

Abstract
Our understanding of the mechanisms underlying process in Alzheimer's disease (AD) is far from completion and new therapeutic targets are urgently needed. Recently, the link between dementia and diabetes mellitus (DM) prompted us to search for new therapeutic strategies from glucose metabolism regulators for neurodegeneration. Previous studies have indicated that fibroblast growth factor 21 (FGF21), an attractive and potential therapeutic treatment for DM, may exert diverse effects in the central nervous system. However, the specific biological function and mechanisms of FGF21 on AD is still largely unknown. We report here a study in vivo and in vitro of the neuroprotective effects of FGF21 on cell apoptosis, tau hyperphosphorylation and oxidative stress induced by amyloid β-peptide 25–35. In the present study, the results also further provided evidence for molecular mechanisms by which FGF21 exerted its beneficial effects in neuron and suggested that the regulation of protein phosphatase 2A / mitogen-activated protein kinases / hypoxia-inducible factor-1α pathway may play a key role in mediating the neuroprotective effects of FGF21 against AD-like pathologies.
Abbreviations: Aβ, amyloid β-peptide; AD, Alzheimer's disease; Bcl-2, B-cell lymphoma-2; Bax, Bcl2-associated X; DM, diabetes mellitus; ERK1/2, extracellular signal-regulated kinases 1/2; FBS, fetal bovine serum; FGF21, fibroblast growth factor 21; HIF-1α, hypoxia-inducible factor-1α; icv, intracerebroventricular; JNK, c-Jun N-terminal kinase; MAPKs, mitogen-activated protein kinases; MWM, Morris water maze; MTT, 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; NFTs, neurofibrillary tangles; 8-OHdG, , 8-hydroxy-2’-deoxyguanosine; p38 MAPK, p38 mitogen activated protein kinase; PP2A, protein phosphatase 2 A; ROS, reactive oxygen species; SCN, suprachiasmatic nucleus; TUNEL, TdT‐mediated dUTP nick end labeling
Keywords: Fibroblast growth factor 21, Alzheimer's disease, Tau, Oxidative stress, Mitogen-activated protein kinases
Graphical abstract
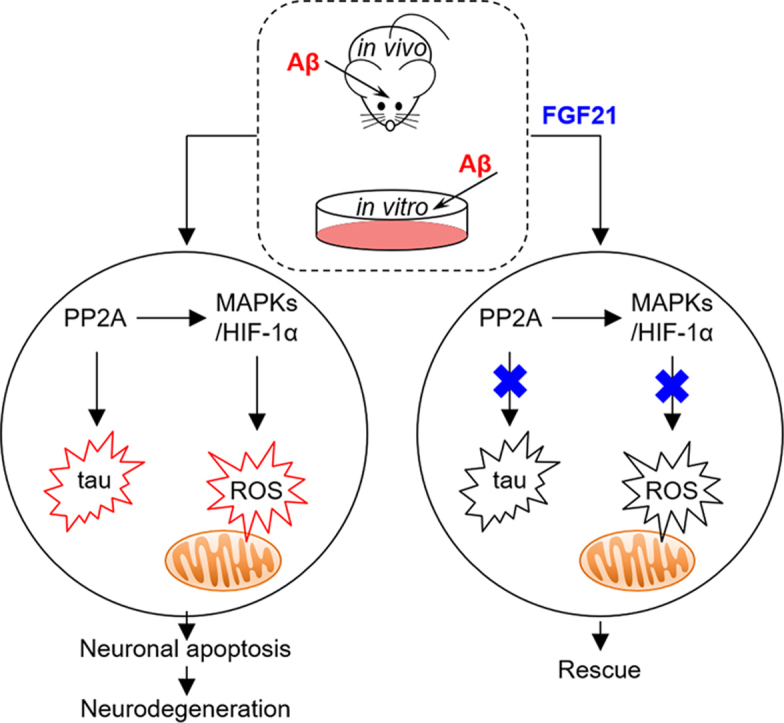
Highlights
-
•
In vivo and in vitro evidence for Aβ -induced neurodegeneration ameliorated by FGF21.
-
•
FGF21 alleviated tau and oxidative stress pathologies in AD rat and cellular models.
-
•
PP2A / MAPKs / HIF-1α pathway was involved in the neuroprotective effect of FGF21.
1. Introduction
Alzheimer's disease (AD), with the main clinical manifestations including impaired cognitive function, decreased learning ability and gradual decline in behavioral ability, is one of the most common neurodegenerative diseases [1], [2]. Since twentieth century, along with the increasing aging of the world population, AD has become a more and more serious threat to the health of the elderly [3], [4]. AD has two notable pathological features including senile plaques caused by the abnormal deposition of amyloid β-peptide (Aβ) and neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau [1], [2]. Aβ may induce toxicity in the central nervous system, which eventually leads to neuron death, and also the neurotoxicity results in tau protein hyperphosphorylation and subsequent aggregation and formation of NFTs, which destroys the microtubule structure stability of neurons, damages the transport function of axons, causes neuronal cell death, and results in cognitive decline. However, the cellular and molecular mechanism underlying AD is still largely unknown, and new therapeutic targets and strategies are urgently needed.
Recently, numerous studies indicated a strong underlying link between dementia and diabetes mellitus (DM) which are two kinds of diseases both with high prevalence in the elderly population [5], [6], [7] and it prompted us to search for pharmacotherapy for AD based on endogenous regulators of glucose metabolism [8], [9], [10], [11], [12]. We were interested in this research area and in recent years focusing on glucose metabolism regulators such as glucagon-like peptide −1 [5], [8], [9], [10], [11] which can be beneficial to neurodegenerative diseases. Fibroblast growth factor 21 (FGF21), a member of the fibroblast growth factor family [13], is also an attractive and potential therapeutic treatment for DM [14]. Previous studies have shown that FGF21 receptors were also expressed in the brain [15], [16], and it has been already confirmed that FGF21 can pass through the blood-brain barrier by simple diffusion [17]. Thus, FGF21 can directly exert effects in the brain [16], [18], [19], [20], [21]. Lan et al. [18] found that FGF21 can regulate body weight and glycemia via the nervous system. Previous report [19] suggested that FGF21 induced sympathetic nerve activity to brown adipose tissue via its actions on corticotropin-releasing factor which is primarily localized in the paraventricular nucleus. Owen et al. [20] demonstrated that FGF21 can act on the suprachiasmatic nucleus (SCN) in the hypothalamus and cause infertility in female mice. Bookout et al. [21] showed that FGF21 can alter circadian behavior and metabolism by acting on the SCN of the hypothalamus and the dorsal vagal complex of the hindbrain. Leng et al. [22] demonstrated that the mood stabilizers lithium and valproic acid may exert synergistic neuroprotective effects through FGF21, and FGF21 can be a potential new therapeutic target for central nervous system disorders. Recent studies also reported that FGF21 can protect animal brain against the effects of high-fat diet [23] and D-galactose [24]. However, our understanding of effects and mechanisms of FGF21 on AD is far from completion. In the present study, we studied the effects as well as the underlying mechanisms of FGF21 on cell apoptosis, tau phosphorylation and oxidative stress induced by amyloid β-peptide 25-35 (Aβ25-35) in vitro and in vivo.
2. Materials and methods
2.1. Materials
RPMI 1640 medium and fetal bovine serum (FBS) were purchased from Gibco Life Technologies (Grand Island, NY, USA). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) and trypsin were purchased from Sigma Chemical Co. (St. Louis, MO, USA). MEK/ERK inhibitor PD98059 was purchased from MedChem Express (Monmouth Junction, NJ, USA). Aβ25–35 was purchased from GL Biochem (Shanghai) Ltd. (Shanghai, China). Aβ25–35 peptide was dissolved in distilled water at the concentration of 1 mg/ml, filtered to remove bacteria and incubated at 37 °C for 4 days to form aggregated Aβ25–35 peptide as previously described by us [25]. Recombinant human FGF21 was expressed and purified from Escherichia coli in our laboratory.
2.2. Cell culture and treatments
SH-SY5Y cells were cultured in a medium consisting of RPMI 1640 medium supplemented with 10% FBS. Cells were grown in humidified 5% CO2/95% air at 37 °C. Cells were digested with 0.25% trypsin and passaged every 2–3 days. The RPMI 1640 medium containing 1% FBS was used for the experimental groups. Unless otherwise indicated, Aβ25–35 (0.125 μM) was added 8 h before FGF21 (1 μM) and cells were incubated in Aβ25-35 with or without FGF21 for 48 h. For experiment using inhibitor, 1 μM PD98059 was added to SH-SY5Y cells 30 min before Aβ25–35.
2.3. Animals and treatments
Adult male Wistar rats (220–250 g) were purchased from Comparative Medicine Centre of Yangzhou University (Yangzhou, China). All rats were randomly divided into the following groups (n = 10 in each group): control, Aβ25–35, Aβ25–35+FGF21 (1 mg·kg−1·d−1), Aβ25–35+FGF21 (2 mg·kg−1·d−1) and Aβ25–35+FGF21 (5 mg·kg−1·d−1) groups. The animals were weighed and anesthetized with an intraperitoneal injection of 10% chloral chloral (300 mg/kg). Then, the anesthetized rats were fixed on the stereotaxic apparatus (RWD Life Science Co., Ltd., Shenzhen, China). Small burr holes were drilled on two sides of the skull (1.0 mm posterior to bregma and 1.5 mm lateral to the midline) to allow intracerebroventricular (icv) injection of Aβ25–35 (5 nmol / 10 μl) / saline at the depth of 3.8 mm. The injection lasted 5 min and the needle was left in place for 3 min after the injection. FGF21 was subcutaneously injected with the dose of 1 mg·kg−1·d−1, 2 mg·kg−1·d−1 or 5 mg·kg−1·d−1 (twice-daily) from the second day after the surgery. The control group and the Aβ25–35 group were treated with saline in the same way. In this study, surgical and animal care procedures were approved by the Institutional Animal Care and Use Committee of China Pharmaceutical University and performed in compliance with the rules in the Guide for the Care and Use of Laboratory Animals.
2.4. Morris water maze (MWM)
After 8 days of administration, MWM test was carried out. The apparatus (ZS Dichuang, Beijing, China) was composed of a circular pool (diameter, 150 cm; height, 60 cm) and an image acquisition system. In the course of the experiment, moderate amount of ink was added into the pool to make the water black and opaque, and the water temperature was controlled between 24 and 26 °C. The round pool was divided into four quadrants and the target platform (diameter, 12 cm; height, 23 cm) was placed in the middle of one of the quadrants 1–2 cm below the water surface. The pool was placed in a test room containing various prominent visual cues so that rat could get a spatial reference memory version of MWM. The rats received two consecutive training trials daily for 5 days of the acquisition training session. The animals were left in the tank facing the wall and allowed to swim freely to the escape platform. If a rat did not find the platform within a period of 90 s, it was gently guided to and remained on the platform for 15 s. The escape latency time and swimming speed were recorded. The probe test was performed on the sixth day. In this test session, the platform was removed from the pool and each rat was allowed to swim for 90 s in the maze. The number of crossing place of the platform and the time spent in the target quadrant were recorded.
2.5. Detection of apoptosis by TdT-mediated dUTP nick-end labeling (TUNEL) assay
Neuronal apoptosis in hippocampus from the various experimental groups was detected by TUNEL assay kit (Roche, Basel, Switzerland) in the paraffin-embedded brain sections according to manufacturer's instructions. Briefly, paraffin-embedded sections (5 µm) were deparaffinized and rehydrated. The sections were permeated with proteinase K solution for 30 min at room temperature, incubated with reaction buffer containing terminal deoxynucleotidyl transferase in a humidified chamber for 1 h at 37 °C, treated with peroxidase-conjugated antibody for 30 min and subsequently developed color in peroxidase substrate. The nuclei were lightly counterstained with hematoxylin. To determine the TUNEL-positive cells, the sections were observed using a light microscopy (Olympus, Tokyo, Japan).
2.6. Assessment of cell viability
Cell viability was measured by a modified MTT assay. Briefly, SH-SY5Y cells were seeded on 96-well plates and incubated with Aβ25–35 (0.015625; 0.03125; 0.0625; 0.125; 0.25; 0.5; 1; 2; 4 μM), or Aβ25–35 (0.125 μM) in the absence / presence of FGF21 (0.0625; 0.125; 0.25; 0.5; 1; 2 μM) for 48 h, and then 500 μg/ml MTT (final concentration) was added to each well and cells were incubated at 37 °C for 4 h. MTT was removed, and 150 μl dimethyl sulfoxide was added to each well to solubilize the formazan crystals formed by live cells. The absorbances at 570 nm and 630 nm (reference) were measured using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
2.7. Detection of apoptosis by Annexin V-FITC/PI assay
Cell apoptosis were determined using Annexin V-FITC/PI assay kit (Beyotime Institute of Biotechnology, Shanghai, China) according to manufacturer's instructions. SH-SY5Y cells were seeded on 6-well plates. After Aβ25–35 treatment with or without drug intervention, cells were digested and centrifuged (400 g, 5 min) to remove the medium. Cells were resuspended with 195 μl binding buffer, stained with 5 μl Annexin V-FITC and 10 μl PI for 15 min, and analysis by a flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
2.8. Western blot analysis
Protein levels in the extracts of SH-SY5Y cells or the hippocampal tissues were analyzed using western blot as previously described [5] with primary antibodies against Thr205-phosphorylated tau-, Tyr307-phosphorylated protein phosphatase 2 A (PP2A)-, total PP2A (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and Thr181-phosphorylated tau-, total tau-, Thr202/Tyr204-phosphorylated extracellular signal-regulated kinases 1/2 (ERK1/2)-, Thr180/Tyr182-phosphorylated p38 mitogen activated protein kinase (p38 MAPK)-, Thr183/Tyr185-phosphorylated c-Jun N-terminal kinase (JNK)-, β-actin-, total ERK1/2-, total p38-, total JNK-, hypoxia-inducible factor-1α (HIF-1α)-, B-cell lymphoma-2 (Bcl-2)-, Bcl2-associated X (Bax)-, cleaved caspase-3-specific antibodies (Cell Signaling Technology, Beverly, MA, USA). Immunoreactive bands were detected with the appropriate horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology) and immunological complexes were visualized by enhanced chemiluminescence reagents (Millipore, Billerica, MA, USA). Western blot signals were analyzed by ImageJ software.
2.9. Immunohistochemistry
8-hydroxy-2’-deoxyguanosine (8-OHdG) in hippocampus from the various experimental groups was detected by immunohistochemistry. The rats were sacrificed and the brains were fixed in 4% paraformaldehyde solution for 24 h at 4 °C. Brain sections were cut at 5 µm thickness and incubated with primary antibodies against 8-OHdG-specific antibody (Santa Cruz Biotechnology). The immunoreaction was detected using the appropriate horseradish peroxidase-conjugated antibody (Cell Signaling Technology) and immunological complexes were visualized by diaminobenzidine tetrahydrochloride (Zymed, San Francisco, CA, USA) and observed with a light microscope (Olympus).
2.10. Morphological detection of mitochondria with transmission electron microscope
The morphological changes of mitochondria were analyzed using transmission electron microscopy. SH-SY5Y cells were seeded on 6-well plates. After Aβ25–35 treatment with or without drug intervention, cells were harvested, fixed in 2.5% glutaraldehyde overnight and then postfixed in 1% OsO4 at 4 °C for 2 h. Afterwards, cells were dehydrated in graded acetone, and embedded in Epon. Ultrathin sections were cut, stained with uranyl acetate followed by lead citrate and observed with a transmission electron microscope (JEOL, Tokyo, Japan).
2.11. Measurement of intracellular reactive oxygen species (ROS)
Intracellular ROS were detected using ROS assay kit (Beyotime Institute of Biotechnology) according to manufacturer's instructions. Briefly, SH-SY5Y cells were incubated with 2′,7′-dichlorofluorescein diacetate (10 μmol/L) at 37 °C for 20 min. Subsequently, cells were washed with PBS three times and observed with a flow cytometry (BD Biosciences). The mean fluorescence intensity was analyzed by FlowJo 10 software.
2.12. Statistical analysis
The mean ± standard error of the mean was determined for each group (n ≥ 3) in a given experiment. Comparison among groups was performed using analysis of variance followed by Tukey's test. P values < 0.05 were considered significant.
3. Results
3.1. The beneficial effect of FGF21 in learning and memory in AD rat models induced by icv-Aβ25–35
First in this study, MWM test was used to investigate spatial learning and memory of rats. In the control group, the average escape latency in searching for the target platform decreased with training. Icv injection of 5 nmol Aβ25–35, however, resulted in longer latency, indicating a significant decline in spatial learning and memory. Meanwhile, the increased escape latency in the AD model rats was attenuated by FGF21 (Fig. 1A and B). The swimming speed among different groups did not show any significant alteration during training period indicating no motor disturbance in the treated animals (Fig. 1C). After five days of training, the spatial probe test (Fig. 1D, E and F) was carried out on the sixth day. The number of crossing the place of the platform (Fig. 1E) and the time spent in the target quadrant (Fig. 1F) in the AD model group were less than those in the control group, and FGF21 increased the crossing number and the swimming time in the target quadrant. These results suggested that FGF21 can improve Aβ25–35-induced cognitive impairment.
Fig. 1.

The beneficial effect of FGF21 in learning and memory in AD rat models induced by icv-Aβ25–35. The MWM was conducted for testing the learning and memory abilities of rats in different groups. n = 10. The representative swim paths (A), the escape latency (B), and average swimming speed (C) in training trials; and the representative swim paths (D), the number of crossing the place of the platform (E), and time spent in the target quadrant (F) in spatial probe test were shown. #P < 0.05, ##P < 0.01 and ###P < 0.001 compared with the control group; * P < 0.05, * * P < 0.01 and * ** P < 0.001 compared with the Aβ25–35 group.
3.2. FGF21 can protect neuron against apoptosis and tau pathology in Aβ25–35-induced rat and cell models
In the present study, we evaluated the effects of FGF21 on neuron apoptosis and tau pathology both in vivo and in vitro models. As the in vivo data showed (Fig. 2A and B), Aβ25–35 induced neuronal apoptosis in the model group; while FGF21 could prevent the apoptosis of hippocampal neurons induced by Aβ25–35 (Fig. 2A). And as shown by the results in Fig. 2B, FGF21 treatment reduced the levels of the phosphorylated tau at Thr181 and Thr205 induced by Aβ25–35 in rats’ hippocampus (Fig. 2B). In the in vitro experiments, first we assessed the effects of Aβ25–35 on cell viability in SH-SY5Y cells. Cells were treated with Aβ25–35 (0.015625–4 μM) for 48 h, and cell viabilities were then analyzed (Fig. 2C). The cell viabilities were reduced significantly by Aβ25–35 in a dose-dependent manner (Fig. 2C). FGF21 (0.0625–2 μM) were added after 8 h of injury by 0.125 μM Aβ25–35; and compared with the Aβ25–35 group, FGF21 intervention can increase cell viabilities, especially at high concentrations (1 μM and 2 μM) (Fig. 2D). Based on in vitro cell model, we further confirmed that FGF21 can alleviate apoptosis induced by Aβ25–35 in vitro (Fig. 2E) and ameliorate tau pathology in Aβ25–35-induced cell models (Fig. 2F).
Fig. 2.

The effects of FGF21 against neuron apoptosis and tau pathology in Aβ25–35-induced cell and rat models. (A) Effect of FGF21 against Aβ25–35-induced neuronal apoptosis in rats’ hippocampus was evaluated by TUNEL assay. Magnified views of the boxed areas in the upper images (Scale bar, 200 µm) were shown in the lower row of images (Scale bar, 50 µm). (B) The levels of Thr181-phosphorylated tau, Thr205-phosphorylated tau, total tau in rats’ hippocampus were measured by western blotting. (C) Effect of Aβ25–35 on SH-SY5Y cell viability. (D) Neuroprotective effect of FGF21 against Aβ25–35-induced decline in SH-SY5Y cell viability. (E) Effect of FGF21 against Aβ25–35-induced apoptosis of SH-SY5Y cells. (F) The levels of Thr181-phosphorylated tau, Thr205-phosphorylated tau, total tau in SH-SY5Y cells were measured by western blotting. All experiments were repeated at least three times. #P < 0.05, ##P < 0.01 and ###P < 0.001 compared with the control group; * P < 0.05, * * P < 0.01 and * ** P < 0.001 compared with the Aβ25–35 group.
3.3. Oxidative stress pathology induced by Aβ25–35 in cell and rat models can be ameliorated via FGF21 treatment
Mitochondria are the primary targets for oxidative damage attacks, and in this study, as shown in the results, Aβ25–35 caused abnormal morphologies such as vacuolation of mitochondria in SH-SY5Y cells, and the abnormalities were ameliorated by FGF21 treatment (Fig. 3A). Then we detected the ROS in cells; and it was shown that the intracellular ROS was significantly increased after Aβ25–35 treatment and decreased by FGF21 treatment, indicating that FGF21 could reduce Aβ25–35 induced up-regulation of intracellular ROS (Fig. 3B). Bcl-2, Bax and cleaved caspase-3 are directly related to mitochondrial related cell apoptosis. Thus we detected the expression levels of Bcl-2, Bax and cleaved caspase-3 under different treatments in SH-SY5Y cells. It was shown that the expression ratio of Bcl-2/Bax decreased and the expression level of cleaved caspase-3 increased in the Aβ25–35 group; and FGF21 could rescue the abnormal expressions of these proteins (Fig. 3C). Excessive ROS causes oxidative damage, and we further detected the level of 8-OHdG, a sensitive marker of DNA oxidation induced by ROS, in brain sections. We found that 8-OHdG was increased under Aβ25–35 treatment, and FGF21 prevented the accumulation of 8-OHdG (Fig. 3D). We also confirmed the changing of Bcl-2, Bax and cleaved caspase-3 in the rat model in this study, and the abnormal expressions of these proteins can be regulated by FGF21 treatment (Fig. 3E). The in vivo and in vitro evidence showed that FGF21 was proved to be protective factor against the oxidative damage caused by Aβ25–35.
Fig. 3.

The neuroprotective effects of FGF21 against mitochondrial oxidative damage pathology in vivo and in vitro. (A) Effect of FGF21 on Aβ25–35-induced morphologic changes of mitochondria in SH-SY5Y cells (Left column: scale bar, 5 µm; right column: scale bar, 2 µm). (B) Effect of FGF21 on Aβ25–35-induced excessive production of ROS in SH-SY5Y cells. (C) Effects of Aβ25–35 and FGF21 on the expression levels of Bcl-2, Bax, cleaved caspase-3 in SH-SY5Y cells. (D) The levels of 8-OHdG in rats’ hippocampus under different treatments (Upper column: scale bar, 200 µm; Lower column: scale bar, 50 µm). (E) Effects of Aβ25–35 and FGF21 on the expression levels of Bcl-2, Bax, cleaved caspase-3 in rats’ hippocampus. All experiments were repeated at least three times. #P < 0.05, ##P < 0.01 and ###P < 0.001 compared with the control group; * P < 0.05 and * * P < 0.01 compared with the Aβ25–35 group.
3.4. PP2A/mitogen-activated protein kinases (MAPKs)/HIF-1α pathway was involved in the neuroprotective effect of FGF21
To investigate the molecular mechanisms of FGF21 mediated amelioration of AD-like pathologies, we analyzed the phosphorylation of PP2A, MAPKs and protein levels of HIF-1α in vivo (Fig. 4A) and in vitro (Fig. 4B). The results showed that FGF21 normalized the phosphorylation levels of PP2A, ERK1/2, p38 and JNK upregulated by Aβ25–35 in vivo and in vitro. FGF21 also decreased the abnormal expression of HIF-1α, which was induced by the treatment of Aβ25–35. Base on the results of ERK1/2, p38 and JNK in this study, it seems that ERK1/2 may play a key role in MAPKs mediated FGF21 effects. Thus we further focused on ERK1/2 in the present study; and results of MEK/ERK inhibitor (PD98059) experiments demonstrated that the amelioration effects of FGF21 on both tau pathology (Fig. 4C) and oxidative stress pathology (Fig. 4D) were at least partially mediated by ERK1/2.
Fig. 4.

The role of PP2A/MAPKs/HIF-1α pathway in the neuroprotective effect of FGF21 against tau pathology and oxidative stress damage. The phosphorylation and total levels of PP2A, ERK1/2, p38 and JNK, and the expression levels of HIF-1α in rats’ hippocampus (A) and in SH-SY5Y cells (B) were measured by western blotting. The effects of inhibitor PD98059 on the phosphorylated-ERK1/2, total ERK1/2, Thr181-phosphorylated tau, Thr205-phosphorylated tau, total tau (C) and ROS production (D) in different groups with or without Aβ25–35 /FGF21 were shown. All experiments were repeated at least three times. #P < 0.05, ##P < 0.01 and ###P < 0.001 compared with the control group; * P < 0.05, * * P < 0.01 and * ** P < 0.001 compared with the Aβ25–35 group.
4. Discussion
AD is one of the most common neurodegenerative diseases [1], however, our understanding for mechanism underlying development of AD is far from completion and new therapeutic targets and strategies are urgently needed. Based on the link between AD and diabetes [5], [6], [7], [26], we are interested in searching for pharmacotherapy for AD from endogenous regulators of glucose metabolism including FGF21. It has been already reported that FGF21's potential receptors were expressed in the central nervous system [15], [16]. And also the property of crossing the blood-brain barrier of FGF21 [17] makes that FGF21 seems promising as a potential new therapeutic target for central nervous system disorders. In addition, previous studies indicated that FGF-21 can ameliorate the brain injury induced by high-fat diet [23] and D-galactose [24]. However, more direct evidence is still needed in order to elucidate the protective effect of FGF21 against AD, and also further studies are also needed to explore the related underlying mechanisms of FGF21 effects on neuron.
Although our understanding of mechanisms underlying the pathology of AD is still limited, the abnormal deposition of Aβ, hyperphosphorylated tau and oxidative stress are considered as key factors for AD [27], [28]. NFTs, which are filamentous inclusions in the hippocampus and the cerebral cortex and mainly composed of abnormally hyperphosphorylated tau proteins, occur in AD and also some other neurodegenerative disorders [29]. Although the phosphorylation of tau is just a normal regulation pattern for the ability of tau to bind and stabilize microtubules, the hyperphosphorylated tau may become toxic for neuron [30]. As to mitochondrial oxidative damage, it is also closely related to the pathogenesis of AD [31], and it is even considered as one of the earliest manifestations of AD [32], [33]. Studies have shown that the excessive production of ROS will cause obvious biological damage to mitochondria when oxidative stress occurs [34], [35]. Meanwhile, the impaired mitochondria will further promote the formation of oxygen free radicals [36]. The two factors are mutually reinforcing and promoting, which eventually lead to synaptic loss, apoptosis and memory impairment. In this study, we demonstrated that FGF21 treatment markedly reduced the cognitive impairments, neuron apoptosis, tau hyperphosphorylation, mitochondrial morphologic abnormalities and oxidative damage, and excessive ROS generation caused by Aβ25–35. Furthermore, we also investigated the possible molecular mechanisms mediating the protective effects of FGF21 against Aβ25–35-induced AD-like pathologies.
Members of the MAPKs family, mainly including ERK1/2, JNK, and p38, widely exist in mammalian cells and participate in many physiological processes. The MAPKs/HIF-1α/mitochondrial oxidative damage pathway may play key role in the neuron apoptosis [37], and abnormal activation of MAPKs induced by Aβ has already been described [25], [37], [38]. PP2A is one of the major phosphatases that negatively regulate phosphorylation of MAPK [39], [40]. Furthermore, PP2A activity can be suppressed by Aβ [25], [41]. These evidences indicated that PP2A/MAPKs may play a critical role in the occurrence and development of AD. On another aspect, numerous previous studies have already indicated that abnormal PP2A is responsible for the hyperphosphorylation of tau [25], [41], [42], [43], [44]. In APP/PS1 transgenic mice, Giraldo et al. [45] reported that they have found an abnormal high level of p-p38 MAPK in the hippocampus; and other studies showed that regulation of MAPK and HIF-1α signaling pathways can alleviate AD-related phenotypes in APP/PS1 mice [37]. Recently, McKenzie-Nickson et al. [46] reported that PP2A was abnormal in APP/PS1 mice, and targeting PP2A can rescue disease phenotype. With regard to human AD brain, there is also evidence showing that MAPK activity was altered [47]; and Liu et al. [48] have indicated that in human AD brain the tau pathology is partially caused by a downregulation of PP2A activity. So as to the molecular mechanism exploration in the present study, first we checked the PP2A phosphorylation levels in vivo and in vitro models. Then we further investigated the PP2A/MAPKs/HIF-1α pathway, and the results indicated that at least to a certain extent all the three classical pathways of MAPKs were involved in the neuroprotective effects of FGF21. The MAPKs, including ERK1/2, JNK, and p38, are downstream molecules of FGF signaling, however, stress effect associated with MAPK activation may make the situation more complicated because of the FGF / dual specificity phosphatase pathway-induced feed back inhibition by dephosphorylating ERK [49]. Dollet et al. [50] found that seipin knockdown cell-line showed activation of p38 MAPK and FGF21 downregulated p-p38 MAPK levels. Base on in vivo and in vitro experiments we can see that ERK1/2 activity variation was much more significant than the other two in response to the Aβ25–35 and FGF21 treatments. So we focused on ERK1/2 in this study, and PD98059 was used to inhibit the phosphorylation of ERK1/2. The inhibitor experiment results suggested that the FGF21 exerted its neuron protective effects at least partially through ERK1/2 pathway. However, mechanism underlying the occurrence and development of AD is complicated by multi-pathway and multi-target involved, and the neuroprotective effects of FGF21 may also be exerted through multiple pathways. As a metabolism regulator, the effects of FGF21 on glucose homeostasis and insulin sensitivity have been shown in previous reports [51], [52]. Meanwhile, according to the view considering AD as "type 3 diabetes", brain energy metabolism may play key role in the development of AD, and brain glucose metabolism and insulin sensitivity may be closely related to Alzheimer-like pathologies [53], [54], [55], [56]. With regard to this one aspect, the potential action of FGF21 on brain glucose metabolism may also be one of the underlying mechanisms of FGF21-mediated neuroprotection.
These data confirmed that Aβ25–35 caused oxidative damage in addition to increasing cell apoptosis and tau phosphorylation levels in rats’ hippocampus and SH-SY5Y cell, and strengthened the link between oxidative stress and AD-like pathologies. More importantly, the results in this study suggested that FGF21 protected against cognitive deficits and oxidative stress. The data showed that FGF21 attenuated Aβ25–35-induced AD-like pathologies and mitochondrial damage. We demonstrated that FGF21 ameliorated abnormal neuronal apoptosis and tau hyperphosphorylation induced by Aβ25–35 in vivo and in vitro. We also found that ROS accumulation, mitochondrial morphologic abnormalities and 8-OHdG accumulation were induced by Aβ25–35 in the AD models, and meanwhile these can be prevented by FGF21 treatment. Results in this study further provided potential molecular mechanisms by which FGF21 exerted its protective effects in neuron and suggested that regulation of PP2A/MAPKs/HIF-1α pathway was a potential target mediated the beneficial effects of FGF21 for AD-like pathologies.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Nos. 81673435, 81872850 and 81430082), the “Double First-Class” University Project (CPU2018GF08), the Open Project of State Key Laboratory of Natural Medicines (No. SKLNMZZCX201822), the “111 Project” from the Ministry of Education of China and the State Administration of Foreign Expert Affairs of China (No. 111-2-07) and PAPD (A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions).
Acknowledgments
Conflict of interest
The authors declare no conflicts of interest.
Contributor Information
Wen-Bing Yao, Email: wbyao@cpu.edu.cn.
Xiang-Dong Gao, Email: xdgao@cpu.edu.cn.
References
- 1.Goedert M., Spillantini M.G. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 2.Querfurth H.W., LaFerla F.M. Alzheimer's disease. N. Engl. J. Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 3.Malik G.A., Robertson N.P. Treatments in Alzheimer's disease. J. Neurol. 2017;264:416–418. doi: 10.1007/s00415-017-8395-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alzheimer's Association Alzheimer's disease facts and figures. Alzheimers Dement. 2018;14:367–429. [Google Scholar]
- 5.Chen S., Liu A.R., An F.M., Yao W.B., Gao X.D. Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related Alzheimer's disease by exendin-4. Age. 2012;34:1211–1224. doi: 10.1007/s11357-011-9303-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narayanan K.L., Prafulla S., Shilpa S., Shankara B.B., Brahmadevarahalli K.S., Meera P., Thangaraju S.P., Sanjeev J., Mathew V., Srikala B. Dementia and diabetes mellitus: association with apolipoprotein E4 polymorphism from a hospital in Southern India. Int. J. Alzheimers Dis. 2014;2012:702972. doi: 10.1155/2012/702972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vagula M. Cognitive impairment and dementia in type 2 diabetes mellitus. U. S. Pharm. 2014;39:33–37. [Google Scholar]
- 8.Chen S., An F.M., Yin L., Liu A.R., Yin D.K., Yao W.B., Gao X.D. Glucagon-like peptide-1 protects hippocampal neurons against advanced glycation end product-induced tau hyperphosphorylation. Neuroscience. 2014;256:137–146. doi: 10.1016/j.neuroscience.2013.10.038. [DOI] [PubMed] [Google Scholar]
- 9.An F.M., Chen S., Xu Z., Yin L., Wang Y., Liu A.R., Yao W.B., Gao X.D. Glucagon-like peptide-1 regulates mitochondrial biogenesis and tau phosphorylation against advanced glycation end product-induced neuronal insult: studies in vivo and in vitro. Neuroscience. 2015;300:75–84. doi: 10.1016/j.neuroscience.2015.05.023. [DOI] [PubMed] [Google Scholar]
- 10.Chen S., Yin L L., X Z., F.M, An, Liu A.R., Wang Y., Yao W.B., Gao X.D. Inhibiting receptor for advanced glycation end product (AGE) and oxidative stress involved in the protective effect mediated by glucagon-like peptide-1 receptor on AGE induced neuronal apoptosis. Neurosci. Lett. 2016;612:193–198. doi: 10.1016/j.neulet.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y., Chen S., X Z., Chen S.T., Yao W.B., Gao X.D. GLP-1 receptor agonists downregulate aberrant GnT-III expression in Alzheimer's disease models through the Akt/GSK-3β/β-catenin signaling. Neuropharmacology. 2017;131:190–199. doi: 10.1016/j.neuropharm.2017.11.048. [DOI] [PubMed] [Google Scholar]
- 12.Duarte A.I., Santos M.S., Oliveira C.R., Moreira P.I. Brain insulin signalling, glucose metabolism and females' reproductive aging: a dangerous triad in Alzheimer's disease. Neuropharmacology. 2018;136:223–242. doi: 10.1016/j.neuropharm.2018.01.044. [DOI] [PubMed] [Google Scholar]
- 13.Kharitonenkov A., Adams A.C. Inventing new medicines: the FGF21 story. Mol. Metab. 2014;3:221–229. doi: 10.1016/j.molmet.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dostalova I., Haluzikova D., Haluzik M. Fibroblast growth factor 21: a novel metabolic regulator with potential therapeutic properties in obesity/type 2 diabetes mellitus. Physiol. Res. 2009;58:1–7. doi: 10.33549/physiolres.931610. [DOI] [PubMed] [Google Scholar]
- 15.Fon T.K., Bookout A.L., Ding X., Hiroshi K., John G.B., Wang L., Regina G., Moosa M., Makoto K., Mangelsdorf D.J., Kliewer S.A. Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 2010;24:2050–2064. doi: 10.1210/me.2010-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanguanmoo P., Chattipakorn N., Chattipakorn S.C. Potential roles of fibroblast growth factor 21 in the brain. Metab. Brain Dis. 2016;31:239–248. doi: 10.1007/s11011-015-9789-3. [DOI] [PubMed] [Google Scholar]
- 17.Hsuchou H., Pan W., Kastin A.J. The fasting polypeptide FGF21 can enter brain from blood. Peptides. 2007;28:2382–2386. doi: 10.1016/j.peptides.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan T., Morgan D.A., Rahmouni K., Sonoda J., Fu X., Burgess S.C., Holland W.L., Kliewer S.A., Mangelsdorf D.J. FGF19, FGF21, and an FGFR1/β-Klotho-activating antibody act on the nervous system to regulate body weight and glycemia. Cell Metab. 2017;26:709–718.e3. doi: 10.1016/j.cmet.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owen B.M., Ding X., Morgan D.A., Coate K.C., Bookout A.L., Rahmouni K., Kliewer S.A., Mangelsdorf D.J. Vol. 20. 2014. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss; pp. 670–677. (Cell Metab.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Owen B.M., Bookout A.L., Ding X., Lin V.Y., Atkin S.D., Gautron L., Kliewer S.A., Mangelsdorf D.J. Vol. 19. 2013. FGF21 contributes to neuroendocrine control of female reproduction; pp. 1153–1156. (Nat. Med.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bookout A.L., de Groot M.H., Owen B.M., Lee S., Gautron L., Lawrence H.L., Ding X., Elmquist J.K., Takahashi J.S., Mangelsdorf D.J., Kliewer S.A. Vol. 19. 2013. FGF21 regulates metabolism and circadian behavior by acting on the nervous system; pp. 1147–1152. (Nat. Med.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leng Y., Wang Z., Tsai L.K., Leeds P., Fessler E.B., Wang J., Chuang D.M. FGF-21, a novel metabolic regulator, has a robust neuroprotective role and is dramatically elevated in neurons by mood stabilizers. Mol. Psychiatry. 2015;20:215–223. doi: 10.1038/mp.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Q., Yuan J., Yu Z., Lin L., Jiang Y., Cao Z., Zhuang P., Whalen M.J., Song B., Wang X.J., Li X., Lo E.H., Xu Y., Wang X. FGF21 attenuates high-fat diet-induced cognitive impairment via metabolic regulation and anti-inflammation of obese mice. Mol. Neurobiol. 2018;55:4702–4717. doi: 10.1007/s12035-017-0663-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu Y., Bai F., Wang W., Liu Y., Yuan Q., Qu S., Zhang T., Tian G., Li S., Li D., Ren G. Fibroblast growth factor 21 protects mouse brain against D-galactose induced aging via suppression of oxidative stress response and advanced glycation end products formation. Pharmacol. Biochem. Behav. 2015;133:122–131. doi: 10.1016/j.pbb.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 25.Xu Z., Chen S., Wang Y., Chen S.T., Yao W.B., Gao X.D. Neuroprotective effects of silk fibroin hydrolysate against Aβ25–35, induced cytotoxicity in SH-SY5Y cells and primary hippocampal neurons by regulating ROS inactivation of PP2A. J. Funct. Foods. 2018;45:100–109. [Google Scholar]
- 26.Yang Y., Song W. Molecular links between Alzheimer's disease and diabetes mellitus. Neuroscience. 2013;25:140–150. doi: 10.1016/j.neuroscience.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 27.Huang H.C., Jiang Z.F. Accumulated amyloid-beta peptide and hyperphosphorylated tau protein: relationship and links in Alzheimer's disease. J. Alzheimers Dis. 2009;16:15. doi: 10.3233/JAD-2009-0960. [DOI] [PubMed] [Google Scholar]
- 28.Markesbery W.R. Oxidative stress hypothesis in Alzheimer's disease. Free Radic. Biol. Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 29.Lamb B. The roles of fractalkine signaling in neurodegenerative disease. Mol. Neurodegener. 2012;7:L21. [Google Scholar]
- 30.Cowan C.M., Bossing T., Page A., Shepherd D., Mudher A. Soluble hyperphosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 2010;120:593–604. doi: 10.1007/s00401-010-0716-8. [DOI] [PubMed] [Google Scholar]
- 31.Beck S.J., Guo L., Phensy A., Tian J., Wang L., Tandon N., Gauba E., Lu L., Pascual J.M., Kroener S., Du H. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer's disease. Nat. Commun. 2016;7:11483. doi: 10.1038/ncomms11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nunomura A., Perry G., Aliev G., Hirai K., Takeda A., Balraj E.K., Jones P.K., Ghanbari H., Wataya T., Shimohama S., Chiba S., Atwood C.S., Petersen R.B., Smith M.A. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 33.Castellani R., Hirai K., Aliev G., Drew K.L., Nunomura A., Takeda A., Cash A.D., Obrenovich M.E., Perry G., Smith M.A. Role of mitochondrial dysfunction in Alzheimer's disease. J. Neurosci. Res. 2002;70:357. doi: 10.1002/jnr.10389. [DOI] [PubMed] [Google Scholar]
- 34.Mancuso M., Coppede F., Migliore L., Siciliano G., Murri L. Mitochondrial dysfunction, oxidative stress and neurodegeneration. J. Alzheimers Dis. 2006;10:59–73. doi: 10.3233/jad-2006-10110. [DOI] [PubMed] [Google Scholar]
- 35.Yu T., Robotham J.L., Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA. 2006;103:2653. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raha S., Robinson B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000;25:502–508. doi: 10.1016/s0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 37.Fang W.L., Zhao D.Q., Wang F., Li M., Fan S.N., Liao W., Zheng Y.Q., Liao S.W., Xiao S.H., Luan P., Liu J. Neurotropin® alleviates hippocampal neuron damage through a HIF-1α/MAPK pathway. Cns. Neurosci. Ther. 2017;23:428–437. doi: 10.1111/cns.12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y., Cao D.H., Wu G.M., Hou X.Y. Involvement of P38MAPK activation by NMDA receptors and non-NMDA receptors in amyloid-β peptide-induced neuronal loss in rat hippocampal CA1 and CA3 subfields. Neurosci. Res. 2014;85:51–57. doi: 10.1016/j.neures.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 39.Grethe S., Porn-Ares M.I. p38 MAPK regulates phosphorylation of Bad via PP2A-dependent suppression of the MEK1/2-ERK1/2 survival pathway in TNF-alpha induced endothelial apoptosis. Cell. Signal. 2006;18:531–540. doi: 10.1016/j.cellsig.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 40.Xu C., Wang X., Zhu Y., Dong X., Liu C., Zhang H., Liu L., Huang S., Chen L. Rapamycin ameliorates cadmium-induced activation of MAPK pathway and neuronal apoptosis by preventing mitochondrial ROS inactivation of PP2A. Neuropharmacology. 2016;105:270–284. doi: 10.1016/j.neuropharm.2016.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu R., Zhou X.W., Tanila H., Bjorkdahl C., Wang J.Z., Guan Z.Z., Cao Y., Gustafsson J.A., Winblad B., Pei J.J. Phosphorylated PP2A (tyrosine 307) is associated with Alzheimer neurofibrillary pathology. J. Cell Mol. Med. 2008;12:241–257. doi: 10.1111/j.1582-4934.2008.00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khoury N.B.E., Papon M.A., Marcouiller F., Julien C., Morin F., Bretteville A., Petry F.R., Gaudreau S., Amrani A., Mathews P.M., Hebert S.S., Planel E. Deregulation of PP2A and hyperphosphorylation of tau protein following onset of diabetes in NOD mice. Diabetes. 2013;62:609–617. doi: 10.2337/db12-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arif M., Wei J., Zhang Q., Liu F., Basurto-Islas G., Grundke-Iqbal I., Iqbal K. Cytoplasmic retention of protein phosphatase 2A inhibitor 2 (I2PP2A) induces Alzheimer-like abnormal hyperphosphorylation of tau. J. Biol. Chem. 2014;289:27677–27691. doi: 10.1074/jbc.M114.565358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang C.C., Kuai X.X., Gao W.B., Yu J.C., Wang Q., Li L., Zhang L. Morroniside-induced PP2A activation antagonizes tau hyperphosphorylation in a cellular model of neurodegeneration. J. Alzheimers Dis. 2016;51:33–44. doi: 10.3233/JAD-150728. [DOI] [PubMed] [Google Scholar]
- 45.Giraldo E., Lloret A., Fuchsberger T., Viña J. Aβ and tau toxicities in Alzheimer's are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. 2014;2:873–877. doi: 10.1016/j.redox.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKenzie-Nickson S., Chan J., Perez K., Hung L.W., Cheng L., Sedjahtera A., Gunawan L., Adlard P.A., Hayne D.J., McInnes L.E., Donnelly P.S., Finkelstein D.I., Hill A.F., Barnham K.J. Modulating protein phosphatase 2A rescues disease phenotype in neurodegenerative Tauopathies. ACS Chem. Neurosci. 2018;9:2731–2740. doi: 10.1021/acschemneuro.8b00161. [DOI] [PubMed] [Google Scholar]
- 47.Swatton J.E., Sellers L.A., Faull R.L., Holland A., Iritani S., Bahn S. Increased MAP kinase activity in Alzheimer's and Down syndrome but not in schizophreniahuman brain. Eur. J. Neurosci. 2004;19:2711–2719. doi: 10.1111/j.0953-816X.2004.03365.x. [DOI] [PubMed] [Google Scholar]
- 48.Liu F., Grundke-Iqbal I., Iqbal K., Gong C.X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005;22:1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 49.Friesel R.E. Fibroblast Growth Factors: Biology and Clinical Application. 2017. Chapter 2: regulation of FGF signaling; pp. 41–72. [Google Scholar]
- 50.Dollet L., Levrel C., Coskun T., Le Lay S., Le May C., Ayer A., Venara Q., Adams A.C., Gimeno R.E., Magré J., Cariou B., Prieur X. FGF21 Improves the adipocyte dysfunction related to seipin deficiency. Diabetes. 2016;65:3410–3417. doi: 10.2337/db16-0327. [DOI] [PubMed] [Google Scholar]
- 51.Xu J., Lloyd D.J., Hale C., Stanislaus S., Chen M., Sivits G., Vonderfecht S., Hecht R., Li Y.S., Lindberg R.A., Chen J.L., Jung D.Y., Zhang Z., Ko H.J., Kim J.K., Véniant M.M. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin Z., Tian H., Lam K.S., Lin S., Hoo R.C., Konishi M., Itoh N., Wang Y., Bornstein S.R., Xu A., Li X. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013;17:779–789. doi: 10.1016/j.cmet.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 53.Baker L.D., Cross D.J., Minoshima S., Belongia D., Watson G.S., Craft S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011;68:51–57. doi: 10.1001/archneurol.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de la Monte S.M. Type 3 diabetes is sporadic Alzheimer׳s disease: mini-review. Eur. Neuropsychopharmacol. 2014;24:1954–1960. doi: 10.1016/j.euroneuro.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu C.C., Hu J., Tsai C.W., Yue M., Melrose H.L., Kanekiyo T., Bu G. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J. Neurosci. 2015;35:5851–5859. doi: 10.1523/JNEUROSCI.5180-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sato N., Morishita R. The roles of lipid and glucose metabolism in modulation of β-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci. 2015;7:199. doi: 10.3389/fnagi.2015.00199. [DOI] [PMC free article] [PubMed] [Google Scholar]