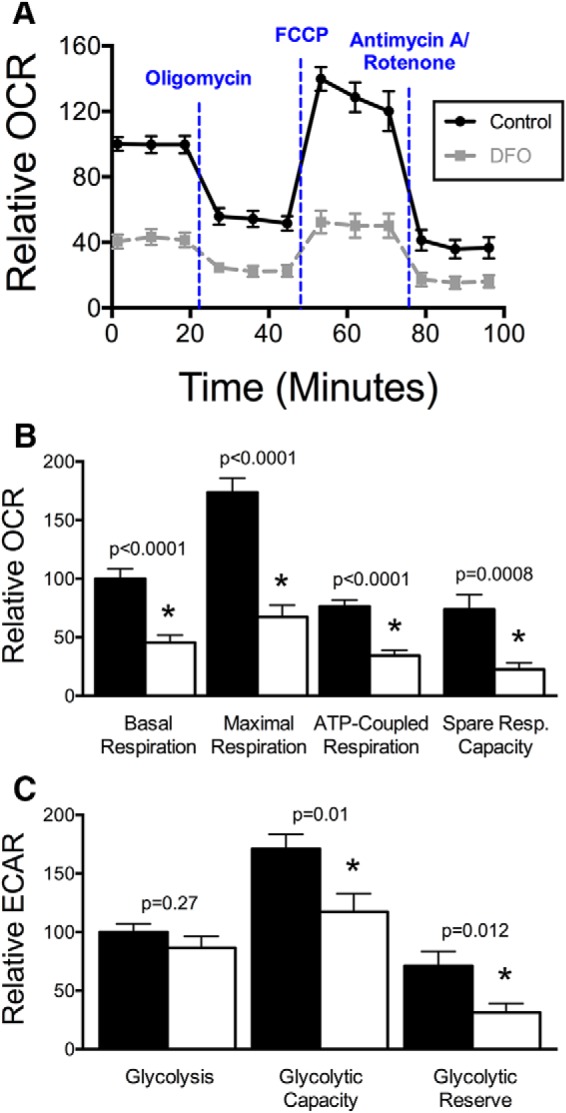
Iron chelation impairs mitochondrial respiration and glycolytic capacity in 11 DIV neurons. Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3 DIV. A, Real-time OCR for 11 DIV cultures (relative to initial control OCR) was measured following treatment with oligomycin (ATP synthase inhibitor), FCCP (uncouples oxygen consumption from ATP production), and antimycin A/rotenone (electron transport chain complex I and III inhibitors). B, Relative OCR contributing to basal respiration (t(38) = 5.13), maximal respiration (t(38) = 6.65), ATP-coupled respiration (t(38) = 5.83), and spare respiratory capacity (t(38) = 3.62) were calculated. C, Relative ECAR contributing to basal glycolysis (t(38) = 1.12), glycolytic capacity (t(38) = 2.71), and glycolytic reserve (t(38) = 2.65) were calculated. The data from three independent cultures were pooled and are presented as mean ± SEM. Student's unpaired t test was performed for each parameter with α = 0.05. *Statistical comparison with a p value that meets the threshold for a significant “discovery” after FDR analysis. Black line/bars represent control (n = 21). Gray dashed line/white bars represent DFO (n = 19).
Dendritic mitochondrial motility
The trafficking of mitochondria in neuronal dendrites is driven by microtubule-associated ATPase motor proteins and thus depends on adequate cellular ATP levels (Hirokawa et al., 2010). We hypothesized that neuronal ID would impair this process, leading to reduced mitochondrial motility in developing dendrites. To test this hypothesis, we performed real-time live cell fluorescent microscopy of mitochondria in terminal dendrite segments of iron-sufficient and iron-deficient 11 DIV hippocampal neuron cultures. Kymograph analysis of the resulting movies with FIJI and KymoAnalyzer allowed us to compare >30 parameters of mitochondrial motility (Neumann et al., 2017) (Table 1). Representative movies for two dendrite segments per group are shown in Movie 1, and the associated mitochondrial images and kymographs are shown in Figure 2A,B.
Figure 2.

Dendritic mitochondrial motility analysis with KymoAnalyzer. Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3 DIV. At 11 DIV, time-lapse live cell imaging of mCherry-Mito-7 transfected neurons was performed for 3 min. Two representative 63× images (t = 0 min) of fluorescently labeled mitochondria within terminal dendrites and the corresponding kymographs are shown for (A) control and (B) DFO neurons. For better visualization, the mitochondria images were straightened using the ImageJ “Straighten” command. Bottom, The resulting manually traced tracks for individual stationary (blue), anterograde (green), retrograde (red), and reversing (yellow) mitochondria. The full 3 min movies for each representative dendrite are shown in Movie 1. Scale bars, 5 μm.
Representative movies of mitochondrial trafficking in terminal dendrites. Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3 DIV. At 11 DIV, time-lapse live cell imaging of mCherry-Mito-7 transfected neurons was performed for 3 min. Two representative 63× movies (1 s imaging interval, 30 fps) of fluorescently labeled mitochondria within terminal dendrites are shown for control (top) and DFO (bottom) neurons. For better visualization, the movies were straightened using the ImageJ “Straighten” command.
One of the most striking effects of DFO treatment was an increased percentage (11.6 ± 1.9% vs 3.7 ± 1.5%, p = 0.005) of the entire mitochondrial population that were characterized as small, round mitochondria with lengths <0.5 μm. Qualitatively, these small mitochondria moved erratically and with greater speeds than the larger, elongated mitochondria. This may constitute a distinct population of mitochondria/mitochondrial fragments with a unique purpose (e.g., mitochondria vesicles) (Sugiura et al., 2014). Thus, the motility data presented are limited to mitochondria >0.5 μm in length. DFO treatment did not significantly alter the overall dendritic density of mitochondria, or the density of moving or stationary mitochondria (data not shown). There was a trend toward a reduced density of mitochondria moving in the anterograde direction, but no change in the density of retrograde or reversing mitochondria in dendrites of DFO-treated neurons (data not shown).
Consistent with our hypothesis, the average speed of moving mitochondria (including pauses, anterograde and retrograde motion) was reduced by 42% following DFO treatment (Fig. 3A). The net velocity of moving mitochondria was significantly lower in DFO-treated neurons, indicating the net distance traveled during the 3 min movie was reduced (Fig. 3A). This was true for net anterograde but not net retrograde (trend with p = 0.055) mitochondria. Interestingly, dendritic mitochondria from DFO-treated neurons were actually moving 15% faster (Control: 0.26 ± 0.01 μm/s vs DFO: 0.30 ± 0.02 μm/s) when the velocity analysis was restricted to only segments during which mitochondria were moving (segmental velocity not including pauses) (Fig. 3B). This was mainly driven by increased velocity in the retrograde direction, as there was not a significant difference in anterograde segmental velocities between control and DFO-treated neurons. The p value for the anterograde segmental velocity comparison was close to meeting the threshold for a significant “discovery” after FDR analysis. Thus, we further examined directional velocities differences with frequency distributions of segmental velocities for anterograde and retrograde segments (Fig. 3C,D). For controls, >60% of both anterograde and retrograde segmental velocities fell in the 0–0.3 μm/s range. DFO treatment did not alter the distribution of anterograde segmental velocities in the 0–0.9 μm/s range, with possibly a slight increase in the percentage of segments with velocities >0.9 μm/s (Fig. 3C, inset). In contrast, DFO treatment decreased the percentage of retrograde segmental velocities in the 0–0.3 μm/s range and increased the percentage of retrograde segmental velocities >0.3 μm/s (Fig. 3D, inset).
Figure 3.

DFO decreases overall but increases segmental mitochondrial speeds. A, The average speed (t(194) = 4.09, Welch's correction) of individual dendritic mitochondria (regardless of direction or pausing) was calculated. The net velocity (t(131.6) = 4.35, Welch's correction) of all mitochondria that displayed net movement was determined and subdivided into velocities of mitochondria with net anterograde (t(70.4) = 4.07, Welch's correction) or retrograde (t(59.6) = 1.96, Welch's correction) movement (Control: n = 120 total moving, n = 99 net moved, n = 58 net anterograde, and n = 41 net retrograde mitochondria; DFO: n = 77 total moving, n = 66 net moved, n = 32 net anterograde, and n = 34 net retrograde mitochondria). B, The average individual segment speed of mitochondria when moving, regardless of direction (t(429.7) = 1.99, Welch's correction), was calculated and subdivided into anterograde (t(299) = 0.87) and retrograde segmental (t(214.4) = 2.03, Welch's correction) velocities (Control: n = 374 total, n = 190 anterograde, and n = 184 retrograde segments; DFO: n = 233 total, n = 111 anterograde, and n = 122 retrograde segments). The data from two independent cultures were pooled and are presented as mean ± SEM. Student's t test was performed for each parameter with α = 0.05. *Statistical comparison with a p value that meets the threshold for a significant “discovery” after FDR analysis. C, D, The relative frequency distributions are shown for (C) anterograde or (D) retrograde mitochondrial segmental velocities. Black bars represent Control. Gray bars represent DFO. Bins are ±0.05 μm/s. For inset frequency distribution, bins are ±0.3 μm/s.
Focusing on the moving mitochondria, the percent time spent moving was decreased following DFO treatment with mitochondria moving in the anterograde direction more severely affected than those moving in the retrograde direction (Fig. 4A). This same trend was also observed for average run lengths (data not shown). The average percent time that moving mitochondria spent pausing was increased by 57% in dendrites of DFO-treated neurons compared with controls (Fig. 4A). This was due to an increase in pause frequency for DFO mitochondria compared with controls and was especially true when mitochondria reversed direction following the pause (Fig. 4B). There was a trend toward increased pause frequency in mitochondria that continued moving in either the anterograde or retrograde directions after the pause (Fig. 4B). There were no differences in average pause duration (data not shown).
Figure 4.

DFO decreases mitochondrial motility and increases pausing. A, The percent time spent moving in total (t(195) = 4.08), in the anterograde direction (t(191.3) = 3.59, Welch's correction), or in the retrograde direction (t(195) = 0.94) was calculated along with the percent time spent pausing (t(195) = 4.08) for all moving dendritic mitochondria (Control: n = 120; DFO: n = 77 moving mitochondria). B, The average pause frequency per moving mitochondria is shown for the total moving mitochondria population (t(195) = 3.50; Control: n = 120; DFO: n = 77 mitochondria) and subdivided into mitochondria with anterograde (t(147) = 1.78; Control: n = 97; DFO: n = 52 mitochondria), retrograde (t(143) = 1.85; Control: n = 85; DFO: n = 60 mitochondria), and reversal (t(95) = 3.35; Control: n = 62; DFO: n = 35 mitochondria) pauses. The data from two independent cultures were pooled and are presented as mean ± SEM. Student's t test was performed for each parameter with α = 0.05. *Statistical comparison with a p value that meets the threshold for a significant “discovery” after FDR analysis.
In addition to increasing the percentage of small, round mitochondria with diameters <0.5 μm, DFO treatment decreased the average length of moving dendritic mitochondria that were >0.5 μm in length (Control: 1.93 ± 0.08 μm vs DFO: 1.54 ± 0.07 μm; t(190.1) = 3.55, p = 0.0005, unpaired t test with Welch's correction). The frequency distribution highlights the striking increase in shorter-length mitochondria and decrease in longer-length mitochondria in DFO-treated neurons (Fig. 5A, inset). These data suggest a possible explanation for the increased segmental velocity of DFO mitochondria, as others have shown a weak, but significant, inverse relationship between mitochondrial size and velocity (Gerencser and Nicholls, 2008; Narayanareddy et al., 2014). We confirmed this inverse correlation between mitochondrial size and velocity when all control and DFO mitochondria were pooled (Fig. 5B, blue line). When considered separately, there was a significant correlation between size and velocity for all moving mitochondria in dendrites of DFO neurons, but not for control neurons. However, there was not a significant difference between groups. For anterograde moving mitochondria, there was a trend toward a significant correlation in DFO, but not control neurons (Fig. 5C). Interestingly, the inverse correlation between size and velocity was strongest when mitochondria were moving in the retrograde direction and was similar in DFO and control neurons (Fig. 5D). Mitochondrial length did not correlate with other motility parameters, such as average speed, pause frequency, or pause duration for either treatment group (data not shown), suggesting that differential size may contribute to the increased segmental speed for mitochondria in DFO-treated neurons but not the increased pausing or overall decreased speed.
Figure 5.

Effect of DFO treatment on mitochondrial size distribution and its relation to speed. A, The relative frequency distribution is shown for the lengths of moving mitochondria. Black bars represent Control. Gray bars represent DFO. Bins are ±0.1 μm/s. For inset frequency distribution, bins are ±0.5 μm/s. The relationship between mitochondrial length and average segment speeds is shown for (B) all moving mitochondria, regardless of direction, (C) mitochondria with anterograde segments, and (D) mitochondria with retrograde segments. The data were pooled from two independent cultures. Linear regression with Pearson's correlation was performed with R2 and correlation p values displayed. Black circles and line represent Control. Red squares and line represent DFO. Blue line indicates combined all mitochondria.
Gene expression indexing mitochondrial fusion and fission
Our mitochondrial length findings suggest a potential shift in mitochondrial fusion/fission processes toward fission in DFO neurons. As a first step to explore this, we quantified mRNA levels for several genes that code for proteins involved in fusion or fission (Table 2). At 11 DIV, when mitochondrial length and motility were impaired, DFO treatment significantly reduced mRNA levels for Opa1, Mfn1, and Mfn2 (Fig. 6A), suggesting impaired mitochondrial fusion. Expression for Drp1, but not Mff or Fis1, genes important for mitochondrial fission, was also reduced in DFO-treated neurons (Fig. 6A). At 18 DIV, Drp1 and Opa1 mRNA levels remained significantly lower in DFO-treated neurons (Fig. 6B). Mfn1 and Mfn2 mRNA levels returned to control levels by 18 DIV, whereas Mff and Fis1 were now significantly lower in DFO-treatment neurons, suggesting continued disruption of mitochondrial fusion and fission.
Figure 6.

The effect of DFO treatment on mitochondrial gene expression indexing fusion and fission. Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3 DIV. At (A) 11 DIV and (B) 18 DIV, cells were collected, total RNA was extracted, and cDNA was synthesized. qRT-PCR was performed for genes indexing fusion (i.e., Opa1, Mfn1, Mfn2) and fission (i.e., Drp1, Mff, and Fis1). Relative mRNA levels are calculated relative to an internal control cDNA sample and a reference gene (i.e., Tbp). The data from two or three independent cultures were pooled and are presented as mean ± SEM. Statistical difference between groups for an individual gene at a given age was determined by unpaired Student's t test with α = 0.05. 11 DIV t values: Opa1 (t(22) = 2.99), Mfn1 (t(22) = 2.24), Mfn2 (t(22) = 3.30), Drp1 (t(22) = 2.11), Mff (t(22) = 1.67), Fis1 (t(22) = 0.21). 18 DIV t values: Opa1 (t(17) = 3.27), Mfn1 (t(17) = 0.93), Mfn2 (t(17) = 0.43), Drp1 (t(11.3) = 2.75, Welch's correction), Mff (t(17) = 5.05), Fis1 (t(17) = 2.83). *Statistical comparison with a p value that meets the threshold for a significant “discovery” after FDR analysis. 11 DIV: n = 12; 18 DIV: n = 9 or 10.
Dendritic mitochondrial density
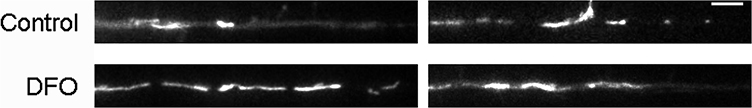
To determine whether the reduced mitochondrial motility during early development ultimately affected long-term trafficking of mitochondria into growing/branching dendrites, we assessed mitochondrial density in terminal dendrite branches of 18 DIV neurons. Representative images of hrGFP-labeled dendrites and mCherry-labeled mitochondria from Control and DFO-treated cultures are shown (Fig. 7A). We used adaptive image thresholding and particle analysis to determine the ratio of total mitochondrial area relative to the total dendritic area. Chronic DFO treatment decreased mitochondrial density in dendrite terminal branches by 35% in 18 DIV neurons.
Figure 7.

DFO decreases mitochondrial density in dendrite tips of 18 DIV neurons. Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3 DIV. At 18 DIV, mCherry-Mito-7 and GFP-expressing neurons were imaged with wide-field fluorescent microscopy and a 100× objective. A, Representative images of fluorescently labeled mitochondria (magenta) within terminal dendrites (green) and the corresponding merged images are shown for Control (left) and DFO (right) neurons. B, C, Dendrite and mitochondria images were thresholded and total area of each determined. The percentage of dendritic area taken up by mitochondrial area was calculated and expressed as the average percent area per neuron. The data from two independent cultures were pooled and are presented as mean ± SEM. Control: n = 36; DFO: n = 33 neurons. Student's t test was performed with α = 0.05; t(67) = 5.50.
Discussion
Figure 8 summarizes the major findings of this study and provides a conceptual model. Consistent with previous in vitro and in vivo findings in more mature developing neurons (de Deungria et al., 2000; Bastian et al., 2016), we demonstrated that chronically restricting neuronal access to iron creates a hypometabolic state, impairing mitochondrial oxygen consumption and reducing intracellular ATP levels in 11 DIV hippocampal neurons. At the same time, chronic iron chelation altered dendritic mitochondrial motility in three main ways: (1) the frequency of pauses was increased (Fig. 8, blue mitochondria), resulting in decreased overall mitochondrial velocity and movement; (2) average mitochondrial size was decreased, with a higher percentage of small, round mitochondria (Fig. 8, purple mitochondria); and (3) overall anterograde movement was decreased (Fig. 8, green mitochondria) and retrograde mitochondria moved with increased velocity (Fig. 8, red mitochondria and arrow). Trafficking of mitochondria into maturing dendrites plays a critical role in both dendritic outgrowth and spine/synapse formation during neuron development (Li et al., 2004; Dickey and Strack, 2011; Fukumitsu et al., 2015, 2016; López-Doménech et al., 2016; Das Banerjee et al., 2017). Thus, this model (Fig. 8) of ID-induced mitochondrial function impairments also provides a plausible developmental mechanism for the long-term neuronal structural abnormalities documented following early-life ID (Jorgenson et al., 2003; Carlson et al., 2009; Brunette et al., 2010; Fretham et al., 2012; Bastian et al., 2016).
Figure 8.

Model illustration summarizing the effect of iron restriction on mitochondrial motility in developing hippocampal neurons. A, Model of mitochondrial motility in terminal dendrite branches from an 11 DIV neuron with sufficient iron. B, Model summary of relative changes in mitochondrial motility in terminal dendrite branches caused by insufficient iron availability. Size of arrows indicates the relative segmental velocities of anterograde (green) and retrograde (red) mitochondria. Number of mitochondria in each color-coded category indicates the relative time spent in each type of motion. DFO treatment causes intracellular iron deficiency (Bastian et al., 2016), impairs mitochondrial respiration, and lowers intracellular ATP levels at 11 DIV, consistent with chronic neuronal energy insufficiency. 11 DIV iron-deficient neurons have an overall decrease in mitochondrial length with an increase in the percentage of small, round mitochondria (magenta), increased mitochondrial pause frequency (blue), decreased overall anterograde motion (green), and increased retrograde segmental velocity with no change in overall retrograde motion (red). Total number of mitochondria per unit length of dendrite was not changed in iron-deficient neurons. These early changes were associated with a decrease in areal mitochondrial density at 18 DIV and decreased dendritic arbor complexity throughout this period of development (Bastian et al., 2016).
Neuronal ID blunts hippocampal neuron dendritic growth/branching leading to a less complex dendritic arbor at both 11 and 18 DIV (Bastian et al., 2016). Because we did not observe a difference in dendritic mitochondrial density at 11 DIV, we concluded that overall mitochondrial respiratory capacity/ATP production in dendrites, and not the actual density of mitochondria, is likely the critical factor underlying the DFO-induced dendritic complexity impairments at this stage (Bastian et al., 2016). The impaired mitochondrial motility at 11 DIV ultimately resulted in decreased mitochondrial density in terminal dendrites of 18 DIV iron-deficient neurons, which also have a persistent reduction in mitochondrial respiratory capacity (Bastian et al., 2016). However, the relative deficit in dendritic arbor complexity are similar at 11 and 18 DIV (Bastian et al., 2016). This suggests that reduced dendritic mitochondrial density does not exacerbate the effects of impaired mitochondrial respiration on dendritic arbor maturation. These findings are consistent with studies by Fukumitsu et al. (2015, 2016) showing that Purkinje neuron dendritic outgrowth can be recovered by supplying exogenous creatine (which restores neuronal ATP levels through the phosphocreatine shuttle) despite persistent mitochondrial trafficking deficits. We hypothesize that the alterations in mitochondrial motility may be part of a larger homeostatic response to reduced energy status, coordinating dendritic growth demands and local ATP supply with energy substrate availability.
Several studies have demonstrated decreased dendritic mitochondrial velocity during conditions of acutely impaired mitochondrial energy production, including hypoxia or treatment with mitochondrial respiration inhibitors such as oligomycin, antimycin A, FCCP, cyanide, or nitric oxide (Rintoul et al., 2003, 2006; Müller et al., 2005; Chang et al., 2006; Zanelli et al., 2006; Mironov, 2007; Cai et al., 2012). However, how dendritic mitochondrial motility/trafficking is altered under conditions where neurons have to adapt to chronic energy impairment was unknown. Our detailed analysis of mitochondrial motility parameters, using KymoAnalyzer software (Neumann et al., 2017), identified increased pause frequency as the causative factor underlying slower mitochondrial speeds in dendrites of neurons with impaired energy metabolism due to chronic ID. Given that the kinesin and dynein motor proteins use ATPase activity to carry mitochondria (Hirokawa et al., 2010), we hypothesize that increased mitochondrial pausing during chronic energy depletion is caused by intermittent exhaustion of local ATP supply due to an inability of mitochondrial respiration to consistently keep up with ATP demand. In dendrites of medulla oblongata respiratory neurons, local ATP reductions are correlated with decreased mitochondrial velocity and increased mitochondrial pausing near active synapses and following hypoxia (Mironov, 2006, 2007). Locally induced axonal ATP depletion, through micropipette administration of uncoupling drugs, also causes local mitochondria stopping (Miller and Sheetz, 2004). Whether local ATP depletion is the direct molecular driving force behind the excessive mitochondrial pausing observed in the dendrites of DFO-treated neurons will need to be specifically tested.
The shift toward smaller mitochondria, with an increased percentage of mitochondria <0.5 μm, may indicate a disruption in mitochondrial fusion and fission. To that end, DFO treatment decreased mRNA expression of the fusion genes Opa1, Mfn1, and Mfn2 at 11 DIV, with Mfn1 and Mfn2 expression restored by 18 DIV. Chronic iron chelation had the opposite effect on fission gene expression with Dnm1l, Mff, and Fis1 mRNA levels decreased at 18 DIV but only Dnm1l expression reduced at 11 DIV. There are no known iron-dependent proteins directly involved in regulating fusion or fission, suggesting this effect is more likely downstream of DFO-induced energy deficits. Indeed, energy depletion and mitochondrial uncoupling can reduce mitochondrial size by inhibiting Opa1-mediated fusion (Baricault et al., 2007; Mishra et al., 2014) and stimulating Drp1-mediated fission (Cereghetti et al., 2008; Toyama et al., 2016). In addition, mitochondrial recruitment of Drp1, and subsequent fission, depends on cellular energy status as the mitochondrial Drp1 receptors Mff and Mid51 are regulated by AMPK and ADP, respectively (Losón et al., 2014; Toyama et al., 2016). Low ATP levels could also indirectly alter fusion/fission homeostasis through impairing the GTPase activity of fusion/fission proteins since GTP is formed from ATP and GDP (Pedersen, 1973).
Altered cellular energy status can also lead to increased mitophagy (Melser et al., 2015). In neurons, mitophagy preferentially targets unhealthy mitochondria with depolarized membranes (Cai et al., 2012; Ye et al., 2015). Although mitophagy can occur locally in distal axons (Ashrafi et al., 2014), it likely occurs preferentially in the soma over the axons or dendrites (Cai et al., 2012; Ye et al., 2015; McWilliams et al., 2016), requiring retrograde transport for distal mitochondria. This is consistent with the increased retrograde movement observed for mitochondria with reduced membrane potential (Miller and Sheetz, 2004; Cai et al., 2012; Ye et al., 2015). Together, these studies support the hypothesis that retrograde mitochondrial motility may be spared (and even augmented) in the dendrites of iron-deficient neurons to traffic damaged/unhealthy mitochondria back to the soma for recycling.
In proximal dendrites, microtubule polarity is mixed (Baas et al., 1988); thus, dynein and kinesin motor proteins can be associated with either anterograde or retrograde mitochondrial transport (Hirokawa et al., 2010). Our study focused on distal dendrites, where microtubule polarity is uniform with plus ends toward the distal tip (Baas et al., 1988). Therefore, the distinct effects of DFO on anterograde and retrograde mitochondrial motility may also involve differential regulation of kinesin and dynein motor proteins and their adaptors.
From a human health perspective, ID affects an estimated 40%–50% of pregnant women and children and results in long-term neurological impairments (e.g., learning and memory, gross and fine motor skills, and psychosocial behaviors) despite iron repletion (Yip, 1994; McLean et al., 2009; Georgieff, 2011). Gestational ID also increases the risk of significant psychopathologies, including schizophrenia, autism spectrum disorders, attention deficit hyperactivity disorder, bipolar disorder, and depression in the offspring later in life (Insel et al., 2008; Chen et al., 2013; Schmidt et al., 2014; Doom et al., 2015). Early-life mitochondrial dysfunction is hypothesized as one of the potential factors underlying these psychiatric disorders (Manji et al., 2012; Streck et al., 2014). Thus, the mitochondrion is an attractive candidate for designing alternative therapeutic interventions, beyond timely iron repletion alone, to prevent long-term neuropathologies in this large population of children.
Footnotes
This work was supported by National Institutes of Health Grants R01 HD029421 to M.K.G., R01 HD094809 to M.K.G., and F32 HD085576 to T.W.B. We thank Justin Campagna and Lanka Dasanayaka for invaluable assistance with culture preparation and image analysis. A portion of the live microscopic imaging was performed at the University Imaging Centers at the University of Minnesota.
The authors declare no competing financial interests.
References
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and parkin. J Cell Biol 206:655–670. 10.1083/jcb.201401070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Deitch JS, Black MM, Banker GA (1988) Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci U S A 85:8335–8339. 10.1073/pnas.85.21.8335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baricault L, Ségui B, Guégand L, Olichon A, Valette A, Larminat F, Lenaers G (2007) OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp Cell Res 313:3800–3808. 10.1016/j.yexcr.2007.08.008 [DOI] [PubMed] [Google Scholar]
- Bastian TW, von Hohenberg WC, Mickelson DJ, Lanier LM, Georgieff MK (2016) Iron deficiency impairs developing hippocampal neuron gene expression, energy metabolism, and dendrite complexity. Dev Neurosci 38:264–276. 10.1159/000448514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian TW, Duck KA, Michalopoulos GC, Chen MJ, Liu ZJ, Connor JR, Lanier LM, Sola-Visner MC, Georgieff MK (2017) Eltrombopag, a thrombopoietin mimetic, crosses the blood–brain barrier and impairs iron-dependent hippocampal neuron dendrite development. J Thromb Haemost 15:565–574. 10.1111/jth.13602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Krieger AM, Yekutieli D (2006) Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93:491–507. 10.1093/biomet/93.3.491 [DOI] [Google Scholar]
- Brunette KE, Tran PV, Wobken JD, Carlson ES, Georgieff MK (2010) Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus. Dev Neurosci 32:238–248. 10.1159/000314341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Zakaria HM, Simone A, Sheng ZH (2012) Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 22:545–552. 10.1016/j.cub.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ES, Stead JD, Neal CR, Petryk A, Georgieff MK (2007) Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus 17:679–691. 10.1002/hipo.20307 [DOI] [PubMed] [Google Scholar]
- Carlson ES, Tkac I, Magid R, O'Connor MB, Andrews NC, Schallert T, Gunshin H, Georgieff MK, Petryk A (2009) Iron is essential for neuron development and memory function in mouse hippocampus. J Nutr 139:672–679. 10.3945/jn.108.096354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, Martins de Brito OM, Chang CR, Blackstone C, Bernardi P, Scorrano L (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A 105:15803–15808. 10.1073/pnas.0808249105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Honick AS, Reynolds IJ (2006) Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci 26:7035–7045. 10.1523/JNEUROSCI.1012-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MH, Su TP, Chen YS, Hsu JW, Huang KL, Chang WH, Chen TJ, Bai YM (2013) Association between psychiatric disorders and iron deficiency anemia among children and adolescents: a nationwide population-based study. BMC Psychiatry 13:161. 10.1186/1471-244X-13-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallman PR. (1986) Biochemical basis for the manifestations of iron deficiency. Annu Rev Nutr 6:13–40. 10.1146/annurev.nu.06.070186.000305 [DOI] [PubMed] [Google Scholar]
- Das Banerjee T, Dagda RY, Dagda M, Chu CT, Rice M, Vazquez-Mayorga E, Dagda RK (2017) PINK1 regulates mitochondrial trafficking in dendrites of cortical neurons through mitochondrial PKA. J Neurochem 142:545–559. 10.1111/jnc.14083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Deungria M, Rao R, Wobken JD, Luciana M, Nelson CA, Georgieff MK (2000) Perinatal iron deficiency decreases cytochrome c oxidase (CytOx) activity in selected regions of neonatal rat brain. Pediatr Res 48:169–176. 10.1203/00006450-200008000-00009 [DOI] [PubMed] [Google Scholar]
- Dickey AS, Strack S (2011) PKA/AKAP1 and PP2A/B 2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J Neurosci 31:15716–15726. 10.1523/JNEUROSCI.3159-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doom JR, Georgieff MK, Gunnar MR (2015) Institutional care and iron deficiency increase ADHD symptomology and lower IQ 2.5–5 years post-adoption. Dev Sci 18:484–494. 10.1111/desc.12223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N (2014) Advanced methods of microscope control using μManager software. J Biol Methods 1:e10. 10.1444/jbm.2014.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fretham SJ, Carlson ES, Wobken J, Tran PV, Petryk A, Georgieff MK (2012) Temporal manipulation of transferrin-receptor-1-dependent iron uptake identifies a sensitive period in mouse hippocampal neurodevelopment. Hippocampus 22:1691–1702. 10.1002/hipo.22004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumitsu K, Fujishima K, Yoshimura A, Wu YK, Heuser J, Kengaku M (2015) Synergistic action of dendritic mitochondria and creatine kinase maintains ATP homeostasis and actin dynamics in growing neuronal dendrites. J Neurosci 35:5707–5723. 10.1523/JNEUROSCI.4115-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumitsu K, Hatsukano T, Yoshimura A, Heuser J, Fujishima K, Kengaku M (2016) Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol Cell Neurosci 71:56–65. 10.1016/j.mcn.2015.12.006 [DOI] [PubMed] [Google Scholar]
- Georgieff MK. (2011) Long-term brain and behavioral consequences of early iron deficiency. Nutr Rev 69 [Suppl 1]:S43–S48. 10.1111/j.1753-4887.2011.00432.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser AA, Nicholls DG (2008) Measurement of instantaneous velocity vectors of organelle transport: mitochondrial transport and bioenergetics in hippocampal neurons. Biophys J 95:3079–3099. 10.1529/biophysj.108.135657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross GG, Junge JA, Mora RJ, Kwon HB, Olson CA, Takahashi TT, Liman ER, Ellis-Davies GC, McGee AW, Sabatini BL, Roberts RW, Arnold DB (2013) Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron 78:971–985. 10.1016/j.neuron.2013.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y (2010) Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68:610–638. 10.1016/j.neuron.2010.09.039 [DOI] [PubMed] [Google Scholar]
- Insel BJ, Schaefer CA, McKeague IW, Susser ES, Brown AS (2008) Maternal iron deficiency and the risk of schizophrenia in offspring. Arch Gen Psychiatry 65:1136–1144. 10.1001/archpsyc.65.10.1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgenson LA, Wobken JD, Georgieff MK (2003) Perinatal iron deficiency alters apical dendritic growth in hippocampal CA1 pyramidal neurons. Dev Neurosci 25:412–420. 10.1159/000075667 [DOI] [PubMed] [Google Scholar]
- Jorgenson LA, Sun M, O'Connor M, Georgieff MK (2005) Fetal iron deficiency disrupts the maturation of synaptic function and efficacy in area CA1 of the developing rat hippocampus. Hippocampus 15:1094–1102. 10.1002/hipo.20128 [DOI] [PubMed] [Google Scholar]
- Kuzawa CW. (1998) Adipose tissue in human infancy and childhood: an evolutionary perspective. Am J Phys Anthropol Suppl 27:177–209. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119:873–887. 10.1016/j.cell.2004.11.003 [DOI] [PubMed] [Google Scholar]
- Lin MY, Sheng ZH (2015) Regulation of mitochondrial transport in neurons. Exp Cell Res 334:35–44. 10.1016/j.yexcr.2015.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZD, Hider RC (2002) Design of clinically useful iron(III)-selective chelators. Med Res Rev 22:26–64. 10.1002/med.1027 [DOI] [PubMed] [Google Scholar]
- López-Doménech G, Higgs NF, Vaccaro V, Ros H, Arancibia-Cárcamo IL, MacAskill AF, Kittler JT (2016) Loss of dendritic complexity precedes neurodegeneration in a mouse model with disrupted mitochondrial distribution in mature dendrites. Cell Rep 17:317–327. 10.1016/j.celrep.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losón OC, Liu R, Rome ME, Meng S, Kaiser JT, Shan SO, Chan DC (2014) The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure 22:367–377. 10.1016/j.str.2014.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji H, Kato T, Di Prospero NA, Ness S, Beal MF, Krams M, Chen G (2012) Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci 13:293–307. 10.1038/nrn3229 [DOI] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60:748–766. 10.1016/j.neuron.2008.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean E, Cogswell M, Egli I, Wojdyla D, de Benoist B (2009) Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public Health Nutr 12:444–454. 10.1017/S1368980008002401 [DOI] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, Ganley IG (2016) mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 214:333–345. 10.1083/jcb.201603039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melser S, Lavie J, Bénard G (2015) Mitochondrial degradation and energy metabolism. Biochim Biophys Acta 1853:2812–2821. 10.1016/j.bbamcr.2015.05.010 [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP (2004) Axonal mitochondrial transport and potential are correlated. J Cell Sci 117:2791–2804. 10.1242/jcs.01130 [DOI] [PubMed] [Google Scholar]
- Mironov SL. (2006) Spontaneous and evoked neuronal activities regulate movements of single neuronal mitochondria. Synapse 59:403–411. 10.1002/syn.20256 [DOI] [PubMed] [Google Scholar]
- Mironov SL. (2007) ADP regulates movements of mitochondria in neurons. Biophys J 92:2944–2952. 10.1529/biophysj.106.092981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Chan DC (2016) Metabolic regulation of mitochondrial dynamics. J Cell Biol 212:379–387. 10.1083/jcb.201511036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Carelli V, Manfredi G, Chan DC (2014) Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab 19:630–641. 10.1016/j.cmet.2014.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Mironov SL, Ivannikov MV, Schmidt J, Richter DW (2005) Mitochondrial organization and motility probed by two-photon microscopy in cultured mouse brainstem neurons. Exp Cell Res 303:114–127. 10.1016/j.yexcr.2004.09.025 [DOI] [PubMed] [Google Scholar]
- Narayanareddy BR, Vartiainen S, Hariri N, O'Dowd DK, Gross SP (2014) A biophysical analysis of mitochondrial movement: differences between transport in neuronal cell bodies versus processes. Traffic 15:762–771. 10.1111/tra.12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Chassefeyre R, Campbell GE, Encalada SE (2017) KymoAnalyzer: a software tool for the quantitative analysis of intracellular transport in neurons. Traffic 18:71–88. 10.1111/tra.12456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olenych SG, Claxton NS, Ottenberg GK, Davidson MW (2007) The fluorescent protein color palette. Curr Protoc Cell Biol 21:21.5. 10.1002/0471143030.cb2105s36 [DOI] [PubMed] [Google Scholar]
- Pedersen PL. (1973) Coupling of adenosine triphosphate formation in mitochondria to the formation of nucleoside triphosphates: involvement of nucleoside diphosphokinase. J Biol Chem 248:3956–3962. [PubMed] [Google Scholar]
- Petry CD, Eaton MA, Wobken JD, Mills MM, Johnson DE, Georgieff MK (1992) Iron deficiency of liver, heart, and brain in newborn infants of diabetic mothers. J Pediatr 121:109–114. 10.1016/S0022-3476(05)82554-5 [DOI] [PubMed] [Google Scholar]
- Pisansky MT, Wickham RJ, Su J, Fretham S, Yuan LL, Sun M, Gewirtz JC, Georgieff MK (2013) Iron deficiency with or without anemia impairs prepulse inhibition of the startle reflex. Hippocampus 23:952–962. 10.1002/hipo.22151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ (2003) Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci 23:7881–7888. 10.1523/JNEUROSCI.23-21-07881.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintoul GL, Bennett VJ, Papaconstandinou NA, Reynolds IJ (2006) Nitric oxide inhibits mitochondrial movement in forebrain neurons associated with disruption of mitochondrial membrane potential: NO inhibits mitochondrial movement. J Neurochem 97:800–806. 10.1111/j.1471-4159.2006.03788.x [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RJ, Tancredi DJ, Krakowiak P, Hansen RL, Ozonoff S (2014) Maternal intake of supplemental iron and risk of autism spectrum disorder. Am J Epidemiol 180:890–900. 10.1093/aje/kwu208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q (2012) Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13:77–93. 10.1038/nrn3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streck EL, Gonçalves CL, Furlanetto CB, Scaini G, Dal-Pizzol F, Quevedo J (2014) Mitochondria and the central nervous system: searching for a pathophysiological basis of psychiatric disorders. Braz J Psychiatr 36:156–167. 10.1590/1516-4446-2013-1224 [DOI] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, McBride HM (2014) A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 33:2142–2156. 10.15252/embj.201488104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ (2016) AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351:275–281. 10.1126/science.aab4138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng Q. (2015) AdaptiveThreshold: ImageJ plugins. Available at https://sites.google.com/site/qingzongtseng/adaptivethreshold. Accessed May 16, 2018.
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA (2007) Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol 292:C125–C136. 10.1152/ajpcell.00247.2006 [DOI] [PubMed] [Google Scholar]
- Ye X, Sun X, Starovoytov V, Cai Q (2015) Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer's disease patient brains. Hum Mol Genet 24:2938–2951. 10.1093/hmg/ddv056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip R. (1994) Iron deficiency: contemporary scientific issues and international programmatic approaches. J Nutr 124:1479S–1490S. 10.1093/jn/124.suppl_8.1479S [DOI] [PubMed] [Google Scholar]
- Zanelli SA, Trimmer PA, Solenski NJ (2006) Nitric oxide impairs mitochondrial movement in cortical neurons during hypoxia: mitochondrial movement during hypoxia. J Neurochem 97:724–736. 10.1111/j.1471-4159.2006.03767.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative movies of mitochondrial trafficking in terminal dendrites. Hippocampal neurons cultured from E16 mice were treated with DFO and 5-FU beginning at 3 DIV. At 11 DIV, time-lapse live cell imaging of mCherry-Mito-7 transfected neurons was performed for 3 min. Two representative 63× movies (1 s imaging interval, 30 fps) of fluorescently labeled mitochondria within terminal dendrites are shown for control (top) and DFO (bottom) neurons. For better visualization, the movies were straightened using the ImageJ “Straighten” command.