

Abstract
Aging is a risk factor for certain forms of kidney injury due to normal physiological changes, but the role of aging in cisplatin-induced kidney injury is not well defined in humans or animal models of the disease. To improve on current knowledge in this field, we treated 8- and 40-wk-old FVB/n mice with one high dose of cisplatin as a model of acute kidney injury or with repeated low doses of cisplatin (7 mg/kg cisplatin once a week for 4 wk) as a clinically relevant model of chronic kidney disease to determine if aging exacerbates cisplatin-induced kidney injury. Levels of acute kidney injury were comparable in 8- and 40-wk-old mice. In 40-wk-old mice, fibrotic markers were elevated basally, but treatment with cisplatin did not exacerbate fibrosis. We concluded that this may be the result of a decreased inflammatory response in 40-wk-old cisplatin-treated mice compared with 8-wk-old mice. Despite a decreased inflammatory response, the level of immune cell infiltration was greater in 40-wk-old cisplatin-treated mice than 8-wk-old mice. Our data highlight the importance of examining age as a risk factor for cisplatin-induced kidney injury.
Keywords: AKI, cisplatin, CKD, fibrosis, nephrotoxicity
INTRODUCTION
By 2030, senior citizens will comprise 21% of the US population (42). Elderly patients, in general, have more health problems, and acute kidney injury (AKI) in this population is a major concern. The average age of an in-hospital patient with AKI is 75 yr, and the incidence of AKI is 3.5–5 times higher in patients >70 yr old (28). This higher rate of AKI in patients >70 yr old may be due to comorbidities that predispose them to AKI, such as cardiovascular disease and diabetes (7, 8, 23). Experimental data in aged rats and mice have shown that normal aging is associated with chronic inflammation, increased oxidative stress, and decreased mitochondrial function (34, 35, 39, 42). These physiological components are also affected during AKI, contributing to increased cell death and damage to the vasculature, indicating that basal activation of these pathways may increase susceptibility to cisplatin-induced AKI (12, 42). Furthermore, experimental data have also shown that aging is associated with poor recovery after AKI (35). In renal epithelial cells, proliferation and basal apoptosis decrease, and growth factors involved in cellular maintenance decrease with age. Increased susceptibility to AKI in combination with decreased recovery after AKI suggests that aging alone, in the absence of comorbidities, may be associated with worsened renal injury.
It is important to note that most of what is known about AKI and aging has been concluded from mouse ischemia-reperfusion (IR) models of kidney injury (9, 12, 15, 39). These studies indicate increased mortality, renal dysfunction, and more interstitial fibrosis with IR injury in aged than young mice. However, susceptibility to injury from cisplatin nephrotoxicity is less clear. Many of the same processes that change with normal aging are also activated during cisplatin-induced AKI, and it is commonly believed that susceptibility to cisplatin nephrotoxicity is increased in patients >50 yr old (43). Patient data are often clouded by age-associated comorbidities that increase risk of cisplatin nephrotoxicity. Similarly, these patients are more likely to have nonrenal complications from cisplatin treatment. Studies have indicated that patients >70 yr old are more likely to have an adverse reaction to cisplatin, developing neutropenia or paresthesia (11, 24). However, these complications developed with no apparent renal failure, and only 14% of patients developed nephrotoxicity.
Conclusions from experimental studies of cisplatin-induced AKI in the context of aging are also limited. Typically, these studies utilize mice or rats at the end of their lifespan (18–24 mo old) (42, 43). This is problematic, inasmuch as cancer impacts a large percentage of middle-aged patients: 85% of all cancer diagnoses occur in individuals >55 yr old (21). For this reason, the effects of moderate aging in the context of cisplatin-induced kidney injury are an important field of study. To further clarify the relationship between cisplatin-induced AKI and aging, we utilized middle- to upper-middle-aged (40-wk-old) FVB/n mice to model a middle-aged human population that would be healthy enough to receive cisplatin for their malignancy. We hypothesized that these mice, when treated with one high dose of cisplatin (25 mg/kg), would still have exacerbated AKI compared with young (8-wk-old) mice treated with the same dose.
Furthermore, previous rodent studies that focused on the impact of aging on cisplatin-induced AKI utilized a single high dose of cisplatin that can cause death of the mouse after a few days. This high-dose cisplatin model is perhaps not clinically relevant, inasmuch as most patients receive low doses of cisplatin repeatedly to maintain therapeutic efficacy of the drug and, simultaneously, curtail nephrotoxic side effects. Thus it is difficult to draw conclusions with regard to the susceptibility of aged humans to cisplatin-induced kidney injury from studies that employ this high-dose model of cisplatin. To model the repeated, low-dosing regimen of cisplatin prescribed for many cancer types in the clinic, we and others developed repeated low-dose cisplatin mouse models (17, 32, 36, 41). Data indicate that repeated low doses of cisplatin in mice induce renal fibrosis and chronic kidney disease (CKD) (17, 36, 37, 41). Aging is associated with an increase in cellular senescence, which may predispose the normal aging kidney to development of fibrosis and CKD (12, 39). In addition, aging is an independent risk factor for development of CKD (18, 30). Using this dosing regimen, we wanted to determine if age increases susceptibility to cisplatin-induced fibrosis and CKD. To test this hypothesis, cohorts of 8- and 40-wk-old mice were treated with saline or 7 mg/kg cisplatin once a week for 4 wk and euthanized at day 24.
Data from this study indicate that 40-wk-old mice did not have worsened cisplatin-induced AKI compared with 8-wk-old mice. However, 40-wk-old mice had basally elevated myofibroblasts and higher total collagen deposition than 8-wk-old mice. Repeated administration of low-dose cisplatin slightly elevated collagen deposition, but there was no statistically significant difference in overall collagen deposition between cisplatin-treated 8- and 40-wk-old mice. Furthermore, the inflammatory cytokine response was less in 40-wk-old (aged) cisplatin-treated mice than young mice. The immune system plays an important role in kidney pathology, and chronic immune cell infiltration is heavily involved in the development of fibrosis. Immunohistochemistry staining for T cells and macrophages indicated more immune cell infiltration into the kidney with cisplatin treatment in 40-wk-old mice, despite the lower inflammatory cytokine response. Together, the data suggest that while there were differences in the inflammatory and immune cell responses during aging in response to cisplatin, this does not translate to worsened AKI or fibrosis indicative of CKD.
MATERIALS AND METHODS
Reagents and antibodies.
The following antibodies were purchased from Cell Signaling Technology (Beverly, MA): cleaved caspase-3 (CC3; catalog no. 9664), C/EEBP homologous protein (CHOP; catalog no. 2895), and transforming growth factor-β (TGFβ; catalog no. 3711). Fibronectin (catalog no. F7387) and β-actin (catalog no. A2228) were obtained from Sigma-Aldrich (St. Louis, MO), and tubulin (catalog no. SC-23948) was obtained from Santa Cruz Biotechnology (Dallas, TX). Pharmaceutical-grade cisplatin [purchased directly from the University of Louisville Hospital pharmacy; 1 mg/ml in normal saline (Teva Pharmaceuticals)] was used for all experiments.
Animals.
Eight- and 40-wk-old FVB/n mice were maintained on a 12:12-h light-dark cycle under standard laboratory conditions, with food and water provided ad libitum. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Louisville (protocol ID 16593) and followed the guidelines of the American Veterinary Medical Association. Briefly, mice were intraperitoneally injected with saline vehicle (0.9% saline in double-distilled H2O) or cisplatin (7 mg/kg) once a week for 4 wk and euthanized at day 24. Another cohort was intraperitoneally injected with saline vehicle (0.9% saline in double-distilled H2O) or cisplatin (25 mg/kg) once and euthanized at day 3. Mice were monitored daily for weight loss or high levels of discomfort/stress. Upon euthanasia, serum was prepared and stored at −80°C. Kidneys were flash-frozen in liquid nitrogen or fixed in 10% neutral buffered formalin.
Blood urea nitrogen determination.
Blood urea nitrogen (BUN) was determined using a kit from AMS Diagnostics (catalog no. 80146) according to the manufacturer’s instructions (AMS Diagnostics, Weston, FL).
ELISAs.
Urinary neutrophil gelatinase-associated lipocalin (NGAL) levels were measured by ELISA (catalog no. DY1857, R&D Systems, Minneapolis, MN) according to the manufacturer’s protocols.
Gene expression.
Total RNA was isolated using the EZNA total RNA kit (catalog no. R6834-02, Omega Bio-tek, Weston, FL) according to the manufacturer’s protocol. cDNA was made from 1 μg of total RNA using high-capacity reverse transcriptase (catalog no. 4368814, Life Technologies, Grand Island, NY) according to the manufacturer’s instructions. Gene-specific cDNA was quantified by real-time quantitative RT-PCR using predesigned TaqMan assays or self-designed SYBR assays. The following predesigned TaqMan assays (Life Technologies) were used: tumor necrosis factor-α (Tnfα; Mm00443258_m1), interleukin-6 (Il6; Mm00446190_m1), chemokine (C-X-C motif) ligand 1 (Cxcl1; Mm04207460_m1), monocyte chemoattractant protein 1 (Mcp1; Mm00441242_m1), and β2-microglobulin (B2m; Mm00437762_m1). Kidney injury molecule-1 (Kim-1) primers were self-designed. Real-time quantitative RT-PCR was performed using iTaq universal probes supermix (catalog no. 172-5134, Bio-Rad, Hercules, CA) or iTaq universal SYBR green supermix (catalog no. 172-5124, Bio-Rad).
Protein isolation/quantification and Western blot analysis.
Protein was isolated and quantified as previously described (36). Western blot analysis was performed as previously described, with 1:5,000 dilutions for primary antibodies and 1:40,000 dilutions for secondary antibodies. Proteins of interest were detected by chemiluminescence substrate. For TGFβ, densitometry was performed using ImageJ software, and TGFβ was normalized to tubulin.
Immunohistochemistry.
Kidney sections (5 μm thick) were rehydrated with Histo-Clear (catalog no. HS-200, National Diagnostics) followed by an ethanol gradient. α-Smooth muscle actin (α-SMA) and F4/80 immunohistochemistry was performed as previously described (36). For cluster of differentiation 3 (CD3), deparaffinization, antigen retrieval, and avidin/biotin blocking were performed as previously described for α-SMA (36). After avidin/biotin blocking, slides were blocked with 5% normal goat serum in PBS (catalog no. 31873, Invitrogen) for 1 h at room temperature. Rat anti-human CD3 (catalog no. MCA1477, Bio-Rad) was diluted 1:100 in normal goat serum and applied to tissue sections. Slides were incubated in primary antibody overnight at 4°C. On the following morning, slides were washed three times with PBS for 5 min each. Goat anti-rat biotinylated secondary antibody was diluted 1:250 in PBS and applied to slides for 1 h. During this time, ABC reagent (catalog no. PK-4001, Vectastain) was prepared. After incubation in secondary antibody, slides were washed twice for 5 min in PBS. ABC reagent was then applied to slides for 30 min, and slides were processed as previously described for α-SMA immunohistochemistry (36).
Sirius red/fast green staining for total collagen.
Serial kidney sections (5 μm thick) were rehydrated in Histo-Clear followed by an ethanol gradient. Sirius red/fast green (SR/FG) staining was performed as previously described (36). Images of whole kidney were taken using an Aperio slide scanner. MetaMorph software was used to determine total collagen, as previously published (36). Briefly, one representative whole kidney section was chosen from the serial sections that had been stained for SR/FG for further analysis. From a nontreated kidney tissue section, red pixels (threshold area) and green pixels (acellular area) were selected using MetaMorph software and stored for use as a filter for further analysis of all other samples. Thus, total collagen is presented as “percent Sirius red-positive pixels,” determined as follows: threshold area/(total area − acellular area).
Statistical analysis.
Data are expressed as means ± SE for all experiments. Multiple comparisons of normally distributed data sets were analyzed by two-way ANOVA, as appropriate, and group means were compared using Tukey’s post tests and GraphPad Prism statistical analysis software. P < 0.05 was considered statistically significant.
RESULTS
Changes in overall survival and weight loss in 8- and 40-wk-old mice treated with the standard-dosing regimen of cisplatin.
AKI is associated with a high rate of mortality, and with the experimental model of cisplatin-induced AKI, mice lose a significant amount of weight likely due to cisplatin’s effects on the gastrointestinal tract. We hypothesized that 40-wk-old (aged) mice treated with high-dose cisplatin would have decreased survival and greater weight loss than 8-wk-old (young) cisplatin-treated mice. Young mice that received 0 or 25 mg/kg cisplatin survived to day 3, at which point the young, cisplatin-treated cohort had an average weight loss of 17.4% (Fig. 1, A and B). Although survival for aged mice that received 0 mg/kg cisplatin was 100%, survival for aged mice treated with high-dose cisplatin was 80% (Fig. 1A). Aged mice (average starting weight of 39.3 g) treated with cisplatin had lost an average of 11.9% of total body weight at day 3, but weight loss was not significantly different from young mice (average starting weight of 26.6 g) treated with cisplatin (Fig. 1B). These data indicate decreased survival of aged cisplatin-treated mice and may allude to worsened kidney injury.
Fig. 1.
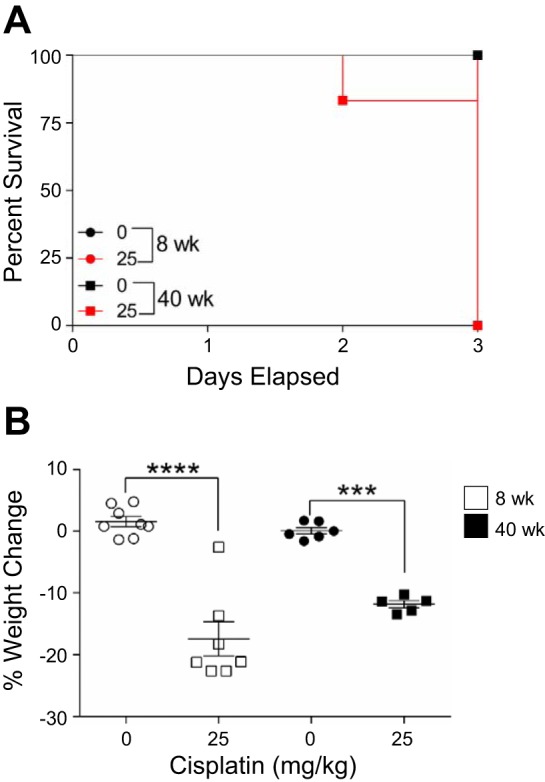
Survival and weight loss with the standard-dosing regimen of cisplatin (0 or 25 mg/kg once) in 8- and 40-wk-old male FVB mice. A: percent survival until day 3. B: percent weight change in mice. Values are means ± SE; n = 5–10. ***P < 0.001, ****P < 0.0001 (by 2-way ANOVA followed by Tukey’s post test).
BUN, urinary NGAL, and kidney Kim-1 mRNA levels increase in 8- and 40-wk-old mice treated with the standard-dosing regimen of cisplatin.
During AKI, a rapid decline in kidney function leads to buildup of waste products in the blood that can be measured by serum creatinine or BUN. Because kidney function declines with normal aging, we hypothesized that aged mice would have increased baseline BUN levels compared with young mice. We further hypothesized that, when treated with the standard-dosing regimen of cisplatin, loss of kidney function would be more severe in aged mice than young mice treated with the same dosing regimen. BUN levels significantly increased in young and aged cisplatin-treated mice, but BUN levels were not significantly different between these two age groups following treatment (Fig. 2A). In addition, BUN was not basally elevated in old mice compared with young mice (Fig. 2A).
Fig. 2.
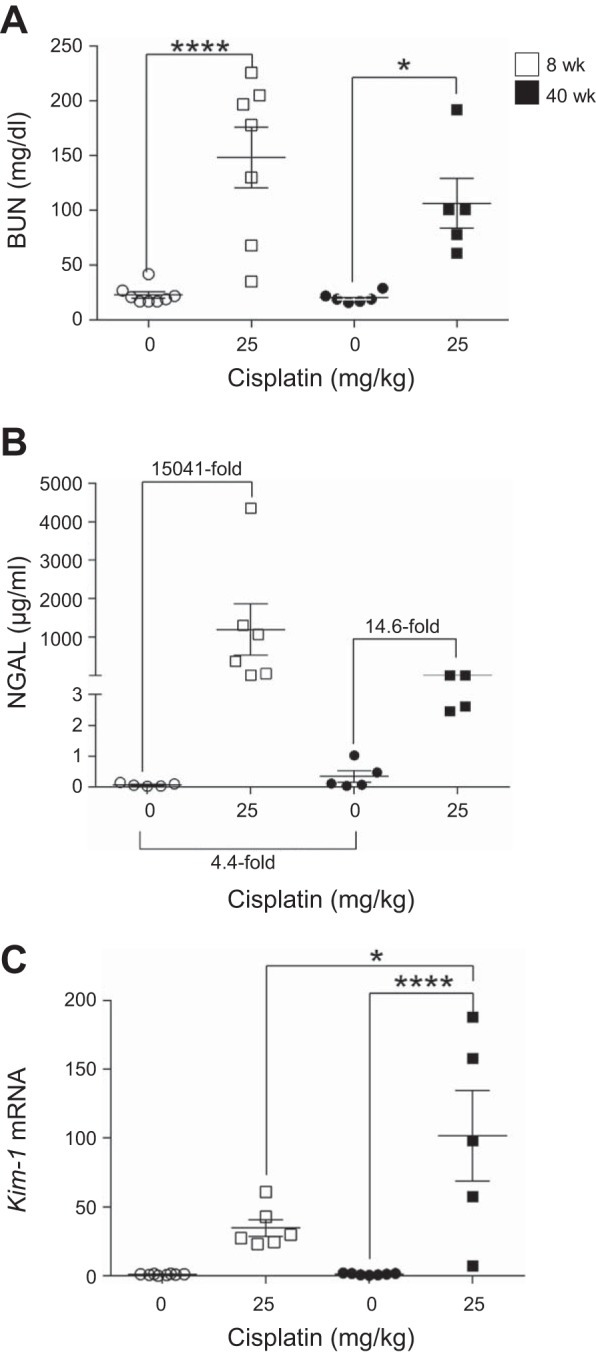
Changes in kidney function and injury markers with the standard-dosing regimen of cisplatin (0 or 25 mg/kg once) in 8- and 40-wk-old male FVB mice. A: blood urea nitrogen (BUN) in plasma. B: neutrophil gelatinase-associated lipocalin (NGAL) in urine. C: kidney injury molecule-1 (Kim-1) mRNA in kidney cortex measured by real-time quantitative RT-PCR. Values are means ± SE; n = 5–10. *P < 0.05, ****P < 0.0001 (by 2-way ANOVA followed by Tukey’s post test).
BUN is elevated only when there is ≥50% loss of kidney function and, thus, is not a sensitive indicator of kidney injury or function. Without overt changes in BUN levels, kidney injury can be detected by NGAL or KIM-1 levels. Urinary NGAL levels increased ~15,000-fold in young mice treated with cisplatin, but only 14.6-fold in aged mice, compared with their respective controls (Fig. 2B). Furthermore, NGAL levels were slightly elevated in aged mice compared with young mice treated with 0 mg/kg cisplatin (Fig. 2B). Kim-1 mRNA levels were elevated in cisplatin-treated young mice and further increased in cisplatin-treated aged mice (Fig. 2C). Together, these data indicate that treatment of aged and young mice with the standard-dosing regimen of cisplatin leads to a comparable loss of function and higher levels of NGAL in young mice but higher levels of Kim-1 in aged mice. Since NGAL is associated with inflammation and KIM-1 is associated with proximal tubule damage, this may indicate a difference in inflammation and injury in the groups examined.
Inflammatory cytokines and chemokines increase in 8- and 40-wk-old mice treated with the standard-dosing regimen of cisplatin.
The activation of inflammatory cytokines and chemokines plays a major role in the pathogenesis of cisplatin-induced AKI. TNFα increases early in kidney injury and initiates the inflammatory response. Tnfα mRNA levels increased ~10-fold in young mice treated with cisplatin, but only 4.7-fold in aged mice (Fig. 3A). Il6, another proinflammatory cytokine, increased 30.8-fold in young mice, but only 11.3-fold in aged mice (Fig. 3B). Cxcl1 and Mcp1, chemokines involved in recruitment of immune cells to the site of injury, increased in young and aged mice treated with cisplatin (Fig. 3, C and D). Interestingly, Mcp1 levels were basally elevated in aged mice compared with young mice (Fig. 3D). Thus these data suggest that aged mice have a decreased inflammatory cytokine response to cisplatin treatment.
Fig. 3.

Changes in inflammatory cytokine and chemokine levels with the standard-dosing regimen of cisplatin (0 or 25 mg/kg once) in 8- and 40-wk-old male FVB mice. Tumor necrosis factor-α (Tnfa; A), interleukin-6 (Il6; B), chemokine (C-X-C motif) ligand 1 (Cxcl1; C), and monocyte chemoattractant protein 1 (Mcp1; D) were measured in the kidney cortex by real-time quantitative RT-PCR. Values are means ± SE; n = 5–10. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (by 2-way ANOVA followed by Tukey’s post test).
Endoplasmic reticulum stress and apoptosis occur in the kidney in 8- and 40-wk-old mice treated with the standard-dosing regimen of cisplatin.
A hallmark of AKI is the high level of apoptotic cell death in both tubule and endothelial cells of the kidney. In our laboratory, treatment of mice with the high-dose cisplatin model is accompanied by endoplasmic reticulum (ER) stress. Aged mice had high basal protein levels of CHOP compared with young mice (Fig. 4). Treatment with cisplatin increased CHOP levels in young and aged mice (Fig. 4). CC3 is an established marker of apoptosis. Basally, CC3 levels were elevated in aged mice, and treatment with cisplatin led to a comparable increase in CC3 levels in aged and young mice (Fig. 4). Together, these data indicate that while aged mice have high basal levels of ER stress, the levels of apoptosis that occur with cisplatin treatment in young and aged mice are comparable, indicating that aged mice do not have higher levels of cell death.
Fig. 4.
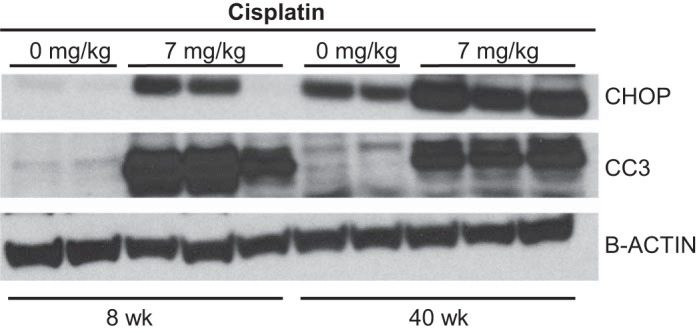
Endoplasmic reticulum stress and apoptosis with the standard-dosing regimen of cisplatin (0 or 25 mg/kg once) in 8- and 40-wk-old male FVB mice. Protein levels of C/EEBP homologous protein (CHOP), cleaved caspase 3 (CC3), and β-actin were determined in kidney cortex homogenate by Western blotting.
Changes in overall survival and weight in 8- and 40-wk-old mice treated with the repeated-dosing regimen of cisplatin.
We previously showed that 8-wk-old mice treated with repeated low-dose cisplatin (7 mg/kg once a week for 4 wk) have a 100% survival rate at day 24 and that these mice can be aged out ≥6 mo after treatment with minimal weight loss. Both young and aged mice treated with repeated doses of 0 or 7 mg/kg cisplatin had a 100% survival rate at day 24 (Fig. 5A). In addition, total weight loss for cisplatin-treated mice in both age groups was <10% (Fig. 5B). These data indicate that there is no difference in survival or weight loss in young and aged mice treated with the repeated-dosing regimen of cisplatin.
Fig. 5.
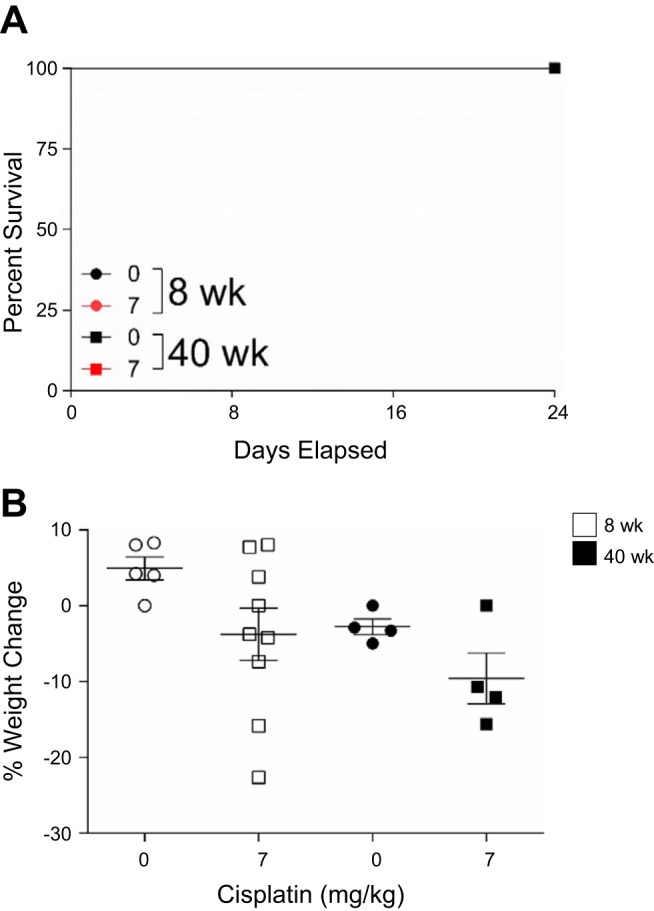
Survival and weight loss with the repeated-dosing regimen of cisplatin (0 or 7 mg/kg once a week for 4 wk) in 8- and 40-wk-old male FVB mice. A: percent survival until day 24. B: percent weight change. Values are means ± SE; n = 4–10. Statistical significance was determined by 2-way ANOVA followed by Tukey’s post test.
BUN, urinary NGAL, and kidney Kim-1 mRNA levels increase in 8- and 40-wk-old mice treated with the repeated-dosing regimen of cisplatin.
With our established repeated low-dose model, cisplatin treatment causes a slight decline in renal function, as determined by BUN, and an increase in NGAL levels. BUN levels in young and aged mice increased with cisplatin treatment, although this increase was not significant in either age group (Fig. 6A). NGAL levels increased 51.2-fold in young mice and only 9.3-fold in aged mice treated with cisplatin. Basally, NGAL levels were elevated 6.8-fold in aged mice compared with young mice (Fig. 6B). KIM-1 is also a sensitive marker of kidney injury. Kim-1 mRNA levels were increased 130-fold in young mice and only 72-fold in aged mice compared with respective controls (Fig. 6C). Thus the data suggest that while injury markers are already elevated basally in aged mice, there is no significant difference in the level of kidney injury or loss of function caused by repeated cisplatin treatment of young and aged mice.
Fig. 6.
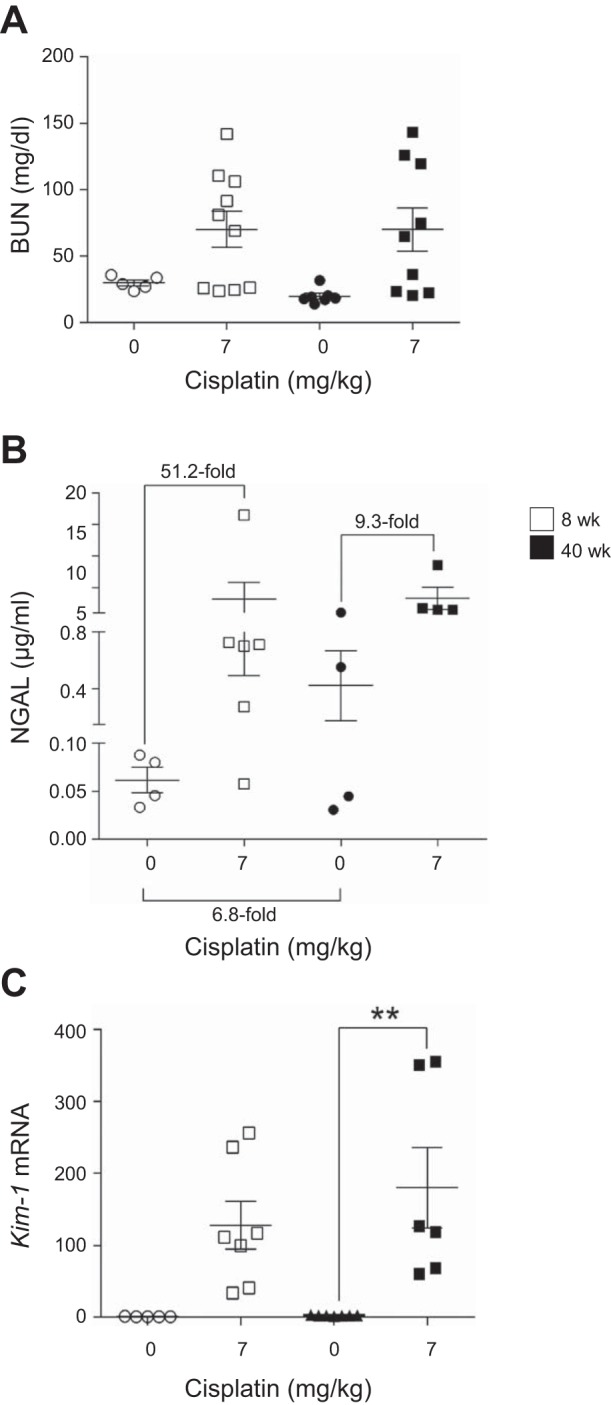
Changes in kidney function and injury markers with the repeated-dosing regimen of cisplatin (0 or 7 mg/kg once a week for 4 wk) in 8- and 40-wk-old male FVB mice. A: blood urea nitrogen (BUN) in plasma. B: neutrophil gelatinase-associated lipocalin (NGAL) in urine. C: kidney injury molecule-1 (Kim-1) in kidney cortex measured by real-time quantitative RT-PCR. Values are means ± SE; n = 4–10. **P < 0.01 (by 2-way ANOVA followed by Tukey’s post test).
Inflammatory cytokines and chemokines increase in 8- and 40-wk-old mice treated with the repeated-dosing regimen of cisplatin.
Chronic inflammation is a major driver of fibrosis. Many of the same inflammatory cytokines and chemokines that play a role in cisplatin-induced AKI are also involved in the progression of cisplatin-induced kidney injury to fibrosis and CKD. Thus we used the repeated low-dose cisplatin regimen to compare inflammatory cytokine expression in young and old mice. Tnfα mRNA levels increased sixfold in young mice treated with cisplatin, but only fourfold in aged mice (Fig. 7A). Il6 levels were elevated 8.8-fold in young mice treated with cisplatin, but only 1.8-fold in aged mice (Fig. 7B). The inflammatory chemokines Cxcl1 and Mcp1 significantly increased in both young and aged mice treated with cisplatin, and Mcp1 was basally elevated in aged mice compared with young mice (Fig. 7, C and D). Thus there is less inflammatory cytokine response in aged mice treated with cisplatin.
Fig. 7.

Changes in inflammatory cytokine and chemokine levels with the repeated-dosing regimen of cisplatin (0 or 7 mg/kg once a week for 4 wk) in 8- and 40-wk-old male FVB mice. Tumor necrosis factor-α (Tnfa; A), interleukin-6 (Il6; B), chemokine (C-X-C motif) ligand 1 (Cxcl1; C), and monocyte chemoattractant protein 1 (Mcp1; D) were measured in the kidney cortex by real-time quantitative RT-PCR. Values are means ± SE; n = 5–10. **P < 0.01, ****P < 0.0001 (by 2-way ANOVA followed by Tukey’s post test).
Fibrosis develops in 8- and 40-wk-old mice treated with the repeated-dosing regimen of cisplatin.
Repeated low-dose administration of cisplatin causes fibrosis. It has been hypothesized that pathways contributing to fibrosis are basally activated with normal renal aging, consequently contributing to the increased rate of CKD in aged populations. Thus we hypothesized that aged mice treated with the repeated-dosing regimen of cisplatin would have an exacerbated fibrotic phenotype compared with young mice treated with cisplatin. TGFβ levels increase after kidney injury, and activation of the TGFβ signaling pathway is one of the major contributors to the development of fibrosis. Furthermore, fibrosis is marked by an increase in extracellular matrix components, namely, fibronectin and collagen. TGFβ and fibronectin levels increased in young and aged mice treated with cisplatin, and quantification of TGFβ showed that levels were not further increased in aged mice compared with young mice (Fig. 8A). Total collagen deposition, as evidenced by SR/FG staining, indicated higher collagen levels basally (3.8-fold increase) in aged than young mice (Fig. 8B). However, treatment with cisplatin led to a 5.3-fold increase in collagen in young mice, but only a 1.5-fold increase in aged mice, although overall collagen deposition levels were comparable between the two groups (~11% Sirius red-positive pixels) (Fig. 8B).
Fig. 8.

Fibrosis develops with the repeated-dosing regimen of cisplatin (0 or 7 mg/kg once a week for 4 wk) in 8- and 40-wk-old male FVB mice. A: protein levels of transforming growth factor-β (TGFβ) and fibronectin in kidney cortex homogenate were determined by Western blotting, and TGFβ protein levels were quantified. B: total collagen levels were determined by Sirius red/fast green staining and quantified as percent Sirius red-positive pixels. C: presence of myofibroblasts was determined by α-smooth muscle actin (α-SMA) immunohistochemistry and quantified as percent α-SMA-positive pixels. Values are means ± SE; n = 3–4. Statistical significance was determined by 2-way ANOVA followed by Tukey’s post test.
Myofibroblasts produce collagen during fibrogenesis. Immunohistochemistry staining for α-SMA indicated more myofibroblasts basally (4.1-fold basal increase) in aged than young mice (Fig. 8C). Myofibroblasts increased 4.2-fold in young mice treated with cisplatin, but only 1.6-fold in aged mice, following a trend similar to that seen with total collagen deposition levels (Fig. 8C). Thus fibrosis was not worsened in aged mice treated with the repeated-dosing regimen of cisplatin compared with young mice, even though α-SMA-positive myofibroblasts and collagen deposition were basally elevated.
Increased immune cell infiltration in the kidneys of aged mice treated with the repeated-dosing regimen of cisplatin.
Immune cell infiltration after insult to the kidney is a driver of injury and initiates adaptive repair responses. Repeated dosing of cisplatin causes an increase in the number of macrophages in the kidney, which is associated with worsening fibrosis. F4/80-positive staining for macrophages increased in aged cisplatin-treated mice compared with young cisplatin-treated mice (Fig. 9A). Arginase-1 (ARG-1) and inducible nitric oxide synthase (iNOS) are markers for M2 and M1 macrophages, respectively. M1 macrophages are proinflammatory and lead to worsened injury, whereas M2 macrophages are involved in adaptive repair due to their anti-inflammatory phenotype. Inos mRNA levels did not increase in either cohort of mice treated with cisplatin; however, Arg-1 mRNA levels increased 5.3-fold in young mice and 1.8-fold in aged mice (Fig. 9, B and C). This indicates that most macrophages in the kidney are of the M2 phenotype.
Fig. 9.

Immune cell populations in the kidney with the repeated-dosing regimen of cisplatin (0 or 7 mg/kg once a week for 4 wk) in 8- and 40-wk-old male FVB mice. A: presence of macrophages was determined by F4/80 immunohistochemistry. B and C: arginase 1 (Arg-1) and inducible nitric oxide synthase (Inos) in the kidney cortex were determined by real-time quantitative RT-PCR. D: total T cells were determined by cluster of differentiation 3 (CD3) immunohistochemistry. Values are means ± SE; n = 5–10. Statistical significance was determined by 2-way ANOVA followed by Tukey’s post test.
T cells are known to contribute to cisplatin-induced AKI and the development of CKD. CD3-positive T cells also increased in aged mice treated with cisplatin compared with young mice (Fig. 9D). Furthermore, these T cells formed clusters in aged mice, but not in young mice. Together, immunochemistry indicates more immune cell infiltration in aged mice treated with cisplatin, even though fibrosis is not worsened in these mice.
DISCUSSION
Renal aging is often marked by the development of glomerulosclerosis, tubular atrophy, increased cellular senescence, interstitial fibrosis, and total nephron loss (12, 39). These physiological changes culminate in decreased glomerular filtration rate (GFR), the inability to properly conserve and secrete sodium, and difficulties in proper concentration or dilution of urine (39). In the context of cisplatin-induced nephrotoxicity, Wen et al. (43) found that lung cancer patients receiving one dose of cisplatin had a 9.2% decrease in GFR and that age was the only risk factor to have a significant relationship with decline in kidney function after the first round of cisplatin treatment. However, this decrease in GFR is minimal and does not meet clinical criteria for AKI diagnosis (33). Furthermore, this change in GFR could be attributed to the fact that these patients also received other chemotherapeutics known to cause kidney injury. Of note, 83 patients that received cisplatin also received gemcitabine, a well-known inducer of nephrotoxicity (29, 43).
Because most experimental studies examining the effects of aging in regard to increased renal injury are performed in old animals toward the end of their lifespan, it is often difficult to translate these findings to the clinical setting, where the median age of all cancer diagnoses is 65 yr (21). In addition, utilization of different mouse strains can alter study outcomes related to aging, inasmuch as median overall survival is different for each mouse strain. For C57BL/6J mice, which are commonly used in renal aging studies, median survival is 866 days, while median survival for the FVB/n strain (mice utilized in our study) is 591 days (45). Along with this variation, renal pathologies also differ. For example, glomerular/matrix expansion is 15.7% in C57BL/6J mice at 20 mo of age, while it is only 8.67% in FVB/n mice (22).
Wen et al. (43) indicated that 18-mo-old C57BL/6J mice developed more severe kidney injury when treated with 15 mg/kg cisplatin than 3-mo-old mice. These aged mice had increased BUN levels when treated with cisplatin. Furthermore, Wen et al. (43) reported an increase in Toll-like receptor 4-mediated signaling, an increase in cisplatin accumulation in the kidney, and a 35% decrease in protein levels of multidrug and toxin extrusion transporter 1, an efflux transporter involved in removal of cisplatin from the kidney. However, this study utilized a relatively old C57BL/6J mouse.
Despite concern for increased susceptibility to cisplatin nephrotoxicity with aging and limited experimental data that support this notion, several studies in patients have indicated that aging alone may not be a major risk factor. First, the rate of nephrotoxicity/AKI in pediatric cancer patients treated with cisplatin is comparable to that in adults (2). Skinner et al. (38) found posttreatment nephrotoxicity in 50% of children treated with either cisplatin or carboplatin and decreased GFR in 36.4% of these patients, indicative of CKD. The rate of cisplatin nephrotoxicity in adult cancer patients ranges from 20 to 30%, and ~20% of all adult patients diagnosed with AKI will progress to CKD (10, 20, 26, 27). Furthermore, in a study by Thyss et al. (40), 80- to 87-yr-old patients without underlying cardiovascular disease did not have an increased rate of cisplatin nephrotoxicity compared with younger populations receiving a similar dosing regimen. This study highlights that comorbidities, such as cardiovascular disease and diabetes, may contribute to kidney function changes that are often associated with normal aging and may be attributed to increased susceptibility to cisplatin nephrotoxicity. This is especially poignant in mouse studies that utilize C57BL/6J mice, as they have poor cardiac function during acute hypoxic challenge via ischemic injury, and thus aging studies utilizing this strain of mouse may be hindered by this cardiovascular susceptibility, especially with the IR model of injury (4).
Furthermore, Hrushesky et al. (13) reported an average 21 ml/min decline in creatinine clearance in 60- to 70-yr-old patients treated with only cisplatin but a 30 ml/min decline in ≤50-yr-old patients. Hrushesky et al. (13) concluded that this 60- to 70-yr-old cohort may be slightly protected from cisplatin nephrotoxicity because the external medulla is highly preserved, despite aging, and that this was the main site of cisplatin concentration in older patients. Finally, with aging there is no change in overall clearance of cisplatin, suggesting that concern for treating patients of advanced age may not be as serious as previously believed (13).
In this study we utilized 40-wk-old FVB mice as our aged cohort. This represents a middle-aged cohort, as the average lifespan of FVB mice is ∼20 mo (45). We believe that this 40-wk-old cohort of mice better reflects the cohort of patients who are diagnosed with cancer and are eligible to receive cisplatin treatment. Our data from treating mice at this age with the standard- and repeated-dosing regimens of cisplatin indicate that there is a difference in both the inflammatory response and immune cell recruitment after injury, without detectable worsening of kidney injury, worsened fibrosis, or an overt change in loss of kidney function.
We saw no age-dependent changes in basal BUN levels, nor did we find differences in BUN between 8- and 40-wk-old mice treated with either dosing regimen of cisplatin. This challenges the belief that normal aging leads to a decline in kidney function and highlights that changes in kidney function in humans may be due to underlying renal or cardiovascular disease. Furthermore, it has been hypothesized that even though nephron mass is lost with normal aging, compensatory hypertrophy of the remaining nephrons may help maintain normal kidney function, especially when kidney injury occurs (5).
NGAL is a standard urinary biomarker for kidney injury (14). We found that aged mice treated with the standard-dosing regimen of cisplatin had much lower levels of NGAL overall and that these levels were comparable to those in 8- and 40-wk-old mice treated with the repeated-dosing regimen of cisplatin. However, basal levels of NGAL were higher in aged than young mice. Furthermore, levels of NGAL in young and aged mice did not correspond to Kim-1 mRNA levels. This may be explained by the fact that NGAL is a more general marker of inflammation and immunomodulation, while KIM-1 is more specific to tubular injury (1). KIM-1 was higher in aged mice treated with 25 mg/kg cisplatin than in young mice treated with cisplatin, indicating potentially increased tubular damage in aged mice, although this trend was less pronounced with the repeated-dosing regimen of cisplatin. NGAL, however, corresponded with the inflammatory response difference between young and aged mice treated with cisplatin. In particular, we found that Tnfa mRNA levels increased with one high dose of cisplatin but that this increase was less pronounced in aged than young mice. As TNFα is one of the mediators of cytokine and chemokine expression with cisplatin injury, we also measured mRNA expression of Il6 and Mcp1, which followed an expression pattern similar to Tnfa (31). To further highlight the difference in the inflammatory response, mRNA levels of Il6 and Mcp1 followed a pattern similar to Tnfa.
Furthermore, in sepsis-induced mouse models of AKI, an increase in NGAL is indicative of cytokine storm, a phenomenon marked by overproduction of cytokines and immune cells (25). Furthermore, it has been shown that NGAL urine and blood levels are higher in patients with sepsis-induced AKI than in patients with non-sepsis-induced AKI (3). In the Awareness During Resuscitation (AWARE) study, NGAL was found to have additive diagnostic value for AKI in pediatric patients. However, patients in this study were enrolled based on Kidney Disease: Improving Global Outcomes criteria for AKI only and were not stratified based on sepsis- vs. non-sepsis-induced AKI (16). Thus the additive performance of NGAL may be due to patients in this cohort who had sepsis-induced AKI (16). Therefore, we hypothesize that the higher NGAL levels in young mice treated with 25 mg/kg cisplatin than in similarly treated aged mice are a consequence of increased Tnfα mRNA levels, since it has been shown that TNFα can induce NGAL expression (1). Together, our data suggest that aged mice have a decreased inflammatory response to cisplatin treatment compared with young mice.
Surprisingly, even though there was less inflammatory cytokine expression with cisplatin treatment of aged mice, more immune cells (CD3-positive T cells and F4/80-positive macrophages) were present in 40-wk-old mice treated with the repeated-dosing regimen of cisplatin. This may seem contradictory, as there was no difference in the message levels of chemoattractant markers (MCP-1 and CXCL1) in young and aged mice treated with cisplatin. Furthermore, even though there were more macrophages in aged kidneys after cisplatin treatment, Arg-1 mRNA levels, a marker for M2 macrophage activity, were lower in aged than young mice. Thus these data may allude to an exacerbation of immune cell recruitment that is dependent on cisplatin treatment, since CD3 T cells and F4/80 macrophages were not altered in aged mice receiving 0 mg/kg cisplatin. However, with normal aging, general immunosuppression makes elderly patients more susceptible to infection and cancer development (44). In the context of kidney function, the immune system plays a critical role in both kidney injury and repair (27). It has been reported that normal kidney aging is associated with an immunosenescent phenotype marked by altered function of immune cells (12, 35). In particular, T cells are highly affected (6, 19). In aged cisplatin-treated mice, T cells formed distinct, clustered patterns within the whole kidney tissue, whereas T cells in young cisplatin-treated mice were fewer in number and were found isolated throughout the kidney. This “clustering” of T cells may indicate either enhanced proliferation of T cells or alteration of chemokines involved in recruitment of T cells to the kidney. Although determining the true cause of T cell clustering is beyond the scope of the present study, future studies will focus on detailed classification of the specific types of T cells present and their activity/function. These future studies will be important for further classification of immune cell alterations in response to cisplatin treatment in the context of aging.
Interestingly, a lack of a robust inflammatory response and potentially altered immune cell activity in the kidney corresponded to differences in fibrosis development in aged mice treated with our repeated-dosing regimen of cisplatin compared with young mice. Forty-week-old mice had increased collagen deposition and more myofibroblasts than 8-wk-old mice basally. However, repeated dosing of cisplatin did not lead to overt collagen deposition or the presence of more myofibroblasts in aged mice than in age-matched vehicle-treated mice. This highlights the importance of a normal inflammatory and immune cell response in fibrogenesis, indicating that the altered inflammatory response to cisplatin treatment in aged mice may be somewhat protective against the development of fibrosis.
Together, the dosing regimens discussed here represent both AKI and CKD, two disease states that are thought to be more likely to occur in aged populations and lead to worsened kidney outcomes. Overall, this study indicates that advanced age does not greatly alter kidney injury or kidney function with both the standard- and repeated-dosing regimens of cisplatin. Thus our data suggest that, in older patients, treatment with cisplatin may not result in a more severe kidney injury phenotype or increased rate of nephrotoxicity. Furthermore, worsened injury and higher susceptibility to injury may be the result of comorbidities aside from age, such as underlying cardiovascular disease, diabetes, polypharmacy, or genetic polymorphisms (12, 42, 43).
GRANTS
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK-093462.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.N.S., L.J.B., and L.J.S. conceived and designed research; C.N.S. and M.D. performed experiments; C.N.S. and M.D. analyzed data; C.N.S., L.J.B., and L.J.S. interpreted results of experiments; C.N.S. and M.D. prepared figures; C.N.S. drafted manuscript; C.N.S., M.D., T.V.D., L.J.B., and L.J.S. edited and revised manuscript; C.N.S., M.D., T.V.D., L.J.B., and L.J.S. approved final version of manuscript.
REFERENCES
- 1.Abella V, Scotece M, Conde J, Gómez R, Lois A, Pino J, Gómez-Reino JJ, Lago F, Mobasheri A, Gualillo O. The potential of lipocalin-2/NGAL as biomarker for inflammatory and metabolic diseases. Biomarkers 20: 565–571, 2015. doi: 10.3109/1354750X.2015.1123354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arga M, Oguz A, Pinarli FG, Karadeniz C, Citak EC, Emeksiz HC, Duran EA, Soylemezoglu O. Risk factors for cisplatin-induced long-term nephrotoxicity in pediatric cancer survivors. Pediatr Int 57: 406–413, 2015. doi: 10.1111/ped.12542. [DOI] [PubMed] [Google Scholar]
- 3.Bagshaw SM, Bennett M, Haase M, Haase-Fielitz A, Egi M, Morimatsu H, D’Amico G, Goldsmith D, Devarajan P, Bellomo R. Plasma and urine neutrophil gelatinase-associated lipocalin in septic versus non-septic acute kidney injury in critical illness. Intensive Care Med 36: 452–461, 2010. doi: 10.1007/s00134-009-1724-9. [DOI] [PubMed] [Google Scholar]
- 4.Barnabei MS, Palpant NJ, Metzger JM. Influence of genetic background on ex vivo and in vivo cardiac function in several commonly used inbred mouse strains. Physiol Genomics 42A: 103–113, 2010. doi: 10.1152/physiolgenomics.00071.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolton WK, Benton FR, Maclay JG, Sturgill BC. Spontaneous glomerular sclerosis in aging Sprague-Dawley rats. I. Lesions associated with mesangial IgM deposits. Am J Pathol 85: 277–302, 1976. [PMC free article] [PubMed] [Google Scholar]
- 6.Cambier J. Immunosenescence: a problem of lymphopoiesis, homeostasis, microenvironment, and signaling. Immunol Rev 205: 5–6, 2005. doi: 10.1111/j.0105-2896.2005.00276.x. [DOI] [PubMed] [Google Scholar]
- 7.Chao C-T, Tsai H-B, Lin Y-F, Ko W-J. Acute kidney injury in the elderly: only the tip of the iceberg. J Clin Gerontol Geriatr 5: 7–12, 2014. doi: 10.1016/j.jcgg.2013.04.002. [DOI] [Google Scholar]
- 8.Chao CT, Tsai HB, Wu CY, Lin YF, Hsu NC, Chen JS, Hung KY. Cumulative cardiovascular polypharmacy is associated with the risk of acute kidney injury in elderly patients. Medicine (Baltimore) 94: e1251, 2015. doi: 10.1097/MD.0000000000001251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen G, Bridenbaugh EA, Akintola AD, Catania JM, Vaidya VS, Bonventre JV, Dearman AC, Sampson HW, Zawieja DC, Burghardt RC, Parrish AR. Increased susceptibility of aging kidney to ischemic injury: identification of candidate genes changed during aging, but corrected by caloric restriction. Am J Physiol Renal Physiol 293: F1272–F1281, 2007. doi: 10.1152/ajprenal.00138.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, Van Lente F, Levey AS. Prevalence of chronic kidney disease in the United States. JAMA 298: 2038–2047, 2007. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 11.Cubillo A, Cornide M, López JL, Molina R, Feliu J, Espinosa E, Zamora P, de Castro J, Ordoñez A, González Barón M. Renal tolerance to cisplatin in patients 70 years and older. Am J Clin Oncol 24: 192–197, 2001. doi: 10.1097/00000421-200104000-00018. [DOI] [PubMed] [Google Scholar]
- 12.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11: 264–276, 2015. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hrushesky WJ, Shimp W, Kennedy BJ. Lack of age-dependent cisplatin nephrotoxicity. Am J Med 76: 579–584, 1984. doi: 10.1016/0002-9343(84)90280-8. [DOI] [PubMed] [Google Scholar]
- 14.Huang HF, Zhou JY, Chen JH. Biomarkers for early diagnosis of acute kidney injury: current progress and clinical prospects. Curr Protein Pept Sci 18: 1205–1210, 2017. doi: 10.2174/1389203717666160909152205. [DOI] [PubMed] [Google Scholar]
- 15.Jang HR, Park JH, Kwon GY, Park JB, Lee JE, Kim DJ, Kim YG, Kim SJ, Oh HY, Huh W. Aging has small effects on initial ischemic acute kidney injury development despite changing intrarenal immunologic micromilieu in mice. Am J Physiol Renal Physiol 310: F272–F283, 2016. doi: 10.1152/ajprenal.00217.2015. [DOI] [PubMed] [Google Scholar]
- 16.Kaddourah A, Basu RK, Bagshaw SM, Goldstein SL; AWARE Investigators . Epidemiology of acute kidney injury in critically ill children and young adults. N Engl J Med 376: 11–20, 2017. doi: 10.1056/NEJMoa1611391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katagiri D, Hamasaki Y, Doi K, Negishi K, Sugaya T, Nangaku M, Noiri E. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int 89: 374–385, 2016. doi: 10.1038/ki.2015.327. [DOI] [PubMed] [Google Scholar]
- 18.Mallappallil M, Friedman EA, Delano BG, McFarlane SI, Salifu MO. Chronic kidney disease in the elderly: evaluation and management. Clin Pract (Lond) 11: 525–535, 2014. doi: 10.2217/cpr.14.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller RA. Aging and immune function. Int Rev Cytol 124: 187–215, 1991. doi: 10.1016/S0074-7696(08)61527-2. [DOI] [PubMed] [Google Scholar]
- 20.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel) 2: 2490–2518, 2010. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.National Cancer Institute Surveillance, Epidemiology, and End Results Program. Stat Facts: Lung and Bronchus Cancer: NCI (Online) https://seer.cancer.gov/statfacts/html/lungb.html [17 December 2018].
- 22.Noordmans GA, Caputo CR, Huang Y, Sheehan SM, Bulthuis M, Heeringa P, Hillebrands JL, van Goor H, Korstanje R. Genetic analysis of mesangial matrix expansion in aging mice and identification of Far2 as a candidate gene. J Am Soc Nephrol 24: 1995–2001, 2013. doi: 10.1681/ASN.2012080838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okusa MD, Molitoris BA, Palevsky PM, Chinchilli VM, Liu KD, Cheung AK, Weisbord SD, Faubel S, Kellum JA, Wald R, Chertow GM, Levin A, Waikar SS, Murray PT, Parikh CR, Shaw AD, Go AS, Chawla LS, Kaufman JS, Devarajan P, Toto RM, Hsu CY, Greene TH, Mehta RL, Stokes JB, Thompson AM, Thompson BT, Westenfelder CS, Tumlin JA, Warnock DG, Shah SV, Xie Y, Duggan EG, Kimmel PL, Star RA. Design of clinical trials in acute kidney injury: a report from an NIDDK workshop—prevention trials. Clin J Am Soc Nephrol 7: 851–855, 2012. doi: 10.2215/CJN.12811211. [DOI] [PubMed] [Google Scholar]
- 24.Oshita F, Kurata T, Kasai T, Fakuda M, Yamamoto N, Ohe Y, Tamura T, Eguchi K, Shinkai T, Saijo N. Prospective evaluation of the feasibility of cisplatin-based chemotherapy for elderly lung cancer patients with normal organ functions. Jpn J Cancer Res 86: 1198–1202, 1995. doi: 10.1111/j.1349-7006.1995.tb03315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otto GP, Busch M, Sossdorf M, Claus RA. Impact of sepsis-associated cytokine storm on plasma NGAL during acute kidney injury in a model of polymicrobial sepsis. Crit Care 17: 419, 2013. doi: 10.1186/cc12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res Int 2014: 967826, 2014. doi: 10.1155/2014/967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007, 2008. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 28.Pascual J, Orofino L, Liaño F, Marcén R, Naya MT, Orte L, Ortuño J. Incidence and prognosis of acute renal failure in older patients. J Am Geriatr Soc 38: 25–30, 1990. doi: 10.1111/j.1532-5415.1990.tb01592.x. [DOI] [PubMed] [Google Scholar]
- 29.Perazella MA. Onco-nephrology: renal toxicities of chemotherapeutic agents. Clin J Am Soc Nephrol 7: 1713–1721, 2012. doi: 10.2215/CJN.02780312. [DOI] [PubMed] [Google Scholar]
- 30.Prakash S, O’Hare AM. Interaction of aging and chronic kidney disease. Semin Nephrol 29: 497–503, 2009. doi: 10.1016/j.semnephrol.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramesh G, Reeves WB. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110: 835–842, 2002. doi: 10.1172/JCI200215606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ravichandran K, Wang Q, Ozkok A, Jani A, Li H, He Z, Ljubanovic D, Weiser-Evans MC, Nemenoff RA, Edelstein CL. CD4 T cell knockout does not protect against kidney injury and worsens cancer. J Mol Med (Berl) 94: 443–455, 2016. doi: 10.1007/s00109-015-1366-z. [DOI] [PubMed] [Google Scholar]
- 33.Ricci Z, Cruz D, Ronco C. The RIFLE classification for acute kidney injury definition. Am J Surg 198: 152–153, 2009. doi: 10.1016/j.amjsurg.2008.06.033. [DOI] [PubMed] [Google Scholar]
- 34.Rodwell GE, Sonu R, Zahn JM, Lund J, Wilhelmy J, Wang L, Xiao W, Mindrinos M, Crane E, Segal E, Myers BD, Brooks JD, Davis RW, Higgins J, Owen AB, Kim SK. A transcriptional profile of aging in the human kidney. PLoS Biol 2: e427, 2004. doi: 10.1371/journal.pbio.0020427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmitt R, Cantley LG. The impact of aging on kidney repair. Am J Physiol Renal Physiol 294: F1265–F1272, 2008. doi: 10.1152/ajprenal.00543.2007. [DOI] [PubMed] [Google Scholar]
- 36.Sharp CN, Doll MA, Dupre TV, Shah PP, Subathra M, Siow D, Arteel GE, Megyesi J, Beverly LJ, Siskind LJ. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Renal Physiol 310: F560–F568, 2016. doi: 10.1152/ajprenal.00512.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharp CN, Siskind LJ. Developing better mouse models to study cisplatin-induced kidney injury. Am J Physiol Renal Physiol 313: F835–F841, 2017. doi: 10.1152/ajprenal.00285.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skinner R, Parry A, Price L, Cole M, Craft AW, Pearson AD. Persistent nephrotoxicity during 10-year follow-up after cisplatin or carboplatin treatment in childhood: relevance of age and dose as risk factors. Eur J Cancer 45: 3213–3219, 2009. doi: 10.1016/j.ejca.2009.06.032. [DOI] [PubMed] [Google Scholar]
- 39.Sturmlechner I, Durik M, Sieben CJ, Baker DJ, van Deursen JM. Cellular senescence in renal ageing and disease. Nat Rev Nephrol 13: 77–89, 2017. doi: 10.1038/nrneph.2016.183. [DOI] [PubMed] [Google Scholar]
- 40.Thyss A, Saudes L, Otto J, Creisson A, Gaspard MH, Dassonville O, Schneider M. Renal tolerance of cisplatin in patients more than 80 years old. J Clin Oncol 12: 2121–2125, 1994. doi: 10.1200/JCO.1994.12.10.2121. [DOI] [PubMed] [Google Scholar]
- 41.Torres R, Velazquez H, Chang JJ, Levene MJ, Moeckel G, Desir GV, Safirstein R. Three-dimensional morphology by multiphoton microscopy with clearing in a model of cisplatin-induced CKD. J Am Soc Nephrol 27: 1102–1112, 2016. doi: 10.1681/ASN.2015010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Bonventre JV, Parrish AR. The aging kidney: increased susceptibility to nephrotoxicity. Int J Mol Sci 15: 15358–15376, 2014. doi: 10.3390/ijms150915358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wen J, Zeng M, Shu Y, Guo D, Sun Y, Guo Z, Wang Y, Liu Z, Zhou H, Zhang W. Aging increases the susceptibility of cisplatin-induced nephrotoxicity. Age (Dordr) 37: 112, 2015. doi: 10.1007/s11357-015-9844-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wick G. The aging immune system: primary or secondary changes in immunity in the elderly. Wien Klin Wochenschr 109: 755–757, 1997. [PubMed] [Google Scholar]
- 45.Yuan R, Tsaih SW, Petkova SB, Marin de Evsikova C, Xing S, Marion MA, Bogue MA, Mills KD, Peters LL, Bult CJ, Rosen CJ, Sundberg JP, Harrison DE, Churchill GA, Paigen B. Aging in inbred strains of mice: study design and interim report on median lifespans and circulating IGF1 levels. Aging Cell 8: 277–287, 2009. doi: 10.1111/j.1474-9726.2009.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]