

Abstract
Identifying pathways related to renal cold storage (CS) that lead to renal damage after transplantation (Tx) will help us design novel pathway-specific therapies to improve graft outcome. Our recent report showed that mitochondrial function was compromised after CS alone, and this was exacerbated when CS was combined with Tx (CS/Tx). The goal of this study was to determine whether the proteasome exacerbates mitochondrial dysfunction after CS/Tx. We exposed the kidneys of male Lewis rats (in vivo) and rat renal proximal tubular (NRK) cells (in vitro) to CS/Tx or rewarming (CS/RW), respectively. To compare CS-induced effects, in vivo kidney Tx without CS exposure (autotransplantation; ATx) was also used. Our study provides the first evidence that the chymotrypsin-like (ChT-L) peptidase activity of the proteasome declined only after CS/Tx or CS/RW, but not after CS or ATx. Interestingly, key mitochondrial proteins involved with respiration [succinate dehydrogenase complex, subunit A (SDHA), a complex II subunit, and ATP5B, an ATP synthase/complex V subunit] were detected in the detergent-insoluble fraction after CS/Tx or CS/RW, with compromised complex V activity. Pharmacological inhibition of ChT-L activity in NRK cells decreased the activity of mitochondrial complexes I, II, and V and also increased the levels of SDHA and ATP5B in the insoluble fraction. On the other hand, inhibiting mitochondrial respiration in NRK cells with antimycin A compromised ChT-L function and increased the amounts of SDHA and ATP5B in the insoluble fraction. Our results suggest that mitochondrial respiratory dysfunction during CS precedes compromised ChT-L function after CS/Tx and proteasome dysfunction further alters mitochondrial protein homeostasis and decreases respiration in the kidneys after CS/Tx. Therefore, therapeutics that preserve mitochondrial and proteasome function during CS may provide beneficial outcomes following transplantation.
Keywords: cold storage, mitochondria, transplantation, ubiquitin-proteasome system
INTRODUCTION
End-stage kidney disease (ESKD) affects more than 661,000 Americans; with 47,000 annual deaths, this disease is the ninth leading cause of death in the United States (https://www.niddk.nih.gov/health-information/health-statistics/kidney-disease). Kidney transplantation (Tx) increases quality of life and reduces the mortality rate of people with ESKD. Due to the shortage of transplantable kidneys (94,956 waiting-list candidates vs. 8,483 transplants as of June 2018; https://www.unos.org), 7 of 10 patients with ESKD will remain on dialysis, and many will die while on the waiting list. Furthermore, the long-term function of transplanted kidneys, particularly those from deceased (brain-dead) donors, continues to be suboptimal (7, 23, 25, 44, 62, 72, 73) because of the cold-storage (CS) process that these kidneys undergo before Tx. During this process, donor kidneys are flushed with and stored in CS solutions, preferably Viaspan [University of Wisconsin (UW) solution] (7, 24, 61, 63), until a recipient is identified.
Extended CS is detrimental to graft function after Tx (7, 15, 25, 44, 62, 72, 73); however, the molecular mechanisms that cause CS-mediated damage are poorly understood. We and others have reported an increase in reactive oxygen species (ROS) and mitochondrial damage during CS (45, 46, 50, 60). Our recent reports using rat renal transplant models suggest that mitochondrial dysfunction and renal damage are exacerbated in kidney Txs that were exposed to CS compared with those without CS (54, 71). Thus it is imperative to elucidate the mechanisms of CS-mediated mitochondrial and renal damage after Tx so that new therapies can be designed to target these pathways and improve graft outcome.
Mitochondrial dysfunction depletes ATP and increases ROS, leading to protein oxidation and inducing a number of cellular pathways, including the ubiquitin-proteasome system (UPS), which plays a crucial role in degrading modified, misfolded, or damaged proteins to maintain intracellular protein homeostasis (79, 81). The 26S proteasome is composed of 20S proteasome and 19S regulatory particles, both of which participate in selectively degrading ubiquitin-tagged proteins (18, 79, 81). The 20S proteasome has three to seven protease active sites (β-catalytic subunits) that hydrolyze peptide bonds in a chymotrypsin (β5)-, trypsin (β2)-, or caspase (β1)-like fashion. Evidence from in vitro models suggests that oxidized proteins are removed by the 20S proteasome (2, 8, 13, 21, 31, 32, 69, 70). Interestingly, higher levels of or chronic exposure to ROS, commonly observed in models of aging or neurodegenerative diseases, can inhibit the proteasome, suggesting that ROS play a complex role in modulating proteasome activity. Excessive ROS generation is implicated in the pathogenesis of ischemia-reperfusion-induced renal damage (52), but no studies, to our knowledge, have considered the contribution of the proteasome to renal injury during CS plus Tx (CS/Tx).
The relationship between the UPS and mitochondrial function is intricate (40, 41, 48, 49, 77) and may include a functional interdependent relationship in renal proximal tubules. However, no studies have explored whether aberrant activation of the proteasome system can directly compromise mitochondrial protein homeostasis and function and whether mitochondrial dysfunction can alter proteasome activity in the context of Tx. The main goal of this study was to determine whether CS/Tx induces aberrant proteasome activity and whether this abnormal proteasome function contributes to altered mitochondrial protein homeostasis after CS/Tx. Here, we demonstrate for the first time that CS-induced mitochondrial dysfunction contributes to abnormal proteasome function after Tx, exacerbating mitochondrial dysfunction.
METHODS
Animals.
Male Lewis rats (200–250 g) were used as transplant donors and recipients. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Arkansas for Medical Sciences (UAMS), and all animal experiments were performed in compliance with institutional and National Institutes of Health guidelines.
Orthotopic renal transplant surgery.
Surgeries were performed as described previously (54, 71). For donor surgeries, rats were anesthetized with isoflurane, and the left and right kidneys were removed and flushed with and stored in UW solution at 4°C for 18 h. The right kidneys of donor rats were referred to as the CS group (n = 8). For recipient surgeries, rats were anesthetized with isoflurane, the native left kidney was removed, and the donor left kidney (exposed to CS) was transplanted by end-to-end anastomosis (54, 71). The surgical ischemia time was less than 45 min. The native right kidney was immediately removed so that renal function was entirely dependent on the transplanted left kidney. The ureter was anastomosed end-to-end over a 5-mm PE-50 polyethylene stent. Postoperatively, animals were given 0.9% (wt/vol) NaCl in the abdominal cavity and placed under a heating lamp to recover from the anesthesia; they were given buprenorphine (0.06 mg/kg sc) for pain. After 24 h of reperfusion, the transplanted left kidney and blood were collected under anesthesia and saved as the 18-h CS/Tx group (n = 8). The survival rate 1 day after surgery was >95% for the CS/Tx groups.
Autotransplant surgery.
Autotransplant (ATx) surgery was included in these studies so that the impact of CS could be isolated from the impact of transplant surgery alone. ATx was performed as described for orthotopic renal transplant surgeries, except that the left kidney was removed, flushed with saline, and immediately transplanted back into the same rat without CS exposure; a right nephrectomy followed (54, 71). After 24 h, the transplanted kidney was harvested under anesthesia; these kidneys were referred to as the ATx group (n = 8).
Sham surgery.
Rats underwent the same procedure for right nephrectomy but without renal Tx (sham operation); the right kidney was saved as a control kidney (n = 8). The left kidney and blood were harvested 24 h later and saved as the sham group (n = 8). Kidneys were immediately flash-frozen for biochemistry assays and Western blotting.
Cell culture and treatment.
Normal rat kidney proximal tubular cell line cultures (NRK-52E; American Type Culture Collection CRL-1571) were maintained in warm growth medium (DMEM plus 5% fetal calf serum and 1% penicillin/streptavidin), and treatment was performed as follows. For CS plus rewarming (CS/RW) treatment, NRK cells were exposed to cold storage solution (UW or Viaspan solution) as reported previously (46). Briefly, NRK cells were washed two times with cold PBS and incubated with cold UW solution (4°C) for 18 h (CS condition). Rewarming/reperfusion was initiated by washing cells three times with cold PBS and then adding cold growth medium; finally, cells were incubated at 37°C for 6 h. Cell morphology images were collected with an EVOS XL Core (Life Technologies, Waltham, MA) microscope (×20). For bortezomib (BTZ) treatment, NRK cells were treated with BTZ (10 nM for 24 h) in normal growth medium. NRK cells treated with the same concentration of DMSO (no BTZ) were used as the vehicle control. For antimycin A treatment, growth medium was removed, NRK cells were washed with warm PBS, treated with the antimycin A (5 or 10 nM) for 30 min in serum- and glucose-free growth medium, and washed and cultured in glucose-free medium for 3.5 h. NRK cells treated with the same concentration of ethanol (no antimycin A) were used as the vehicle control.
Kidney morphology based on periodic acid-Schiff staining.
Paraffin sections (5 µm) from control, CS, sham, ATx, and CS/Tx kidneys were assessed for tissue injury using the periodic acid-Schiff (PAS) reaction as described previously (52). Analyses were conducted in a blinded fashion based on the following criteria: tubular and glomerular necrosis, loss of tubular brush border, interstitial edema, tubular degeneration, and casts in lumen. All parameters culminating in a tubular injury score were graded as 0, no lesion; 1, minimal change (sporadic tubular damage); 2, moderate change (small group of tubular damage); 3, severe change (confluent tubular damage but limited to outer stripe of outer medulla); 4, very severe change (confluent tubular damage, extending to inner cortex); or 5, excessive change (necrosis of tubules, glomeruli, and blood vessels). Comparisons were made between the untreated control, CS, sham, ATx, and CS/Tx groups. All images were taken with a Nikon Eclipse TE2000-U microscope and Nikon Elements software.
Chymotrypsin-like proteasome function.
Chymotrypsin-like (ChT-L) peptidase activity was measured in renal tissue or NRK cell extracts by hydrolysis of its fluorogenic peptide substrate Suc-LLVY-7-amino-4-methylcoumarin (specific for β5/PSMB5). Kidney tissue extracts were prepared with proteasome extraction buffer (50 mM Tris·HCl, pH 7.5; 250 mM sucrose; 5 mM MgCl2; 1 mM EDTA; 1 mM DTT; 0.025% digitonin) (37). NRK cell extracts were prepared by scraping cells into the proteasome extraction buffer and passing the cells through a 27-gauge needle five times, followed by 20 min of centrifugation at 16,000 g at 4°C. Renal tissue (12.5 µg) or NRK cell extracts (5 µg) were diluted in proteasome assay buffer (50 mM Tris·HCl, pH 7.5; 40 mM KCl; 5 mM MgCl2; 1 mM DTT; 0.5 mg/ml BSA) with or without epoxomicin (20 µM) and incubated with the peptide substrate (Suc-LLVY-AMC; 100 µM for renal tissue extract and 50 µm for NRK cell extract). After 30 min of incubation at 37°C, fluorescence was measured (excitation, 380 nm; emission, 460 nm) with an F-2500 fluorescence spectrophotometer (Hitachi). Because our CS models exhibit low ATP levels (71), the assay was performed without exogenous ATP in the assay buffer.
High-resolution respirometry of NRK cells.
The activity of mitochondrial respiratory complexes in digitonin-permeabilized NRK cells was measured with high-resolution respirometry (Oxygraph-2k, Oroboros Instruments, Innsbruck, Austria) according to the substrate-inhibitor-titration protocol previously described (50, 55, 56). Briefly, NRK cells (5 × 106) were incubated for 10 min with digitonin (50 µg/ml) prepared in MiRO5 buffer (60 mM K-lactobionate; 0.5 mM EDTA; 3 mM MgCl2; 20 mM taurine; 10 mM KH2PO4; 20 mM HEPES; 110 mM BSA; 1 g/l sucrose, pH 7.0) under nutating conditions, followed by three washes with MiRO5 buffer. Mitochondrial respiration was initiated by adding 2 mM malate and 10 mM glutamate, and maximum active respiration was achieved by adding 5 mM ADP. Rotenone (0.5 µM) was then added to completely inhibit complex I respiration. To measure complex II and complex III respiration, 10 mM succinate was added, followed by 2 mM malonic acid to inhibit complex II respiration (complex II activity) and 5 µM antimycin A to inhibit complex III respiration (complex III activity). Inhibitor concentrations were selected based on experimental determinations of doses needed to maximally reduce substrate-induced respiration. Finally, data analysis was performed with DATLAB 4.2 software (Oroboros), and cellular respiration was expressed as oxygen flux (pmol·mg−1·s−1).
Mitochondrial ATP synthase (complex V) activity assay.
The ATPase activity of complex V was measured in total renal extracts using a commercially available MitoCheck Complex V Activity Assay kit (Cayman, Ann Arbor, MI). Briefly, renal extracts were prepared by using dounce homogenization of kidney tissues (flash frozen) or NRK cells in a sucrose-based buffer (250 mM sucrose; 20 mM HEPES; 10 mM KCl; 1.5 mM MgCl2; 1 mM EDTA; 1 mM PMSF; 1 mM DTT; pH 7.5), followed by 10 min of centrifugation at 10,000 g at 4°C. Ten micrograms of the renal extract (in the presence or absence of oligomycin (20 μM) to inhibit ATP synthase) was used to monitor kinetic rate of oligomycin-sensitive ADP-dependent NADH oxidation to NAD at 340 nm for up to 30 min (using a BioTek Gen5 microplate reader; BioTek, Winooski, VT), according to the manufacturer’s instructions. The rate of complex V ATPase activity was determined with the formula: ATPase activity rate·min−1·mg−1 = [(slope/min)/10] × 1,000.
Renal extract preparation for Western blotting.
Renal extracts from whole-kidney homogenates and NRK cells were prepared with 1% Triton X-100 lysis buffer (53) [1 mM PMSF; 1.2 mM Na3VO4; 2.5 mM NaF; and 1 mM DTT (Sigma-Aldrich); and protease inhibitor cocktail (Pierce)]. After lysis, the extracts were centrifuged (16,000 g for 20 min at 4°C), and the supernatant was saved as the soluble fraction. The pellet was resuspended in lysis buffer (8 M urea; 3% SDS; and 1% Triton X-100) and exposed to three freeze-thaw cycles, followed by sonication on ice (3 cycles of 10-s sonication plus 10-s pause). The sonicated extract was centrifuged (16,000 g for 20 min at 4°C), and the resulting supernatant was collected and labeled as the insoluble fraction. To obtain the total whole-cell protein extract, the renal tissue homogenate or cells were boiled with lysis buffer (8 M urea; 3% SDS; and 1% RIPA) and centrifuged (16,000 g for 20 min at 4°C). Protein concentrations were determined with the BCA Protein Assay kit (Pierce). Renal extracts (20 µg cells, 30 µg tissues; soluble or insoluble fraction) were separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were incubated with antibodies to succinate dehydrogenase (SDHA; 1:1,000; 459200, Invitrogen), ATP5B (1:1,000; A21351, Invitrogen), or β-actin (loading control, 1:1,000; A5441, Sigma-Aldrich). Probed membranes were washed three times, incubated with horseradish peroxidase-conjugated secondary antibodies (1:30,000; Seracare KPL), and assayed for enhanced chemiluminescence (ThermoFisher Scientific). Densitometry was performed with AlphaEase FC software (Alpha Innotech).
Native gel Western blot analysis.
Renal extracts from whole-kidney homogenates were prepared with 0.9% digitonin lysis buffer (50 mM Tris·HCl, pH = 7.5; 250 mM sucrose; 1 mM EDTA; 5 mM MgCl2; 1 mM DTT; 0.9% digitonin; 1.2 mM sodium orthovanadate; 2.5 mM NaF; and Halt Proteasome Inhibitor Cocktail, 78430, ThermoFisher Scientific). The homogenate was mixed with lysis buffer at a 1:1 ratio and then incubated on ice for 20 min with occasional mixing. The lysate was centrifuged (16,000 g for 20 min at 4°C), and the supernatant was collected and saved as the digitonin extract. Protein concentrations were determined with Coomassie Plus Protein Assay Reagent (Pierce). Renal extracts (20 µg) were resolved onto a Bis-Tris (4–12%) gel and transferred to a PVDF membrane (54). Western blot analysis was performed with antibodies for PSMB5/β5 (1:1.000; ab3330, Abcam), SDHA (1:1,000; 459200, Invitrogen, ATP5B (1:1,000; A21351, Invitrogen), and NDUFS3 (1:1,000; ab110246, Abcam). Chemiluminescence-based detection and densitometry were performed as described above.
Statistical analysis.
Results are presented as means ± SE (GraphPad Prism software). Data were analyzed with a one-way ANOVA and Tukey’s post hoc test for multiple group comparisons, and an unpaired Student's t-test was used when differences between the means of two groups (Control vs. CS) at a 95% level of confidence were compared. Differences with P < 0.05 were considered statistically significant. Control kidneys were compared with CS kidneys because both groups were harvested from healthy rats. Sham kidneys served as controls for both transplant models (ATx and CS/Tx) because all underwent a nephrectomy (removal of right kidney).
RESULTS
Proteasome function is compromised after CS/RW or CS/Tx.
We recently reported that CS increases mitochondrial dysfunction in renal tissues, and this dysfunction increased after cold-stored kidneys were transplanted (54, 71). This prompted us to determine whether renal CS also alters the function of the proteasome. In our renal models (in vivo and in vitro), we used the fluorogenic peptide substrate Suc-LLVY-AMC to investigate peptidase activity from the proteasome. The CS condition had no effect on ChT-L peptidase activity in NRK cells (in vitro, Fig. 1A) or in rat kidney lysates (ex vivo, Fig. 1B). Interestingly, ChT-L activity was significantly lower in NRK CS/RW cells than in untreated (control) NRK cells (Fig. 1A). Similarly, ChT-L activity was significantly lower in kidneys exposed to CS/Tx than in sham kidneys or ATx kidneys (i.e., transplanted without CS) (Fig. 1B). Trypsin- and caspase-like activities, assayed with specific fluorogenic peptide substrates, did not differ among the groups (data not shown). To determine whether this decrease in ChT-L activity occurred because the amount of enzyme decreased, we performed Western blotting with a nondenaturing gel (nonreducing condition). We did not observe any changes in the β5/PSMB5 subunit in fully assembled 26S or 20S proteasome complexes after CS/Tx compared with sham or ATx kidneys (Fig. 1C, arrows), suggesting that the decline in activity was not due to a decline in enzyme.
Fig. 1.
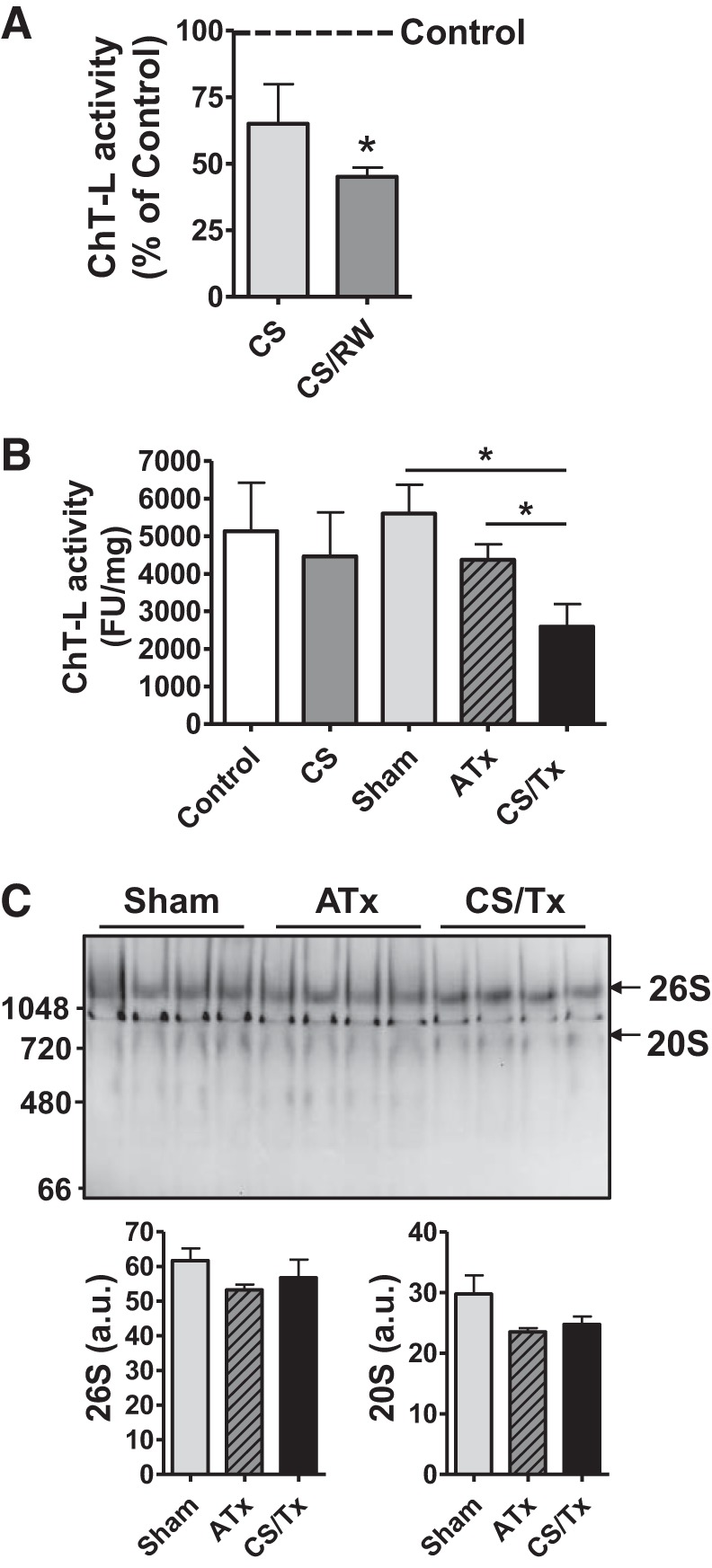
Proteasome function declines after cold storage plus rewarming (CS/RW) or transplantation (CS/Tx). A: proteasome-based chymotrypsin-like (ChT-L) peptidase activity was evaluated in NRK cells after CS (18-h) or 18-h CS plus 6-h RW (CS/RW). Cell extracts were prepared and incubated for 30 min with the fluorogenic peptide substrate Suc-LLVY-AMC (to assess ChT-L peptidase activity) in the presence or absence of epoxomicin. Epoxomicin-inhibitable catalytic activity in renal extracts was determined by measuring fluorescence with a fluorometer (380-nm excitation; 440-nm emission). Results are representative of 3 independent analyses for each group. Values were expressed as percent of control, presented as means ± SE (bars) of 3 independent experiments. Differences between the means of the groups were compared with a 1-way ANOVA followed by Tukey’s post hoc test for multiple group comparisons (control, CS, and CS/RW). *Means are significantly different (P < 0.05) between untreated (control) and CS/RW. B: ChT-L peptidase activity in rat kidneys. Five experimental groups were considered: 1) control (untreated), 2) CS (18-h CS), 3) sham (right nephrectomy with 1-day reperfusion), 4) CS/Tx (18-h CS plus Tx with 1-day reperfusion), and 5) autotransplantation (ATx; Tx with no CS plus 1-day reperfusion). Renal extracts were prepared from kidney homogenates, and ChT-L peptidase activity was evaluated as in A. Results are expressed as means ± SE (bars; a.u., arbitrary units) and are representative of 7 independent analyses for each group. Differences between the means of the groups were compared with a 1-way ANOVA followed by Tukey’s post hoc test for multiple group comparisons (control, CS, sham, ATx, and CS/Tx). *Means are significantly different (P < 0.05) between sham and CS/Tx or between ATx and CS/Tx. C: native Western blot of 26S and 20S proteasomes in digitonin extracts of kidney homogenates from sham, ATx, and CS/Tx groups; 20 µg protein/well was loaded in a Bis-Tris gel under nonreducing conditions followed by Western blotting with PSMB5/β5 antibody. Arrows indicate molecular weights of 26S/20S native proteasomes. Densitometric values are expressed as means ± SE (bars; a.u., arbitrary units); results are representative of n = 4 rats/group. Differences between means were compared with a 1-way ANOVA for multiple group comparisons (sham, ATx, and CS/Tx).
CS/RW or CS/Tx induces severe renal damage.
Previously, we published that 24 h of CS followed by 6 h of rewarming leads to 40% NRK cell death (46). In the current study, we found that 18-h CS plus 6-h rewarming led to ~24% NRK cell death. After CS exposure (18 h), NRK cells exhibited a rounded and constricted cell morphology (Fig. 2A, magnified inset). Similarly, CS/RW significantly altered cell morphology, as indicated by rounded, stretched, or swollen cells (Fig. 2A, magnified inset).
Fig. 2.

Cold storage followed by rewarming (CS/RW) or transplantation (CS/Tx) induces renal injury. A: NRK cell morphology was assessed after CS (18-h) or CS followed by rewarming (6-h). Untreated cells were used as a control. Images were taken with an EVOS microscope; ×200 magnification. Representative images (n = 4) are shown with zoomed inset. Scale bar indicates 1 mm. B: renal injury was assessed in periodic acid-Schiff (PAS)-stained renal sections after cold storage plus transplantation (CS/Tx). Representative micrographs (×300) of the PAS staining of renal cortex and medulla regions after control (untreated), cold storage (CS; 18-h), sham (1-day reperfusion post-right nephrectomy), ATx (no CS; 1-day reperfusion posttransplant), or CS/Tx (18-h CS plus 1-day reperfusion posttransplant). CS/Tx sections showed distorted tubular/glomerular morphology (thick arrow, necrosis; thin arrow, brush border loss; double thin arrow, casts in lumen; arrowhead, cell swelling). Scale bar indicates 50 μm. Graphs show pathology scoring for cumulative tubular injury score. Data are means ± SE (n = 6 for all conditions; P < 0.05 considered significant).
Given our prior observations of extensive graft dysfunction after CS/Tx (54), further studies used PAS staining of renal sections to assess renal tubular and glomerular damage after CS/Tx. We found that CS alone induced mild changes within the kidneys, which was evident by brush border loss (thin arrow), tubular degeneration/necrosis (thick arrow), and cell swelling (arrowhead) (Fig. 2B). As anticipated, ATx induced sporadic tubular damage as indicated by tubular necrosis (thick arrow), brush border loss (thin arrow), cell swelling (arrowhead), and casts (double thin arrow) in the lumen (Fig. 2B). Interestingly, a greater extent of renal damage was noted after CS/Tx than after CS alone or ATx (Fig. 2B). CS/Tx induced profound renal damage, as indicated by severe necrosis (thick arrow), massive brush border loss (thin arrow), cell swelling (arrowhead), and substantial casts formation (double thin arrow) (Fig. 2B). Semiquantitative scoring of PAS-stained sections (see methods) indicated significant overall renal damage in CS/Tx kidneys compared with sham or ATx (Fig. 2B).
CS/RW or CS/Tx shifts key mitochondrial respiratory proteins to the insoluble fraction.
We recently demonstrated that the function of mitochondrial respiratory complexes I–III is compromised after CS/Tx and key mitochondrial proteins involved in fission and fusion are altered after CS/Tx (54). Here, we attempted to evaluate the expression of other mitochondrial proteins, specifically those involved in respiration, using our in vitro and in vivo renal CS models. Because we found that CS/Tx compromised the proteasome function (ChT-L; Fig. 1) and mitochondrial respiration (54), we used Western blotting to evaluate the effects of CS/Tx or CS/RW on the solubility of respiratory complex proteins as an indicator of altered mitochondrial protein homeostasis. We solubilized NRK cells or kidney homogenates with 1% Triton X-100 to extract respiratory-complex membrane proteins out of potentially aggregated membrane proteins present in the Triton-insoluble fraction. NRK cell extracts (soluble and insoluble) obtained after CS or CS/RW were evaluated for the expression of respiratory complex subunits. Western blots of soluble NRK cell extracts indicated no change in the levels of SDHA (complex II subunit) and ATP5B (ATP synthase/complex V subunit) after CS or CS/RW (Fig. 3A). Furthermore, levels of NDUFS3 (complex I subunit), Core 2 (complex III subunit), and COXI (complex IV subunit) were unaltered after CS or CS/RW (data not shown). However, CS/RW increased the abundance of SDHA and ATP5B proteins within the insoluble fraction (Fig. 3B). In addition, the Western blot data using NRK cell total extract (prepared after boiling with SDS/urea/RIPA buffer) did not show any changes in total ATP5B or SDHA protein levels among all conditions (Fig. 3C), suggesting that these proteins may have been altered and impacted the function of mitochondrial complexes.
Fig. 3.

Cold storage followed by rewarming (CS/RW) impairs mitochondrial respiratory complex protein homeostasis in NRK cells. Western blot of succinate dehydrogenase (SDHA)4 and ATP5B from NRK cells exposed to 18-h of CS or 18-h CS plus 6-h of rewarming (CS/RW). A: Triton-soluble fraction. B: Triton-insoluble fraction. C: total extracts. Representative blots of 3 experiments are shown. Densitometry values were normalized to β-actin loading control and expressed as mean density ± SE (bars; a.u., arbitrary units) of 3 independent experiments. Differences between the means were compared with a 1-way ANOVA and Tukey’s post hoc tests. *Means are significantly different (P < 0.05) compared with untreated control or CS. Con, control; CS, cold storage; RW, rewarming.
We also evaluated the levels of respiratory complex subunits within kidney extracts (soluble and insoluble) with Western blotting. In contrast to the results with NRK cells exposed to CS/RW, soluble fractions of kidney extracts had less SDHA and ATP5B after CS/Tx than did sham or ATx kidneys (Fig. 4A). Excitingly, there was significantly more SDHA and ATP5B in the insoluble fraction after CS/Tx (Fig. 4B); this was similar to the CS/RW treatment for NRK cells. Levels of NDUFS3, Core 2, and COXI in the soluble and insoluble fractions were unchanged in all groups (data not shown). Of note, we reported previously that Core 2 abundance did not change after CS/Tx (54). Similarly, we did not observe any changes in total levels of ATP5B or SDHA with Western blotting (Fig. 4C), suggesting that these proteins were likely altered (conformation change or misfolded) by CS/Tx, and therefore shifted to the detergent-insoluble fraction.
Fig. 4.
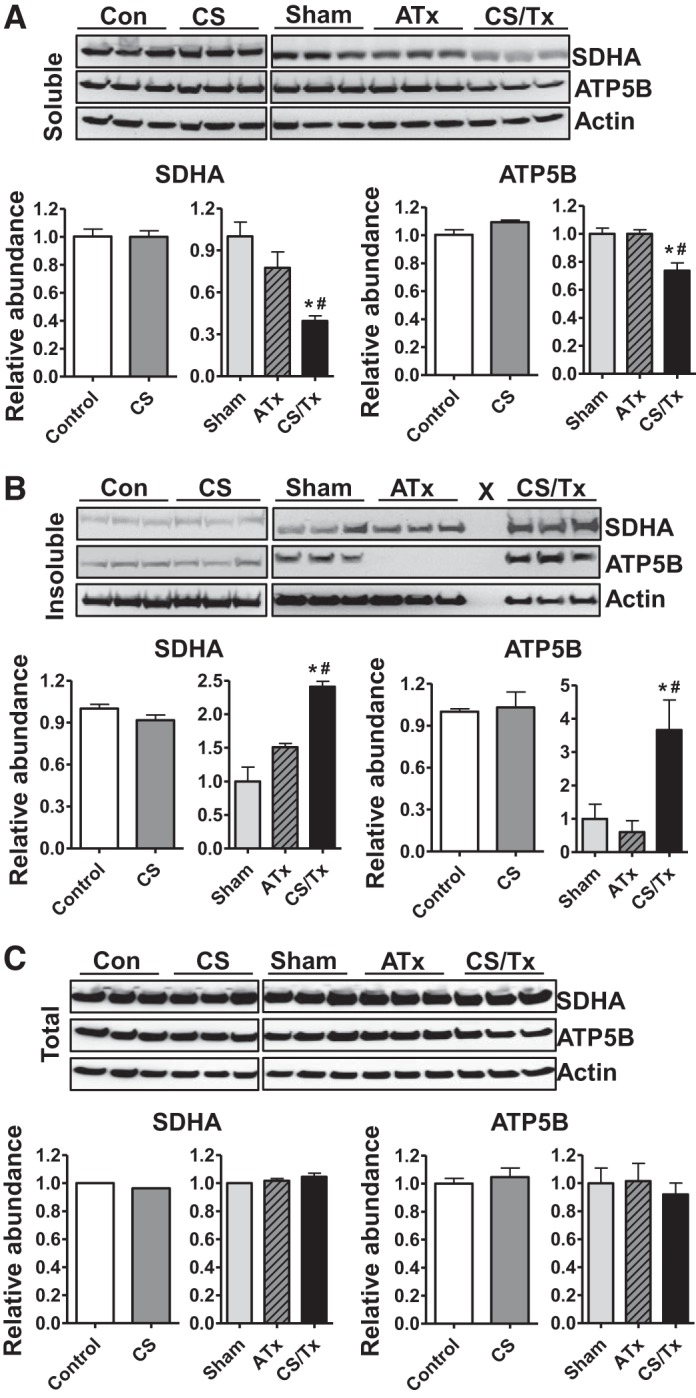
Renal cold storage followed by transplantation (CS/Tx) alters mitochondrial respiratory complex protein homeostasis in rat kidneys. A: Triton-soluble fraction. B: Triton-insoluble fraction. C: total extracts were prepared from rat kidney homogenates from control (untreated), cold storage (CS; 18-h), sham (1-day reperfusion post-right nephrectomy), ATx (no CS; 1-day reperfusion posttransplant), or CS/Tx (18-h CS plus 1-day reperfusion posttransplant). Renal extracts (30 µg) from Triton-soluble and Triton-insoluble fractions and total extracts were resolved on SDS-PAGE gel followed by Western blotting for SDHA and ATP5B. Representative blots of 3 experiments are shown. X in B indicates empty lane. Densitometry values were normalized to the β-actin loading control and are expressed as relative abundance of control (for CS) or sham (for ATx and CS/Tx; bar graphs, n = 3). Differences between groups were compared with a Student’s t-test (control vs. CS) or 1-way ANOVA with Tukey’s post hoc test for multiple group comparisons (sham, ATx, and CS/Tx). *Means are significantly different (P < 0.05) between sham and CS/Tx. #Means are significantly different (P < 0.05) between ATx and CS/Tx.
CS/Tx compromises mitochondrial ATP synthase ATPase activity.
Because we observed increased levels of ATP5B in the Triton-insoluble fraction, we were prompted to evaluate the functional status of mitochondrial ATP synthase (complex V). To our knowledge, no ATP synthase activity assays are commercially available. Thus we used a commercial kit (see methods) that measures the ATPase activity of ATP synthase. This kit is based on assaying the conversion of ATP to ADP by ATPase of the ATP synthase, which is then coupled to the oxidation reaction of NADH to NAD with a reduction in absorbance at 340 nm. Indeed, the rate of ATPase activity (oligomycin-inhibitable) was significantly compromised after CS/Tx compared with sham or ATx kidneys (Fig. 5).
Fig. 5.
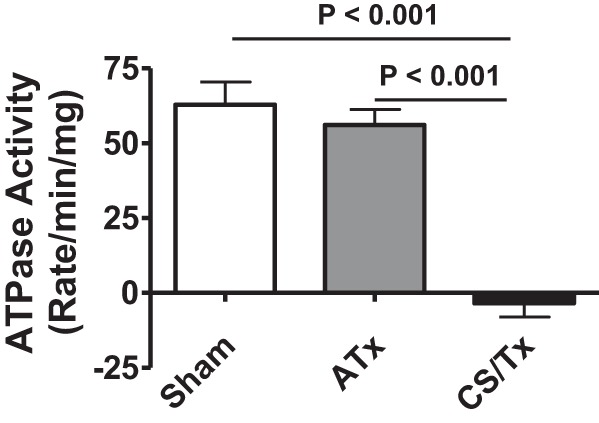
Renal cold storage followed by transplantation (CS/Tx) reduces the ATPase activity of mitochondrial complex V (ATP synthase). Renal tissue extracts were prepared by dounce-homogenizing of rat kidney homogenates from sham (1-day reperfusion post-right nephrectomy), ATx (no CS; 1-day reperfusion post-transplant), or CS/Tx (18-h CS plus 1-day reperfusion post-transplant) (see methods). Renal extracts (10 μg) with or without oligomycin (20 μM) were incubated with the enzyme mix in the presence of ATP and NADPH (MitoCheck Complex V Activity Assay kit, Cayman). The kinetic rate of NADH oxidation was monitored at 340 nm for up to 30 min with a BioTek Gen5 microplate reader. The graph shows the oligomycin-sensitive ATPase activity expressed as means ± SE (n = 3). Differences between group means were compared with a 1-way ANOVA and Tukey’s post hoc test. P < 0.05 was considered significant.
Inhibiting proteasomal ChT-L activity induces mitochondrial respiratory dysfunction.
The ChT-L activity of the proteasome can be inhibited with pharmacological agents that target the β5-catalytic subunits (38). BTZ, a clinically relevant reversible proteasome inhibitor, is highly effective at inhibiting ChT-L activity. To investigate the relationship between proteasome activity and mitochondrial respiratory function, we treated NRK cells (without CS) with BTZ (10 nM, 24 h) and used high-resolution respirometry to assay the activity of mitochondrial complexes I–III. Interestingly, the activity of mitochondrial complexes I and II decreased after exposure to BTZ (Fig. 6A). Similarly, BTZ compromised the ATPase activity of ATP synthase (Fig. 6B).
Fig. 6.
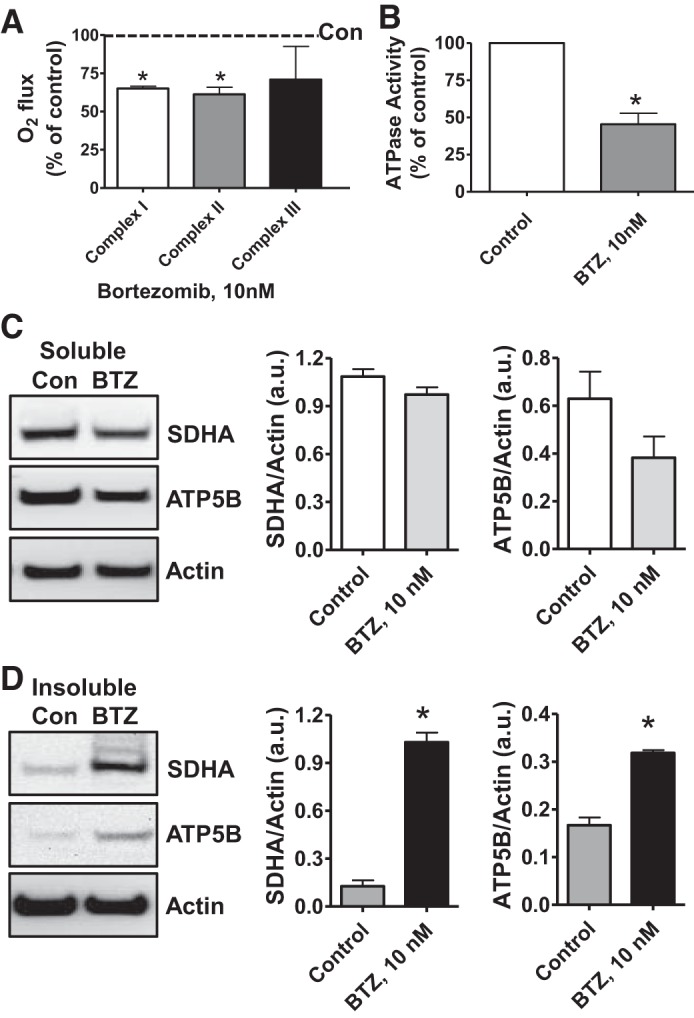
Proteasome inhibition decreases mitochondrial respiration and impairs mitochondrial respiratory complex protein homeostasis in NRK cells. A: digitonin-permeabilized NRK cells (5 × 106) were treated with vehicle control (DMSO) or the ChT-L proteasome inhibitor bortezomib (BTZ; 10 nM) for 24 h. High-resolution respirometry was used to determine oxygen flux through complexes I, II, and III. Values are expressed as % of control of 4 independent experiments. Differences between groups were compared with an unpaired Student’s t-test. *Means are significantly different (P < 0.05) compared with control. B: ATPase activity of ATP synthase (complex V) was assayed in NRK cells treated with DMSO (vehicle control) or BTZ for 24 h. Renal cell extracts (10 μg) with or without oligomycin (20 μM) were incubated with the enzyme mix in the presence of ATP and NADPH (MitoCheck Complex V Activity Assay kit, Cayman). NADH oxidation rate was monitored at 340 nm for up to 30 min with a BioTek Gen5 microplate reader. Bar graph shows oligomycin-inhibitable ATPase activity expressed as % of control (n = 3). Differences between groups were compared with a Student’s t-test. *Means are significantly different (P < 0.001). C and D: Western blots of SDHA and ATP5B in soluble (C) and insoluble fractions (D) of NRK cell extracts after treatment with vehicle control (Con; DMSO) or BTZ (10 nM) for 24 h. Representative blots of 4 independent experiments are shown. Densitometry values (normalized to β-actin loading control) are means ± SE (bars). Differences between groups were compared with an unpaired Student’s t-test. *Means are significantly different (P < 0.05) compared with respective control (Con).
Inhibiting ChT-L activity impairs the degradation of key mitochondrial respiratory proteins.
Inhibiting the proteasome with BTZ is known to increase the accumulation of damaged proteins. Because we found that BTZ altered mitochondrial respiration in NRK cells (Fig. 6, A and B), we used Western blotting to evaluate the effects of BTZ (10 nM, 24 h) on the solubility of respiratory complex proteins as an indicator of altered mitochondrial protein homeostasis. The levels of soluble SDHA and ATP5B were not affected by BTZ treatment (Fig. 6C); however, BTZ increased the levels of insoluble SDHA and ATP5B (Fig. 6D). These results are similar to those for NRK cells under CS/RW (Fig. 3B). Furthermore, BTZ did not affect the levels of soluble or insoluble NDUFS3, Core 2, or COXI (data not shown).
Inhibiting mitochondrial respiration compromises the ChT-L activity of proteasomes.
We previously reported that the function of mitochondrial respiratory complexes I and II in rat kidneys was impaired after 18 h of CS alone, and the function of complexes I–III declined after 18 h of CS combined with transplantation (CS/Tx) (54). Here, we show that proteasome function was compromised only after CS/Tx or CS/RW (Fig. 1, A and B). Because BTZ-mediated inhibition of proteasomal ChT-L activity increased the levels of SDHA and ATP5B respiratory complex subunits in the insoluble fraction, we sought to determine whether mitochondrial dysfunction also alters proteasome activity in renal tubule cells. We treated NRK cells with antimycin A, a potent inhibitor of mitochondrial respiration that depletes ATP, and evaluated proteasome (ChT-L) function. Antimycin A (10 nM, 4 h) significantly decreased ChT-L peptidase activity (Fig. 7A). As with CS/RW (Fig. 3A) or BTZ treatment (Fig. 6A), antimycin A did not alter the levels of SDHA and ATP5B in the soluble fraction, as determined with Western blotting (Fig. 7B); instead, SDHA and ATP5B accumulated in the insoluble fraction (Fig. 7C). Consistent across all conditions, the levels of NDUFS3, Core2, and COXI subunits were not altered in the soluble or insoluble fractions.
Fig. 7.
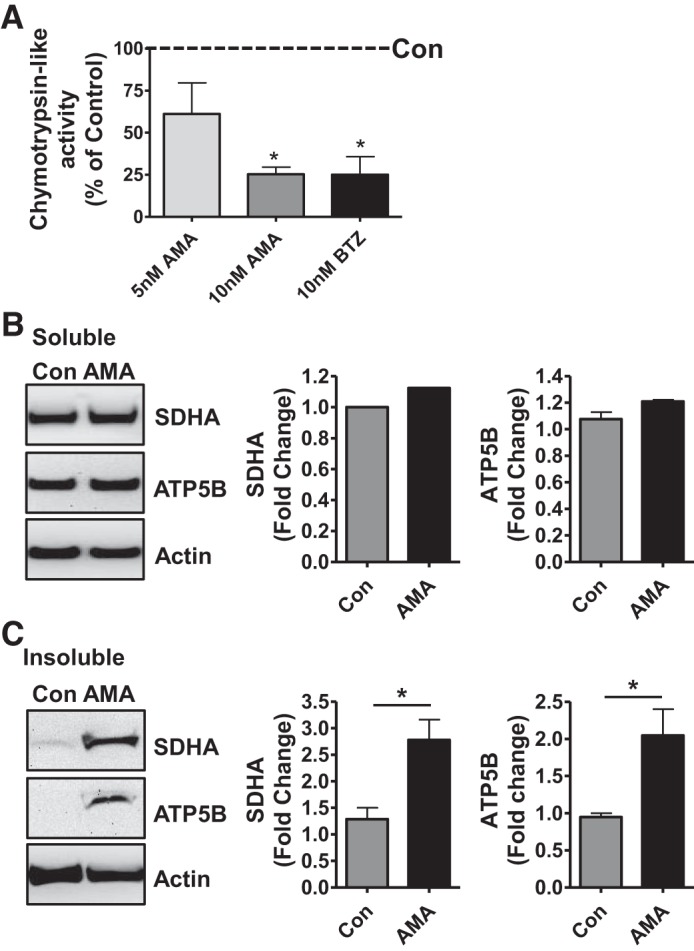
Antimycin A compromises chymotrypsin-like (ChT-L) proteasome function and mitochondrial respiratory complex homeostasis. A: ChT-L activity in extracts of NRK cells treated with antimycin A (5 or 10 nM) for 0.5 h in glucose-free medium (ATP-depletion model) followed by culturing of the cells in glucose-free medium for additional 3.5 h (see methods). Ethanol-treated NRK cells were the vehicle control; BTZ-treated NRK cells were the positive control. Values (% of control) are the mean ± SE (bars) of 3 independent experiments. Differences between groups were compared by one-way ANOVA and Tukey’s post hoc test for multiple group comparisons. *Means are significantly different (P < 0.05) compared with vehicle-treated control. B and C: Western blots of extracts of NRK cells treated with antimycin A (10 nM) as described in A. SDHA and ATP5B in soluble (B) and insoluble fractions (C) of extracts of NRK cells treated with antimycin A. Ethanol-treated NRK cells were the vehicle control (Con). Representative blots of 5 independent experiments are shown. Densitometry values were normalized to β-actin loading control. Fold-change was based on the mean of the untreated control and expressed as means ± SE (bars). Differences between groups were compared with an unpaired Student’s t-test. *Means are significantly different (P < 0.05) compared with respective control (Con).
DISCUSSION
Here, we demonstrated for the first time that the ChT-L peptidase activity of the proteasome is significantly compromised after renal CS/Tx (Fig. 1), and this is in agreement with our previous report of severe renal dysfunction in this model (54, 71). Interestingly, ChT-L peptidase activity remained unaltered after CS or ATx (Tx with no CS), which suggests that the CS condition may have primed renal tissues for the alteration of ChT-L activity in the proteasome. In contrast, studies of the heart and liver showed that inhibiting proteasome function during CS actually improves organ function after transplantation (4, 19, 43, 84); this suggests that proteasome activity during heart and liver CS may contribute to organ damage after transplantation. However, this does not appear to be the case with renal CS/Tx or CS/RW. Our observation is also supported by a report that pharmacological inhibition of ChT-L activity in the proteasome during warm ischemia-reperfusion injury could aggravate renal damage (29).
In an experimental model of Parkinson’s disease, the simultaneous inhibition of the proteasome and mitochondrial complex I produces severe neuronal toxicity (26). We propose that impaired mitochondrial respiratory function [complex I–III (54) and V (Fig. 5)] and proteasome function (Fig. 1) resulting from CS/Tx exacerbates renal damage (Fig. 2B) and dysfunction in this model (54). In addition, mitochondrial respiratory function (complex I and II activities) was impaired by renal CS alone (54), clearly suggesting that mitochondrial respiratory dysfunction precedes proteasome dysfunction in the CS/Tx model. Another key finding from the current study is that proteasome-based ChT-L activity appears to be necessary to maintain the integrity of renal SDHA (subunit of complex II) and ATP5B (subunit of ATP synthase). Interestingly, all treatments (CS/Tx, CS/RW, BTZ, and antimycin A) compromised renal proteasome function and increased the accumulation of SDHA and ATP5B in the detergent-insoluble fraction. These two respiratory-complex subunits are encoded by the nuclear genome and processed in the cytosol as preproteins before being transported into mitochondria. In general, a functional UPS is required to maintain the integrity of many mitochondrial outer membrane proteins, specifically those involved with mitochondrial fission and fusion (75, 76, 82, 83). For inner membrane (including respiratory complexes) or matrix proteins, such maintenance is primarily carried out by mitochondrial proteases (6, 58, 66) or the retrospective translocation of damaged mitochondrial proteins to the outer membrane, where they are cleared by the UPS (75, 82). The current study provides evidence that compromised ChT-L activity directly impacts the homeostasis of SDHA and ATP5B subunits, suggesting a possible mechanism by which respiratory complex and renal dysfunction are exacerbated during renal CS/Tx.
Triton, a non-ionic (mild) detergent, at low concentration has been widely used to solubilize mitochondrial complexes in its native form (3, 17, 64) and to make cellular fractionation (39). In contrast, RIPA buffer denatures the protein and, in the presence of SDS and urea, helps extract total proteins, including that with altered conformation. We opted to use Triton solubility as a measure of altered conformation of these multimeric mitochondrial complexes (22). In particular, a shift from Triton-soluble to -insoluble fraction without changing the amount of total ATP5B and SDHA protein levels after CS/Tx implies that these subunits lost their protein conformation or folding. In the context of our CS/Tx model, we showed previously that the activity of mitochondrial complexes I, II, and III decrease after renal CS/Tx (54). We now have data showing that the ATPase activity of complex V is also compromised after CS/Tx (Fig. 5), which lends support to the notion that localization to the insoluble fraction equates with loss of function. Future studies are warranted to evaluate posttranslational modifications to these subunits that may provide further insight with regard to precise mechanisms. Numerous disease models, including neurodegeneration and aging, exhibit compromised proteasome function and elevated levels of protein aggregates within the insoluble fraction (5, 36, 67, 68, 80). Furthermore, these insoluble aggregates are suggested to enhance the pathology of these disease models. We also propose that ATP5B and SDHA proteins within the insoluble fraction represent altered or modified proteins that accumulate following proteasome dysfunction in the CS/Tx model.
Chen et al. (9) suggested that the relocation of lysosomal chymotrypsin to the cytosol induces Drp1-mediated mitochondrial fission in apoptotic cells. This is interesting because our recent report demonstrated a significant loss of fission/fusion molecules with disrupted mitochondrial dynamics after CS/Tx (54). Interestingly, the chymotrypsin-like activity of an NRK-enriched lysosomal fraction showed negligible activity and did not change with CS or CS/RW (data not shown), clearly suggesting that the chymotrypsin-like activity was contributed by the proteasome. However, future studies are warranted to evaluate the localization of lysosomal chymotrypsin, particularly in the cytosol, and whether it has any downstream effects on mitochondrial dynamics in the CS/Tx model.
Here, we provide the first evidence of BTZ-mediated changes to SDHA and ATP5B subunits and impairment of mitochondrial complexes I, II, and V in renal proximal tubular (NRK) cells (Fig. 6). BTZ-mediated ChT-L inhibition is known to increase protein ubiquitination (both cytosolic and mitochondrial) (42). At lower concentrations, BTZ specifically inhibits the ChT-L activity of the proteasome, but at higher concentrations it can modestly inhibit chymotrypsin- or trypsin-like peptidase activity (1, 78). The BTZ concentration (10 nM) used in this study is just sufficient to inhibit ChT-L activity and has negligible effects on the trypsin- or chymotrypsin-like peptidase activity of the proteasome (data not shown). Taken together, these data indicate that SDHA and ATP5B proteins, under physiological conditions, are maintained by the ChT-L activity of the proteasome. Surprisingly, BTZ treatment reduced complex I activity, but NDUFS3, a subunit of complex I, remained unaltered. It is possible that the BTZ-mediated impairment of complex II induces reverse electron transport to alter complex I activity, as described by Chouchani et al. (11).
Our model of renal CS/Tx exhibits mitochondrial dysfunction (54, 71), oxidative stress (47), and energy (ATP) depletion (71). Mitochondrial respiratory dysfunction directly contributes to ATP depletion and increases ROS, which further oxidize proteins. ROS and ATP are considered critical determinants for proteasome function (19, 27, 57). Our finding that certain mitochondrial proteins (i.e., SDHA and ATP5B) accumulate within the insoluble fraction after antimycin A treatment suggests that severe ATP depletion enhances their rate of aggregation. Antimycin A, a potent inhibitor of mitochondrial respiration, not only severely depletes ATP in NRK cells, but also increases mitochondrial ROS (12) and significantly decreases ChT-L activity (Fig. 7). Antimycin A also induces the accumulation of ubiquitinated proteins (cytosolic and mitochondrial) (28, 42), suggesting possible alterations in proteasome function. In fact, Huang et al. (28) reported that antimycin A-induced mitochondrial inhibition impairs the proteasome in rat cortical neuronal cells. Excitingly, our in vitro data provided direct evidence that antimycin A-induced inhibition of mitochondrial respiration inhibits proteasomal ChT-L activity in NRK cells. Indeed, Chou et al. (10) showed that inhibiting complex I with rotenone inhibits the proteasome. These observations, combined with ours, indicate a tight and balanced relationship between mitochondrial function and normal proteasome health, and vice versa, in NRK cells. Our in vitro and in vivo data further suggest that interrupting this balance could cause renal tubular damage.
The posttranslational modification of key mitochondrial proteins has been implicated in the decline of respiratory complex function (33, 68, 74). Although we did not evaluate such modifications, it is plausible that SDHA and ATP5B subunits could be targets of oxidative posttranslational modifications that alter these subunits and increase respiratory dysfunction (54, 71) after CS/Tx. Because proteasome function is also impaired, these modified proteins cannot be cleared, causing them to be localized to the insoluble fraction. In a rodent model of a high-fat, high-fructose diet, impaired complex II activity was associated with a reversible cysteine oxidative posttranslational modification of complex II (cysteines 89 and 231 in SDHA and cysteines 100, 103, and 115 in SDHB) that also depleted ATP (74). Similarly, a murine model of Alzheimer’s disease exhibiting ATP depletion also showed evidence of ATP5B subunit nitration in brain tissue (33, 68). Future studies are needed to determine whether mitochondrial respiratory function is altered by oxidative posttranslational modifications (e.g., to SDHA or ATP5B) during renal CS/Tx.
Interestingly, CS/Tx decreased proteasomal ChT-L activity without altering the levels of the native enzyme (Fig. 1). Emerging evidence suggests that posttranslational modifications to proteasome subunits may significantly impact proteasome function (65). It is possible that the β5 catalytic subunit is targeted specifically during renal CS, in which case therapies that target the β5 subunit could be beneficial after transplantation. Future studies are needed to determine whether CS/Tx induces such posttranslational modifications of the proteasome.
The maintenance of proteome integrity is extremely important for renal cell viability during stressful conditions, especially during CS/Tx. Misfolded or damaged proteins should be monitored by the protein quality-control machinery, which refolds, sequesters, or degrades damaged proteins. It is equally important to consider the proteostatic role of chaperone proteins because this class of proteins, in conjunction with the UPS and autophagy/mitophagy, is crucial for removing misfolded or aggregated substrates (20, 34, 35, 59). There are conflicting reports on the role of autophagy (having both protective and detrimental properties) during renal warm ischemia-reperfusion injury (16), and there are no conclusive data for the renal CS/Tx model (14, 30). Given the extent of graft damage and dysfunction, we hypothesize that the autophagic flux is interrupted during prolonged CS with Tx. The focus of this study was to establish the role of the UPS; however, thorough and succinct research is required to define the status and role of autophagy in the context of mitochondrial and graft dysfunction after renal CS/Tx.
Mitigating the injury associated with renal CS could help us better utilize donor kidneys, prevent graft failure, improve long-term graft survival, and decrease the mortality rate of patients with end-stage kidney disease. Here, we demonstrated for the first time that CS/Tx decreases the ChT-L activity of the proteasome. We propose a sequence of events (Fig. 8) in which CS initiates defects in mitochondrial respiration (via complexes I and II). This compromises the proteasome function (ChT-L activity) after Tx, which exacerbates mitochondrial injury. The end result is a vicious cycle of proteasome dysfunction and mitochondrial damage that leads to severe renal damage (54, 71). Therefore, both the mitochondrion and the proteasome should be considered novel targets for renal preservation to increase the success of transplants.
Fig. 8.
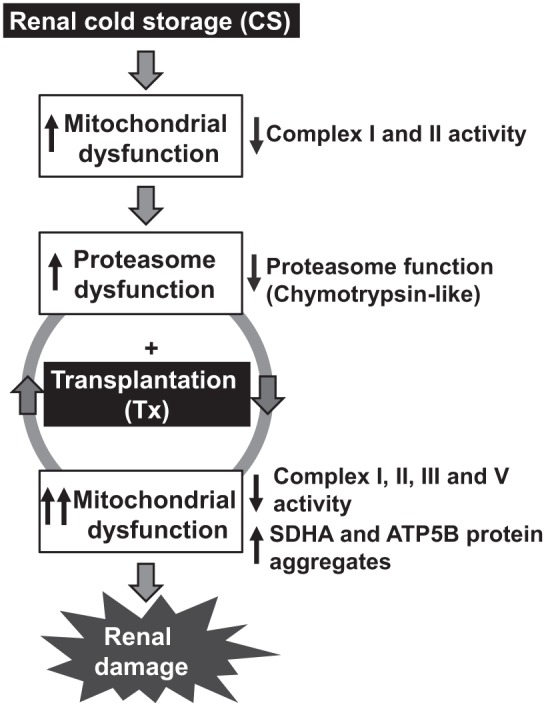
Proposed sequence of events during renal cold storage plus transplantation (CS/Tx). CS compromises the function of mitochondrial respiratory complexes I and II. This decreases the ChT-L activity of the proteasome after CS/Tx, which potentially exacerbates mitochondrial damage (inhibition of respiration and aggregation of mitochondrial proteins). The result is a vicious cycle of mitochondrial and proteasomal damage resulting in renal tissue damage.
GRANTS
This research was supported, in part, by the American Heart Association (16SDG27600026), the National Institutes of Health COBRE Center for Translational Pediatric Research (P20GM121293), the Arkansas Children’s Research Institute, the Arkansas Biosciences Institute, the Center for Translational Pediatric Research at the Arkansas Children’s Research Institute, and the University of Arkansas for Medical Sciences Barton Pilot award (17-DN-08).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.P. conceived and designed research; S.L. performed experiments; S.L., L.A.M.-C., and N.P. analyzed data; L.A.M.-C. and N.P. interpreted results of experiments; S.L., L.A.M.-C., and N.P. prepared figures; S.L., L.A.M.-C., and N.P. drafted manuscript; S.L., L.A.M.-C., and N.P. edited and revised manuscript; S.L., L.A.M.-C., and N.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the University of Arkansas for Medical Sciences Translational Pathology Shared Resource for excellent service in processing paraffin-embedded tissue blocks. We also thank the Science Communication Group at the University of Arkansas for Medical Sciences for editorial assistance during the preparation of this manuscript.
REFERENCES
- 1.Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma YT, Plamondon L, Stein RL. Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg Med Chem Lett 8: 333–338, 1998. doi: 10.1016/S0960-894X(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 2.Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteomics 10: R110.006924, 2011. doi: 10.1074/mcp.M110.006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold I, Pfeiffer K, Neupert W, Stuart RA, Schägger H. Yeast mitochondrial F1F0-ATP synthase exists as a dimer: identification of three dimer-specific subunits. EMBO J 17: 7170–7178, 1998. doi: 10.1093/emboj/17.24.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker TA, Geng Q, Romero J, Picken MM, Gamelli RL, Majetschak M. Prolongation of myocardial viability by proteasome inhibition during hypothermic organ preservation. Biochem Biophys Res Commun 401: 548–553, 2010. doi: 10.1016/j.bbrc.2010.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basso M, Samengo G, Nardo G, Massignan T, D’Alessandro G, Tartari S, Cantoni L, Marino M, Cheroni C, De Biasi S, Giordana MT, Strong MJ, Estevez AG, Salmona M, Bendotti C, Bonetto V. Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS One 4: e8130, 2009. doi: 10.1371/journal.pone.0008130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohovych I, Chan SS, Khalimonchuk O. Mitochondrial protein quality control: the mechanisms guarding mitochondrial health. Antioxid Redox Signal 22: 977–994, 2015. doi: 10.1089/ars.2014.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bon D, Chatauret N, Giraud S, Thuillier R, Favreau F, Hauet T. New strategies to optimize kidney recovery and preservation in transplantation. Nat Rev Nephrol 8: 339–347, 2012. doi: 10.1038/nrneph.2012.83. [DOI] [PubMed] [Google Scholar]
- 8.Breusing N, Grune T. Regulation of proteasome-mediated protein degradation during oxidative stress and aging. Biol Chem 389: 203–209, 2008. doi: 10.1515/BC.2008.029. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Zhang J, Zhao K, Li W, Miao Q, Sun Y, Zhao X, Wei T, Yang F. Lysosomal chymotrypsin induces mitochondrial fission in apoptotic cells by proteolytic activation of calcineurin. Protein Cell 5: 643–647, 2014. doi: 10.1007/s13238-014-0085-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chou AP, Li S, Fitzmaurice AG, Bronstein JM. Mechanisms of rotenone-induced proteasome inhibition. Neurotoxicology 31: 367–372, 2010. doi: 10.1016/j.neuro.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab 23: 254–263, 2016. doi: 10.1016/j.cmet.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Cruthirds DL, Saba H, MacMillan-Crow LA. Overexpression of manganese superoxide dismutase protects against ATP depletion-mediated cell death of proximal tubule cells. Arch Biochem Biophys 437: 96–105, 2005. doi: 10.1016/j.abb.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 13.Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie 83: 301–310, 2001. doi: 10.1016/S0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 14.De Deken J, Rex S, Lerut E, Martinet W, Monbaliu D, Pirenne J, Jochmans I. Postconditioning effects of argon or xenon on early graft function in a porcine model of kidney autotransplantation. Br J Surg 105: 1051–1060, 2018. doi: 10.1002/bjs.10796. [DOI] [PubMed] [Google Scholar]
- 15.Debout A, Foucher Y, Trebern-Launay K, Legendre C, Kreis H, Mourad G, Garrigue V, Morelon E, Buron F, Rostaing L, Kamar N, Kessler M, Ladriere M, Poignas A, Blidi A, Soulillou JP, Giral M, Dantan E. Each additional hour of cold ischemia time significantly increases the risk of graft failure and mortality following renal transplantation. Kidney Int 87: 343–349, 2015. [DOI] [PubMed] [Google Scholar]
- 16.Decuypere JP, Ceulemans LJ, Agostinis P, Monbaliu D, Naesens M, Pirenne J, Jochmans I. Autophagy and the kidney: implications for ischemia-reperfusion injury and therapy. Am J Kidney Dis 66: 699–709, 2015. doi: 10.1053/j.ajkd.2015.05.021. [DOI] [PubMed] [Google Scholar]
- 17.Dibley MG, Ryan MT, Stroud DA. A novel isoform of the human mitochondrial complex I subunit NDUFV3. FEBS Lett 591: 109–117, 2017. doi: 10.1002/1873-3468.12527. [DOI] [PubMed] [Google Scholar]
- 18.Eytan E, Ganoth D, Armon T, Hershko A. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc Natl Acad Sci USA 86: 7751–7755, 1989. doi: 10.1073/pnas.86.20.7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geng Q, Romero J, Saini V, Baker TA, Picken MM, Gamelli RL, Majetschak M. A subset of 26S proteasomes is activated at critically low ATP concentrations and contributes to myocardial injury during cold ischemia. Biochem Biophys Res Commun 390: 1136–1141, 2009. doi: 10.1016/j.bbrc.2009.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grune T, Catalgol B, Licht A, Ermak G, Pickering AM, Ngo JK, Davies KJ. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic Biol Med 51: 1355–1364, 2011. doi: 10.1016/j.freeradbiomed.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grune T, Shringarpure R, Sitte N, Davies K. Age-related changes in protein oxidation and proteolysis in mammalian cells. J Gerontol A Biol Sci Med Sci 56: B459–B467, 2001. doi: 10.1093/gerona/56.11.B459. [DOI] [PubMed] [Google Scholar]
- 22.Gurtubay JI, Goñi FM, Gómez-Fernández JC, Otamendi JJ, Macarulla JM. Triton X-100 solubilization of mitochondrial inner and outer membranes. J Bioenerg Biomembr 12: 47–70, 1980. doi: 10.1007/BF00745012. [DOI] [PubMed] [Google Scholar]
- 23.Hanf W, Codas R, Meas-Yedid V, Berthiller J, Buron F, Chauvet C, Brunet M, Giroud A, McGregor BC, Olivo-Marin JC, Hadj-Aissa A, Faure A, Petruzzo P, Martin X, Badet L, Morelon E. Kidney graft outcome and quality (after transplantation) from uncontrolled deceased donors after cardiac arrest. Am J Transplant 12: 1541–1550, 2012. doi: 10.1111/j.1600-6143.2011.03983.x. [DOI] [PubMed] [Google Scholar]
- 24.Hoeger S, Lueg G, Tsagogiorgas C, Schneider M, Theisinger S, Theisinger B, Fontana J, Waldherr R, Krämer BK, Schnuelle P, Yard B. UW is superior compared with HTK after prolonged preservation of renal grafts. J Surg Res 170: e149–e157, 2011. doi: 10.1016/j.jss.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 25.Hoeger S, Petrov K, Reisenbuechler A, Fontana J, Selhorst J, Hanusch C, Beck G, Seelen MA, van Son WJ, Waldherr R, Schnuelle P, Yard BA. The additional detrimental effects of cold preservation on transplantation-associated injury in kidneys from living and brain-dead donor rats. Transplantation 87: 52–58, 2009. doi: 10.1097/TP.0b013e318191b2ca. [DOI] [PubMed] [Google Scholar]
- 26.Höglinger GU, Carrard G, Michel PP, Medja F, Lombès A, Ruberg M, Friguet B, Hirsch EC. Dysfunction of mitochondrial complex I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J Neurochem 86: 1297–1307, 2003. doi: 10.1046/j.1471-4159.2003.01952.x. [DOI] [PubMed] [Google Scholar]
- 27.Huang H, Zhang X, Li S, Liu N, Lian W, McDowell E, Zhou P, Zhao C, Guo H, Zhang C, Yang C, Wen G, Dong X, Lu L, Ma N, Dong W, Dou QP, Wang X, Liu J. Physiological levels of ATP negatively regulate proteasome function. Cell Res 20: 1372–1385, 2010. doi: 10.1038/cr.2010.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Q, Wang H, Perry SW, Figueiredo-Pereira ME. Negative regulation of 26S proteasome stability via calpain-mediated cleavage of Rpn10 subunit upon mitochondrial dysfunction in neurons. J Biol Chem 288: 12161–12174, 2013. doi: 10.1074/jbc.M113.464552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huber JM, Tagwerker A, Heininger D, Mayer G, Rosenkranz AR, Eller K. The proteasome inhibitor bortezomib aggravates renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 297: F451–F460, 2009. doi: 10.1152/ajprenal.90576.2008. [DOI] [PubMed] [Google Scholar]
- 30.Jain S, Keys D, Nydam T, Plenter RJ, Edelstein CL, Jani A. Inhibition of autophagy increases apoptosis during re-warming after cold storage in renal tubular epithelial cells. Transpl Int 28: 214–223, 2015. doi: 10.1111/tri.12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung T, Grune T. The proteasome and its role in the degradation of oxidized proteins. IUBMB Life 60: 743–752, 2008. doi: 10.1002/iub.114. [DOI] [PubMed] [Google Scholar]
- 32.Jung T, Höhn A, Grune T. The proteasome and the degradation of oxidized proteins: Part II - protein oxidation and proteasomal degradation. Redox Biol 2: 99–104, 2014. doi: 10.1016/j.redox.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kane LA, Van Eyk JE. Post-translational modifications of ATP synthase in the heart: biology and function. J Bioenerg Biomembr 41: 145–150, 2009. doi: 10.1007/s10863-009-9218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kästle M, Reeg S, Rogowska-Wrzesinska A, Grune T. Chaperones, but not oxidized proteins, are ubiquitinated after oxidative stress. Free Radic Biol Med 53: 1468–1477, 2012. doi: 10.1016/j.freeradbiomed.2012.05.039. [DOI] [PubMed] [Google Scholar]
- 35.Kettern N, Dreiseidler M, Tawo R, Höhfeld J. Chaperone-assisted degradation: multiple paths to destruction. Biol Chem 391: 481–489, 2010. doi: 10.1515/bc.2010.058. [DOI] [PubMed] [Google Scholar]
- 36.Kimura T, Fukuda T, Sahara N, Yamashita S, Murayama M, Mizoroki T, Yoshiike Y, Lee B, Sotiropoulos I, Maeda S, Takashima A. Aggregation of detergent-insoluble tau is involved in neuronal loss but not in synaptic loss. J Biol Chem 285: 38692–38699, 2010. doi: 10.1074/jbc.M110.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kisselev AF, Goldberg AL. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol 398: 364–378, 2005. doi: 10.1016/S0076-6879(05)98030-0. [DOI] [PubMed] [Google Scholar]
- 38.Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol 19: 99–115, 2012. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ. Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem 277: 5411–5417, 2002. doi: 10.1074/jbc.M105326200. [DOI] [PubMed] [Google Scholar]
- 40.Livnat-Levanon N, Glickman MH. Ubiquitin-proteasome system and mitochondria - reciprocity. Biochim Biophys Acta 1809: 80–87, 2011. doi: 10.1016/j.bbagrm.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 41.Livnat-Levanon N, Kevei É, Kleifeld O, Krutauz D, Segref A, Rinaldi T, Erpapazoglou Z, Cohen M, Reis N, Hoppe T, Glickman MH. Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Reports 7: 1371–1380, 2014. doi: 10.1016/j.celrep.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 42.Maharjan S, Oku M, Tsuda M, Hoseki J, Sakai Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci Rep 4: 5896, 2014. doi: 10.1038/srep05896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Majetschak M, Patel MB, Sorell LT, Liotta C, Li S, Pham SM. Cardiac proteasome dysfunction during cold ischemic storage and reperfusion in a murine heart transplantation model. Biochem Biophys Res Commun 365: 882–888, 2008. doi: 10.1016/j.bbrc.2007.11.092. [DOI] [PubMed] [Google Scholar]
- 44.McAnulty JF. Hypothermic organ preservation by static storage methods: current status and a view to the future. Cryobiology 60, Suppl: S13–S19, 2010. doi: 10.1016/j.cryobiol.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Mitchell T, Rotaru D, Saba H, Smith RA, Murphy MP, MacMillan-Crow LA. The mitochondria-targeted antioxidant mitoquinone protects against cold storage injury of renal tubular cells and rat kidneys. J Pharmacol Exp Ther 336: 682–692, 2011. doi: 10.1124/jpet.110.176743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitchell T, Saba H, Laakman J, Parajuli N, MacMillan-Crow LA. Role of mitochondrial-derived oxidants in renal tubular cell cold-storage injury. Free Radic Biol Med 49: 1273–1282, 2010. doi: 10.1016/j.freeradbiomed.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nafar M, Sahraei Z, Salamzadeh J, Samavat S, Vaziri ND. Oxidative stress in kidney transplantation: causes, consequences, and potential treatment. Iran J Kidney Dis 5: 357–372, 2011. [PubMed] [Google Scholar]
- 48.Neutzner A, Benard G, Youle RJ, Karbowski M. Role of the ubiquitin conjugation system in the maintenance of mitochondrial homeostasis. Ann NY Acad Sci 1147: 242–253, 2008. doi: 10.1196/annals.1427.012. [DOI] [PubMed] [Google Scholar]
- 49.Neutzner A, Youle RJ, Karbowski M. Outer mitochondrial membrane protein degradation by the proteasome. Novartis Found Symp 287: 4–14, 2007. [PubMed] [Google Scholar]
- 50.Parajuli N, Campbell LH, Marine A, Brockbank KG, Macmillan-Crow LA. MitoQ blunts mitochondrial and renal damage during cold preservation of porcine kidneys. PLoS One 7: e48590, 2012. doi: 10.1371/journal.pone.0048590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parajuli N, MacMillan-Crow LA. Role of reduced manganese superoxide dismutase in ischemia-reperfusion injury: a possible trigger for autophagy and mitochondrial biogenesis? Am J Physiol Renal Physiol 304: F257–F267, 2013. doi: 10.1152/ajprenal.00435.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parajuli N, Müller-Holzner E, Böck G, Werner ER, Villunger A, Doppler W. Infiltrating CD11b+CD11c+ cells have the potential to mediate inducible nitric oxide synthase-dependent cell death in mammary carcinomas of HER-2/neu transgenic mice. Int J Cancer 126: 896–908, 2010. doi: 10.1002/ijc.24805. [DOI] [PubMed] [Google Scholar]
- 54.Parajuli N, Shrum S, Tobacyk J, Harb A, Arthur JM, MacMillan-Crow LA. Renal cold storage followed by transplantation impairs expression of key mitochondrial fission and fusion proteins. PLoS One 12: e0185542, 2017. doi: 10.1371/journal.pone.0185542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patil NK, Parajuli N, MacMillan-Crow LA, Mayeux PR. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am J Physiol Renal Physiol 306: F734–F743, 2014. doi: 10.1152/ajprenal.00643.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pesta D, Gnaiger E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol 810: 25–58, 2012. doi: 10.1007/978-1-61779-382-0_3. [DOI] [PubMed] [Google Scholar]
- 57.Powell SR, Davies KJ, Divald A. Optimal determination of heart tissue 26S-proteasome activity requires maximal stimulating ATP concentrations. J Mol Cell Cardiol 42: 265–269, 2007. doi: 10.1016/j.yjmcc.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pryde KR, Taanman JW, Schapira AH. A LON-ClpP proteolytic axis degrades complex I to extinguish ROS production in depolarized mitochondria. Cell Reports 17: 2522–2531, 2016. doi: 10.1016/j.celrep.2016.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reeg S, Jung T, Castro JP, Davies KJA, Henze A, Grune T. The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Radic Biol Med 99: 153–166, 2016. doi: 10.1016/j.freeradbiomed.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salahudeen AK. Free radicals in kidney disease and transplantation. Saudi J Kidney Dis Transpl 10: 137–143, 1999. [PubMed] [Google Scholar]
- 61.Salahudeen AK. Consequences of cold ischemic injury of kidneys in clinical transplantation. J Investig Med 52: 296–298, 2004. doi: 10.1136/jim-52-05-27. [DOI] [PubMed] [Google Scholar]
- 62.Salahudeen AK, Haider N, May W. Cold ischemia and the reduced long-term survival of cadaveric renal allografts. Kidney Int 65: 713–718, 2004. doi: 10.1111/j.1523-1755.2004.00416.x. [DOI] [PubMed] [Google Scholar]
- 63.Salahudeen AK, May W. Reduction in cold ischemia time of renal allografts in the United States over the last decade. Transplant Proc 40: 1285–1289, 2008. doi: 10.1016/j.transproceed.2008.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schägger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J 19: 1777–1783, 2000. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scruggs SB, Zong NC, Wang D, Stefani E, Ping P. Post-translational modification of cardiac proteasomes: functional delineation enabled by proteomics. Am J Physiol Heart Circ Physiol 303: H9–H18, 2012. doi: 10.1152/ajpheart.00189.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seo JH, Rivadeneira DB, Caino MC, Chae YC, Speicher DW, Tang HY, Vaira V, Bosari S, Palleschi A, Rampini P, Kossenkov AV, Languino LR, Altieri DC. The mitochondrial unfoldase-peptidase complex ClpXP controls bioenergetics stress and metastasis. PLoS Biol 14: e1002507, 2016. doi: 10.1371/journal.pbio.1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shaw BF, Lelie HL, Durazo A, Nersissian AM, Xu G, Chan PK, Gralla EB, Tiwari A, Hayward LJ, Borchelt DR, Valentine JS, Whitelegge JP. Detergent-insoluble aggregates associated with amyotrophic lateral sclerosis in transgenic mice contain primarily full-length, unmodified superoxide dismutase-1. J Biol Chem 283: 8340–8350, 2008. doi: 10.1074/jbc.M707751200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shin SJ, Lee SE, Boo JH, Kim M, Yoon YD, Kim SI, Mook-Jung I. Profiling proteins related to amyloid deposited brain of Tg2576 mice. Proteomics 4: 3359–3368, 2004. doi: 10.1002/pmic.200400961. [DOI] [PubMed] [Google Scholar]
- 69.Shringarpure R, Grune T, Davies KJ. Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell Mol Life Sci 58: 1442–1450, 2001. doi: 10.1007/PL00000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shringarpure R, Grune T, Mehlhase J, Davies KJ. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem 278: 311–318, 2003. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- 71.Shrum S, MacMillan-Crow LA, Parajuli N. Cold storage exacerbates renal and mitochondrial dysfunction following transplantation. J Kidney 2: 114, 2016. [PMC free article] [PubMed] [Google Scholar]
- 72.Stubenitsky BM, Booster MH, Brasile L, Green EM, Hermens FH, Stroosma OB, Kootstra G. I: Negative effect of cold ischemia on initial renal function. ASAIO J 46: 60–61, 2000. doi: 10.1097/00002480-200001000-00016. [DOI] [PubMed] [Google Scholar]
- 73.Stubenitsky BM, Brasile L, Booster MH, Haisch CE, Kootstra G. Deletrious effect of prolonged cold ischemia on renal function. Transpl Int 14: 256–260, 2001. doi: 10.1111/j.1432-2277.2001.tb00054.x. [DOI] [PubMed] [Google Scholar]
- 74.Sverdlov AL, Elezaby A, Behring JB, Bachschmid MM, Luptak I, Tu VH, Siwik DA, Miller EJ, Liesa M, Shirihai OS, Pimentel DR, Cohen RA, Colucci WS. High fat, high sucrose diet causes cardiac mitochondrial dysfunction due in part to oxidative post-translational modification of mitochondrial complex II. J Mol Cell Cardiol 78: 165–173, 2015. doi: 10.1016/j.yjmcc.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380, 2010. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tar K, Dange T, Yang C, Yao Y, Bulteau AL, Salcedo EF, Braigen S, Bouillaud F, Finley D, Schmidt M. Proteasomes associated with the Blm10 activator protein antagonize mitochondrial fission through degradation of the fission protein Dnm1. J Biol Chem 289: 12145–12156, 2014. doi: 10.1074/jbc.M114.554105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taylor EB, Rutter J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem Soc Trans 39: 1509–1513, 2011. doi: 10.1042/BST0391509. [DOI] [PubMed] [Google Scholar]
- 78.Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS-341 in cancer therapy. Clin Cancer Res 5: 2638–2645, 1999. [PubMed] [Google Scholar]
- 79.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem 68: 1015–1068, 1999. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 80.White Z, White RB, McMahon C, Grounds MD, Shavlakadze T. High mTORC1 signaling is maintained, while protein degradation pathways are perturbed in old murine skeletal muscles in the fasted state. Int J Biochem Cell Biol 78: 10–21, 2016. doi: 10.1016/j.biocel.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 81.Wilkinson KD. The discovery of ubiquitin-dependent proteolysis. Proc Natl Acad Sci USA 102: 15280–15282, 2005. doi: 10.1073/pnas.0504842102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu S, Peng G, Wang Y, Fang S, Karbowski M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol Biol Cell 22: 291–300, 2011. doi: 10.1091/mbc.e10-09-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 286: 19630–19640, 2011. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zaouali MA, Bardag-Gorce F, Carbonell T, Oliva J, Pantazi E, Bejaoui M, Ben Abdennebi H, Rimola A, Roselló-Catafau J. Proteasome inhibitors protect the steatotic and non-steatotic liver graft against cold ischemia reperfusion injury. Exp Mol Pathol 94: 352–359, 2013. doi: 10.1016/j.yexmp.2012.12.005. [DOI] [PubMed] [Google Scholar]