

Abstract
Aims
Cyclic adenosine monophosphate (cAMP) regulates cardiac excitation–contraction coupling by acting in microdomains associated with sarcolemmal ion channels. However, local real time cAMP dynamics in such microdomains has not been visualized before. We sought to directly monitor cAMP in a microdomain formed around sodium–potassium ATPase (NKA) in healthy and failing cardiomyocytes and to better understand alterations of cAMP compartmentation in heart failure.
Methods and results
A novel Förster resonance energy transfer (FRET)-based biosensor termed phospholemman (PLM)-Epac1 was developed by fusing a highly sensitive cAMP sensor Epac1-camps to the C-terminus of PLM. Live cell imaging in PLM-Epac1 and Epac1-camps expressing adult rat ventricular myocytes revealed extensive regulation of NKA/PLM microdomain-associated cAMP levels by β2-adrenoceptors (β2-ARs). Local cAMP pools stimulated by these receptors were tightly controlled by phosphodiesterase (PDE) type 3. In chronic heart failure following myocardial infarction, dramatic reduction of the microdomain-specific β2-AR/cAMP signals and β2-AR dependent PLM phosphorylation was accompanied by a pronounced loss of local PDE3 and an increase in PDE2 effects.
Conclusions
NKA/PLM complex forms a distinct cAMP microdomain which is directly regulated by β2-ARs and is under predominant control by PDE3. In heart failure, local changes in PDE repertoire result in blunted β2-AR signalling to cAMP in the vicinity of PLM.
Keywords: cAMP, Phospholemman, Phosphodiesterase, FRET, Heart failure
1. Introduction
3′,5′-Cyclic adenosine monophosphate (cAMP) as an ubiquitous second messenger regulates cardiomyocyte function by acting in discrete subcellular microdomains. Such microdomains are formed for example around ion channels participating in excitation–contraction (EC) coupling and involve a set of differentially localized receptors, cAMP-dependent protein kinase (PKA) with its anchoring proteins, and cAMP hydrolyzing enzymes phosphodiesterases (PDEs) which are crucial for spatial confinement of cyclic nucleotide gradients in cells.1,2 Functionally distinct cAMP microdomains have previously been described in caveolin-rich membrane structures,3 various subsarcolemmal locations,4 and around sarcoplasmic reticulum (SR) calcium ATPase.5 However, local cAMP dynamics in any microdomain directly associated with a sarcolemmal ion channel involved in EC coupling, and its changes in cardiac disease have not been studied before.
An important cAMP microdomain might be associated with the sodium–potassium ATPase (NKA), a sarcolemmal pump which, as the predominant Na+ efflux mechanism in cardiomyocytes, maintains the transmembrane Na+ gradient. This gradient is essential not only for a variety of Na-dependent membrane transporters but also for the rapid upstroke of action potential upon depolarization.6,7 Subsequent elevation of intracellular Ca2+ triggers cell contraction followed by the diastolic Ca2+ extrusion via the sodium–calcium exchanger (NCX) in its forward mode which in turn is promoted by the transmembrane Na+ gradient.8 Therefore, NKA activity orchestrates calcium cycling and EC coupling. In heart failure, an increase in intracellular Na+, in part due to a decline in NKA activity, dissipates this Na+ gradient and hence contributes to altered Ca cycling, diastolic dysfunction, arrhythmias, and changes in mitochondrial metabolism.9–12
Phospholemman (PLM, also known as FXYD1) is a 72 amino acid transmembrane muscle cell protein13–15 which forms a stable complex with NKA.16,17 The cytosolic C-terminus of PLM bears at least three different phosphorylation sites, one of them Ser68 can be phosphorylated by PKA and protein kinase C.18–20 PLM tonically inhibits NKA, and PLM phosphorylation by PKA leads, via a conformational change occurring without a dissociation, to an increase of NKA activity (its sodium affinity and transport kinetics), thereby lowering intracellular sodium load during increases in heart rate under β-adrenoceptor (β-AR) stimulation.21 Conversely, hypo-phosphorylation of PLM, as occurs in heart failure, exacerbates contractile dysfunction, increases Na+ overload, and promotes myocardial remodelling.22 Despite the importance of PLM phosphorylation in both normal and pathophysiology, local cAMP dynamics in this critical microdomain has not been directly investigated.
Here, we developed a novel targeted Förster resonance energy transfer (FRET)-based cAMP biosensor to monitor real time cAMP dynamics in the vicinity of the NKA/PLM complex. FRET measurements uncovered tight regulation of this microdomain by the β2-AR/cAMP/PKA and local pools of PDE3, which was lost in heart failure. These data provide a molecular basis for better understanding of disease-associated local signalling changes and may help develop new therapies to prevent sodium overload in heart failure.
2. Methods
Detailed Section 2 is available in the Supplementary material online.
2.1 Chemicals
BAY 60-7550 was from Santa Cruz. Cilostamide, rolipram, (-) isoproterenol (ISO), forskolin, CGP20712A, and ICI118551 were purchased from Sigma. 3-isobutyl-1-methylxanthine (IBMX) was from Applichem. All other chemicals were from Sigma, unless stated otherwise.
2.2 Sensor construction
Full-length canine PLM sequence lacking the stop codon was fused via a flexible linker encoding the KRSRAQASNSAVDGTPVATG amino acid sequence to the N-terminus of Epac1-camps.23 The adenovirus generation is performed using the Gateway® Cloning system (Life Technologies) and in accordance with the manufacturer’s instructions.
2.3 FRET-based cAMP measurements in cardiomyocytes
Rats were anaesthetized by isoflurane inhalation and sacrificed by cervical dislocation. Adult rat ventricular myocytes (ARVMs) were isolated by enzymatic Langendorff perfusion, transduced for 48 h with Epac1-camps or PLM-Epac1 adenoviruses, and subjected to FRET measurements as described.24 Imaging was performed at room temperature in the buffer A containing in mmol/L: NaCl 144, KCl 5.4, MgCl2 1, CaCl2 1, HEPES 10, pH = 7.3. Compounds were diluted in the same buffer and applied onto cells cumulatively if not stated otherwise.
2.4 Myocardial infarction chronic heart failure model
All animal experiments were performed in accordance with Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health under assurance number A5634-01. All surgical procedures and perioperative management conformed to the UK Animals (Scientific Procedures) Act 1986 and were approved by the Animal Welfare and Ethical Review Body (AWERB) Committee of Imperial College London. Adult male Sprague-Dawley rats (250–300 g) underwent proximal coronary ligation to induce MI under isoflurane anaesthesia as previously described.25,26 Righting reflex was used to monitor the adequacy of anaesthesia. For analgesia, metamizole (300 mg/kg/day in drinking water 3 days prior and 7 days after surgery) and buprenorphine (0.05 mg/kg subcutaneously, 30 min prior to and every 8 h during 7 days after surgery) were used. Rats were functionally characterized by echocardiography and sacrificed 16 weeks after MI by cervical dislocation in isoflurane anaesthesia for cardiomyocyte isolation. Echocardiographic and functional parameters of the rats used in this study have been previously described.27
2.5 Statistics
Normal distribution was tested by the Kolmogorov–Smirnov test. Differences were analysed using OriginPro8.6 (OriginLab, Northampton) or R software (R Foundation for Statistical Computing) and one-way ANOVA or Mann–Whitney U tests, followed by Bonferroni’s post hoc test, as appropriate. When several cells isolated from the same animal were considered, mixed ANOVA followed by Wald χ2-test was used. Data are presented as means ± standard error of the mean (SE) from the indicated number of experiments (animals and cells) per condition.
3. Results
3.1 Generation and characterization of a novel targeted PLM-Epac1 biosensor
To enable real time measurements of cAMP directly in the NKA/PLM microdomain, we sought to develop a targeted version of the highly sensitive FRET biosensor Epac1-camps.23 Epac1-camps is a cytosolic cAMP sensor which contains a cyclic nucleotide binding domain derived from Epac1 which is sandwiched between two green fluorescent protein (GFP) mutants, namely enhanced cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP). In absence of cAMP, CFP and YFP are located in close proximity, generating a high degree of FRET signal. Binding of cAMP to Epac1 domain leads to a conformational change in the biosensor molecules which brings both fluorophores far apart, decreasing the FRET signal.27 Based on a previously developed functional PLM-YFP construct containing a flexible 20 amino acid linker between canine PLM and YFP sequence,28 we fused our cAMP sensor sequence in a similar way to the PLM C-terminus (Figure 1A). When introduced into ARVMs by adenoviral gene transfer (at the multiplicity of infection 300), this targeted biosensor was found in the surface membrane and co-localized well with the endogenous NKA α1-subunits, similar to native PLM (Supplementary material online, Figure S1A–D). At least some considerable fraction of the sensor protein was detected at the cell surface using membrane impermeable biotinylation reagent (Supplementary material online, Figure S1B). It could also inhibit NKA activity in transfected cells and be co-immunoprecipitated with endogenous cardiomyocyte NKA α1-subunits in both directions (Supplementary Figure S2A–C), confirming PLM functionality inside the PLM-Epac1 biosensor. Fluorescence in PLM-Epac1 expressing cardiomyocytes showed clear membrane localization in outer sarcolemma and a strong intracellular striated pattern which was clearly distinct from localization pattern of cytosolic proteins such as GFP and Epac1-camps (Figure 1B,C; Supplementary material online, Figure S3). Since this intracellular pattern was highly reminiscent of overexpressed junctional sarcoplasmic reticulum (jSR) proteins retained in the biosynthetic pathway including perinuclear space and SR,29 these areas were excluded from image data analysis. Instead of collecting intensity from the whole myocyte, we measured FRET responses solely in the region encompassing outer sarcolemma (Figure 1D). Stimulation of cells with the β-adrenergic agonist ISO led to a rapid decrease in YFP and a concomitant increase in CFP fluorescence in the sarcolemmal membrane cell region, indicative of a decrease in YFP/CFP ratio and an increase of local cAMP levels (Figure 1D). Interestingly, no detectable change of FRET signal could be recorded in the perinuclear region with strong overexpression, suggesting that the sensor molecules might be still immature in this intracellular compartment and should not be affecting recordings from the functionally relevant biosensor pool. Likewise, no detectable change of FRET signal in the membrane regions could be observed in cells stimulated with saturating concentrations (10 µmol/L) of protein kinase C activator phorbol 12-myristate 13-acetate (change in FRET 0.09 ± 0.11%, followed by a positive control 10 µmol/L forskolin plus 100 µmol/L IBMX which caused 9.9 ± 0.6% decrease of FRET ratio, number of animals/cells was 4/10).
Figure 1.

Generation of a targeted PLM-Epac1 biosensor. (A) Schematic representation of the PLM-Epac1 sensor construct that includes full-length phospholemman (PLM) fused via a flexible linker to the parental cytosolic cAMP biosensor Epac1-camps comprised of the cAMP-binding domain sandwiched between enhanced yellow fluorescent protein (YFP) and enhanced cyan fluorescent protein (CFP). (B) Representative confocal images of adult rat ventricular myocytes (ARVMs) transduced with adenoviral constructs to express Epac1-camps or PLM-Epac1. Fluorescence in YFP and CFP channels reveals sarcolemmal and partially striated intracellular membrane localization pattern for PLM-Epac1, as compared to the cytosolic Epac1-camps. See Supplementary material online, Figure S3 for autofluorescence image. (C) Representative confocal image (n = 10) showing live cell membrane staining of PLM-Epac1 expressing ARVM. Scale bar 2 µm. (D) Representative FRET recording (n = 10) of a PLM-Epac1 expressing ARVM stimulated with the β-adrenergic agonist isoproterenol (ISO 100 nmol/L). Left, YFP, and CFP intensities measured in a red-marked membrane region of interest to exclude strongly fluorescent perinuclear overexpression show concomitant changes, indicative of a decreasing YFP/CFP FRET ratio (here normalized to the basal ratio values) which represents an increase of local cAMP levels upon ISO treatment. In contrast, no detectable FRET change could be observed in perinuclear location (marked black) with sensor overexpression (right). Scale bars 10 µm. TM, transmission image.
3.2 FRET imaging shows considerable β2-AR/cAMP signals in the vicinity of PLM
Next, we performed FRET measurements under selective stimulation of β1- and β2-ARs, the major cAMP stimulating adrenergic receptor subtypes expressed in cardiomyocytes. Selective β1-AR stimulation performed as previously established3,5,25,30 led to comparable cAMP signals in PLM-Epac1 and Epac1-camps expressing myocytes, suggesting similar degree of local cAMP increase as in the bulk cytosol (Figure 2A,B). In sharp contrast, β2-AR/cAMP responses were nearly doubled in the PLM/NKA microdomain, as compared to the cytosol (Figure 2C–E), suggesting close functional association of this receptor subtype with PLM. We directly compared the affinities of both biosensors for cAMP and their sensitivities to ISO stimulation in intact ARVMs, using a previously established protocol.24 Interestingly, the affinity of PLM-Epac1 was slightly lower than that of Epac1-camps (Supplementary material online, Figure S4A), suggesting that the actual deference in β2-AR/cAMP between bulk cytosol and PLM/NKA microdomain might be even greater. Using these sensor calibration data, we recalculated FRET values into absolute cAMP concentrations which were 0.73 ± 0.04 and 2.95 ± 0.41 µM for global (cytosolic) vs. local β2-AR stimulated cAMP, respectively. Interestingly, the potencies derived from concentration-response dependencies for ISO measured with both sensors were comparable (Supplementary material online, Figure S4B). This suggests that β-adrenergic stimulation in general leads to higher cAMP levels at NKA/PLM as compared to the bulk cytosol, which is compatible with the idea of privileged receptor—microdomain communication previously observed also for the phospholamban microdomain.5
Figure 2.

Strong local β2-AR/cAMP signals in the PLM/NKA microdomain. (A, B) Representative FRET traces for Epac1-camps and PLM-Epac1 expressing ARVMs upon β1-AR-selective stimulation (100 nmol/L ISO plus 50 nmol/L of the β2-AR blocker ICI118551). (C, D) Representative FRET traces for β2-AR-selective stimulation (100 nmol/L ISO plus 100 nmol/L of the β1-AR blocker CGP20712A). Maximal stimulation of cAMP was subsequently achieved by the PDE2 selective inhibitor (BAY 60-7550, 100 nmol/L used in C and D, see Figure 4), the unselective PDE inhibitor IBMX (100 μmol/L), and the adenylyl cyclase activator forskolin (10 μmol/L). (E) Quantification of FRET responses to selective β1- and β2-AR stimulations shown in A–D. Number of cells (n) and hearts (N) per condition were as follows—n/N = 40/6 and n/N = 33/6 for β1-AR and n/N = 31/9 and n/N = 38/8 for β2-AR, respectively. *—significant difference, P < 0.05 by mixed ANOVA followed by Wald χ2-test.
3.3 Role of PDEs in shaping cAMP signals in the NKA/PLM microdomain
To understand how cAMP dynamics is regulated at the local level, we next studied the effects of various PDE inhibitors at basal state and after β1-AR- or β2-AR-selective receptor stimulations in healthy cells. The concentrations of PDE inhibitors used have been previously established in FRET and PDE activity assays to be selective for the respective PDE families.3,5 In the absence of prestimulation with β-AR ligands, there were no significant differences found in the effects of PDE3 and PDE4 selective inhibitors between bulk cytosol and the NKA/PLM microdomain. However, the local contribution of PDE2 was significantly higher in the vicinity of PLM (Figure 3). After β1-AR stimulation, all three PDE family inhibitors showed no significant differences in FRET responses between the two sensors (Figure 4C). In sharp contrast, the contribution of PDE3 was strikingly much more pronounced in the NKA/PLM microdomain after β2-AR prestimulation (Figure 4A,B,D). These FRET data correspond to 1.2 ± 0.1 and 31.0 ± 12.2 µM of global (cytosolic) vs. local cAMP, respectively, after cilostamide application in presence of β2-AR stimulation. These data indicate that local cAMP dynamics at PLM is tightly controlled by PDE2 at basal state and by PDE3 under β2-AR stimulation, while PDE4 effects are present but not significantly different in both locations.
Figure 3.

PDE contributions to cAMP hydrolysis under basal conditions. Representative FRET traces recorded in ARVMs expressing either cytosolic (A) or NKA/PLM microdomain specific (B) biosensor upon PDE2 inhibition with 100 nmol/L BAY 60-7550, followed by 100 µmol/L of the unselective PDE inhibitor IBMX and by 100 nmol/L ISO and 10 µmol/L forskolin to achieve maximal stimulation. (C) Quantification of the FRET ratio change induced by the specific PDE inhibitors (10 µmol/L cilostamide for PDE3 and 10 µmol/L rolipram for PDE4 were used, respectively) related to the maximum stimulation uncovered PDE2 as the PDE family crucial for confining the PLM/NKA compartment from the bulk cytosol at basal state. Means ± SE, number of cells (n), and hearts (N) per condition were as follows—n/N = 10/4 and n/N = 10/4 for PDE2, n/N = 12/4 and n/N = 24/6 for PDE3, and n/N = 12/5 and n/N = 12/5 for PDE4, respectively. *—significant differences, P < 0.05 by Mann–Whitney U test. P < 0.05 by mixed ANOVA followed by Wald χ2-test. n.s., not significant.
Figure 4.

PDE regulation of cytosolic and local cAMP levels under subtype specific β-AR stimulations. Representative FRET ratio traces for Epac1-camps (A) and PLM-Epac1 (B) expressing ARVMs recorded during selective β2-AR stimulation with 100 nmol/L ISO plus 50 nmol/L CGP20712A followed by the PDE3 inhibitor cilostamide (Cilo, 10 µmol/L) and the unselective PDE inhibitor IBMX (100 µmol/L). Forskolin (10 µmol/L) was used to fully stimulate the sensors. (C) Quantification of the PDE inhibitor responses in cells with prestimulated β1-AR. 100 nmol/L BAY 60-7550 and 10 µmol/L rolipram were used to inhibit PDE2 and PDE4, respectively. Shown are means ± SE, number of cells (n), and hearts (N) per condition were as follows—n/N = 13/4 and n/N = 14/5 for PDE2, n/N = 13/4 and n/N = 16/5 for PDE3, and n/N = 19/3 and n/N = 15/9 for PDE4, respectively. (D) Quantification of the PDE inhibitor responses in cells with prestimulated β2-AR. Means ± SE, number of cells (n), and hearts (N) per condition were as follows—n/N = 12/4 and n/N = 13/4 for PDE2, n/N = 11/4 and n/N = 16/4 for PDE3, and n/N = 13/4 and n/N = 14/4 for PDE4, respectively. *—significant differences, P < 0.05 by mixed ANOVA followed by Wald χ2-test. P 0.05 by the same test. n.s., not significant.
3.4 Heart failure blunts local β2-AR control and alters PDE2/PDE3 balance in the NKA/PLM microdomain
Since cardiac disease has been associated with changes in cAMP signalling, we next investigated how the NKA/PLM microdomain is affected in the rat model of chronic heart failure induced by myocardial infarction (MI). Sixteen weeks after MI when the chronic failing phenotype has been established, ARVMs were isolated and transduced with either cytosolic Epac1-camps or with targeted PLM-Epac1 biosensor. At this stage of disease, as expected, β1-AR levels were down-regulated whereas the expression of β2-AR, regulatory PKA subunits and adenylyl cyclases 5 and 6 were not significantly changed (Supplementary material online, Figure S5). Importantly, heart failure did not lead to detectable changes of PLM-Epac1 sensor co-localization with endogenous NKA α1-subunit, nor was this co-localization affected by various pharmacological treatments used in our study (Supplementary material online, Figure S6). Heart failure also did not visibly affect the subcellular localization of the sensor which could be found in intracellular and membrane regions expressing caveolin 3 (Supplementary material online, Figures S7 and S8). Strikingly, heart failure led to a loss of the local β2-AR/cAMP signal compartmentation, leading to FRET responses which were indistinguishable from those measured in the cytosol (Figure 5A–E). These FRET values correspond to 0.8 ± 0.1 and 1.9 ± 0.2 µM of cytosolic vs. local β2-AR stimulated cAMP, respectively. Interestingly, while there were no major disease-driven changes in PDE effects detected in the cytosol (Supplementary material online, Figure S9), measurements with PLM-Epac1 sensor in failing cells after β2-AR stimulation revealed a significant increase in local PDE2 inhibitor effects accompanied by a decrease in microdomain-specific PDE3 effects (Figure 5A–D,F). Finally, using immunofluorescence, we detected a clear co-localization of PDE3 with endogenous PLM which was significantly reduced in heart failure (Supplementary material online, Figure S10). Importantly, as expected from the FRET data (see Figures 2E and5E), β2-AR selective stimulation led to a clear phosphorylation of PLM at the Ser68, and this effect was abolished in failing cells (Figure 6).
Figure 5.

Changes in local cAMP dynamics at PLM in heart failure. (A–D) Representative FRET traces for ARVMs isolated from either age-matched controls (AMC) or from rats with chronic heart failure 16 weeks after MI and transduced to express PLM-Epac1 sensor. Cells were first stimulated with 100 nmol/L ISO plus 100 nmol/L CGP20712A to activate β2-AR followed by the PDE2 and PDE3 inhibitors and forskolin. (E) β2-AR stimulation shows a significantly attenuated cAMP response in the microdomain, indicating a loss of β2-AR/cAMP compartmentation. Data from cells expressing the cytosolic sensor Epac1-camps are also provided for comparison. Shown are means ± SE, number of cells (n), and hearts (N) per condition were as follows—n/N = 27/4 and n/N = 26/4 for AMC, and n/N = 23/4 and n/N = 27/4 for MI groups, respectively. (F) Data analysis for the experiments shown in A–D. In heart failure, there is a pronounced increase in local PDE2 and a decrease in local PDE3 inhibitor effects. Compound concentrations were as in Figure 4. Means ± SE, number of cells (n), and hearts (N) per condition were as follows—n/N = 7/4 and n/N = 6/3 for PDE2, and n/N = 12/4 and n/N = 9/3 for PDE3, respectively. *—significant differences, P < 0.05 by mixed ANOVA followed by Wald χ2-test.
Figure 6.

Immunoblot analysis of PLM phosphorylation. (A) Representative immunoblots for ARVMs isolated from either controls (AMC) or from rats with chronic heart failure after MI. Cells were stimulated for 5 min with 100 nmol/L ISO plus 100 nmol/L CGP20712A to selectively activate β2-AR. PLM phosphorylation was analysed using a Ser68 phosphoantibody. In healthy cells, β2-AR stimulates PLM phosphorylation, this effect is blunted in heart failure. Data analysis is in (B). Means ± SE, number of blots/hearts were 6/3 and 5/3 for control and stimulated groups, respectively. *—significant difference, P < 0.05 by one-way ANOVA.
4. Discussion
Despite a clear evidence that cAMP modulates cardiac EC coupling by acting in microdomains associated with sarcolemmal ion channels, local compartmentalized cAMP dynamics in their close proximity has not been analysed before. In this study, we directly monitored local cAMP dynamics in the vicinity of NKA by creating a novel PLM-targeted FRET biosensor and using it in healthy and failing ARVMs.
Based on biochemical evidence and co-localization experiments, PLM-Epac1 sensor was found in the microdomain of interest and retained functional properties of unmodified PLM in terms of physical association with α1-subunits and inhibition of NKA function (see Supplementary material online, Figures S1 and S2). Similar to endogenous PLM, it localized in outer sarcolemma, while a clear intracellular pattern was also observed which was excluded from the FRET analysis (Figure 1B–D; Supplementary material online, Figures S7 and S8). Sarcolemmal localization of the PLM-Epac1 biosensor was independently confirmed using a biochemical biotinylation assay (see Supplementary material online, Figure S1B). However, adenoviral overexpression of the sensor also led to a strong perinuclear overexpression artefact and several lower molecular weight bands of immunoblots (see Supplementary material online, Figure S2) which may result from immature and/or degraded sensor protein. Importantly, there was no detectable change in FRET upon stimulation of cAMP production when recorded from perinuclear overexpression areas (see Figure 1D), suggesting that these irrelevant protein species do not interfere with specific cAMP recordings. Another clearly striated and partially dotted intracellular pattern was observed (see Figure 1B,D, Supplementary material online, Figures S1D, S6A, S7, and S8) which is reminiscent of overexpressed jSR proteins retained in the biosynthetic pathway.29 Since optical microscopy techniques with its resolution close to half of the wavelength of light do not allow to properly resolve T-tubular membrane from jSR which are just 10–30 nm apart, we cannot conclude on the localization of the biosensor pool or even a part of it in the T-tubular membrane. Silverman et al.31 have shown that around 75% of endogenous PLM are localized on sarcolemmal membrane while a minor residual fraction might be located in T-tubuli of guinea pig ventricular myocytes. On the other hand, rat cardiomyocytes were reported to show somewhat more of a striated or dotted, presumably T-tubular staining.32–34 Since we could not definitely resolve the exact location of the intracellular striated sensor pools, to this end, we analysed FRET responses from the regions of interest encompassing outer sarcolemma.
Interestingly, our FRET-based analysis revealed much stronger β2-AR/cAMP responses in the NKA/PLM microdomain, as compared to the bulk cytosol (see Figure 2). This suggests an important functional coupling of β2-AR with this microdomain, which, in addition to much more abundant β1-AR stimulated cAMP, might critically regulate NKA function. To selectively activate β1- and β2-AR subtypes, we used a combination of the unselective β-adrenergic agonist ISO together with β1- and β2-AR subtype-selective antagonists CGP20712A and ICI118551 which we established to be selective at the respective concentrations using β1- and β2-AR knockout mice.24 In a previous study, using scanning ion conductance microscopy in combination with FRET-based cAMP recordings, we could detect β2-AR/cAMP signals originating from functional receptors exclusively located in T-tubules of healthy ARVMs.25 This T-tubular localization of the β2-AR leads to local cAMP confinement by PKA-dependent buffering and activation of PDE4. In contrast, β1-AR as the predominant β-AR subtype was evenly distributed across the sarcolemma. β1-AR modulation leads to changes in cAMP signals in both cytosolic and sarcolemmal membrane microdomains, to strong contractile responses and promotes cardiac remodelling. In contrast, functional β2-ARs are exclusively located in T-tubules, thereby generating highly localized cAMP responses which do not significantly diffuse into the deep cytosol containing sarcoplasmic reticulum and contractile proteins but may protect from pathological remodelling.25,35 Therefore, one may expect a functional interaction of β2-AR with NKA/PLM. It is also well documented that β2-ARs are localized in caveolin-rich membrane domains which may include sarcolemmal caveolae.36 Obviously, it is difficult to conclude for our data whether PLM is co-localized with T-tubuli. However, FRET recordings indicate a close functional association of β2-AR/cAMP pools with the NKA/PLM microdomain.
It is tempting to speculate that β2-AR could also play a cardioprotective role by inducing a local pool of cAMP which leads to phosphorylation of PLM and protection from Ca2+ overload. However, in chronic heart failure, this cAMP microdomain becomes disrupted (see Figure 5E) which may limit the potentially beneficial effects of β2-AR. Previously, Juel et al. showed that β2-AR activation modulates NKA activity in human skeletal muscle,37 although PLM phosphorylation was not examined in this study. Two other reports38,39 showed that pharmacological β2-AR agonists (terbutaline and formoterol) both increased PLM Ser68 phosphorylation in human skeletal muscle. Here, we report for the first time that β2-AR signalling modulates local cAMP gradients around the NKA/PLM microdomain, leading to PKA-mediated PLM phosphorylation in cardiac myocytes which is changed in disease. Our FRET data go in line with immunoblot analysis which shows that selective β2-AR stimulation leads to an increase of PLM phosphorylation, and that this effect is clearly abolished in heart failure (see Figure 6). The drop of β2-AR/cAMP signal in the vicinity of NKA/PLM could be, at least in part, a result of β2-AR redistribution from T-tubules to detubulated membrane areas which has been previously described for failing cardiomyocytes isolated the same MI-induced disease model.25 Potentially, detubulation could promote also PDE3 redistribution, thereby affecting local cAMP degradation.
Equally important could be changes in PDE composition of the NKA/PLM microdomain detected in this study. By analysing the effects of the selective inhibitors for the major cardiomyocyte cAMP hydrolyzing PDEs (2, 3, and 4), we found that the NKA/PLM microdomain is under stringent PDE2 dependent control at basal state, while after β2-AR stimulation, PDE3 becomes dominant (see Figures 3and4). This is comparable with the data from adult mouse cardiomyocytes obtained using a targeted version of the Epac1-camps sensor localized in the caveolin-rich membrane microdomains including T-tubules (pmEpac).3 Using immunofluorescence, significant co-localization of PDE3A with endogenous PLM could be observed, which may result from PDE3A association with sarcolemma and SR membrane, with a clear reduction in heart failure (see Supplementary material online, Figure S10). Likewise, functional effect of the PDE3 inhibitor cilostamide on cAMP levels in the NKA/PLM microdomain was dramatically reduced in failing cells (see Figure 5C,D,F). Instead, the local effect of PDE2 was significantly increased (see Figure 5A,B,F). This important PDE has been previously shown to be up-regulated at the mRNA and protein level in various heart failure models40 and seems to affect especially the local microdomain signalling (Figure 7), while no significant disease-driven alterations could be detected in the bulk cytosol (Supplementary material online, Figure S9). Previously, using an early disease model of compensated cardiac hypertrophy induced by transverse aortic constriction in pmEpac expressing mice, we uncovered a PDE2 and PDE3 redistribution between β1- and β2-AR-associated caveolar and non-caveolar membrane microdomains. In this case, PDE3 as the predominant PDE family at the β2-AR was substituted by PDE2, leading to changes in cGMP/cAMP cross-talk engaged by atrial natriuretic peptide receptors.3 Obviously, a similar change in PDE repertoire in the NKA/PLM microdomain can be now detected also in a chronic rat heart failure model (see Figure 5). This change of the microdomain-specific PDE composition should definitely affect the rates of cAMP hydrolysis resulting in a change of responsiveness to β2-AR stimulation. The modulation of the local cAMP response by cGMP associated with atrial and brain natriuretic peptide signalling is unlikely to play a role in this decompensated late disease model because of the desensitization of their common NPR1 receptor which is typical for chronic heart failure.41
Figure 7.
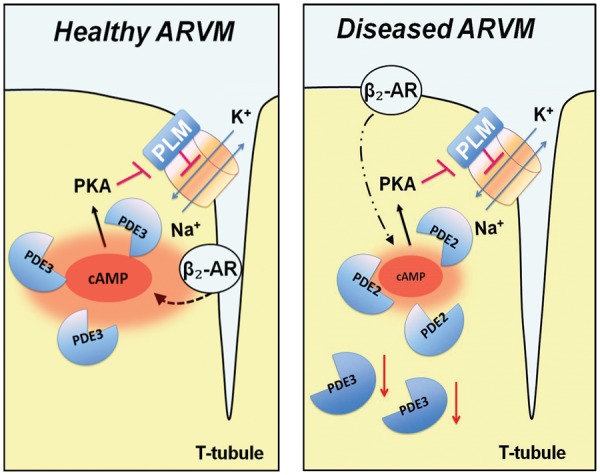
Schematics depicting local regulation of cAMP in the NKA/PLM microdomain and its changes in heart failure. In healthy ARVMs, NKA/PLM is in close functional proximity to the β2-AR and PDE3. In disease, receptor and PDE redistribution disrupts the proper β2-AR signalling to cAMP/PKA/PLM. Local increase of PDE2 mediated effects and a decrease of PDE3 co-localization with the microdomain can be detected in disease.
In summary, we provide the first direct visualization of local cAMP signalling at a sarcolemmal cardiomyocyte ion channel regulated in a microdomain formed around the NKA/PLM complex. FRET-based live cell imaging indicates tight regulation of this microdomain by the β2-AR and local pools of PDE3 and its dramatic alteration in heart failure. This knowledge could provide a molecular basis for novel therapeutic strategies to prevent disease-associated sodium overload and impaired EC coupling.
Supplementary Material
Acknowledgments
We thank Tobias Goldak and Karina Schlosser for ARVM isolation and technical assistance.
Conflict of interest: none declared.
Footnotes
Time for primary review: 43 days
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (IRTG1816 to V.O.N. and M.J.S.), Wellcome Trust (109604/Z/15/Z to D.P.), British Heart Foundation (RG/12/18/30088 to J.G.), and the Gertraud und Heinz-Rose Stiftung to V.O.N.
References
- 1. Perino A, Ghigo A, Scott JD, Hirsch E.. Anchoring proteins as regulators of signaling pathways. Circ Res 2012;111:482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perera RK, Nikolaev VO.. Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol (Oxf) 2013;207:650–662. [DOI] [PubMed] [Google Scholar]
- 3. Perera RK, Sprenger JU, Steinbrecher JH, Hubscher D, Lehnart SE, Abesser M, Schuh K, El-Armouche A, Nikolaev VO.. Microdomain switch of cGMP-regulated phosphodiesterases leads to ANP-induced augmentation of beta-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ Res 2015;116:1304–1311. [DOI] [PubMed] [Google Scholar]
- 4. Rochais F, Vandecasteele G, Lefebvre F, Lugnier C, Lum H, Mazet JL, Cooper DM, Fischmeister R.. Negative feedback exerted by cAMP-dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes: an in vivo study using adenovirus-mediated expression of CNG channels. J Biol Chem 2004;279:52095–52105. [DOI] [PubMed] [Google Scholar]
- 5. Sprenger JU, Perera RK, Steinbrecher JH, Lehnart SE, Maier LS, Hasenfuss G, Nikolaev VO.. In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat Commun 2015;6:6965.. [DOI] [PubMed] [Google Scholar]
- 6. Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta 1957;23:394–401. [DOI] [PubMed] [Google Scholar]
- 7. Blaustein MP. Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am J Physiol 1977;232:C165–1C173. [DOI] [PubMed] [Google Scholar]
- 8. Bers DM, Despa S.. Na+ transport in cardiac myocytes: implications for excitation-contraction coupling. IUBMB Life 2009;61:215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pogwizd SM. Clinical potential of sodium-calcium exchanger inhibitors as antiarrhythmic agents. Drugs 2003;63:439–452. [DOI] [PubMed] [Google Scholar]
- 10. Verdonck F, Volders PG, Vos MA, Sipido KR.. Intracellular Na+ and altered Na+ transport mechanisms in cardiac hypertrophy and failure. J Mol Cell Cardiol 2003;35:5–25. [DOI] [PubMed] [Google Scholar]
- 11. Pieske B, Maier LS, Piacentino V 3rd, Weisser J, Hasenfuss G, Houser S.. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation 2002;106:447–453. [DOI] [PubMed] [Google Scholar]
- 12. Liu T, O'Rourke B.. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res 2008;103:279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Palmer CJ, Scott BT, Jones LR.. Purification and complete sequence determination of the major plasma membrane substrate for cAMP-dependent protein kinase and protein kinase C in myocardium. J Biol Chem 1991;266:11126–11130. [PubMed] [Google Scholar]
- 14. Bogaev RC, Jia LG, Kobayashi YM, Palmer CJ, Mounsey JP, Moorman JR, Jones LR, Tucker AL.. Gene structure and expression of phospholemman in mouse. Gene 2001;271:69–79. [DOI] [PubMed] [Google Scholar]
- 15. Wetzel RK, Sweadner KJ.. Phospholemman expression in extraglomerular mesangium and afferent arteriole of the juxtaglomerular apparatus. Am J Physiol Renal Physiol 2003;285:F121–F129. [DOI] [PubMed] [Google Scholar]
- 16. Crambert G, Fuzesi M, Garty H, Karlish S, Geering K.. Phospholemman (FXYD1) associates with Na,K-ATPase and regulates its transport properties. Proc Natl Acad Sci USA 2002;99:11476–11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khafaga M, Bossuyt J, Mamikonian L, Li JC, Lee LL, Yarov-Yarovoy V, Despa S, Bers DM.. Na(+)/K(+)-ATPase E960 and phospholemman F28 are critical for their functional interaction. Proc Natl Acad Sci USA 2012;109:20756–20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walaas SI, Czernik AJ, Olstad OK, Sletten K, Walaas O.. Protein kinase C and cyclic AMP-dependent protein kinase phosphorylate phospholemman, an insulin and adrenaline-regulated membrane phosphoprotein, at specific sites in the carboxy terminal domain. Biochem J 1994;304:635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han F, Bossuyt J, Despa S, Tucker AL, Bers DM.. Phospholemman phosphorylation mediates the protein kinase C-dependent effects on Na+/K+ pump function in cardiac myocytes. Circ Res 2006;99:1376–1383. [DOI] [PubMed] [Google Scholar]
- 20. Fuller W, Howie J, McLatchie LM, Weber RJ, Hastie CJ, Burness K, Pavlovic D, Shattock MJ.. FXYD1 phosphorylation in vitro and in adult rat cardiac myocytes: threonine 69 is a novel substrate for protein kinase C. Am J Physiol Cell Physiol 2009;296:C1346–13C1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Despa S, Tucker AL, Bers DM.. Phospholemman-mediated activation of Na/K-ATPase limits [Na]i and inotropic state during beta-adrenergic stimulation in mouse ventricular myocytes. Circulation 2008;117:1849–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boguslavskyi A, Pavlovic D, Aughton K, Clark JE, Howie J, Fuller W, Shattock MJ.. Cardiac hypertrophy in mice expressing unphosphorylatable phospholemman. Cardiovasc Res 2014;104:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse MJ.. Novel single chain cAMP sensors for receptor-induced signal propagation. J Biol Chem 2004;279:37215–37218. [DOI] [PubMed] [Google Scholar]
- 24. Börner S, Schwede F, Schlipp A, Berisha F, Calebiro D, Lohse MJ, Nikolaev VO.. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat Protoc 2011;6:427–438. [DOI] [PubMed] [Google Scholar]
- 25. Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J.. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010;327:1653–1657. [DOI] [PubMed] [Google Scholar]
- 26. Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J.. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci USA 2009;106:6854–6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schobesberger S, Tokar S, Wright P, Bhargava A, Glukhov AV, Poulet C, Buzuk A, Monszpart A, Sikkel MB, Harding SE, Nikolaev VO, Lyon AR, Gorelik J.. T-tubule remodelling disturbs localised β2-adrenergic signalling in rat ventricular myocyte during the progression of heart failure. Cardiovasc Res 2017;113:770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tulloch LB, Howie J, Wypijewski KJ, Wilson CR, Bernard WG, Shattock MJ, Fuller W.. The inhibitory effect of phospholemman on the sodium pump requires its palmitoylation. J Biol Chem 2011;286:36020–36031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sleiman NH, McFarland TP, Jones LR, Cala SE.. Transitions of protein traffic from cardiac ER to junctional SR. J Mol Cell Cardiol 2015;81:34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nikolaev VO, Bünemann M, Schmitteckert E, Lohse MJ, Engelhardt S.. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ Res 2006;99:1084–1091. [DOI] [PubMed] [Google Scholar]
- 31. Silverman B, Fuller W, Eaton P, Deng J, Moorman JR, Cheung JY, James AF, Shattock MJ.. Serine 68 phosphorylation of phospholemman: acute isoform-specific activation of cardiac Na/K ATPase. Cardiovasc Res 2005;65:93–103. [DOI] [PubMed] [Google Scholar]
- 32. Cheung JY, Zhang XQ, Song J, Gao E, Rabinowitz JE, Chan TO, Wang J.. Phospholemman: a novel cardiac stress protein. Clin Transl Sci 2010;3:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wypijewski KJ, Howie J, Reilly L, Tulloch LB, Aughton KL, McLatchie LM, Shattock MJ, Calaghan SC, Fuller W.. A separate pool of cardiac phospholemman that does not regulate or associate with the sodium pump: multimers of phospholemman in ventricular muscle. J Biol Chem 2013;288:13808–13820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang XQ, Qureshi A, Song J, Carl LL, Tian Q, Stahl RC, Carey DJ, Rothblum LI, Cheung JY.. Phospholemman modulates Na+/Ca2+ exchange in adult rat cardiac myocytes. Am J Physiol Heart Circ Physiol 2003;284:H225–H233. [DOI] [PubMed] [Google Scholar]
- 35. Xiao RP. Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci STKE 2001;2001:re15.. [DOI] [PubMed] [Google Scholar]
- 36. Head BP, Patel HH, Roth DM, Lai NC, Niesman IR, Farquhar MG, Insel PA.. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J Biol Chem 2005;280:31036–31044. [DOI] [PubMed] [Google Scholar]
- 37. Juel C, Hostrup M, Bangsbo J.. The effect of exercise and beta2-adrenergic stimulation on glutathionylation and function of the Na, K-ATPase in human skeletal muscle. Physiol Rep 2015;3:e12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hostrup M, Kalsen A, Onslev J, Jessen S, Haase C, Habib S, Ortenblad N, Backer V, Bangsbo J.. Mechanisms underlying enhancements in muscle force and power output during maximal cycle ergometer exercise induced by chronic beta2-adrenergic stimulation in men. J Appl Physiol (1985) 2015;119:475–486. [DOI] [PubMed] [Google Scholar]
- 39. Kalsen A, Hostrup M, Backer V, Bangsbo J.. Effect of formoterol, a long-acting beta2-adrenergic agonist, on muscle strength and power output, metabolism, and fatigue during maximal sprinting in men. Am J Physiol Regul Integr Comp Physiol 2016;310:R1312–R1321. [DOI] [PubMed] [Google Scholar]
- 40. Mehel H, Emons J, Vettel C, Wittkopper K, Seppelt D, Dewenter M, Lutz S, Sossalla S, Maier LS, Lechene P, Leroy J, Lefebvre F, Varin A, Eschenhagen T, Nattel S, Dobrev D, Zimmermann WH, Nikolaev VO, Vandecasteele G, Fischmeister R, El-Armouche A.. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta-adrenergic responses in cardiomyocytes. J Am Coll Cardiol 2013;62:1596–1606. [DOI] [PubMed] [Google Scholar]
- 41. Kuhn M. Structure, regulation, and function of mammalian membrane guanylyl cyclase receptors, with a focus on guanylyl cyclase-A. Circ Res 2003;93:700–709. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.