

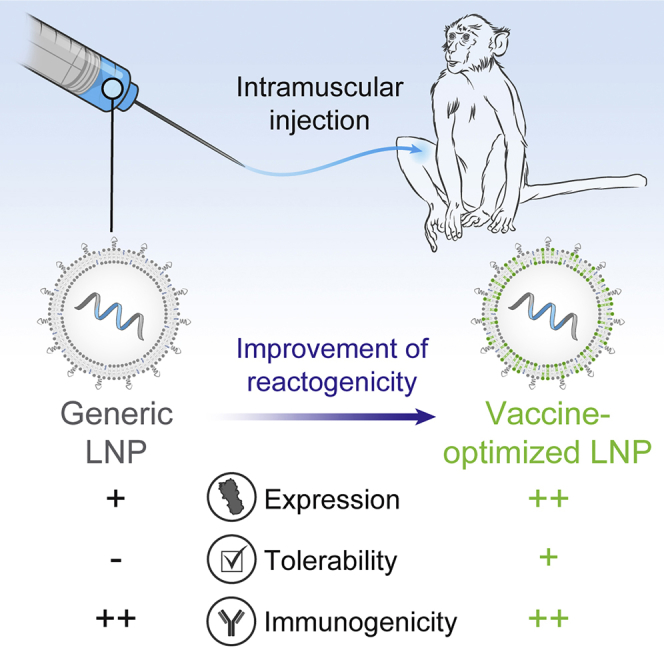
Abstract
mRNA vaccines have the potential to tackle many unmet medical needs that are unable to be addressed with conventional vaccine technologies. A potent and well-tolerated delivery technology is integral to fully realizing the potential of mRNA vaccines. Pre-clinical and clinical studies have demonstrated that mRNA delivered intramuscularly (IM) with first-generation lipid nanoparticles (LNPs) generates robust immune responses. Despite progress made over the past several years, there remains significant opportunity for improvement, as the most advanced LNPs were designed for intravenous (IV) delivery of siRNA to the liver. Here, we screened a panel of proprietary biodegradable ionizable lipids for both expression and immunogenicity in a rodent model when administered IM. A subset of compounds was selected and further evaluated for tolerability, immunogenicity, and expression in rodents and non-human primates (NHPs). A lead formulation was identified that yielded a robust immune response with improved tolerability. More importantly for vaccines, increased innate immune stimulation driven by LNPs does not equate to increased immunogenicity, illustrating that mRNA vaccine tolerability can be improved without affecting potency.
Keywords: mRNA, vaccines, LNP, intramuscular, lipids, tolerability, immunogenicity, formulation
Graphical Abstract
Introduction
Since the first active immunization, vaccines have provided increased life expectancy and improved public health, saving countless lives.1, 2 Today, a variety of technologies exist for vaccine development, including live and attenuated viruses, recombinant proteins, synthetic peptides, glycoconjugates, and nucleic acids.1 Nucleic-acid (DNA and mRNA)-based vaccines offer several advantages over other technologies. They can be rapidly produced with reduced development time and costs by using a common manufacturing platform and purification methods regardless of the antigen. Unlike manufacturing for other vaccines, these methods would not include propagation of viruses or purification of a recombinant protein. The antigen would be expressed in situ, allowing for transmembrane domains to be present, if needed, and multimeric complexes to be formed.3 Additionally, nucleic acids do not suffer from anti-vector immunity like viral vectored vaccines do. Lastly, proteins produced by nucleic-acid-based vaccines can provide a more natural presentation to the immune system, yielding better T cell responses.4 Even so, more than two decades after the first proof-of-concept report,5 no nucleic-acid-based vaccine has been approved for use in humans.
A key factor hampering both DNA and mRNA vaccine development is the lack of a potent, well-tolerated delivery system. Because DNA requires delivery to the nucleus, an inherently inefficient process, high doses (1–2 mg) and an electroporation device are required to generate robust immune responses. Although recent advances in DNA electroporation have shown promise, the broad adoption of the technology will likely be limited due to the necessity of a specialized device and the pain associated with electroporation.6, 7, 8 An advantage of mRNA over DNA is that mRNA only requires cytosolic delivery. In rodents, early studies showed that intramuscular administration of buffer-formulated mRNA can lead to measurable levels of immunogenicity.9 However, a recent phase I trial of a rabies mRNA vaccine administered in Ringer’s buffer yielded no immunogenicity unless delivered with a high-pressure intra-dermal injection device.10
Although promising, these results highlight the need for more potent intracellular delivery technologies for mRNA vaccines. One such technology is lipid nanoparticles (LNPs). LNPs are typically composed of an ionizable lipid, cholesterol, PEGylated lipid, and a helper lipid such as distearoylphosphatidylcholine (DSPC). Early work with small interfering RNA (siRNA) identified the ionizable lipid as the primary driver of potency.11, 12, 13 The most clinically advanced LNP contains the ionizable lipid MC3 and has been shown to be safe in humans after intravenous (IV) administration of siRNA.14 Our own vaccine trials with MC3-based LNPs for influenza gave 100% seroconversion with a 100-μg dose of modified mRNA. However, consistent with other vaccines,15, 16 we did observe mild to moderate local and systemic adverse events.17 As healthy individuals ranging from day-old newborns to the elderly receive vaccines, critical features for broad vaccine adoption are minimal injection site reactivity and high tolerability. To date, the only LNPs evaluated for intramuscular (IM) mRNA vaccine delivery were originally optimized for IV delivery of siRNA to the liver.18, 19 Although there are preclinical reports of novel LNPs being evaluated for vaccines, no rationale has been provided regarding formulation composition or selection.20, 21, 22
Here, we describe rational evolution and selection of an improved formulation for IM administration of mRNA, focusing on the impact of the ionizable lipid component as the primary driver of expression and tolerability. Our previous experience with IV administration of the proprietary ionizable lipids showed rapid clearance compared to MC3,23 resulting in improved systemic tolerability. Our work here illustrates that the ideal formulation for IV expression is not necessarily ideal for IM expression. Additionally, we also show that increased innate immune stimulation driven by the LNP is not necessary for increased immunogenicity, illustrating that we have an opportunity to improve vaccine tolerability without affecting vaccine potency.
Results
Observations of mild to moderate adverse events in our clinical work with MC317 and data showing slow MC3 clearance after IV administration23 fueled a hypothesis that the adverse events might be related to the extended presence of MC3 at the injection site. Mass spectrometry analysis of muscle tissue revealed that, 24 h after IM injection, the MC3 concentration only decreased by 50% compared to Cmax (Figure 1A). Further, MC3 was also detectable in liver and spleen 24 h post-IM injection (Figures 1B and 1C). Thus, IM administration of MC3-formulated mRNA LNPs resulted in extended local and systemic lipid exposure.
Figure 1.

Pharmacokinetics of LNPs containing MC3 after IM administration in mice
Lipid concentration (nanograms per gram) after IM administration of modified mRNA encoding luciferase formulated in LNPs containing MC3 (gray triangles) in muscle, liver, and spleen up to 24 h post-injection (n = 3 per group per time point).
The goal of the work described here was to identify a new ionizable lipid with improved tolerability and a potency equal or better than that of MC3. To do so, we screened 30 novel LNPs, each containing a different ionizable lipid in place of MC3. Each LNP formulation maintained the same lipid-nitrogen-to-phosphate ratio (N:P) and molar composition of lipid components (ionizable lipid, cholesterol, phospholipid, and polytheylene glycol [PEG] lipid). Co-formulation of mRNAs encoding firefly luciferase and the H10N8 influenza hemagglutinin (HA) antigen allowed both protein expression and immunogenicity to be evaluated in the same study. Luciferase activity was measured by whole-body imaging 6 h post-IM injection of the first dose. Immunogenicity was evaluated by quantifying α-H10 immunoglobulin (Ig)G titers 2 weeks after the second dose, which was administered 3 weeks after the first. The ionizable lipids screened here all contain a tertiary amine with ester-containing lipid tails to enable rapid in vivo metabolism.23 In addition, we also tested the quaternary ammonium containing lipid N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium (DOTAP).
Consistent with our previous publications, MC3-formulated mRNA yielded robust titers and protein expression at a low dose (0.001 mg per kg).17, 24 In contrast, we observed no detectable protein expression or immunogenicity for DOTAP-containing LNPs (Figure 2A). Many of our novel biodegradable lipids proved superior to MC3 for both protein expression and immunogenicity upon IM administration. However, there was no strong relationship between protein expression and immunogenicity (r = 0.54). Of the 14 lipids yielding higher α-H10 IgG titers than MC3, four lipids yielded significantly less luciferase expression relative to MC3, whereas four lipids yielded significantly greater luciferase activity (Figure 2B). The two lipids with the highest α-H10 IgG titers were only 1.3-fold better than MC3 with regard to protein expression, illustrating that protein expression upon IM administration was a poor predictor of immunogenicity.
Figure 2.

Expression and Immunogenicity from LNPs Containing Novel Ionizable Lipids in Mice
(A) Thirty novel lipid LNPs, A through E′ were compared to a D (MC3) LNP control for expression and immunogenicity. Lipids are arranged left to right in order of pKa from low (A) to high (DOTAP). Expression measured by luminescence in flux (photons per second) 6 h after administration of modified mRNA encoding luciferase delivered at 0.5 mg/kg IV in CD-1 mice, 0.01 mg/kg IM or 0.001 mg/kg IM in BALB/c mice (n = 5 per group). Immunogenicity measured by H10-specific IgG titers measured 2 weeks after two doses administered 3 weeks apart delivered IM at 0.001 mg/kg IM in BALB/c mice (n = 5 per group). Data are represented as log2 fold change compared to MC3. Squares containing an X indicate >4-fold change (log2) lower than for MC3. (B) Log2 fold increase in expression was compared to the log2 fold change in immunogenicity at the low dose level administered IM (0.001 mg/kg). The five lead novel lipids and MC3 LNPs are labeled accordingly: MC3 (gray triangles), lipid H (green circles), lipid M (orange squares), lipid P (purple diamonds), lipid Q (tan inverted triangles), and lipid N (yellow hexagons). (C) Lipid pKa versus fold increase in immunogenicity at 0.001 mg/kg IM for lipids A through E′. (D) Circulating IgG antibody (micrograms per milliliter of serum) 6 h after administration of 0.2 mg/kg modified mRNAs encoding the heavy chain and light chain of an influenza monoclonal antibody formulated at a 2:1 mass ratio in LNPs containing MC3 or novel lipids (n = 5 per group). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, ordinary one-way ANOVA with Dunnett’s multiple comparisons test of each novel lipid versus MC3.
We also found little correspondence in rank between the LNPs with regard to IM versus IV expression (Figure 2A), illustrating that formulations can behave differently when administered locally versus systemically. A possible explanation for the lack of correlation between IM and IV performance could be that the optimal physical or chemical properties differ between the two routes. One strong determinant of immunogenicity was the lipid pKa, with a range of 6.6–6.9 being optimal for IM immunogenicity (Figure 2C). This differs from the optimal pKa range for IV delivery of siRNAs and mRNAs, which has been reported as 6.2–6.6.11, 23 mRNA encapsulation efficiencies and LNP sizes ranged from 69% to 100% and from 50 to 142 nm, respectively. While there was no relationship between encapsulation efficiency and either IM protein expression or immunogenicity, there was a relationship between both readouts and LNP size, with the best performing formulations being 75–95 nm (Figures S1A and S1B).
For further study, we picked the five ionizable lipids exhibiting the greatest increase in α-H10 IgG titers compared to MC3 (colored symbols in Figure 2; structures in Figure 3A). Notably, the pKa for all five lipids was very close to 6.75 (Figure 2C). As an additional measure of potency, we compared the ability of each lead LNP to drive the expression of a secreted IgG antibody after IM administration in mice (Figure 2D). With the exception of lipid Q, the other four lipids yielded higher IgG serum concentrations than MC3 (p < 0.05).
Figure 3.

Chemical Structure and Pharmacokinetics of Lead Lipids
(A) Chemical structures and pKa of MC3 and novel lipids. (B–D) Lipid concentration (nanograms per gram) after IM administration of modified mRNA encoding luciferase formulated in LNPs containing lipid H (green circles), lipid M (orange squares), lipid P (purple diamonds), lipid Q (tan inverted triangles), and lipid N (yellow hexagons) in (B) muscle, (C) liver, and (D) spleen up to 48 h post-injection (n = 3 per group per time point).
To understand the biodegradability of these lipids, we measured lipid levels after IM administration. As expected, IM delivery of these LNPs in CD-1 mice was followed by rapid clearance (Figures 3B–3D). All lead lipids degraded faster than MC3 in muscle (Figure 3B), spleen (Figure 3C), and liver (Figure 3D). 24 h post-injection, the amount of lipid present in muscle dropped considerably from peak levels for all formulations tested, though lipids H and Q did not return to baseline levels by 48 h. Liver and spleen lipid levels closely followed IM lipid levels, though lipid H showed a peak at 6 h that dropped by 24 h in the spleen and liver.
Immunogenicity in non-human primates (NHPs) was evaluated after IM injections of H10N8 mRNA formulated with the five lead lipids as LNPs. ELISA antibody titers (Figure 4A) and HAI titers (Figure 4B) were not statistically different for any group (one-way ANOVA, p > 0.05), except lipid P was significantly lower than MC3 after the first dose (one-way ANOVA, p < 0.01) by ELISA and after the second dose (one-way ANOVA, p < 0.001) by HAI titer. Immune responses were measurable after a single dose by ELISA. After a second dose, both HAI and ELISA titers boosted considerably, indicating strong immune priming.
Figure 4.

Expression and Immunogenicity in Non-human Primates
(A and B) Immunogenicity measured by H10-specific (A) ELISA or (B) HAI at days 0, 21 (3 weeks after the first dose), and 42 (3 weeks after the second dose). Each dose in cynomolgus monkeys contained 5 μg modified mRNA encoding H10N8 formulated in LNPs containing either MC3 (gray triangles), lipid H (green circles), lipid M (orange squares), lipid P (purple diamonds), lipid Q (tan inverted triangles), or lipid N (yellow hexagons) (n = 3 per group). (C) Circulating IgG levels (in micrograms per milliliter) after a 500-μg IM administration in cynomolgus monkeys of modified mRNA encoding heavy- and light-chain antibodies in a 2:1 weight ratio formulated in LNPs containing MC3 or novel lipids (n = 3 per group). (D and E) Site of injection was monitored for (D) edema and (E) erythema 1 and 3 days after injection. (F) Circulating IL-6 levels (in picograms per milliliter) 6 h after administration. #p > 0.05; ##p > 0.001, two-way ANOVA with Dunnett’s multiple comparison test of each lipid versus MC3 at each time point. **p > 0.01; ****p > 0.0001, z test of areas under the curve (AUCs) for each novel lipid versus MC3.
We also tested protein expression of the five lead lipids in NHPs. 500 μg IgG mRNA formulated in LNPs was injected IM, and serum antibody expression levels were monitored for 2 weeks. While three out of the five selected lipids yielded expression comparable to that of MC3-based LNPs, lipid H (p < 0.001) and lipid M (p = 0.05) showed significantly more expression over time then MC3 (Figure 4C). For lipid H, the maximum antibody concentration measured 24 h post-injection was three times the antibody concentration measured with MC3-formulated material.
To assess tolerability in NHPs, the site of injection was monitored for edema (Figure 4D) and erythema (Figure 4E) 1 and 3 days after injection and was rated based on severity. Despite enhanced protein expression, NHPs injected with lipid-H-based LNPs exhibited no signs of swelling or redness 1 or 3 days post-injection, with all NHPs receiving a score of 0 for both edema and erythema. All other novel lipids evaluated elicited mild to moderate scores for edema and erythema in at least 1 animal dosed. The MC3 group had one NHP receive a score of 3 for edema on day 1 post-injection, resolving to a score of 1 on day 3 post-injection. All lipids tested, except for lipid H, elicited an erythema score of 1 in at least one NHP. Serum interleukin (IL)-6 levels were comparable for all lipids based on one-way ANOVA (Figure 4F). The NHPs in the MC3 group with the highest level of IL-6 also showed the highest level of edema, indicating a strong innate immune response in that individual animal.
To assess and compare the local tolerability of the different ionizable lipid LNPs, we administered 0.01 mg or 0.1 mg mRNA expressing prM-E from the Zika virus formulated in either MC3, lipid H, lipid M, lipid P, lipid Q, or lipid N in Sprague-Dawley rats IM. Serum cytokines in rats receiving both the high and low doses were measured 6 h after administration, using a 22-plex Luminex panel. Changes were observed in eotaxin, GRO-alpha, IP-10, RANTES, and MCP-1 (Figures 5A–5E). With the exception of IP-10 at the 0.01 mg dose, lipid H induced the lowest systemic cytokine production.
Figure 5.

Tolerability in Rats
Serum concentrations (in picograms per milliliter) of cytokines (A) eotaxin, (B) GRO-alpha, (C) IP-10, (D) RANTES, and (E) MCP-1 were measured 6 h after a single IM administration of 0.01 mg or 0.1 mg modified mRNA encoding prM-E from Zika virus formulated in LNPs containing MC3 (gray), lipid H (green), lipid M (orange), lipid P (purple), lipid Q (tan), or lipid N (yellow) (n = 3 per group). (F–I) Representative histology sections stained with H&E 2 days after a single IM administration of 0.1 mg of modified mRNA encoding prM-E from Zika virus formulated in LNPs containing MC3 or lipid H in the (F and H) muscle and (G and I) skin. (F) MC3 muscle; (G) MC3 skin; (H) lipid H muscle; (I) lipid H skin.
Forty-eight hours after administration, animals were sacrificed, and the injection sites were collected, paraffin embedded, sectioned, H&E stained, and blindly reviewed by a pathologist (Table 1). To evaluate, compare, and rank the local tolerability of each LNP, various endpoints were evaluated and graded, including mixed-cell inflammation at the injection site and in the dermis, myofiber necrosis, and relative number of degenerated neutrophils. MC3-formulated mRNA was the worst tolerated lipid tested, whereas lipid H was the best tolerated lipid tested (Figures 5F–5I).
Table 1.
Pathology Summary
| Formulation and Dose | Muscle Fiber Necrosis | Mixed-Cell Inflammation | Degenerate Neutrophils | Mixed-Cell Inflammation | Degenerate Neutrophils |
|---|---|---|---|---|---|
| MC3 | |||||
| 0.01 mg | 2.3 | 2.4 | 1.7 | 2 | 0 |
| 0.1 mg | 2.3 | 2.7 | 3.3 | 2 | 1 |
| Lipid H | |||||
| 0.01 mg | 1 | 1.8 | 1 | 0 | 0 |
| 0.1 mg | 1.3 | 2.9 | 2.3 | 1.3 | 0 |
| Lipid M | |||||
| 0.01 mg | 2 | 2 | 1.3 | 1.7 | 0 |
| 0.1 mg | 1.7 | 2.7 | 2 | 2 | 0 |
| Lipid P | |||||
| 0.01 mg | 2.3 | 2.2 | 1.7 | 1.3 | 0 |
| 0.1 mg | 2.3 | 2.8 | 2.3 | 2.3 | 0.7 |
| Lipid Q | |||||
| 0.01 mg | 2.3 | 2.2 | 2 | 0.7 | 0 |
| 0.1 mg | 2 | 2.9 | 3 | 2.5 | 1 |
| Lipid N | |||||
| 0.01 mg | 0.7 | 1.4 | 2 | 0 | 0 |
| 0.1 mg | 1.3 | 2 | 2.3 | 0 | 0 |
Rats (n = 3 per group) were injected IM with 0.01 or 0.1 mg modified mRNA encoding prM-E from the Zika virus formulated in LNPs containing MC3 or lipid H. Average histopathology scores on a 0–4 scale were recorded for events occurring in the muscle and skin.
Rats dosed with MC3 formulations at both the high and low doses displayed a dose-dependent mixed-cell inflammation characterized by edema; numerous intact and degenerate neutrophils; macrophages; and a few lymphocytes distending endomysium, epimysium, and adjacent connective tissue of the muscle and compressing myofibers at the injection site (Figures 5F and S3A). A dose-dependent multifocal degeneration and/or necrosis of individual myofibers, infiltrated by inflammatory cells at times, was also observed. The mixed inflammation observed in the muscle extended into the subcutaneous portion of the skin (Figures 5G and S3B). The subcutaneous tissue was expanded by edema and numerous intact and degenerate neutrophils, macrophages, and a few lymphocytes.
The dose-related mixed-cell inflammation observed in rats administered lipid H was lower in magnitude and severity when compared to the rats given MC3 (Figure 5H). The relative amount of degenerate neutrophils was also lower, and it is worth noticing that there was less degeneration and/or regeneration and/or necrosis in the myofibers. The extension and spillage of the inflammation from the muscular injection site into the subcutaneous tissue was also less severe and with much less edema than in animals given MC3 (Figure 5I).
Discussion
mRNA vaccines delivered with LNPs have the potential to address numerous unmet medical needs not accessible with current vaccine technologies. Multiple reports from the siRNA field have shown that the ionizable lipid is the primary driver of LNP potency.11, 12, 13 In this work, we observed the impact of ionizable lipid identity on expression, immunogenicity, and tolerability when delivered IM. Our working hypothesis was that the inclusion of a biodegradable lipid within an LNP would lead to vaccines with improved tolerability, as the lipid would be cleared quickly from the site of injection following mRNA delivery, and other tissues would also have minimal exposure to the lipid due to metabolic breakdown and clearance. Interestingly, throughout our initial screening, we noticed little correlation between expression and vaccine immunogenicity, indicating that expression alone is insufficient to identify improved mRNA vaccine formulations. We also observed a divergence in the best expressing formulations between the IV and IM routes of administration.
Ionizable lipid pKa is thought to affect the protein opsonization of the particles, cellular uptake, and endosomal escape efficiency. The optimal lipid pKa for siRNA-mediated knockdown in the liver has been reported to be between 6.2 and 6.5, in line with our finding of the optimal pKa for mRNA delivery to and expression in the liver as between 6.2 and 6.8.11, 13, 23 However, the best lipids with respect to protein expression after IV administration generally had lower pKas than the best lipids for protein expression after IM administration. Lipids such as V (pKa = 6.87) and AC (pKa = 7.09) show little to no expression after IV administration yet were some of the highest expressing lipids after IM administration, indicating a yet-to-be-elucidated difference between these two routes of administration. Different cell types have shown variations in endosome acidification, demonstrating the need for additional work to better understand the performance of LNPs in the context of mRNA delivery across multiple tissues.25, 26 We also found that optimal lipid pKa for immunogenicity was between 6.6 and 6.8. Independent of cytosolic mRNA delivery, lipid pKa may also play a role in formulation interactions with the immune system. Although this research area has not been thoroughly explored, a recent report illustrates how ionizable lipids can drive uptake and transfection in immune cells, demonstrating potential areas of research for LNP-mediated delivery of mRNA vaccines.27 Although lipid pKa was found to be an important factor for driving immunogenicity, it was not the only factor, as many lipids fell within that pKa range and were no better than the MC3 control. In addition to differences in pKa, lipid H also showed an improvement in endosomal escape efficiency, consistent with our previously published report on this class of lipids (Figure S4).23
Multiple previous reports speak to the need for a balance between expression and immune stimulation for optimal mRNA vaccine potency.28, 29 Pollard et al. documented the negative impact of interferon signaling on the magnitude of mRNA expression.29 The mRNAs we used all contained a base modification on uridine to minimize innate immune activation.24, 30 As the mRNA is immune silent compared with canonical uridine-containing mRNA, both antigen selection and delivery system are important to generate potent immune responses. LNPs have been shown to be effective adjuvants for protein subunit vaccines, but it is unclear how important that adjuvant mechanism is for inducing immune responses from an mRNA vaccine. We previously showed that MC3-based LNPs generated innate immune activation and a potent cellular infiltrate.31 The histopathology presented here for lipid H, compared to that for MC3, is consistent with improved tolerability and reduced innate immune stimulation. The reduction in inflammatory cell infiltrate, myofiber damage, and systemic cytokines support the hypothesis that mRNA vaccines may not require a strong adjuvant response for potent immune responses.
The improved tolerability and safety mediated by the inclusion of biodegradable lipids within LNPs correlate well with lipid half-life after IV delivery.23, 32 The lead ionizable lipids in this study showed improved biodegradability while maintaining immune titers compared to MC3. The tolerability data suggest that this increased biodegradability leads to a reduction in injection site inflammation. Our data also show that extended residence time of the ionizable lipid post-transfection is not required for a robust immune response. Indeed, clearance is preferred to extended residence, which results in undesirable inflammation at the site of injection beyond when the protein antigen is cleared. Interestingly, the data also indicate that biodegradability is not the only factor in tolerability—lipid H was the best tolerated lipid yet showed a biodegradability similar to that of the other lead lipids tested. Degradation and tolerability of the lipid metabolites likely contribute to the tolerability of any formulation.
Other components, such as PEG, may play a role in vaccine potency due to the impact of anti-PEG responses that have been well described for IV-administered liposomal therapeutics. To date, there is no published information on the impact of anti-PEG responses across other routes of administration. The field of viral vector delivery has described how anti-vector immunity can substantially reduce immune response and can even completely prevent vaccine boosting when a homologous vector is used for both priming and boosting.33 Given that we see a substantial increase in immune titers after a second dose, we do not believe that a neutralizing anti-PEG response affects the LNP-based vaccines we describe here.
The tolerability of any new vaccine is a key performance criterion, as vaccines are given to healthy individuals throughout different stages of life, from 1-day-old neonates to the elderly. Here, we have described the identification, performance, and tolerability assessment of novel ionizable lipids for inclusion in mRNA vaccine formulations. We focused on the ionizable lipid component of the LNP, as it has been previously demonstrated to be the primary driver of LNP potency and tolerability. Given their improved tolerability and increased antigen expression, the formulations we identified have the potential for both active and passive immunization applications.
Materials and Methods
mRNA Synthesis and Formulation
UTR sequences and mRNA production processes were performed as previously described.19 Briefly, mRNA was synthesized in vitro by T7 RNA polymerase-mediated transcription from a linearized DNA template, which incorporates the 5′ and 3′ UTRs and a poly(A) tail. The final mRNA utilizes Cap1 and full replacement of uridine with N1-methyl-pseudouridine. mRNA encoding influenza HA genes originated from the H10N8 strain34, and the mRNA encoding prM-E from Zika utilized the signal sequences from human IgE (MDWTWILFLVAAATRVHS) and the prM and E genes from an Asian ZIKV strain (Micronesia 2007; GenBank: EU545988), which is >99% identical to circulating American strains.35 All coding sequences were generated using a proprietary algorithm.
LNP formulations were prepared using a modified procedure of a method previously described.17 Briefly, lipids were dissolved in ethanol at molar ratios of 50:10:38.5:1.5 (ionizable lipid:DSPC:cholesterol:PEG lipid). LNPs formulated with the ionizable lipid MC3 were used as a control throughout these studies and were produced as previously described.11 Novel ionizable lipids were synthesized as described elsewhere.36 The lipid mixture was combined with an acidification buffer of 50 mM sodium citrate (pH 4.0) or 25 mM sodium acetate (pH 5.0) containing mRNA at a volume ratio of 3:1 (aqueous:ethanol) using a microfluidic mixer (Precision Nanosystems, Vancouver, BC, Canada). The ratio of nitrogen present on the ionizable N:P ratio was set to 5.67 for each formulation. Formulations were dialyzed against PBS (pH 7.2) or 20 mM Tris (pH 7.4) with 8% sucrose in Slide-A-Lyzer dialysis cassettes (Thermo Scientific, Rockford, IL, USA) for at least 18 h. Formulations were concentrated using Amicon ultra-centrifugal filters (EMD Millipore, Billerica, MA, USA), if needed, and then passed through a 0.22-μm filter and stored at 4°C (PBS) or −20°C (20 mM Tris-8% sucrose) until use. Formulations were tested for particle size, RNA encapsulation, and endotoxin. All LNPs were found to be between 50 and 142 nm in size by dynamic light scattering and with greater than 69% encapsulation and <3 EU/mL endotoxin. Lead lipids selected for further evaluation were between 66 and 107 nm, with greater than 72% encapsulation.
pKa Analysis
Assay buffers (buffers containing 150 mM sodium chloride, 10 mM sodium phosphate, 10 mM sodium borate, and 10 mM sodium citrate) were pH adjusted with sodium hydroxide or hydrochloric acid to create buffers with pH ranges from pH 3 to pH 11.5. In a black-bottom, 96-well plate, 300 μM 6-(p-toluidino)-2-naphthalenesulfonic acid sodium salt in DMSO (TNS reagent) (Sigma-Aldrich, St. Louis, MO, USA), LNP, and assay buffer were combined. Each pH unit of buffer was repeated in triplicate with TNS reagents and LNPs. Fluorescent measurements were taken using a Synergy H1 microplate reader (BioTek Instruments, Winooski, VT, USA), with excitation set to 325 nm and emission collected at 435 nm. Fluorescence intensity was plotted against the pH of the assay buffer. The log of the inflection point was assigned the apparent pKa of the LNP.
Expression and Immunogenicity Screening Studies in a Murine Model
All animal experiments and husbandry followed guidelines from NIH (NIH publication #8023, eighth edition) and the U.S. National Research Council. Female BALB/c mice 5–8 weeks old were purchased from Charles River Laboratories (Wilmington, MA, USA) and housed at Moderna Therapeutics (Cambridge, MA, USA). Mice were acclimated for at least 3 days before the initiation of a study. Initial murine screening studies evaluated expression and immunogenicity in the same study, as previous work showed that co-formulation of the two mRNAs did not affect individual results (data not shown). On days 1 and 22, mice were injected in the quadriceps with 50 μL lipid nanoparticle formulations encapsulating an equal amount of luciferase and H10N8 mRNAs. 6 h post-dose, animals received an intraperitoneal injection of 3 mg luciferin and were imaged on an in vivo imaging system (IVIS Spectrum, PerkinElmer, Waltham, MA, USA). On days 21 and 36, mice were bled through the submandibular cavity. Serum was separated from the blood by centrifugation and then used to evaluate immunogenicity by ELISA. Group geometric means were calculated for each LNP evaluated and compared to the geometric mean of the MC3 group in the same study (expression) or of all MC3 groups tested (immunogenicity).
Lipid Clearance in a Murine Model
Female CD-1 mice were purchased from and housed at Charles River Laboratories. Mice were acclimated for at least 3 days before the initiation of a study. Mice were injected IM with 50 μL containing 2 μg of luciferase mRNA formulated in LNPs. At 1, 2, 4, 8, and 24 h post-injection, 3 mice were sacrificed and the plasma, spleen, liver, site of injection muscle, and draining lymph nodes were harvested. Tissues were frozen and sent to Agilux (Worcester, MA, USA) for evaluation of the remaining lipid by mass spectroscopy.
Quantification of Lipid by LC-MS/MS
Tissue samples were homogenized by Omni probe following the addition of 19 equivalents (w/v) of water. Lipid and proteins were precipitated and analyzed against calibration standards prepared in a matching blank. Chromatographic separation and quantification was accomplished with a liquid chromatography-tandem mass spectroscopy (LC-MS/MS) system. Samples were separated on a Clipeus C8 column (Higgins Analytical, Mountain View, CA, USA) equilibrated with 35% solvent A containing 5 mM formic acid in 50% methanol (H2O:MeOH:FA, 50:50:1) and 65% solvent B containing 5 mM formic acid in methanol (MeOH:FA, 100:1; Thermo Fisher Scientific). A triple-quadrupole MS/MS system (Applied Biosystems, API 5500) operated in positive ion mode was used for signal detection.
Tolerability in a Rat Model
Female Sprague-Dawley rats were purchased from Charles River Laboratories and housed at Moderna Therapeutics, Cambridge MA, USA. Rats were injected with 100 μL containing either 10 or 100 μg of mRNA formulated in LNPs. 6 h post-injection, blood was drawn, and serum was used for Luminex cytokine analysis (Austin, TX, USA). 48 h post-injection, rats were sacrificed, and the liver, site of injection, muscle, and skin were collected. Tissues were sectioned, stained with H&E, evaluated by a blinded board-certified pathologist, and graded on a scale from 0 to 5 based on severity for myofiber necrosis, mixed-cell infiltration within muscle and skin, and degenerate neutrophils in muscle and skin.
Expression and Immunogenicity in NHPs
NHP studies were conducted at Charles River Laboratories (Sherbrooke, QC, Canada) using naive cynomolgus monkeys, 2–5 years old and weighing 2–3 kg. Animals were housed in stainless steel, perforated-floor cages, in a temperature- and humidity-controlled environment (21–26°C and 30–70%, respectively), with an automatic 12-h/12-h dark/light cycle. Animals were fed PMI Nutrition Certified Primate Chow No. 5048 twice daily. Tuberculin tests were carried out on arrival at the test facility. The study plan and procedures were approved by pre-clinical services Sherbrook (PCS-SHB) IACUC. Animal experiments and husbandry followed NIH (Publication no. 8023, eighth edition), U.S. National Research Council, and Canadian Council on Animal Care (CCAC) guidelines.
To evaluate expression, cynomolgus NHPs were injected IM with 300 μL containing a total of 500 μg mRNA (heavy chain and light chain in a 2:1 weight:weight ratio) encoding an antibody formulated in LNPs. The site of injection was monitored for erythema and edema and graded for severity from 0 (no reaction) to 4 (severe reaction). Blood was collected 6 h before dosing and then 2, 6, 24, 48, 96, 168, 264, and 336 h post-injection to measure antibody levels. Blood from −6, 48, and 336 h was used to measure hematology, coagulation, D-dimer, and clinical chemistry markers.
To evaluate immunogenicity, cynomolgus monkeys received IM injections of 5 μg H10N8 mRNA-formulated LNP in 100 μL on days 1 and 22. 0.5 mL blood was collected on day 22 and day 43 post-dosing from a peripheral vein and centrifuged at 1200 × g for 10 min at 4°C for separation of serum. Serum was stored at −80°C until analysis by hemagglutination inhibition assay (HAI) and ELISA.
HAI Assay
The HAI titers of serum samples were determined using a protocol described previously.17 Sera were first treated with receptor-destroying enzyme (RDE) to inactivate nonspecific inhibitors. The RDE was inactivated by incubation at 56°C for 30 min. Treated sera were serially diluted in 96-well plates, mixed with a standardized amount of recombinant HA (8 HA units of H10N8; Medigen, Frederick, MD, USA), and incubated for 30 min at room temperature. Turkey red blood cells (RBCs) (Lampire Biological Laboratories, Everett, PA, USA) were then added to the wells of the 96-well plates, mixed, and incubated at room temperature for 45 min. The most dilute serum sample that completely inhibited HA was the reported titer for that replicate. Each serum sample was analyzed in triplicate, and the results are reported as the geometric mean of the 3 results.
Anti-H10N8 ELISA
Nunc MaxiSorp 96-well plates (Thermo Fisher, Rochester, NY, USA) were coated at 100 μL per well with 1 μg/mL H10 protein in PBS overnight at 4°C. Plates were washed three times with PBS containing 0.1% Tween 20 (wash buffer). 200 μL Superblock (Pierce, Rockford, IL, USA) was added to each well and incubated at 37°C for at least 1.5 h and then washed three times with wash buffer. In each well, 100 μL PBS containing 5% goat serum (GIBCO, Gaithersburg, MD, USA) with 0.1% Tween 20 was added, and serum was serially diluted and incubated for 2 h at 37°C. Plates were washed three times, and 100 μL horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibody (Southern Biotech, Birmingham, AL, USA) diluted 1:20,000 in PBS containing 5% goat serum with 0.1% Tween 20 was added and incubated for 1 h at 37°C. Plates were washed three times, and 100 μL SureBlue TMB Microwell Peroxidase substrate (Kirkegaard & Perry Labs, Milford, MA, USA) was added to each well and incubated for 15 min. 100 μL TMB Stop Solution (Kirkegaard & Perry Labs, Milford, MA, USA) was added to each well, and the plates were read at 450 nm. The average blank value was subtracted from each sample. Titers were defined as the reciprocal serum dilution at approximately OD450 nm (optical density 450 nm) = 0.6 (normalized to a standard included on every plate).
Monoclonal Antibody Detection
QUICKPLEX 96-well plates (MSD) were coated with 100 μg of 1 μg/mL capture protein in PBS per well and incubated overnight at 4°C. Plates were washed with PBS with 0.5% Tween 20 three times. Serial dilutions for a reference standard and samples were performed into a 100-μL final volume in the plate and then were incubated at room temperature for 1.5 h, with shaking at 120 rpm. Plates were washed with PBS with 0.5% Tween 20 three times. 50 μL affinity-purified goat anti-human IgG (sulfo-tagged) at 0.5 μg/mL was added to each well and incubated for 1 h at room temperature, with shaking at 120 rpm. After incubation, plates were washed six times, and 150 μL MSD Read Buffer T was added to each well. The plates were read on an MSD instrument (Meso Scale Diagnostics, Rockville, MD, USA).
Author Contributions
Conceptualization, G.C. and L.A.B.; Methodology, K.J.H., K.E.B., E.J., and L.A.B.; Formal analysis, K.J.H. and I.M.; Investigation, K.J.H., E.J., A. Lee, A.W., O.Y., S.H., J.D., B.M.G., T.K., and A. Lynn; Writing – Original Draft, K.J.H., and L.A.B.; Writing – Review and Editing, K.J.H., M.J.M., and L.A.B.; Visualization, K.J.H., I.M., and L.A.B.; Supervision, K.E.B., C.M., J.J.S., M.G.S., O.A., and G.C.
Conflicts of Interest
All authors are either current or previous employees of Moderna Therapeutics and own stock options and/or shares in the company.
Acknowledgments
We would like to thank Moderna’s Preclinical Production group for production of mRNA and Moderna’s Nonclinical Sciences group for in vivo experiments. This work was funded by Moderna Therapeutics.
Footnotes
Supplemental Information includes Supplemental Methods and four figures and can be found with this article online at https://doi.org/10.1016/j.omtn.2019.01.013.
Supplemental Information
References
- 1.Rappuoli R., Mandl C.W., Black S., De Gregorio E. Vaccines for the twenty-first century society. Nat. Rev. Immunol. 2011;11:865–872. doi: 10.1038/nri3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riedel S. Edward Jenner and the history of smallpox and vaccination. Proc. (Bayl. Univ. Med. Cent.) 2005;18:21–25. doi: 10.1080/08998280.2005.11928028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.John S., Yuzhakov O., Woods A., Deterling J., Hassett K., Shaw C.A., Ciaramella G. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine. 2018;36:1689–1699. doi: 10.1016/j.vaccine.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 4.Deering R.P., Kommareddy S., Ulmer J.B., Brito L.A., Geall A.J. Nucleic acid vaccines: prospects for non-viral delivery of mRNA vaccines. Expert Opin. Drug Deliv. 2014;11:885–899. doi: 10.1517/17425247.2014.901308. [DOI] [PubMed] [Google Scholar]
- 5.Ulmer J.B., Donnelly J.J., Parker S.E., Rhodes G.H., Felgner P.L., Dwarki V.J., Gromkowski S.H., Deck R.R., DeWitt C.M., Friedman A. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science. 1993;259:1745–1749. doi: 10.1126/science.8456302. [DOI] [PubMed] [Google Scholar]
- 6.Ferraro B., Morrow M.P., Hutnick N.A., Shin T.H., Lucke C.E., Weiner D.B. Clinical applications of DNA vaccines: current progress. Clin. Infect. Dis. 2011;53:296–302. doi: 10.1093/cid/cir334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roos A.K., Eriksson F., Walters D.C., Pisa P., King A.D. Optimization of skin electroporation in mice to increase tolerability of DNA vaccine delivery to patients. Mol. Ther. 2009;17:1637–1642. doi: 10.1038/mt.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tebas P., Roberts C.C., Muthumani K., Reuschel E.L., Kudchodkar S.B., Zaidi F.I., White S., Khan A.S., Racine T., Choi H. Safety and immunogenicity of an anti-Zika virus DNA vaccine - preliminary report. N. Engl. J. Med. 2017 doi: 10.1056/NEJMoa1708120. Published online October 4, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carralot J.P., Probst J., Hoerr I., Scheel B., Teufel R., Jung G., Rammensee H.G., Pascolo S. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell. Mol. Life Sci. 2004;61:2418–2424. doi: 10.1007/s00018-004-4255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alberer M., Gnad-Vogt U., Hong H.S., Mehr K.T., Backert L., Finak G., Gottardo R., Bica M.A., Garofano A., Koch S.D. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet. 2017;390:1511–1520. doi: 10.1016/S0140-6736(17)31665-3. [DOI] [PubMed] [Google Scholar]
- 11.Jayaraman M., Ansell S.M., Mui B.L., Tam Y.K., Chen J., Du X., Butler D., Eltepu L., Matsuda S., Narayanannair J.K. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love K.T., Mahon K.P., Levins C.G., Whitehead K.A., Querbes W., Dorkin J.R., Qin J., Cantley W., Qin L.L., Racie T. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semple S.C., Akinc A., Chen J., Sandhu A.P., Mui B.L., Cho C.K., Sah D.W., Stebbing D., Crosley E.J., Yaworski E. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 14.Coelho T., Adams D., Silva A., Lozeron P., Hawkins P.N., Mant T., Perez J., Chiesa J., Warrington S., Tranter E. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013;369:819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 15.Roman F., Vaman T., Kafeja F., Hanon E., Van Damme P. AS03(A)-adjuvanted influenza A (H1N1) 2009 vaccine for adults up to 85 years of age. Clin. Infect. Dis. 2010;51:668–677. doi: 10.1086/655830. [DOI] [PubMed] [Google Scholar]
- 16.Reisinger K.S., Baxter R., Block S.L., Shah J., Bedell L., Dull P.M. Quadrivalent meningococcal vaccination of adults: phase III comparison of an investigational conjugate vaccine, MenACWY-CRM, with the licensed vaccine, Menactra. Clin. Vaccine Immunol. 2009;16:1810–1815. doi: 10.1128/CVI.00207-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bahl K., Senn J.J., Yuzhakov O., Bulychev A., Brito L.A., Hassett K.J., Laska M.E., Smith M., Almarsson Ö., Thompson J. Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol. Ther. 2017;25:1316–1327. doi: 10.1016/j.ymthe.2017.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geall A.J., Verma A., Otten G.R., Shaw C.A., Hekele A., Banerjee K., Cu Y., Beard C.W., Brito L.A., Krucker T. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA. 2012;109:14604–14609. doi: 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richner J.M., Himansu S., Dowd K.A., Butler S.L., Salazar V., Fox J.M., Julander J.G., Tang W.W., Shresta S., Pierson T.C. Modified mRNA vaccines protect against Zika virus infection. Cell. 2017;168:1114–1125.e10. doi: 10.1016/j.cell.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pardi N., Hogan M.J., Pelc R.S., Muramatsu H., Andersen H., DeMaso C.R., Dowd K.A., Sutherland L.L., Scearce R.M., Parks R. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature. 2017;543:248–251. doi: 10.1038/nature21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutz J., Lazzaro S., Habbeddine M., Schmidt K.E., Baumhof P., Mui B.L., Tam Y.K., Madden T.D., Hope M.J., Heidenreich R., Fotin-Mleczek M. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines. 2017;2:29. doi: 10.1038/s41541-017-0032-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oberli M.A., Reichmuth A.M., Dorkin J.R., Mitchell M.J., Fenton O.S., Jaklenec A., Anderson D.G., Langer R., Blankschtein D. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017;17:1326–1335. doi: 10.1021/acs.nanolett.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabnis S., Kumarasinghe E.S., Salerno T., Mihai C., Ketova T., Senn J.J., Lynn A., Bulychev A., McFadyen I., Chan J. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 2018;26:1509–1519. doi: 10.1016/j.ymthe.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richner J.M., Himansu S., Dowd K.A., Butler S.L., Salazar V., Fox J.M., Julander J.G., Tang W.W., Shresta S., Pierson T.C. Modified mRNA vaccines protect against Zika virus infection. Cell. 2017;169:176. doi: 10.1016/j.cell.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 25.Kou L., Sun J., Zhai Y., He Z. The endocytosis and intracellular fate of nanomedicines: Implication for rational design. Asian Journal of Pharmaceutical Sciences. 2013;8:1–10. [Google Scholar]
- 26.Rybak S.L., Murphy R.F. Primary cell cultures from murine kidney and heart differ in endosomal pH. J. Cell. Physiol. 1998;176:216–222. doi: 10.1002/(SICI)1097-4652(199807)176:1<216::AID-JCP23>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 27.Fenton O.S., Kauffman K.J., Kaczmarek J.C., McClellan R.L., Jhunjhunwala S., Tibbitt M.W., Zeng M.D., Appel E.A., Dorkin J.R., Mir F.F. Synthesis and biological evaluation of ionizable lipid materials for the in vivo delivery of messenger RNA to B lymphocytes. Adv. Mater. 2017;29:1606944. doi: 10.1002/adma.201606944. [DOI] [PubMed] [Google Scholar]
- 28.Brito L.A., Chan M., Shaw C.A., Hekele A., Carsillo T., Schaefer M., Archer J., Seubert A., Otten G.R., Beard C.W. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol. Ther. 2014;22:2118–2129. doi: 10.1038/mt.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollard C., Rejman J., De Haes W., Verrier B., Van Gulck E., Naessens T., De Smedt S., Bogaert P., Grooten J., Vanham G., De Koker S. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol. Ther. 2013;21:251–259. doi: 10.1038/mt.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karikó K., Buckstein M., Ni H., Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Liang F., Lindgren G., Lin A., Thompson E.A., Ols S., Röhss J., John S., Hassett K., Yuzhakov O., Bahl K. Efficient targeting and activation of antigen-presenting cells in vivo after modified mRNA vaccine administration in rhesus macaques. Mol. Ther. 2017;25:2635–2647. doi: 10.1016/j.ymthe.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maier M.A., Jayaraman M., Matsuda S., Liu J., Barros S., Querbes W., Tam Y.K., Ansell S.M., Kumar V., Qin J. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 2013;21:1570–1578. doi: 10.1038/mt.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J., Ewald B.A., Lynch D.M., Denholtz M., Abbink P., Lemckert A.A., Carville A., Mansfield K.G., Havenga M.J., Goudsmit J., Barouch D.H. Magnitude and phenotype of cellular immune responses elicited by recombinant adenovirus vectors and heterologous prime-boost regimens in rhesus monkeys. J. Virol. 2008;82:4844–4852. doi: 10.1128/JVI.02616-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H., Yuan H., Gao R., Zhang J., Wang D., Xiong Y., Fan G., Yang F., Li X., Zhou J. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: a descriptive study. Lancet. 2014;383:714–721. doi: 10.1016/S0140-6736(14)60111-2. [DOI] [PubMed] [Google Scholar]
- 35.Lanciotti R.S., Kosoy O.L., Laven J.J., Velez J.O., Lambert A.J., Johnson A.J., Stanfield S.M., Duffy M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008;14:1232–1239. doi: 10.3201/eid1408.080287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benenato, K.E., Kumarasinghe, E.S., and Cornebise, M. (2017). Compounds and compositions for intracellular delivery of therapeutic agents. US patent application publication 20170210697 A1, filed March 31, 2017, and published July 27, 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.