

Abstract
Here we have investigated whole-body pharmacokinetics (PK) of exogenously administered T cells in a mouse model of melanoma and have developed a physiologically based pharmacokinetic (PBPK) model to quantitatively characterize the data. T cells were isolated from the spleen of tumor-bearing mice, activated, and labeled with chromium-51 to facilitate the quantification. Labeled T cells were injected in the tumor-bearing mice, and PK was measured in 19 different tissues. It was found that T cells disappear from the blood rapidly after administration and accumulate in the tissues to various extents. Spleen, liver, lung, kidney, bone, and lymph nodes accounted for more than 90% of T cells in the body. The distribution of T cells in solid tumors was found to be very low, hovering below 1%ID/g (percent of injected dose per gram of tissue) during the entire study. However, this observation may differ for targeted TCR-T and CAR-T cells. Observed PK profiles also suggest that T-cell-based therapies may be more successful in treating cancers of the lymphatic system and bone marrow metastases compared to solid tumors. A PBPK model was developed to characterize the whole-body PK of T cells, which incorporated key processes such as extravasation, elimination, and recirculation of T cells via lymph flow. Retention factors were incorporated into the spleen, liver, and kidney compartment to adequately capture the PK profiles. The model was able to characterize observed PK profiles reasonably well, and parameters were estimated with good confidence. The PK data and PBPK model presented here provide unprecedented insight into the biodistribution of exogenously administered T cells.
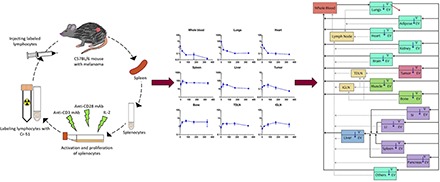
Introduction
Adoptive cell therapy has been gaining interest over the past three decades as a targeted anticancer therapy. Several advancements have been made in this field, which include tumor-infiltrating lymphocytes (TILs) and engineered cells such as T-cell-receptor T cells (TCR-T) and chimeric antigen receptor T cells (CAR-T). In fact, CAR-T cells targeting more than 25 tumor antigens have been investigated in preclinical and clinical studies so far, resulting in recent approvals of Kymriah and Yescarta for the treatment of hematologic cancers such as B cell leukemia and lymphoma (Gross and Eshhar, 2016).
However, the development of T-cell based therapies for solid tumors remains challenging for several reasons, such as unknown cellular trafficking to the site of action, inadequate tumor infiltration, and immunosuppressive tumor microenvironments (Newick et al., 2017; Xia et al., 2017). In addition, despite the clinical success, to date we do not have a comprehensive understanding of the whole-body pharmacokinetics (PK) of T-cell–based therapeutics. Since T cells possess unique properties such as tissue margination, the PK of T cells are bound to be different than traditional small or large molecules and require further investigation to understand and predict the exposure–response relationships for the efficacy and toxicity of T-cell-based therapeutics.
Only a few published studies have investigated the elimination and biodistribution of T cells in various tissues of mice (Table 1). These studies have been limited in the number of sampling time points and tissues being analyzed, and they provided contradictory results. Wallace et al. (1993) were among the first to investigate the disposition of iodine-125 (I-125) PKH95–labeled TILs and activated splenocytes in mice bearing MC38 lung metastases. They observed that 20 hours after administration the activated splenocytes accumulated more in the spleen followed by the lungs and liver, whereas the TILs accumulated more in the lungs followed by spleen and liver. Interestingly, the authors concluded that the higher retention of TILs in the lungs was not because of the presence of lung metastases as a similar disposition pattern was also observed for non–tumor-bearing mice.
TABLE 1.
Comparison of different biodistribution studies of T cells in mice. Methods of T cell isolation, expansion, and labeling, as well as tissue biodistribution at 20-24 hours are compared.
| Method | Wallace et al. (1993) | Melder et al. (2002) | Present Study | |||
|---|---|---|---|---|---|---|
| Labeling | I-125 PKH95–labeled T cells | In-111 oxine–labeled T cells | Cr-51–labeled T cells | |||
| Preparation of T cells | Splenocytes exposed to 6000 IU/ml IL-2 | TILs extracted from MC38 s.c. tumors | Lymphocytes extracted from spleen | Mice sensitized with splenocytes of MCaIV tumor-bearing C3H mice and lymphocytes extracted from spleen | Splenocytes expanded using anti-CD3, anti-CD28, and IL-2 | TILs extracted from B16-BL6 s.c. tumors |
| Tissues listed in descending order of concentrations at 20–24 h | Spleen | Lung | Spleen | Lung | Spleen | Lung |
| Lung | Spleen | Liver | Spleen | Liver | Spleen | |
| Liver | Liver | Lung | Liver | Lung | Liver | |
| Skin | Skin | Tumor | Tumor | Kidney | Kidney | |
| Muscle | Muscle | Heart | Heart | TDLN | Tumor | |
Melder et al. (2002) subsequently presented a more comprehensive investigation, where tumor-antigen–activated and normal (nonactivated) lymphocytes obtained from the spleen were labeled with the residualizing isotope indium-111 (In-111), and the PK of the cells were measured in MCaIV mammary-carcinoma–bearing mice. They observed that the PK of tumor-antigen–activated and normal lymphocytes was the same, and the specificity of the lymphocytes toward the tumor antigen did not result in a significant increase in tumor uptake. They also found that the spleen and the liver were the major tissues that demonstrated lymphocyte accumulation. However, more recently Xu et al. (2013) used 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled CD3+ T cells that were derived from mouse spleen and activated against tumor antigen. They concluded that 1–3 days after the administration, T cells accumulated to a high level in the tumor followed by the liver, kidney, and spleen. As such, the extent of T cell distribution in many tissues is not known, and there is no consensus on the expected PK profiles of exogenously administered T cell therapeutics throughout the body.
In addition, there is also a lack of robust mathematical models that can accurately describe the whole-body PK of T cells. Most of the models developed thus far are based on scant biodistribution data. Stekel et al. (1997) and Ganusov and Auerbach (2014) developed minimal physiologically based pharmacokinetic (PBPK) models to describe T cell disposition in rats. The model by Stekel et al. includes blood, spleen, and lymphatic compartments; and the model by Ganusov et al. includes blood, lung, liver, spleen, Peyer’s patch, mesenteric, and subcutaneous lymph node compartments. More comprehensive models have been developed by Zhu et al. (1996) for mice, rats, and humans; their models include most of the tissues and tumors, and each tissue compartment is divided into vascular and extravascular spaces to account for rolling, adhesion, and extravasation of T cells in tissues. However, these models are based on very sparse data from two to three tissues in each species, and many parameters of the model were fixed to zero due to the lack of experimental data.
To better understand whole-body PK of exogenously administered T cells and to facilitate the development of a more reliable and robust mathematical model, we conducted a comprehensive PK investigation in a mouse model of melanoma. T cells were isolated from mouse spleen then activated and labeled with chromium-51 (Cr-51), which is reported to be a reliable label with no significant elution from the cells or reuptake into other cells (McMillan and Scott, 1968; Rannie and Donald, 1977). In fact, labeling lymphocytes with Cr-51 to investigate their in vivo biodistribution is an old and well-established method (Zatz and Lance, 1970). We injected the labeled T cells into tumor-bearing mice and measured the PK of T cells in blood, tumors, and tissues. The measured PK profiles were quantitatively characterized using a novel PBPK model developed for exogenously administered T cells. The observed PK profiles were also compared with previously reported results.
Materials and Methods
Reagents, Cell Line, and Animal
Mouse melanoma cell line B16-BL6 was obtained from Creative Bioarray (Shirley, NY), and maintained in Dulbecco’s modified Eagle’s medium with 10% heat inactivated FBS and 1% penicillin-streptomycin. Cells were cultured at 37°C in 5% CO2 in a humidified incubator. All animal experiments were performed in accordance with the protocols approved by the institutional animal care and use committee. Eight-week-old male C57BL/6 mice were purchased from Taconic Biosciences (Hudson, NY) and housed individually. The mice were subcutaneously inoculated with 0.5 million B16-BL6 cells to develop the tumor. When the tumor volume reached ∼250 mm3, the mice were subjected to the experimental procedure.
Sodium chromate (1 mCi/ml of 51Cr) was purchased from PerkinElmer (Waltham, MA), and anti-CD3 and anti-CD28 antibodies were purchased from BD Biosciences (San Jose, CA). The reagents for TIL isolation such as the mouse tumor dissociation kit, anti-CD105 antibody-biotin, anti-CD3 antibody-biotin, anti-biotin microbeads ultrapure, LD columns, LS columns, and QuadroMACS separator were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany).
Isolation and Expansion of T Cells from Mouse Spleen
The spleen from a melanoma-bearing mouse was collected and homogenized under aseptic conditions to obtain a mononuclear cell suspension in AIM V medium containing 10% FBS and beta-mercaptoethanol. The cell suspension was centrifuged at 400g and resuspended in red blood cell lysis buffer for 5 minutes at room temperature. AIM V medium was added to dilute the lysis buffer and was centrifuged to remove the buffer and medium, followed by resuspension of the pellet in AIM V medium at a density of 1 to 2 × 107 cells/ml in a nontissue culture treated flask. The flask was precoated with 1 μg/ml anti-CD3 and anti-CD28 antibodies in 5 ml of PBS overnight at 4°C. The PBS solution containing antibodies was removed before adding the mononuclear cell suspension. Interleukin 2 (IL-2) at a concentration of 50 IU/ml was added to cell suspension in the flask. After 48 hours, cells were centrifuged, and the medium was replaced by fresh AIM V medium without IL-2. Four days after isolation of splenocytes, the cells were analyzed using flow cytometry to determine the expression of CD3 and CD8 (Trickett and Kwan, 2003; Koya et al., 2012).
Isolation of Tumor Infiltrating Lymphocytes
To facilitate a pilot study, TILs were isolated for mouse melanoma tumors using a paramagnetic bead system. Melanoma tumors were processed using the tumor dissociation kit to generate a single cell suspension of tumor cells. B16-BL6 cells were depleted from the slurry of cells by passing the cells labeled with biotin-anti-CD105 antibody and anti-biotin microbeads through the LD column. The T cells were then labeled for positive selection using biotin-anti-CD3 antibody and isolated using LS columns.
Cr-51 Labeling and Pharmacokinetic Study
T cells collected from mouse spleen were washed with PBS and incubated with sodium chromate (0.5 mCi of Cr-51 for 200 × 106 cells) for 1 hour at room temperature. After incubation, the cells were washed 3 times with PBS to remove free Cr-51. Radioactivity of the final cell suspension was measured using a gamma counter. We injected 10 μCi of labeled T cells in melanoma-bearing mice via penile vein injections. The mice (n = 3) were sacrificed at predetermined time points (1 hour and 1, 3, 7, and 14 days), and 19 tissues were collected from each mouse including blood, tumor, inguinal lymph nodes (tumor-draining [TDLN] and non–tumor-draining [IGLN]), lungs, heart, kidney, spleen, liver, bone, muscle, skin, brain, thyroid, small intestine (SI), large intestine (LI), pancreas, adipose, and testicles.
The predetermined sampling times were chosen based on several factors. We wanted to investigate the biodistribution of T cells for more than 4 weeks, but the tumor volume reached the permissible institutional animal care and use committee limit within 2 weeks, so we had to design our study for only a period of 2 weeks. Although we could have reduced the initial tumor volume to a value lower than ∼200–250 mm3 to prolong the study duration, it would have affected our ability to quantify T cells in the tumor, so it was not a viable option.
Despite acceptable labeling efficiency, 10–20 million T cells were needed to be injected per animal to attain a radioactive dose of 10 μCi, so we were restricted by the number of T cells available while choosing how many animals we could include in the study. Finally, because most of the previous studies have reported T cell disposition for a short period of time, we wanted to make sure we included at least two early sampling time points (i.e., <24 hours) for comparison with published data, and sufficient later time points to capture the elimination of T cells from the system.
The blood and tissues were collected in preweighed tubes, and their radioactivity was measured using a gamma counter. Radioactive counts were corrected for background and decay. Final counts per minute were converted to percent of injected dose/gram of tissue (%ID/g) by using the following equation:
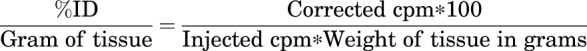 |
PBPK Model Development
Figure 1 describes the PBPK model developed to characterize whole-body PK of T cells. The model includes 12 major tissues and a tumor compartment connected in an anatomical manner via blood and lymph flows. All tissue compartments are divided into vascular and extravascular subcompartments to characterize the process of T cell extravasation. Extravasation of T cells in each tissue is captured using an irreversible first-order transmigration rate process.
Fig. 1.

Schematic of the PBPK model developed to characterize the whole-body disposition of T cells in mice. Tissues are arranged and connected in an anatomical manner by blood flow (black sold lines) and lymph flow (gray dotted lines). The red arrow shows the elimination process via lungs, and blue arrows show the transmigration process in each tissue. The tissue compartments are color coded as follows: red is the whole-blood compartment; yellow compartments are lymph node compartments; purple compartments are tissues emptying blood into the liver compartment; blue is the liver compartment that receives the blood from the hepatic artery and hepatic portal vein (i.e., gut compartments); teal compartments are tissues obtaining blood directly from the whole-blood compartment and draining lymph directly into the lymph node compartment; green compartments are tissues with lymphatic fluid draining into two lymph nodes (TDLN and IGLN); and pink is the tumor compartment with lymphatic fluid draining into the tumor-draining lymph node. V denotes a vascular subcompartment, and EV denotes an extravascular subcompartment.
For most of the tissues it was assumed that the transmigrated T cells in the extravascular space recirculate back into the lymph nodes via the lymphatic flow. For bone and muscle it was assumed that half of the T cells recirculate to the IGLN (left inguinal lymph node) and the rest recirculate to the TDLN (right inguinal lymph node). It is because tibia and thigh muscle were collected as representative bone and muscle tissues and to mimic the physiology of lymph drainage into the inguinal lymph node from the hind legs, we divided the lymph flow from muscle and bone into right and left inguinal lymph nodes. Considering the tumor was implanted in the right flank, lymph drainage from the tumor was added only in the right inguinal lymph node (i.e., TDLN). Lymphatic fluid from all tissues was assumed to drain into lymph node, which was connected to the central blood compartment.
To capture the T cell concentrations in the IGLN and TDLN, the model incorporated two more transit lymph node compartments, which were assumed to receive lymphatic fluids from the bone, muscle, and tumor. Elimination of T cells was assumed to occur only in the vascular compartment of the lungs, based on the model by Zhu et al. (1996). To capture the plateauing concentrations of T cells in the spleen, liver, and kidney over the time, retention factors were incorporated into those tissues. To maintain the mass balance, an “others” compartment was incorporated into the PBPK model. All the physiological parameters for the mouse PBPK model (i.e., blood and lymph flow values, and compartment volumes) were obtained from the literature (Shah and Betts, 2012).
Blood flow for the tumor compartment was obtained from Zhu et al. (1996), and the total tumor volume was based on the average tumor size in our study. Transmigration rates of T cells for each tissue and the retention factors were estimated by fitting the model to the observed whole-body PK data. The equations for the PBPK model are provided in the model equations section, and the description of the model parameters are provided in Table 2.
TABLE 2.
List of model parameters along with their numerical values and sources
| Parameter | Description (unit) | Estimate | CV% |
|---|---|---|---|
| Jlung | Transmigration rate for each tissue (1/h) | 1843.0 | 15.4 |
| Jheart | 34.9 | 17.2 | |
| Jkidney | 87.3 | 37.9 | |
| Jbrain | 1.4 | 21.2 | |
| Jmuscle | 0.5 | 19.7 | |
| Jbone | 82.6 | 16.7 | |
| Jtumor | 0.6 | 18.6 | |
| Jskin | 0.6 | 18.7 | |
| Jfat | 1.6 | 18.5 | |
| JSI | 12.9 | 17.1 | |
| JLI | 5.2 | 18.1 | |
| Jspleen | 114.0 | 33.8 | |
| Jliver | 126.9 | 18.9 | |
| Jpancreas | 10.0 | 20.0 | |
| Jother | 86.8 | 17.7 | |
| Rkidney | Retention factor | 3.9 | 37.9 |
| Rspleen | 9.8 | 34.8 | |
| Rliver | 2.5 | 18.4 | |
| Elung | Elimination rate (1/h) | 0.84 | Fixed (Zhu et al., 1996) |
| Ftissue | Fraction of T cells recirculated to lymph node | 1 | Fixed (assumption) |
| Qtissue, Ltissue, Vtissue | Physiologic blood flows, lymph flows, and tissue volumes for each mouse tissue (ml/h, ml/h, ml) | Fixed (Shah and Betts, 2012) | |
| Qtumor | Tumor blood flow (ml/h) | 6 | Fixed (Zhu et al., 1996) |
| Vtumor | Tumor volume (ml or g) | 0.45 | Fixed (radius∼3.85 mm) |
Model Equations
Whole Blood
 |
(1) |
Lungs
Vascular
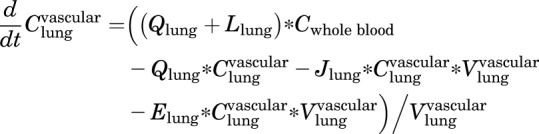 |
(2) |
Extravascular
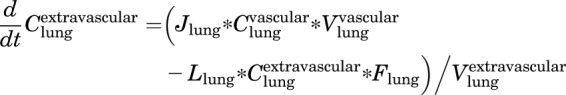 |
(3) |
Typical Tissue
Vascular
 |
(4) |
Extravascular
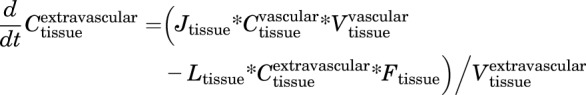 |
(5) |
Tissues with Retention Factor
Vascular
 |
(6) |
Extravascular
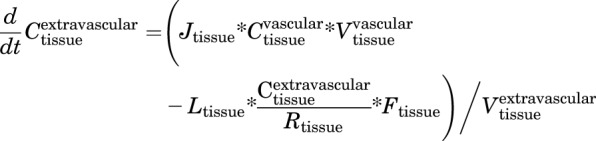 |
(7) |
Liver
Vascular
| (8) |
Extravascular
 |
(9) |
Lymph Node
 |
(10) |
Inguinal Lymph Node
 |
(11) |
Tumor Draining Lymph Node
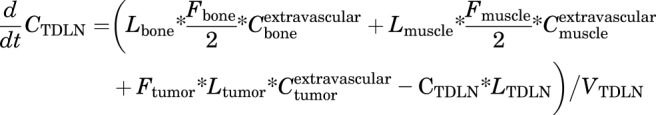 |
(12) |
PK Analysis and PBPK Model Fitting
The area under the concentration versus time curve (AUC) for all the T cell PK profiles and associated variability was calculated using noncompartmental analysis in Phoenix WinNonlin 7.0 (Certara USA, Princeton, NJ). The ratio of T cell AUC in tissues over the AUC in blood was represented as a biodistribution coefficient (BC) value (Shah and Betts, 2013; Li et al., 2016). The PBPK model was fitted to the observed PK data using the maximum likelihood estimation method of ADAPT V software (Biomedical Simulations Resource, University of Southern California, Los Angeles, CA). The following variance model was used for model fitting: Var(t) = ( +
+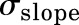 Y(t))2, where
Y(t))2, where 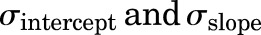 are variance model parameters and Y(t) is the model output.
are variance model parameters and Y(t) is the model output.
Results
Whole-Body Pharmacokinetics of T Cells.
Activation of splenocytes with anti-CD3 antibody, anti-CD28 antibody, and IL-2 led to more than 90% of CD3+ T cells in the final preparation, of which >85% were CD8+ cells. A representative flow cytometry analysis of these cells is provided in (Supplemental Fig. 1). The efficiency of Cr-51 incorporation was ∼50 μCi per 108 lymphocytes, and thus a dose of 10 μCi/mouse comprised ∼2 × 107 T cells administered per mouse.
Figure 2 shows typical PK profiles generated after intravenous administration of Cr-51–labeled T cells in tumor-bearing mice, along with AUC values for each profile. It was found that T cells were eliminated from the blood and the lungs rapidly within 24 hours, followed by their accumulation in spleen and liver. Smith et al. (1980) showed similar results in rats, where exogenous activated lymphocytes accumulated in the lungs up to 4–6 hours and then localized to the spleen and liver over 24 hours. This phenomenon can be attributed to the presence of a reticuloendothelial system within the spleen and liver, which is reported to be significantly involved in the disposition of T cells (Pabst, 1988; Norelli et al., 2016). In addition, lymphoid tissues such as lymph nodes and bone marrow showed high accumulation of T cells over time.
Fig. 2.

Observed concentration (%ID/gram of tissue) versus time (hours) curves of Cr-51–labeled T cells in blood, tumor, and other tissues excised from melanoma-bearing mice. Reported are the AUC (%ID/gram * hour) and biodistribution coefficient (i.e., tissue/blood AUC ratio) for each tissue.
Of note, no significant difference was seen between the PK of T cells in TDLN versus IGLN (AUC: 635% ± 77% versus 712% ± 192%ID/g*h). Interestingly, the T cell concentrations peaked around 24 hours in the TDLN, which was similar to the tumor, whereas T cells accumulated gradually over 3–7 days in the IGLN, which was similar to the bone marrow. The spleen, liver, lung, kidney, bone, and lymph nodes accounted for more than 90% of the T cells in the body over the 2 weeks, with BC value of 448, 148, 39, 36, 27, and 16, respectively (Fig. 2).
The PK of T cells in the heart displayed a similar profile as the lungs, where majority of the T cells were eliminated from the tissue by 72 hours. The intestines, pancreas, testes, muscle, fat, skin, and brain showed the lowest amount of T cell accumulation, with concentrations hovering below 0.5%ID/g for most of the time. T cell concentrations in the thyroid started off lower than 0.5%ID/g, and gradually decreased to below the limit of quantitation after 3 days; two of three mice showed a high T cell concentration on day 14. This peculiar PK profile may stem from lymphocytes recirculating back to the thyroid after 2 weeks, but this hypothesis needs further support.
Importantly, tumors had a BC value of 1.3, and the tumor concentrations of T cells hovered below 1%ID/g during the whole study. This observation may stem from many factors, such as an immunosuppressive tumor microenvironment, insufficient trafficking of T cells into the tumor, and lower tumor uptake of T cells due to less expression of cell adhesion molecules on the tumor cells (Pandolfi et al., 1992). These data bring into question the potential for treating solid tumors using T-cell-based therapy and demand further investigation into the tumor disposition of targeted T cells like TCR-T and CAR-T cells. In addition, our results suggest that T-cell-based therapies may be more successful for treating cancers of the lymphatic system and bone marrow metastases.
Development of the T-Cell PBPK Model.
The PBPK model (Fig. 1) was able to characterize T cell PK profiles in most of the tissues reasonably well (Fig. 3). The model parameters were also estimated with good confidence (Table 2). The estimates of transmigration rates were higher for tissues showing a high accumulation of T cells and BC values greater than 20, such as the lungs, spleen, liver, and kidneys. Retention factors for spleen, liver, and kidney were 9.8, 2.5, and 3.9, respectively.
Fig. 3.

Comparison of observed and model-generated T cell PK profiles for various tissues.
Incorporation of the retention process was necessary to characterize the high concentrations of T cells in the spleen and liver, which may relate to T cell processing by the reticuloendothelial system. High concentrations of T cells in the kidney may stem from the presence of the Cr-51 label in that organ. Elimination of T cells from the body was incorporated only in the lungs, and the parameter for this process was fixed to the value of 0.84 hour−1 obtained from Zhu et al. (1996). Of note, Zhu et al. obtained this value by scaling down the parameter value estimated using human data.
Discussion
With the emergence of activated T cell and T cell targeting antibodies as novel anticancer therapeutics, it has become necessary to quantitatively understand the whole-body disposition of T cells to further advance the clinical potential of these therapeutics. The process of T cell disposition is complex and depends on multiple stimuli-dependent steps such as rolling and adhesion on vascular endothelial cells, chemokine-driven extravasation, and margination to specific tissues (Hogg, 1993; Springer, 1994; Girard and Springer, 1995). This process is further complicated by dynamic regulation of cell adhesion molecules expressed on tissues and changes in cell surface phenotype based on the nature of the T cell (Defilippi et al., 1993; Ager et al., 2016).
Nonetheless, as an important first step toward understanding the whole-body disposition of engineered T cells, here we have conducted a comprehensive PK study of activated T cells in a mouse model of melanoma. We have chosen a xenograft tumor model rather than a spontaneous tumor model to avoid the possibility of developing host tolerance (Lechner et al., 2013). As such, further studies are warranted to assess how the T cell disposition may differ between xenograft, spontaneous, and orthotopic tumors.
The T cells employed in our investigation were isolated from the splenocytes of melanoma-bearing mice and expanded to enrich CD3+ T cells. The cells were incubated with sodium chromate to accomplish Cr-51 labeling. After the labeling, >95% of cells were found to be alive via trypan blue method. An advantage of using Cr-51 as a label is the fact that Cr3+ ions bind to organic ions such as citrate and amino acids within the cells to form complexes, which do not leak out of the cells or nonspecifically transfer to surrounding cells (Sanderson, 1976). However, occasionally macrophages may phagocytose dead/dying T cells and accumulate a small amount of Cr-51, which should be kept in mind while interpreting the biodistribution data from Cr-51–labeled T cells (Sprent, 1976; Thakur, 1985).
Our findings were comparable to previously published sparse PK data (Table 1). Figure 4 shows the comparison of our PK data generated using Cr-51–labeled activated T cells, with previously published PK data generated using T cells labeled with In-111 and I-125 (Wallace et al., 1993; Melder et al., 2002). We have also included data from a pilot PK study that was conducted using Cr-51 labeled TILs for comparison. This study was limited due to the very low number of TILs available from B16-BL6 tumors, and the PK data were collected only up to 24 hours.
Fig. 4.

Superimposition of the observed PK of Cr-51 labeled T cells (red) with previously reported studies of In-111 (green) and I-125 labeled (blue) T cells. Closed symbols represent non–tumor-targeting lymphocytes, and open symbols represent lymphocytes activated against tumor antigen. Please refer to Table 1 for further information.
As shown in Fig. 4, all the studies demonstrate that the spleen has the highest accumulation of T cells followed by the liver. However, it was observed that T cells were eliminated faster from the lungs, heart, and tumors in our study compared with the In-111–labeled T-cell study. This may be due to the residualizing nature of In-111, as the naïve and tumor-antigen-activated T cells demonstrated no difference in the PK profile. Interestingly, when TILs were administered, a higher concentration of T cells was observed in the lungs compared with the spleen.
This observation was similar to the one reported using I-125–labeled TILs (Melder et al., 2002), where the lungs had a higher accumulation of tumor-sensitized T cells compared with normal lymphocytes. This suggests that when T cells are activated against tumor antigens they may exhibit a tendency to accumulate in the lungs at a higher level. Although the mechanism for this phenomenon is not fully elucidated, there is an increase in LFA-1 clustering on TILs and their affinity toward ICAM-1, expressed in the lungs, is also high, leading to a higher accumulation of TILs in lungs (Hamann et al., 2000; Reina and Espel, 2017).
Regardless of activation, lymphocytes are also known to temporarily accumulate in the lungs due to the pressure difference between the right ventricle that pumps the blood to the lungs and the left ventricle that circulates the blood through the rest of the body. During the long transit through the lung, lymphocytes can get trapped and exposed to alveolar macrophages, which can phagocytose a small fraction of the cells (Hall, 1985; Westermann et al., 2003).
Of note, in our study the tumor distribution of TILs was not much different than activated T cells. However, this observation needs further research using targeted TCR-T and CAR-T cells that can recognize tumor cells. In addition, we have only focused on quantitatively understanding the biodistribution of a pooled CD3+ T cell population. Consequently, the biodistribution characteristics of specific T cell subtypes may differ. It also remains to be investigated whether the disposition of T cells throughout the body will be different between tumor-bearing and non–tumor-bearing mice.
In addition to mice, a few short-term lymphocyte migration studies with rats have reported using chromium. Interestingly, these studies were conducted by labeling thoracic duct lymphocytes to track the recirculation of lymphocytes, which showed the highest distribution in superficial lymph nodes within a day. Among organs, the spleen exhibited the second highest accumulation, and the rest of the lymphoid and nonlymphoid tissues showed a pattern similar to our study (Rannie and Donald, 1977; Smith and Ford, 1983; Ganusov and Auerbach, 2014). Smith and Ford passaged the Cr-51–labeled thoracic duct lymphocytes through an intermediate animal before injecting them into final recipient rats, to mimic the physiologic conditions and reduce the effect of cell handling during in vitro incubation. Passaged thoracic duct lymphocytes entered and exited the spleen at a faster rate compared with nonpassaged thoracic duct lymphocytes. Discrepancies such as this could stem from differences in the origin and maturation stage of the cells, cell handling procedures, or the animal species used for the investigation, which may require further investigation (Bainbridge et al., 1966; Heslop and Hardy, 1971; Hamann et al., 2000).
When it comes to evaluation of the whole-body disposition of lymphocytes in humans, little has been published except two studies that used chromium-labeled lymphocytes (McMillan and Scott, 1968; Hersey, 1971). These studies were performed half a century ago, and the techniques used for the isolation and expansion of lymphocytes from peripheral blood mononuclear cells were archaic. Nonetheless, McMillan and Scott (1968) reported high body surface counts of lymphocytes in the liver, followed by the heart, spleen, and lungs over 24 hours. In addition, lymphocytes disappeared from the blood rapidly, and very low Cr-51 counts were detectable 6 hours after the injection. However, Hersey (1971) followed lymphocyte distribution over 8 days in healthy individuals compared with patients with chronic lymphatic leukemia, and he reported high body surface counts in the spleen, bone marrow, and liver. He concluded that the difference between his results and those of the study performed by McMillan and Scott was the fact that the earlier study had been performed using a mixture of leukocytes due to poor separation techniques. Hersey also observed that the lymphocyte percent decreased fast from the blood and lungs of healthy patients, similar to earlier reports. Thus, it seems that in both humans and in mice lymphocytes distribute rapidly to the tissues immediately after administration, and spleen and liver accumulate the majority of the administered dose.
We also have presented the development of a PBPK model to characterize the whole-body PK of exogenously administered T cells. Our PBPK model is more parsimonious than previously published models (Zhu et al., 1996; Stekel et al., 1997; Ganusov and Auerbach, 2014) and was developed based on the dense PK data we generated in this investigation. The PBPK model assumes retention of T cells in the spleen, liver, and kidney and elimination only through the lungs. The retention phenomenon is incorporated using the retention factors, and it is based on several observations.
It has been reported that lymphocytes activated in vitro or in vivo tend to localize to secondary lymphoid organs such as the spleen (Gowans and Knight, 1964; Hamann and Rebstock, 1993), and we observed a similar trend in our exogenous T cell PK study. Previous reports also suggested a slow turnover and very long half-life of T cell subsets in certain tissues (Sprent and Tough, 1994), which may be the reason for the observed stable concentrations of T cells in the spleen and liver over the time. As such, the retention factor in the model represents lymphocyte accumulation in the extravascular region of the spleen, liver, and kidney, which may stem from decreased recycling of lymphocytes from these tissues via the lymph flow. Elimination of T cells from the body was incorporated only in the lungs, based on the reports that T cells undergo relatively faster clearance in the lungs due to the presence of alveolar macrophages (Milik et al., 1997). However, it is important to note that the estimated value of pulmonary clearance for T cells may be inflated, as the model does not take into account the elimination of T cells from tissues like the liver and spleen, which have been shown to demonstrate some degree of T cell elimination.
Our model excludes the well-known processes of reversible rolling and irreversible adhesion of T cells, owing to the lack of sufficient in vitro data to estimate the parameters related to these processes with confidence. Consequently, the extravasation of lymphocytes through the endothelial capillary wall into the tissues is characterized in our model using a first-order process termed the transmigration rate. Each tissue was considered to have a unique transmigration rate due to the differences in endothelial surface area and chemokine stimuli. However, in the future it might be valuable to augment the model with experimentally observed values of T cell extravasation and endothelial cell interaction (Simionescu et al., 1974; Hamann and Rebstock, 1993; Knust et al., 2009), which can enable the PBPK model to predict a priori the T cell PK in other species. At present it is very challenging to incorporate such values due to the lack of quantitative in vitro experiments reported in the literature for transmigration rate of lymphocytes for each tissue.
In summary, our results provide an unprecedented insight into the whole-body disposition of exogenously administered T cells. Immediately after administration, T cells disappear from the blood; while spleen, liver, lungs, kidneys, bone, and lymph nodes accounted for more than 90% of the T cells in the body. There was no difference found between the PK of T cells in TDLN and IGLN. Interestingly, we did not find T cell accumulation in the solid tumor; however, these observations may differ for targeted T cells like TCR-T and CAR-T cells. Our results suggest that T-cell-based therapies may be more successful in treating cancers of the lymphatic system and bone marrow metastases compared with solid tumors. We also found that when T cells are activated against tumor antigens they may accumulate in the lungs at a higher level. We also mathematically characterized the PK data we generated by the use of a novel PBPK model developed for exogenously administered T cells. The data and PBPK model presented here pave the way for the development of a systems pharmacokinetic model for T cells, which can be further combined with the PK model of monoclonal antibodies (Shah and Betts, 2012) to develop translational PK models for T cells engaging and targeting antibodies.
Acknowledgments
We would like to thank Peter Bloomingdale for careful review of the manuscript.
Abbreviations
- AUC
area under the concentration–time curve
- BC
biodistribution coefficient
- CAR-T cells
chimeric antigen receptor T cells
- Cr-51
chromium-51
- I-125
iodine-125
- %ID/g
percent of injected dose per gram of tissue
- IGLN
inguinal lymph node
- IL-2
interleukin 2
- In-111
indium-111
- LI
large intestine
- PBPK model
physiologically based pharmacokinetic model
- PK
pharmacokinetics
- SI
small intestine
- TCR-T cells
T-cell-receptor modified T cells
- TDLN
tumor-draining lymph node
- TILs
tumor-infiltrating lymphocytes
Authorship Contributions
Participated in research design: Khot, Matsueda, Koya, Shah.
Conducted experiments: Khot, Matsueda, Thomas.
Contributed new reagents or analytic tools: Khot, Shah.
Performed data analysis: Khot, Shah.
Wrote or contributed to the writing of the manuscript: Khot, Koya, Shah.
Footnotes
This work was supported by the Centre for Protein Therapeutics at University at Buffalo, and by the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM114179] (to D.K.S.) and National Institute of Allergy and Infectious Diseases Grant [Grant R2-AI138195] (to D.K.S.).
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Ager A, Watson HA, Wehenkel SC, Mohammed RN. (2016) Homing to solid cancers: a vascular checkpoint in adoptive cell therapy using CAR T-cells. Biochem Soc Trans 44:377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge DR, Brent L, Gowland G. (1966) Distribution of allogeneic 51Cr-labelled lymph node cells in mice. Transplantation 4:138–153. [DOI] [PubMed] [Google Scholar]
- Defilippi P, Silengo L, Tarone G. (1993) Regulation of adhesion receptors expression in endothelial cells. Curr Top Microbiol Immunol 184:87–98. [DOI] [PubMed] [Google Scholar]
- Ganusov VV, Auerbach J. (2014) Mathematical modeling reveals kinetics of lymphocyte recirculation in the whole organism. PLOS Comput Biol 10:e1003586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard JP, Springer TA. (1995) High endothelial venules (HEVs): specialized endothelium for lymphocyte migration. Immunol Today 16:449–457. [DOI] [PubMed] [Google Scholar]
- Gowans JL, Knight EJ. (1964) The route of re-circulation of lymphocytes in the rat. Proc R Soc Lond B Biol Sci 159:257–282. [DOI] [PubMed] [Google Scholar]
- Gross G, Eshhar Z. (2016) Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off-tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol 56:59–83. [DOI] [PubMed] [Google Scholar]
- Hall J. (1985) The study of circulating lymphocytes in vivo: a personal view of artifice and artifact. Immunol Today 6:149–152. [DOI] [PubMed] [Google Scholar]
- Hamann A, Klugewitz K, Austrup F, Jablonski-Westrich D. (2000) Activation induces rapid and profound alterations in the trafficking of T cells. Eur J Immunol 30:3207–3218. [DOI] [PubMed] [Google Scholar]
- Hamann A, Rebstock S. (1993) Migration of activated lymphocytes. Curr Top Microbiol Immunol 184:109–124. [DOI] [PubMed] [Google Scholar]
- Hersey P. (1971) The separation and 51 chromium labeling of human lymphocytes with in vivo studies of survival and migration. Blood 38:360–371. [PubMed] [Google Scholar]
- Heslop BF, Hardy BE. (1971) The distribution of 51Cr-labeled syngeneic and allogeneic lymph node cells in the rat. Transplantation 11:128–134. [DOI] [PubMed] [Google Scholar]
- Hogg N. (1993) A model of leukocyte adhesion to vascular endothelium. Curr Top Microbiol Immunol 184:79–86. [DOI] [PubMed] [Google Scholar]
- Knust J, Ochs M, Gundersen HJ, Nyengaard JR. (2009) Stereological estimates of alveolar number and size and capillary length and surface area in mice lungs. Anat Rec (Hoboken) 292:113–122. [DOI] [PubMed] [Google Scholar]
- Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T, et al. (2012) BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res 72:3928–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, Ohlfest JR, Hu P, Epstein AL. (2013) Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother 36:477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Krippendorff BF, Sharma S, Walz AC, Lavé T, Shah DK. (2016) Influence of molecular size on tissue distribution of antibody fragments. MAbs 8:113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan R, Scott JL. (1968) Leukocyte labeling with 51-Chromium. I. Technic and results in normal subjects. Blood 32:738–754. [PubMed] [Google Scholar]
- Melder RJ, Munn LL, Stoll BR, Marecos EM, Baxter LT, Weissleder R, Jain RK. (2002) Systemic distribution and tumor localization of adoptively transferred lymphocytes in mice: comparison with physiologically based pharmacokinetic model. Neoplasia 4:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milik AM, Buechner-Maxwell VA, Sonstein J, Kim S, Seitzman GD, Beals TF, Curtis JL. (1997) Lung lymphocyte elimination by apoptosis in the murine response to intratracheal particulate antigen. J Clin Invest 99:1082–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newick K, O’Brien S, Moon E, Albelda SM. (2017) CAR T cell therapy for solid tumors. Annu Rev Med 68:139–152. [DOI] [PubMed] [Google Scholar]
- Norelli M, Casucci M, Bonini C, Bondanza A. (2016) Clinical pharmacology of CAR-T cells: linking cellular pharmacodynamics to pharmacokinetics and antitumor effects. Biochim Biophys Acta 1865:90–100. [DOI] [PubMed] [Google Scholar]
- Pabst R. (1988) The spleen in lymphocyte migration. Immunol Today 9:43–45. [DOI] [PubMed] [Google Scholar]
- Pandolfi F, Trentin L, Boyle LA, Stamenkovic I, Byers HR, Colvin RB, Kurnick JT. (1992) Expression of cell adhesion molecules in human melanoma cell lines and their role in cytotoxicity mediated by tumor-infiltrating lymphocytes. Cancer 69:1165–1173. [DOI] [PubMed] [Google Scholar]
- Rannie GH, Donald KJ. (1977) Estimation of the migration of thoracic duct lymphocytes to non-lymphoid tissues. A comparison of the distribution of radioactivity at intervals following i.v. transfusion of cells labelled with 3H, 14C, 75Se, 99mTc, 125I and 51Cr in the rat. Cell Tissue Kinet 10:523–541. [PubMed] [Google Scholar]
- Reina M, Espel E. (2017) Role of LFA-1 and ICAM-1 in cancer. Cancers (Basel) 9:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson CJ. (1976) The uptake and retention of chromium by cells. Transplantation 21:526–529. [PubMed] [Google Scholar]
- Shah DK, Betts AM. (2012) Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn 39:67–86. [DOI] [PubMed] [Google Scholar]
- Shah DK, Betts AM. (2013) Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. MAbs 5:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simionescu M, Simionescu N, Palade GE. (1974) Morphometric data on the endothelium of blood capillaries. J Cell Biol 60:128–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ME, Ford WL. (1983) The recirculating lymphocyte pool of the rat: a systematic description of the migratory behaviour of recirculating lymphocytes. Immunology 49:83–94. [PMC free article] [PubMed] [Google Scholar]
- Smith ME, Martin AF, Ford WL. (1980) Migration of lymphoblasts in the rat. Preferential localization of DNA-synthesizing lymphocytes in particular lymph nodes and other sties. Monogr Allergy 16:203–232. [PubMed] [Google Scholar]
- Sprent J. (1976) Fate of H2-activated T lymphocytes in syngeneic hosts. I. Fate in lymphoid tissues and intestines traced with 3H-thymidine, 125I-deoxyuridine and 51chromium. Cell Immunol 21:278–302. [DOI] [PubMed] [Google Scholar]
- Sprent J, Tough DF. (1994) Lymphocyte life-span and memory. Science 265:1395–1400. [DOI] [PubMed] [Google Scholar]
- Springer TA. (1994) Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76:301–314. [DOI] [PubMed] [Google Scholar]
- Stekel DJ, Parker CE, Nowak MA. (1997) A model of lymphocyte recirculation. Immunol Today 18:216–221. [DOI] [PubMed] [Google Scholar]
- Thakur ML. (1985) Radiolabeled Cellular Blood Elements: Pathophysiology, Techniques, and Scintigraphic Applications, Springer Science & Business Media, New York. [Google Scholar]
- Trickett A, Kwan YL. (2003) T cell stimulation and expansion using anti-CD3/CD28 beads. J Immunol Methods 275:251–255. [DOI] [PubMed] [Google Scholar]
- Wallace PK, Palmer LD, Perry-Lalley D, Bolton ES, Alexander RB, Horan PK, Yang JC, Muirhead KA. (1993) Mechanisms of adoptive immunotherapy: improved methods for in vivo tracking of tumor-infiltrating lymphocytes and lymphokine-activated killer cells. Cancer Res 53 (10 Suppl):2358–2367. [PubMed] [Google Scholar]
- Westermann J, Söllner S, Ehlers EM, Nohroudi K, Blessenohl M, Kalies K. (2003) Analyzing the migration of labeled T cells in vivo: an essential approach with challenging features. Lab Invest 83:459–469. [DOI] [PubMed] [Google Scholar]
- Xia AL, Wang XC, Lu YJ, Lu XJ, Sun B. (2017) Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget 8:90521–90531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WL, Li SL, Wen M, Wen JY, Han J, Zhang HZ, Gao F, Cai JH. (2013) Tracking in vivo migration and distribution of antigen-specific cytotoxic T lymphocytes by 5,6-carboxyfluorescein diacetate succinimidyl ester staining during cancer immunotherapy. Chin Med J (Engl) 126:3019–3025. [PubMed] [Google Scholar]
- Zatz MM, Lance EM. (1970) The distribution of chromium 51-labelled lymphoid cells in the mouse. A survey of anatomical compartments. Cell Immunol 1:3–17. [DOI] [PubMed] [Google Scholar]
- Zhu H, Melder RJ, Baxter LT, Jain RK. (1996) Physiologically based kinetic model of effector cell biodistribution in mammals: implications for adoptive immunotherapy. Cancer Res 56:3771–3781. [PubMed] [Google Scholar]