

Abstract
The mechanism that explains the association of APOL1 variants with nondiabetic kidney diseases in African Americans remains unclear. Kidney disease risk is inherited as a recessive trait, and many studies investigating the intracellular function of APOL1 have indicated the APOL1 variants G1 and G2 are associated with cytotoxicity. Whether cytotoxicity results from the absence of a protective effect conferred by the G0 allele or is induced by a deleterious effect of variant allele expression has not be conclusively established. A central issue hampering basic biology studies is the lack of model systems that authentically replicate APOL1 expression patterns. APOL1 is present in humans and a few other primates and appears to have important functions in the kidney, as the kidney is the primary target for disease associated with the genetic variance. There have been no studies to date assessing the function of untagged APOL1 protein under native expression in human or primate kidney cells, and no studies have examined the heterozygous state, a disease-free condition in humans. A second major issue is the chronic kidney disease (CKD)-associated APOL1 variants are conditional mutations, where the disease-inducing function is only evident under the appropriate environmental stimulus. In addition, it is possible there may be more than one mechanism of pathogenesis that is dependent on the nature of the stressor or other genetic variabilities. Studies addressing the function of APOL1 and how the CKD-associated APOL1 variants cause kidney disease are challenging and remain to be fully investigated under conditions that faithfully model known human genetics and physiology.
Keywords: chronic kidney disease, genetics, podocytes
INTRODUCTION
It has been 10 years since the publication of reports associating nondiabetic chronic kidney disease (CKD) in African Americans with a locus on chromosome 22 (21, 24). A gene in this locus, APOL1, was identified 2 years later with several coding single-nucleotide polymorphisms restricted to individuals of recent African ancestry (16, 17, 59). Kidney diseases associated with these polymorphisms are human immunodeficiency virus (HIV)-associated nephropathy (HIVAN), idiopathic focal segmental glomerulosclerosis (FSGS), hypertension-attributed CKD, and severe lupus nephritis (12, 16, 22, 23, 30, 35). The APOL1 polymorphisms not only are associated with the deleterious CKD risk but also are a beneficial protection against African sleeping sickness, a lethal disease caused by infection of trypanosome parasites transmitted by the bite of tsetse flies. The survival advantage conferred by the APOL1 variants from trypanosomes or other infectious agents may have contributed to a selective sweep of the locus, making them common alleles in many communities in sub-Saharan Africa, African Americans, and other recent African diasporas. Genetically, APOL1 is a recent evolutionary adaptation of the primate innate immune system and is present only in humans and a few other nonhuman primates. Although other mammals may have genes named APOL1, they are not orthologs of human APOL1 and do not possess innate immunity against trypanosomes. For more details on the evolution and population frequencies of APOL1 and its variants, we refer the reader to excellent recent reviews (14, 27).
The trypanolytic APOL1 protein is synthesized by the liver (56) and is constitutively secreted into the circulation as part of a minor class of HDL particles that contain additional proteins that aid trypanolysis (54, 61). These unique APOL1-containing HDLs are engulfed as a nutrient by the free-living parasites and kill by forming an ion pore in membranes, disrupting the osmotic integrity of these lipid compartments, leading to parasite death. The reference or common allele, referred to as G0, kills the African trypanosome T. b. brucei, but does not kill the related organisms T. b. rhodesiense and T. b. gambiense, the major causes of sleeping sickness. T. b. rhodesiense evades APOL1 function by producing a novel protein called SRA (serum resistance associated protein). SRA-producing trypanosomes render APOL1 inactive by binding their SRA protein to APOL1, making them resistant to APOL1-mediated killing. The two CKD-associated APOL1 variants, referred to as G1 and G2, contain polymorphisms in the SRA binding region that reduce the ability of SRA to bind APOL1, in part, and thus restore APOL1 killing function and protection against sleeping sickness.
The APOL1 protein responsible for trypanosome killing is abundant in the circulation. However, several cohort studies did not associate circulating APOL1 levels with CKD (3, 25), and kidney transplantation outcomes were poorer if the donor had an APOL1 high-risk genotype, but not the recipient (10, 13, 31, 52). Together, these studies indicate circulating APOL1 is not associated with CKD risk or kidney injury and thus has been largely discounted as a contributor to CKD pathogenesis. APOL1 also is expressed in many other tissues, including the kidney (37–39), and this intracellular APOL1 has become the focus of research to identify the mechanism of pathogenesis. Because APOL1 is limited to humans and a few other primates, basic research has been hampered by the lack of good model systems that accurately replicate human expression patterns, genetics, and biology, most notably the recessive mode of inheritance and the requirement of a second-hit stressor.
MODE OF INHERITANCE
The two traits associated with carriage of APOL1 variant alleles, resistance to sleeping sickness and CKD risk, have different modes of inheritance. Protection against African sleeping sickness is an autosomal dominant trait. Since APOL1 circulating levels are constitutive and abundant, individuals with only one variant allele (heterozygotes) produce sufficient variant APOL1 protein to confer full protection against SRA-expressing trypanosomes. Risk for CKD, however, is an autosomal recessive trait. Population studies consistently associate CKD risk with an APOL1 genotype of any combination of two variant alleles (referred to as a high-risk genotype), with little risk associated with no or one variant allele (referred to as a low-risk genotype) (9, 16, 17, 22, 23, 35, 59).
Phenotypically, protection against sleeping sickness is a gain-of-function event (expanded range of parasite killing), but antithetically results from genetic mutations that are loss-of-function (inability to bind SRA). This apparent incongruity between a genetic loss resulting in a phenotypic gain is not unusual, as a similar process is well known for cancer, where loss-of-function mutations in tumor suppressor genes results in the phenotypic gain of oncogenic functions. Although the genotype-phenotype mechanism for APOL1’s dominant role in sleeping sickness protection is well described (50), we do not have a clear understanding of the recessive mechanisms that underlie CKD risk. Another important caveat is that most individuals with a high-risk APOL1 genotype do not have CKD. The current theory is CKD risk is an example of a gene-environment interaction (discussed below), where the genetic risk is only disease-causing in the setting of an appropriate environmental stressor. This suggests the APOL1 variants are conditional mutations, where the function or dysfunction of the variants is only disease causing when exposed to the appropriate environmental condition.
Gain-of-Function
Despite strong evidence establishing CKD risk as a recessive trait (a mode of inheritance more commonly associated with loss-of-function mutations), most studies examining the disease mechanism have concluded the APOL1 variants produce a new protein function and represent gain-of-function mutations (18, 26, 28, 36, 44, 48, 62). The proposed new function endowed by the variants results in cytotoxicity; however, details of the biochemical mechanism are not fully established. Potential cellular processes implicated to date have included changes in endosomal acidification and trafficking, autophagy induction and flux, endoplasmic reticulum and mitochondrial stress, and inflammation-triggered apoptosis (pyroptosis), all of which have some parallels to the mechanism established for trypanosome killing. Due to the lack of good cell culture systems, many of these in vitro studies have relied on overexpression techniques, also known to result in artifacts that activate these same cellular processes. Protein overexpression can force protein mislocalization, aggregation, and non-native protein-protein interactions causing endoplasmic reticulum stress and the unfolded protein response, frequently also resulting in cytotoxicity via autophagy and apoptosis. Also problematic in these overexpression systems are the observations that G0 is cytotoxic, although several reports find G0 is less cytotoxic than G1 and G2. APOL1 G0, G1, and G2 are all typically membrane- or lipid-associated proteins with an equivalent ability to form an ion pore. If expression levels saturate their normal membrane localization and permit the ectopic localization on other membranes, such as the plasma membrane, cytotoxicity could result. In addition, many of the studies purporting a gain-of-function cytotoxicity used short-term cell viability assays that measure metabolic activity as a surrogate for cell death. Similar studies using a clonogenic assay, a direct measure of cell viability, demonstrated that cytotoxicity was variant independent but protein expression-level dependent (46). Transcription and translation are highly organized, tightly controlled processes that maintain homeostasis in addition to normal responses to environmental stresses. Although many pathogenic events originate with aberrant gene expression, it is unclear whether APOL1 variant-dependent cytotoxicity is the true mechanism of pathogenesis or a methodological artifact.
The most relevant in vivo models examining APOL1 function have transduced mice to express human APOL1. Since mice do not have APOL1, expression of G0 would replicate a human low-risk genotype, and mice expressing either G1 or G2 would replicate a human high-risk genotype. The first model developed by the Raper laboratory used hydrodynamic gene delivery to transiently express a variety of human and primate APOL1s (58). The hydrodynamic gene delivery method primarily targets hepatocytes, and this group was able to show robust production of APOL1 by the liver and secretion into the circulation. These animals did not develop kidney injury, but the authors concluded there was likely little APOL1 expression in the kidney. Our group reported the first transgenic mouse model of APOL1-expressing transgenes for either G0 or G2 under the control of the Nphs1 (Nephrin) promoter to restrict APOL1 expression to podocytes (4). These mice expressed G0 or G2 throughout fetal development and adulthood. Despite establishing multiple transgenic lines with varying levels of expression for both G0 and G2, we did not observe changes in renal function or development of glomerulosclerosis with either transgene up to ~1 yr of age. These mice will be useful to now assess the function of G0 vs. G2 in the setting of a disease-inducing stressor. A second report of APOL1 transgenic mice by the Susztak laboratory also used the Nphs1 promoter to restrict expression to podocytes, but with the addition of a doxycycline-inducible element to regulate the timing and level of APOL1 expression (1). When the transgene was activated in adulthood, de novo G1 and G2 expression reduced podocyte autophagic flux, caused podocyte loss, renal function declines, and glomerulosclerosis, whereas expression of G0 had no effect. Of note, kidney disease was evident in the G1 and G2 mice without the addition of a disease-inducing stressor. The variable outcomes of these mouse models are probably attributable to variability in expression levels, and, similar to the in vitro studies described above, accurately replicating native APOL1 expression levels is critical. It is unclear whether any of the mouse models published to date accurately replicated physiological expression of human APOL1 in the kidney. The ongoing development of knock-in mouse models of the entire human APOL1 gene will provide a more authentic system to evaluate native human APOL1 expression.
The concept that the APOL1 variant proteins possess a new and intrinsically deleterious function is difficult to reconcile with two key observations in humans. First, APOL1 is expressed by many other tissues which do not exhibit pathology. Most notable is the liver, the site of production of the constitutive and abundant circulating APOL1 (56). Not only is APOL1 synthesized and secreted using normal cellular processes in hepatocytes, reverse cholesterol transport returns APOL1-containing HDLs back into the hepatocyte’s endocytic pathways. APOL1 expression in other tissues may contribute to other nonrenal diseases, such as the recent report associating fetal APOL1 high-risk genotypes with preeclampsia (53). Like other genetic diseases with a predominant renal phenotype, the cell type initiating pathology typically has a nonredundant or critical function for the gene product. Thus the reported variant-induced cytotoxicity would have to reflect loss of a unique or highly important G0 function in kidney cells. Second, if APOL1 variants are inherently cytotoxic, it is unclear why a dominant or additive mode of inheritance is not associated with CKD risk, and more importantly, why most individuals with a high-risk genotype do not have CKD. It is possible gain-of-function mutations can have a recessive inheritance pattern if APOL1 function requires the formation of dimers or multimers. A model has been proposed (34) where heterozygotes expressing both G0 and a variant interact in heterodimers in which the variant function is both suppressed by the G0 protein and homodimers containing only the variant APOL1 protein are thermodynamically unfavored. This hypothesis, however, remains to be tested. In fact, there have been no mechanistic studies to date that have examined the heterozygous state, a disease-free condition in humans.
Loss-of-Function
An autosomal recessive inheritance pattern is characteristic of loss-of-function mutations, with disease resulting from the absence of wild-type protein function. For APOL1 and CKD risk, a recessive inheritance pattern may suggest G0 provides a beneficial function to the kidney that is missing with only APOL1 variant expression. APOL1 variants alter only a few amino acids in the SRA binding domain of the G0 protein and retain most G0 function (i.e., G0, G1, and G2 are functional equivalent in ion pore formation and killing trypanosomes). The variants are therefore not true null mutations, but are only partial loss-of-function mutations. Interestingly, a human APOL1 null mutation has been described. An Asian individual with an unusual susceptibility to trypanosomiasis has a truncating mutation in APOL1 resulting in an APOL1 null phenotype (20, 60). This individual does not have CKD and it can be interpreted that the absence of APOL1 alone does not cause CKD. In addition, with the exception of a few other primates, no other mammal has an ortholog of human APOL1, and this also supports the concept that APOL1 is dispensable for normal kidney development and function. However, loss-of-function may not necessarily equate to dispensability of function; that is, its function may be conditionally necessary. For example, the individual with the APOL1 null mutation may not have CKD because he has never been exposed to a CKD-inducing stressor. Also, it is possible that mammalian kidneys can develop and function normally without APOL1, because APOL1 function is only needed in response to stochastic stressors. Taking some perspective from its trypanolytic role, APOL1 is constitutively present in our circulation throughout life and apparently has no function until we are bitten by a tsetse fly. Thus only in the setting of an invading pathogen does the function of circulating APOL1 become apparent and necessary. Extending this concept to intracellular APOL1, maybe APOL1 also has no function in kidney cells until a stressor is encountered.
Few studies have examined the function of intracellular APOL1 in the setting of a stressor. HIVAN is the CKD most strongly associated with the APOL1 high-risk genotype and the only APOL1-associated CKD where the disease-inducing stressor is known. Several in vitro studies have examined the intracellular function of APOL1 in the setting of HIV infection. Monocytic cells with γ-interferon-induced endogenous G0 expression suppress HIV-1 replication by several pre- and postintegration mechanisms (57). Similarly, an unbiased screen to identify host restriction factors for HIV also identified APOL1 as a cellular factor in T cells that suppresses HIV replication (41). In renal cells, one study reported HIV-1 infections of podocytes isolated from individuals with a high-risk and a low-risk genotype were not different with regard to supporting HIV infection (33). However, in another study, APOL1 overexpression in a commonly used podocyte cell line found HIV-1 uptake and persistence were greater with the risk variants compared with G0, suggesting anti-HIV functions observed in monocytes and T cells may also occur in podocytes and the risk variants may have a reduced ability to suppress infection (42).
Establishing the normal intracellular function of G0 will help determine the disease-causing dysfunction of G1 and G2. With regard to trypanosome killing, the G1 and G2 variants differ from G0 only by an altered ability to form protein-protein interactions with the trypanosome SRA protein. Extending this observation to kidney cells, we have shown APOL1 also directly interacts with human proteins, including the vesicular SNAREs VAMP1 and VAMP8 (38). Similar to the SRA protein, the variants G1 and G2 have a similar diminished capacity to bind VAMP8 in human cells. Since the pore-forming function of APOL1 is unchanged in the variants, it seems more likely this difference in protein-protein interaction will be important to the mechanism initiating pathogenesis.
There is functional evidence for perturbations of the APOL1-interacting pathway in model organism studies. In our transgenic mouse model described earlier, constitutive G2 expression causes an accelerated but subclinical age-related loss of podocyte density not observed with G0. Longitudinal projections suggest this enhanced podocyte attrition would not reach a disease threshold during a normal lifespan (45). Thus APOL1 engages a cellular pathway but this pathway is not a critical cellular function in the basal state, yet does have some function or subclinical phenotype. Transgenic flies expressing either G0 or G1 have a normal number of nephrocytes in early adulthood, but also develop a progressive loss of nephrocytes with aging accompanied by nephrocyte hypertrophy with greater effects observed in flies expressing G1 (15). In zebrafish, expression of G1 or G2 but not G0 caused histological changes in endothelial cells and podocytes, but like our APOL1 transgenic mice, no overt kidney phenotype developed (47). In humans, disease-free kidney transplant donors with an APOL1 high-risk genotype have poorer renal function outcomes postdonation compared with donors with a low-risk genotype (8). Similarly, preliminary studies found pretransplant donor kidneys had lower podocyte densities if the donor had a high-risk APOL1 genotype compared with a low-risk genotype donor (5). In aggregate, these model organism studies and human observations suggest APOL1 variants have a mild or subclinical phenotype independent of a second-hit stressor.
In summary, the high-risk and low-risk APOL1 genotypes have different phenotypes under basal and stressed conditions (Fig. 1). Under basal conditions, genotypes with carriage of at least one G0 allele have no apparent phenotype, whereas genotypes with carriage of two risk alleles appear to have a subtle phenotype that remains subclinical and is not disease causing. Under a disease stressor, the disease-triggered phenotype of G0 and the risk variants are revealed. However, it is unclear whether the stress-induced G0 phenotype provides a protective function, endowing resilience to the podocyte to withstand the stress, or if the variant phenotype directly induces pathogenic events and causes CKD.
Fig. 1.
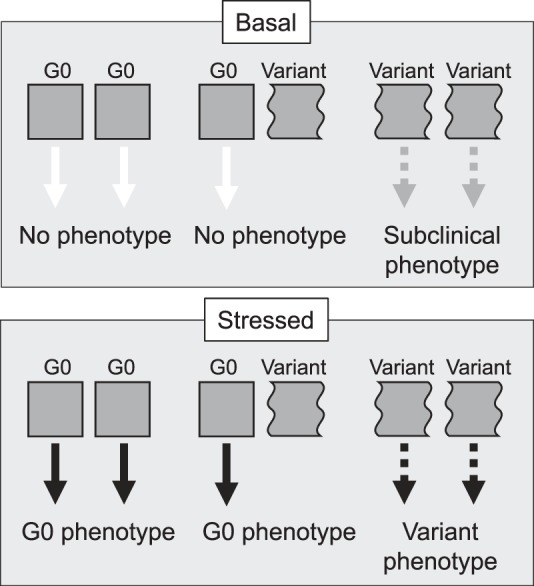
Summary of mode of inheritance and associated phenotypes for APOL1 genotypes. All allelic combinations of homozygous common (G0), heterozygotes, and homozygous variants are shown. In the basal condition, there is no obvious phenotype in any allelic combination, but the homozygous variants produce a subclinical phenotype (accelerated age-related podocyte density losses). Under stressed conditions, the homozygous G0 and heterozygotes have a similar phenotype that is disease free. Under stressed conditions, the homozygous variants produce the disease phenotype. In both basal and stressed conditions, heterozygotes are disease free and the G0 phenotype supersedes or suppresses the variant phenotype.
GENE-ENVIRONMENT INTERACTION AND SECOND-HIT STRESSORS
By definition, conditional mutations are functionally indistinguishable from wild-type alleles or with only a mild phenotype under one set of conditions, but exhibit a strong mutant phenotype under other conditions. Accurate assessment of the abnormal phenotype is therefore dependent on the presence of the environmental condition that reveals mutant protein function. Various models for gene-environment interactions have been proposed (19), differentiated by either a basal increase in disease susceptibility (genetically enhanced model) vs. a lower threshold for disease induction (stress-enhanced model) (Fig. 2). In HIVAN, differences in disease thresholds seem unlikely. Early in the HIV/AIDS epidemic before effective treatment options, the severity of HIV infection and immunodeficiency reached maximal extremes in all infected subjects; however, HIVAN was only diagnosed in African Americans. Thus development of HIVAN is not dependent on the severity of the disease stressor, but an inherent susceptibility to disease by a given stress.
Fig. 2.

Gene-environment interaction (conditional mutation) models. Disease risk (y-axis) increases as a function of stressor severity (x-axis), with disease occurring after a disease threshold (gray area) is reached. At baseline in the genetically enhanced model, the variant generates a phenotype that approaches, but does not reach the disease threshold. With stress, both the normal and variant respond similarly; however, with the baseline difference, the variant reaches the disease threshold while the normal does not. In other words, the effects of the genetic risk and environmental stressor are additive. In the stress-enhanced model, there is no effect or phenotypic difference between the normal and the variant at baseline. With the addition of the stressor, the variant responds much more aggressively compared with the normal, reaching the disease threshold at a lower level of stress. The normal also can reach the disease threshold if the stress is more severe.
Much importance has been placed on the observation that APOL1 gene expression can be induced by various immune mediators such as tumor necrosis factor and interferons. Thus it is possible the disease-causing second hit is an immune response that induces APOL1 expression. CKD initiated by this mechanism may have been replicated in the inducible transgenic mouse model reported by the Susztak laboratory (1). In support of this concept, collapsing glomerulopathy can be caused by interferon therapy, although it is unknown whether the high interferon levels correlated with high APOL1 expression (40). Severe lupus nephritis also is a disease characterized by a chronic type I interferon-like response, but there are no studies yet examining a possible link between interferon levels with APOL1 expression and lupus nephritis risk. A causal relationship between HIVAN and immune-induced APOL1 expression is unclear. HIV infection stimulates a robust immune response that at end stage leads to immunodeficiency, and although HIVAN can develop at any time in the course of HIV infection, it is more common in the later stages of disease (49). HIVAN also is effectively prevented and treated with antiretroviral medication, which functions to both suppress viral replication and restore the immune system. However, human studies that specifically investigated a link between CKD risk and APOL1 expression did not find an association with circulating APOL1 protein levels in both HIV-infected and uninfected cohorts (3, 43). In general, systemic and acutely high levels of interferons routinely occur throughout life with any viral infection, including the common cold and flu. Similarly, long-term elevations of immune mediators are common with other chronic infections, but neither cause CKD nor are associated with an APOL1 high-risk genotype. Alternatively, it is possible the necessary immune second hit is not a systemic event, but a local immune-signaling event within the kidney or glomerulus. In the few human studies examining APOL1 expression in diseased kidney biopsies, APOL1 protein expression levels do not appear to be altered in a disease-dependent or APOL1 genotype-dependent manner (39), and similarly, APOL1 mRNA expression levels also were not different in a comparison of high-risk and low-risk genotype CKD patients (55). Temporal associations, however, may be difficult to establish using archived samples, and further studies examining locally produced immune mediators and podocyte-expressed APOL1 may help clarify the cause-effect role of induced APOL1 gene expression and kidney pathology.
HIVAN may be the simplest paradigm for APOL1-mediated disease with one major genetic risk factor and one major environmental second-hit stressor to initiate pathogenesis. For the other APOL1-associated CKDs, pathogenic mechanisms may be more complex with multiple environmental stressors and multiple genetic modifiers, to either exacerbate or mitigate the effects of APOL1. Additional genetic and environmental contributors could provide a mechanism to explain both the incomplete penetrance of CKD in high-risk genotype subjects and also the clinical and pathological differences in the APOL1-associated CKDs. Preliminary studies have identified polymorphisms in NPHS2, BMP4, SDCCAG4, and UBD that modify APOL1-mediated CDK risk (7, 25, 64). However, a recent meta-analysis of genome-wide association data concluded there are likely few genetic factors that contribute a strong modifying second hit to APOL1-associated CKD risk and concludes the critical second-hit stressors for disease induction are more likely to be environmental (29).
The importance and variabilities in the interaction between the environmental stressor and APOL1 may contribute to the differences in presentation of the various CKDs associated with APOL1 risk alleles (Fig. 3). For example, both hypertension-attributed CKD and HIVAN are strongly associated with a high-risk genotype but have discernable pathologies, and patients with HIVAN typically present without hypertension and have less hypertension than would be expected for an African American population with similarly advanced disease (2, 32, 51). Some stressors may be more acute and severe, whereas others may be chronic and low grade. Similar to additional genetic modifiers described above, other environmental factors may also alter CKD risk. Recent reports identified JC virus infection as reducing CKD risk in the setting of a high-risk genotype (6, 11).
Fig. 3.

Hypothetical models of renal function outcomes comparing APOL1 genotype and various stressors. Graphical representation of the known age-associated declines in renal function and podocyte density through human lifespan or age is shown. In the absence of a stressor, neither a low-risk (LR) nor high-risk (HR) APOL1 genotype is disease causing; however, a HR genotype appears to have mildly accelerated age-related losses in renal function and podocyte density. In the setting of an acute high-grade stressor, such as human immunodeficiency virus (HIV) infection in HIV-associated nephropathy (HIVAN), a LR genotype responds to the stress and is capable of recovering and returning to baseline function. The HR genotype, however, rapidly declines and is incapable of recovering from the stress, quickly reaching renal failure. In the setting of a chronic low-grade stressor, possibly representing a stress pattern associated with hypertension-attributed chronic kidney disease (CKD), the LR genotype adapts to the stresses and renal function is largely preserved. The HR genotype, however, cannot adapt to the stresses and loses renal function, resulting in renal failure within a normal lifespan.
CONCLUSIONS AND PERSPECTIVE
The biology that explains the association of APOL1 variants with nondiabetic kidney diseases in African Americans remains incompletely understood. The mode of inheritance is recessive, and APOL1 variant alleles alone are insufficient to cause disease; however, an understanding of the other required environmental or genetic events needed for disease induction remains largely unknown. Although intense research efforts are underway, an understanding of the normal function of APOL1 in kidney cells and how this function is altered by the risk variants have not been clearly defined. Central issues hampering basic biology studies are the lack of model systems that can authentically replicate human APOL1 expression in concert with appropriate protein interactors, the inclusion of relevant environmental stimuli in experiments, and the use of appropriate kidney cells (i.e., podocytes). APOL1 and the APOL1-mediated CKDs such as HIVAN are essentially a human-only gene causing a human-only disease. Developing model systems to study APOL1 function will always be an approximation balancing their benefits with disadvantages. As with all modeling studies, appropriate verification in human cells and tissues is an essential part of the discovery process.
Whether the APOL1 variants are gain-of-function or loss-of-function events, a pressing question is whether future therapeutic strategies should target the genetic variation or the triggering environmental cofactor. For HIVAN, targeting the environmental factor has been effective. Use of prevention strategies to avoid infection or suppressive antiviral medications to treat existing infections has significantly reduced the incidence and prevalence of HIVAN. For the other APOL1-associated CKDs, especially the common hypertension-attributed CKD, the answer is less clear. For decades, attempts to address environmental or behavioral factors to manage hypertension-attributed CKDs have not made progress in reducing disease progression in African Americans (63). Unlike HIVAN, the key environmental trigger for hypertension-attributed CKD has yet to be identified; thus strategies targeting the genetic predisposition may be a useful alternative. However, if APOL1 is to be a therapeutic target, it is imperative to determine whether the effect of the APOL1 variants are a gain-of-function in which the pathological function of G1 and G2 must be suppressed, or loss-of-function in which therapies must restore the function of G0.
GRANTS
L. A. Bruggeman, J. F. O’Toole, and J. R. Sedor are supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK097836 and DK108329.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.A.B. drafted manuscript and prepared figures; L.A.B., J.F.O., and J.R.S. edited and revised manuscript; L.A.B., J.F.O., and J.R.S. approved final version of manuscript.
REFERENCES
- 1.Beckerman P, Bi-Karchin J, Park AS, Qiu C, Dummer PD, Soomro I, Boustany-Kari CM, Pullen SS, Miner JH, Hu CA, Rohacs T, Inoue K, Ishibe S, Saleem MA, Palmer MB, Cuervo AM, Kopp JB, Susztak K. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 23: 429–438, 2017. doi: 10.1038/nm.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bigé N, Lanternier F, Viard JP, Kamgang P, Daugas E, Elie C, Jidar K, Walker-Combrouze F, Peraldi MN, Isnard-Bagnis C, Servais A, Lortholary O, Noël LH, Bollée G. Presentation of HIV-associated nephropathy and outcome in HAART-treated patients. Nephrol Dial Transplant 27: 1114–1121, 2012. doi: 10.1093/ndt/gfr376. [DOI] [PubMed] [Google Scholar]
- 3.Bruggeman LA, O’Toole JF, Ross MD, Madhavan SM, Smurzynski M, Wu K, Bosch RJ, Gupta S, Pollak MR, Sedor JR, Kalayjian RC. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol 25: 634–644, 2014. doi: 10.1681/ASN.2013070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruggeman LA, Wu Z, Luo L, Madhavan SM, Konieczkowski M, Drawz PE, Thomas DB, Barisoni L, Sedor JR, O’Toole JF. APOL1-G0 or APOL1-G2 transgenic models develop preeclampsia but not kidney disease. J Am Soc Nephrol 27: 3600–3610, 2016. doi: 10.1681/ASN.2015111220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen D, Zaky Z, Poggio E, Herlitz L, El Rifai R, Schold J, Drawz PE, Bruggeman LA, O’Toole JF, Sedor JR. Podocyte density is lower in transplant allografts carrying two APOL1 risk variants (Abstract) J Am Soc Nephrol 27: 31A, 2017. https://www.asn-online.org/api/download/?file=/education/kidneyweek/archives/KW17Abstracts.pdf. [Google Scholar]
- 6.Divers J, Núñez M, High KP, Murea M, Rocco MV, Ma L, Bowden DW, Hicks PJ, Spainhour M, Ornelles DA, Kleiboeker SB, Duncan K, Langefeld CD, Turner J, Freedman BI. JC polyoma virus interacts with APOL1 in African Americans with nondiabetic nephropathy. Kidney Int 84: 1207–1213, 2013. doi: 10.1038/ki.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Divers J, Palmer ND, Lu L, Langefeld CD, Rocco MV, Hicks PJ, Murea M, Ma L, Bowden DW, Freedman BI. Gene-gene interactions in APOL1-associated nephropathy. Nephrol Dial Transplant 29: 587–594, 2014. doi: 10.1093/ndt/gft423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doshi MD, Ortigosa-Goggins M, Garg AX, Li L, Poggio ED, Winkler CA, Kopp JB. APOL1 genotype and renal function of black living donors. J Am Soc Nephrol 29: 1309–1316, 2018. doi: 10.1681/ASN.2017060658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foster MC, Coresh J, Fornage M, Astor BC, Grams M, Franceschini N, Boerwinkle E, Parekh RS, Kao WH. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol 24: 1484–1491, 2013. doi: 10.1681/ASN.2013010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freedman BI, Julian BA, Pastan SO, Israni AK, Schladt D, Gautreaux MD, Hauptfeld V, Bray RA, Gebel HM, Kirk AD, Gaston RS, Rogers J, Farney AC, Orlando G, Stratta RJ, Mohan S, Ma L, Langefeld CD, Hicks PJ, Palmer ND, Adams PL, Palanisamy A, Reeves-Daniel AM, Divers J. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant 15: 1615–1622, 2015. doi: 10.1111/ajt.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freedman BI, Kistler AL, Skewes-Cox P, Ganem D, Spainhour M, Turner J, Divers J, Langefeld CD, Murea M, Hicks PJ, Hemal AK, Snipes JA, Zhao L, Abend JR, Lyles DS, Ma L, Skorecki KL. JC polyoma viruria associates with protection from chronic kidney disease independently from apolipoprotein L1 genotype in African Americans. Nephrol Dial Transplant 33: 1960–1967, 2018. doi: 10.1093/ndt/gfx368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freedman BI, Langefeld CD, Andringa KK, Croker JA, Williams AH, Garner NE, Birmingham DJ, Hebert LA, Hicks PJ, Segal MS, Edberg JC, Brown EE, Alarcón GS, Costenbader KH, Comeau ME, Criswell LA, Harley JB, James JA, Kamen DL, Lim SS, Merrill JT, Sivils KL, Niewold TB, Patel NM, Petri M, Ramsey-Goldman R, Reveille JD, Salmon JE, Tsao BP, Gibson KL, Byers JR, Vinnikova AK, Lea JP, Julian BA, Kimberly RP; Lupus Nephritis–End‐Stage Renal Disease Consortium . End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol 66: 390–396, 2014. doi: 10.1002/art.38220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freedman BI, Pastan SO, Israni AK, Schladt D, Julian BA, Gautreaux MD, Hauptfeld V, Bray RA, Gebel HM, Kirk AD, Gaston RS, Rogers J, Farney AC, Orlando G, Stratta RJ, Mohan S, Ma L, Langefeld CD, Bowden DW, Hicks PJ, Palmer ND, Palanisamy A, Reeves-Daniel AM, Brown WM, Divers J. APOL1 genotype and kidney transplantation outcomes from deceased African American donors. Transplantation 100: 194–202, 2016. doi: 10.1097/TP.0000000000000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman DJ. A brief history of APOL1: a gene evolving. Semin Nephrol 37: 508–513, 2017. doi: 10.1016/j.semnephrol.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Fu Y, Zhu JY, Richman A, Zhang Y, Xie X, Das JR, Li J, Ray PE, Han Z. APOL1-G1 in nephrocytes induces hypertrophy and accelerates cell death. J Am Soc Nephrol 28: 1106–1116, 2017. doi: 10.1681/ASN.2016050550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845, 2010. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Genovese G, Tonna SJ, Knob AU, Appel GB, Katz A, Bernhardy AJ, Needham AW, Lazarus R, Pollak MR. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int 78: 698–704, 2010. doi: 10.1038/ki.2010.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Granado D, Müller D, Krausel V, Kruzel-Davila E, Schuberth C, Eschborn M, Wedlich-Söldner R, Skorecki K, Pavenstädt H, Michgehl U, Weide T. Intracellular APOL1 risk variants cause cytotoxicity accompanied by energy depletion. J Am Soc Nephrol 28: 3227–3238, 2017. doi: 10.1681/ASN.2016111220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunter DJ. Gene-environment interactions in human diseases. Nat Rev Genet 6: 287–298, 2005. doi: 10.1038/nrg1578. [DOI] [PubMed] [Google Scholar]
- 20.Johnstone DB, Shegokar V, Nihalani D, Rathore YS, Mallik L, Ashish, Zare V, Ikizler HO, Powar R, Holzman LB. APOL1 null alleles from a rural village in India do not correlate with glomerulosclerosis. PLoS One 7: e51546, 2012. doi: 10.1371/journal.pone.0051546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kao WH, Klag MJ, Meoni LA, Reich D, Berthier-Schaad Y, Li M, Coresh J, Patterson N, Tandon A, Powe NR, Fink NE, Sadler JH, Weir MR, Abboud HE, Adler SG, Divers J, Iyengar SK, Freedman BI, Kimmel PL, Knowler WC, Kohn OF, Kramp K, Leehey DJ, Nicholas SB, Pahl MV, Schelling JR, Sedor JR, Thornley-Brown D, Winkler CA, Smith MW, Parekh RS; Family Investigation of Nephropathy and Diabetes Research Group . MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet 40: 1185–1192, 2008. doi: 10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasembeli AN, Duarte R, Ramsay M, Mosiane P, Dickens C, Dix-Peek T, Limou S, Sezgin E, Nelson GW, Fogo AB, Goetsch S, Kopp JB, Winkler CA, Naicker S. APOL1 risk variants are strongly associated with HIV-associated nephropathy in black South Africans. J Am Soc Nephrol 26: 2882–2890, 2015. doi: 10.1681/ASN.2014050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, Mokrzycki MH, Kimmel PL, Limou S, Ahuja TS, Berns JS, Fryc J, Simon EE, Smith MC, Trachtman H, Michel DM, Schelling JR, Vlahov D, Pollak M, Winkler CA. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 22: 2129–2137, 2011. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopp JB, Smith MW, Nelson GW, Johnson RC, Freedman BI, Bowden DW, Oleksyk T, McKenzie LM, Kajiyama H, Ahuja TS, Berns JS, Briggs W, Cho ME, Dart RA, Kimmel PL, Korbet SM, Michel DM, Mokrzycki MH, Schelling JR, Simon E, Trachtman H, Vlahov D, Winkler CA. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet 40: 1175–1184, 2008. doi: 10.1038/ng.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozlitina J, Zhou H, Brown PN, Rohm RJ, Pan Y, Ayanoglu G, Du X, Rimmer E, Reilly DF, Roddy TP, Cully DF, Vogt TF, Blom D, Hoek M. Plasma levels of risk-variant APOL1 do not associate with renal disease in a population-based cohort. J Am Soc Nephrol 27: 3204–3219, 2016. doi: 10.1681/ASN.2015101121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kruzel-Davila E, Shemer R, Ofir A, Bavli-Kertselli I, Darlyuk-Saadon I, Oren-Giladi P, Wasser WG, Magen D, Zaknoun E, Schuldiner M, Salzberg A, Kornitzer D, Marelja Z, Simons M, Skorecki K. APOL1-mediated cell injury involves disruption of conserved trafficking processes. J Am Soc Nephrol 28: 1117–1130, 2017. doi: 10.1681/ASN.2016050546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kruzel-Davila E, Wasser WG, Skorecki K. APOL1 nephropathy: a population genetics and evolutionary medicine detective story. Semin Nephrol 37: 490–507, 2017. doi: 10.1016/j.semnephrol.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Lan X, Jhaveri A, Cheng K, Wen H, Saleem MA, Mathieson PW, Mikulak J, Aviram S, Malhotra A, Skorecki K, Singhal PC. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol 307: F326–F336, 2014. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langefeld CD, Comeau ME, Ng MCY, Guan M, Dimitrov L, Mudgal P, Spainhour MH, Julian BA, Edberg JC, Croker JA, Divers J, Hicks PJ, Bowden DW, Chan GC, Ma L, Palmer ND, Kimberly RP, Freedman BI. Genome-wide association studies suggest that APOL1-environment interactions more likely trigger kidney disease in African Americans with nondiabetic nephropathy than strong APOL1-second gene interactions. Kidney Int 94: 599–607, 2018. doi: 10.1016/j.kint.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larsen CP, Beggs ML, Saeed M, Walker PD. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol 24: 722–725, 2013. doi: 10.1681/ASN.2012121180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee BT, Kumar V, Williams TA, Abdi R, Bernhardy A, Dyer C, Conte S, Genovese G, Ross MD, Friedman DJ, Gaston R, Milford E, Pollak MR, Chandraker A. The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant 12: 1924–1928, 2012. doi: 10.1111/j.1600-6143.2012.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lescure FX, Flateau C, Pacanowski J, Brocheriou I, Rondeau E, Girard PM, Ronco P, Pialoux G, Plaisier E. HIV-associated kidney glomerular diseases: changes with time and HAART. Nephrol Dial Transplant 27: 2349–2355, 2012. doi: 10.1093/ndt/gfr676. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Das JR, Tang P, Han Z, Jaiswal JK, Ray PE. Transmembrane TNF-α facilitates HIV-1 infection of podocytes cultured from children with HIV-associated nephropathy. J Am Soc Nephrol 28: 862–875, 2017. doi: 10.1681/ASN.2016050564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Limou S, Dummer PD, Nelson GW, Kopp JB, Winkler CA. APOL1 toxin, innate immunity, and kidney injury. Kidney Int 88: 28–34, 2015. doi: 10.1038/ki.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lipkowitz MS, Freedman BI, Langefeld CD, Comeau ME, Bowden DW, Kao WH, Astor BC, Bottinger EP, Iyengar SK, Klotman PE, Freedman RG, Zhang W, Parekh RS, Choi MJ, Nelson GW, Winkler CA, Kopp JB; SK Investigators . Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int 83: 114–120, 2013. doi: 10.1038/ki.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma L, Chou JW, Snipes JA, Bharadwaj MS, Craddock AL, Cheng D, Weckerle A, Petrovic S, Hicks PJ, Hemal AK, Hawkins GA, Miller LD, Molina AJ, Langefeld CD, Murea M, Parks JS, Freedman BI. APOL1 renal-risk variants induce mitochondrial dysfunction. J Am Soc Nephrol 28: 1093–1105, 2017. doi: 10.1681/ASN.2016050567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma L, Shelness GS, Snipes JA, Murea M, Antinozzi PA, Cheng D, Saleem MA, Satchell SC, Banas B, Mathieson PW, Kretzler M, Hemal AK, Rudel LL, Petrovic S, Weckerle A, Pollak MR, Ross MD, Parks JS, Freedman BI. Localization of APOL1 protein and mRNA in the human kidney: nondiseased tissue, primary cells, and immortalized cell lines. J Am Soc Nephrol 26: 339–348, 2015. doi: 10.1681/ASN.2013091017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madhavan SM, O’Toole JF, Konieczkowski M, Barisoni L, Thomas DB, Ganesan S, Bruggeman LA, Buck M, Sedor JR. APOL1 variants change C-terminal conformational dynamics and binding to SNARE protein VAMP8. JCI Insight 2: e92581, 2017. doi: 10.1172/jci.insight.92581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol 22: 2119–2128, 2011. doi: 10.1681/ASN.2011010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markowitz GS, Nasr SH, Stokes MB, D’Agati VD. Treatment with IFN-α, -β, or -γ is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 5: 607–615, 2010. doi: 10.2215/CJN.07311009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLaren PJ, Gawanbacht A, Pyndiah N, Krapp C, Hotter D, Kluge SF, Götz N, Heilmann J, Mack K, Sauter D, Thompson D, Perreaud J, Rausell A, Munoz M, Ciuffi A, Kirchhoff F, Telenti A. Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology 12: 41, 2015. doi: 10.1186/s12977-015-0165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikulak J, Oriolo F, Portale F, Tentorio P, Lan X, Saleem MA, Skorecki K, Singhal PC, Mavilio D. Impact of APOL1 polymorphism and IL-1β priming in the entry and persistence of HIV-1 in human podocytes. Retrovirology 13: 63, 2016. doi: 10.1186/s12977-016-0296-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nadkarni GN, Galarneau G, Ellis SB, Nadukuru R, Zhang J, Scott SA, Schurmann C, Li R, Rasmussen-Torvik LJ, Kho AN, Hayes MG, Pacheco JA, Manolio TA, Chisholm RL, Roden DM, Denny JC, Kenny EE, Bottinger EP. Apolipoprotein L1 variants and blood pressure traits in African Americans. J Am Coll Cardiol 69: 1564–1574, 2017. doi: 10.1016/j.jacc.2017.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D’Agati V, Markowitz G, Kopp JB, Alper SL, Pollak MR, Friedman DJ. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int 87: 332–342, 2015. doi: 10.1038/ki.2014.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Toole JF, Bruggeman LA, Madhavan S, Sedor JR. The cell biology of APOL1. Semin Nephrol 37: 538–545, 2017. doi: 10.1016/j.semnephrol.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Toole JF, Schilling W, Kunze D, Madhavan SM, Konieczkowski M, Gu Y, Luo L, Wu Z, Bruggeman LA, Sedor JR. ApoL1 overexpression drives variant-independent cytotoxicity. J Am Soc Nephrol 29: 869–879, 2018. doi: 10.1681/ASN.2016121322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olabisi O, Al-Romaih K, Henderson J, Tomar R, Drummond I, MacRae C, Pollak M. From man to fish: what can zebrafish tell us about ApoL1 nephropathy? Clin Nephrol 86, Suppl 1: 114–118, 2016. doi: 10.5414/CNP86S116. [DOI] [PubMed] [Google Scholar]
- 48.Olabisi OA, Zhang JY, VerPlank L, Zahler N, DiBartolo S III, Heneghan JF, Schlöndorff JS, Suh JH, Yan P, Alper SL, Friedman DJ, Pollak MR. APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc Natl Acad Sci USA 113: 830–837, 2016. doi: 10.1073/pnas.1522913113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palau L, Menez S, Rodriguez-Sanchez J, Novick T, Delsante M, McMahon BA, Atta MG. HIV-associated nephropathy: links, risks and management. HIV AIDS (Auckl) 10: 73–81, 2018. doi: 10.2147/HIV.S141978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pays E, Vanhollebeke B, Uzureau P, Lecordier L, Pérez-Morga D. The molecular arms race between African trypanosomes and humans. Nat Rev Microbiol 12: 575–584, 2014. doi: 10.1038/nrmicro3298. [DOI] [PubMed] [Google Scholar]
- 51.Ramsuran D, Bhimma R, Ramdial PK, Naicker E, Adhikari M, Deonarain J, Sing Y, Naicker T. The spectrum of HIV-related nephropathy in children. Pediatr Nephrol 27: 821–827, 2012. doi: 10.1007/s00467-011-2074-8. [DOI] [PubMed] [Google Scholar]
- 52.Reeves-Daniel AM, DePalma JA, Bleyer AJ, Rocco MV, Murea M, Adams PL, Langefeld CD, Bowden DW, Hicks PJ, Stratta RJ, Lin JJ, Kiger DF, Gautreaux MD, Divers J, Freedman BI. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 11: 1025–1030, 2011. doi: 10.1111/j.1600-6143.2011.03513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reidy KJ, Hjorten RC, Simpson CL, Rosenberg AZ, Rosenblum SD, Kovesdy CP, Tylavsky FA, Myrie J, Ruiz BL, Haque S, Mozhui K, Nelson GW, David VA, Yang X, Suzuki M, Jacob J, Reznik SE, Kaskel FJ, Kopp JB, Winkler CA, Davis RL. Fetal-not maternal-APOL1 genotype associated with risk for preeclampsia in those with African ancestry. Am J Hum Genet 103: 367–376, 2018. doi: 10.1016/j.ajhg.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samanovic M, Molina-Portela MP, Chessler AD, Burleigh BA, Raper J. Trypanosome lytic factor, an antimicrobial high-density lipoprotein, ameliorates Leishmania infection. PLoS Pathog 5: e1000276, 2009. doi: 10.1371/journal.ppat.1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sampson MG, Robertson CC, Martini S, Mariani LH, Lemley KV, Gillies CE, Otto EA, Kopp JB, Randolph A, Vega-Warner V, Eichinger F, Nair V, Gipson DS, Cattran DC, Johnstone DB, O’Toole JF, Bagnasco SM, Song PX, Barisoni L, Troost JP, Kretzler M, Sedor JR; Nephrotic Syndrome Study Network . Integrative genomics identifies novel associations with APOL1 risk genotypes in black NEPTUNE subjects. J Am Soc Nephrol 27: 814–823, 2016. doi: 10.1681/ASN.2014111131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shukha K, Mueller JL, Chung RT, Curry MP, Friedman DJ, Pollak MR, Berg AH. Most ApoL1 is secreted by the liver. J Am Soc Nephrol 28: 1079–1083, 2017. doi: 10.1681/ASN.2016040441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taylor HE, Khatua AK, Popik W. The innate immune factor apolipoprotein L1 restricts HIV-1 infection. J Virol 88: 592–603, 2014. doi: 10.1128/JVI.02828-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomson R, Genovese G, Canon C, Kovacsics D, Higgins MK, Carrington M, Winkler CA, Kopp J, Rotimi C, Adeyemo A, Doumatey A, Ayodo G, Alper SL, Pollak MR, Friedman DJ, Raper J. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA 111: E2130–E2139, 2014. doi: 10.1073/pnas.1400699111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, Skorecki K. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 128: 345–350, 2010. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vanhollebeke B, Truc P, Poelvoorde P, Pays A, Joshi PP, Katti R, Jannin JG, Pays E. Human Trypanosoma evansi infection linked to a lack of apolipoprotein L-I. N Engl J Med 355: 2752–2756, 2006. doi: 10.1056/NEJMoa063265. [DOI] [PubMed] [Google Scholar]
- 61.Weckerle A, Snipes JA, Cheng D, Gebre AK, Reisz JA, Murea M, Shelness GS, Hawkins GA, Furdui CM, Freedman BI, Parks JS, Ma L. Characterization of circulating APOL1 protein complexes in African Americans. J Lipid Res 57: 120–130, 2016. doi: 10.1194/jlr.M063453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wen H, Kumar V, Lan X, Shoshtari SSM, Eng JM, Zhou X, Wang F, Wang H, Skorecki K, Xing G, Wu G, Luo H, Malhotra A, Singhal PC. APOL1 risk variants cause podocytes injury through enhancing endoplasmic reticulum stress. Biosci Rep 38: BSR20171713, 2018. doi: 10.1042/BSR20171713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Whelton PK, Einhorn PT, Muntner P, Appel LJ, Cushman WC, Diez Roux AV, Ferdinand KC, Rahman M, Taylor HA, Ard J, Arnett DK, Carter BL, Davis BR, Freedman BI, Cooper LA, Cooper R, Desvigne-Nickens P, Gavini N, Go AS, Hyman DJ, Kimmel PL, Margolis KL, Miller ER III, Mills KT, Mensah GA, Navar AM, Ogedegbe G, Rakotz MK, Thomas G, Tobin JN, Wright JT, Yoon SS, Cutler JA; National Heart, Lung, and Blood Institute Working Group on Research Needs to Improve Hypertension Treatment and Control in African Americans . Research needs to improve hypertension treatment and control in African Americans. Hypertension 68: 1066–1072, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang JY, Wang M, Tian L, Genovese G, Yan P, Wilson JG, Thadhani R, Mottl AK, Appel GB, Bick AG, Sampson MG, Alper SL, Friedman DJ, Pollak MR. UBD modifies APOL1-induced kidney disease risk. Proc Natl Acad Sci USA 115: 3446–3451, 2018. doi: 10.1073/pnas.1716113115. [DOI] [PMC free article] [PubMed] [Google Scholar]