

Abstract
The antifibrotic peptide N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is released from thymosin-β4 (Tβ4) by the meprin-α and prolyl oligopeptidase (POP) enzymes and is hydrolyzed by angiotensin-converting enzyme (ACE). Ac-SDKP is present in urine; however, it is not clear whether de novo tubular release occurs or if glomerular filtration is the main source. We hypothesized that Ac-SDKP is released into the lumen of the nephrons and that it exerts an antifibrotic effect. We determined the presence of Tβ4, meprin-α, and POP in the kidneys of Sprague-Dawley rats. The stop-flow technique was used to evaluate Ac-SDKP formation in different nephron segments. Finally, we decreased Ac-SDKP formation by inhibiting the POP enzyme and evaluated the long-term effect in renal fibrosis. The Tβ4 precursor and the releasing enzymes meprin-α and POP were expressed in the kidneys. POP enzyme activity was almost double that in the renal medulla compared with the renal cortex. With the use of the stop-flow technique, we detected the highest Ac-SDKP concentrations in the distal nephron. The infusion of a POP inhibitor into the kidney decreased the amount of Ac-SDKP in distal nephron segments and in the proximal nephron to a minor extent. An ACE inhibitor increased the Ac-SDKP content in all nephron segments, but the increase was highest in the distal portion. The chronic infusion of a POP inhibitor increased kidney medullary fibrosis, which was prevented by Ac-SDKP. We conclude that Ac-SDKP is released by the nephron and is part of an important antifibrotic system in the kidney.
Keywords: Ac-SDKP, ACE inhibitors, meprin, prolyl oligopeptidase, renal fibrosis
INTRODUCTION
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a natural tetrapeptide with antifibrotic and anti-inflammatory properties. Ac-SDKP, which was originally isolated from calf bone marrow (21), has been proposed to act as a hematopoiesis inhibitor, preventing the entry of pluripotent progenitor cells into the DNA synthesis phase (S) of the cell cycle (21). Ac-SDKP was later shown to inhibit the proliferation of fibroblasts and epithelial cells, including kidney epithelia in vitro (14, 24). In various conditions, the exogenous administration of Ac-SDKP decreases inflammation by reducing macrophage and lymphocyte infiltration into tissues and by decreasing proinflammatory cytokine levels (30, 45). Ac-SDKP is present in several tissues, including the kidney (37). In the kidney, exogenous Ac-SDKP exerts anti-inflammatory and antifibrotic effects, conferring renal protection and antiproteinuric effects in several animal models (6, 22, 23, 45, 46). Ac-SDKP is released from its precursor, thymosin-β4 (Tβ4), through two sequential enzymatic steps. The first step is mediated by meprin-α, and the second step is mediated by prolyl oligopeptidase (POP; Refs. 7, 19). Ac-SDKP is degraded by angiotensin-converting enzyme (ACE), and ACE inhibitors (ACEi) increase its plasma concentrations and urinary excretion (1, 17). There is evidence that the some of the cardiorenal protective effects of ACEi are partly mediated by increased Ac-SDKP concentrations (36, 39).
It has been suggested that in humans, Ac-SDKP is filtered by the glomerulus and hydrolyzed in the proximal tubule by ACE (1). However, data indicate that the kidney may also produce Ac-SDKP (17, 34). Treatment of mice with a neutralizing antibody against Ac-SDKP decreases plasma levels of Ac-SDKP and prevents its glomerular filtration, but the Ac-SDKP level in the urine remains unchanged, indicating that the kidney could produce Ac-SDKP (34). Furthermore, all of the components needed for Ac-SDKP production, including its precursor substrate (Tβ4) and the meprin and POP enzymes, have been detected in the kidney in independent studies (11, 37). Moreover, the upregulation of profibrotic microRNA-324-3p decreases POP activity in the kidney and is associated with low Ac-SDKP content in rat urine (25). All of this evidence suggests that local renal production of Ac-SDKP may be possible. However, the release site(s) and the role of locally produced Ac-SDKP are not known.
Therefore, we hypothesized that Ac-SDKP is released in the kidney via the nephrons and that Ac-SDKP is part of an antifibrotic system that includes Tβ4 and meprin-α and POP enzymes.
METHODS
Animals and reagents.
Male Sprague-Dawley rats (Charles River) weighing 250–350 g were used for all experiments. Intraperitoneal injection of thiobutabarbital was administered at 125 mg/kg body wt (BW) for anesthesia 30 min before surgery. All experiments were approved by the Henry Ford Health System Institutional Animal Care and Use Committee and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Ac-SDKP tissue levels.
Rat tissues were harvested under systemic anesthesia, immediately immersed in liquid nitrogen, and then stored at −80°C until further processing. The tissues were homogenized in lysis buffer supplemented with 10−5 M captopril to prevent Ac-SDKP degradation during sample processing. The total protein concentration was quantified by the Bradford method. Ac-SDKP was measured and expressed as nanograms per milligram total protein. Ac-SDKP was measured with an ELISA kit (SPI-Bio) according to the manufacturer’s instructions.
Differences in venous-to-arterial Ac-SDKP concentration.
Blood samples were collected from the renal vein and the aorta in heparinized prechilled tubes containing freshly prepared captopril at a final concentration of 10−5 M. Ac-SDKP was extracted from the plasma with methanol and measured with an ELISA kit (SPI-Bio) according to the manufacturer’s instructions.
Renal clearance of Ac-SDKP.
A 1% FITC-inulin bolus was injected at 3 µl/g BW followed by the infusion of 1% FITC-inulin in PBS at 0.15 µl·g−1·min−1 for 60 min. Clearances were calculated according to the formula urinary concentration (in nanomoles) times urine flow (in milliliters per minute) divided by plasma concentration (in nanomoles) adjusted by body weight and were expressed as milliliters per minute per 100 g BW. Plasma Ac-SDKP was calculated as the average of the two plasma samples. The Ac-SDKP excretion fraction was calculated using the Ac-SDKP-to-inulin clearance ratio.
Tβ4 and meprin-α and POP enzymes gene expression in the kidney.
The gene expression of Tβ4 (Tmsb4x), the meprin-α subunit (Mep1a), and prolyl oligopeptidase (Prep) was evaluated in 14 separate microdissected renal tubule segments from untreated male Sprague-Dawley rats (200–250 g BW) using the publicly accessible NephronRNAseq database (20). The main product of this work is an extensive database of gene expression along the nephron, provided as a publicly accessible Web page: https://hpcwebapps.cit.nih.gov/ESBL/Database/NephronRNAseq/index.html. The database expresses the whole transcriptome of 14 renal tubules subsegments obtained by classic renal tubule microdissection, from the proximal tubule through the inner medullary collecting duct of rat kidneys. Polyadenylated mRNAs were captured with oligo(dT) primers and processed into adapter-ligated cDNA libraries that were sequenced using the Illumina platform. Transcriptomes were identified to a median depth of 8,261 genes in microdissected renal tubule samples (105 replicates in total) and glomeruli (5 replicates). The expression levels of all genes were reported by the median reads per kilobase of exon model per million mapped reads.
Immunostaining.
Frozen kidney sections were fixed with acetone and immunostained with an antibody against meprin-α (MEP1A Antibody, AF4007, at 1:50 dilution; R&D Systems). A rabbit anti-goat IgG cross-adsorbed secondary antibody conjugated to Alexa Fluor 488 (A-11078, at 1:100 dilution; Invitrogen) was used. 4′,6′-Diamidino-2-phenylindole staining was used to counterstain cellular nuclei. Paraffin-embedded kidney sections were deparaffinized and immunostained with antibodies against human prolyl oligo peptidase (NBP2-33950; Novus Biologicals) at a 1:40 dilution. Then, the sections were incubated with a secondary biotinylated antibody (1:200, goat anti-rabbit IgG). Immunoreactivity was detected with a streptavidin reagent and an ABC peroxidase kit (VECTASTAIN Elite ABC Kit; Vector). Double-staining for the POP enzyme and the distal tubule marker epithelial sodium channel (ENaC) was performed on 8-µm kidney frozen section. Briefly, tissues were fixed in 4% paraformaldehyde and immunostained for POP (monoclonal anti-Prep, H00005550-M04, 1:50 dilution; Abnova). A goat anti-mouse IgG secondary antibody conjugated to Alexa Fluor 647 (ab150115; Abcam) was used. After washing, a primary antibody against ENaC (SPC-4040; StressMarq Biosciences) and a secondary goat anti-rabbit antibody conjugated to Alexa Fluor 488 (ab11426; Abcam) were used.
Prolyl oligopeptidase activity assay.
Meprin-α activity was measured with a fluorogenic substrate, Mca-YVADAPK(Dnp)-OH (19). Kidney homogenates (50 μg) from Sprague-Dawley rats were incubated for 30 min at 37°C and pH 7.4 with 10 μmol/l fluorogenic substrate in PBS containing the following broad-range protease inhibitors: cOmplete EDTA-free cocktail from Roche, 1 μg/ml pepstatin A, 10 μmol/l leupeptin, 1 mmol/l CaCl2, and 1 mmol/l ZnCl2. The fluorescence of the released product was measured using a fluorometer (SpectraMax M2) at an excitation wavelength of 320 nm and an emission wavelength of 405 nm. Relative meprin-α activity is expressed in arbitrary fluorescence units per minute per milligram of total protein. The meprin-α inhibitor actinonin (100 μmol/l) was used as a control. POP enzyme activity was measured as described previously using a fluorogenic substrate, Z-Gly-Pro-AMC (19). Briefly, kidney homogenates from the cortex and the medulla were obtained using a cold Stadie-Riggs microtome. Protein homogenates (50 µg) were incubated with 50 µmol/l fluorogenic substrate in PBS, pH 7.4, for 60 min at 37°C. The fluorescence of the released 7-amino-4-methylcoumarin product was measured at an excitation of 360 nm and an emission of 460 nm and was quantified against AMC standards. Specific POP activity is expressed as picomoles AMC per minute per milligram total protein. A POP inhibitor, S17092 (20 µmol/l), was used as a control.
Kidney stop-flow method.
To evaluate Ac-SDKP production, secretion, and degradation within segments along the nephron, the stop-flow technique was applied (12, 44). Briefly, a femoral vein catheter (PE-50) was used to infuse a bolus of 3 µl/g BW of 1% FITC-inulin followed by 1% FITC-inulin in 0.77 M sodium chloride solution as a continuous infusion. Additionally, 6% mannitol was continuously infused to cause osmotic diuresis. The left ureter was cannulated close to the renal pelvis using PE-10 tubing and was tied with a 4-0 silk suture. After 25 min, the ureteral catheter was occluded, stopping the tubular flow and allowing the secretion, formation, and degradation of various substances to modify the quality of the urine in each segment of the nephron. After 4 min of occlusion, a bolus of p-aminohippuric acid (PAH) was administered at 30 µmol/kg BW. After 6 min of occlusion, the clamp was released, and urine was collected in 22 consecutive 20-µl fractions in glass capillary tubes. The early fractions represent the distal nephron segments (fractions 2–8), and the later fractions (16–22) represent the proximal nephron. Free-flow control, an index of normal values, was obtained by collecting 2 fractions after allowing free urine flow for 2 min (fractions 23–24). Inulin, PAH (a proximal tubule marker), and the enzymatic activity of a distal tubule marker, kallikrein, were measured in each sample (4, 42). Ac-SDKP concentrations corrected by water reabsorption (Ac-SDKP-to-inulin ratio) were calculated for each set of animals to evaluate renal Ac-SDKP handling in different nephron segments. A set of animals received captopril at 50 mg/kg ip 3 h before the experiment. Another set of animals received an infusion of either KYP-2047 (POP inhibitor) at 0.1 mg·kg−1·day−1 in dimethyl sulfoxide (DMSO) or vehicle alone, directly in the kidney, for 7 days before the stop-flow experiments. Local delivery of the POP enzyme inhibitor was performed by implanting a fenestrated polyethylene tubing (PE-10) attached to an osmotic minipump (ALZET). The fenestrated catheter was inserted in the cortical-medullary junction, and the renal capsule was sealed with Vetbond Tissue Adhesive (3M) to fix the catheter and prevent any leak outside of the kidney.
The dose of the inhibitors was calculated based on previous studies (15, 34).
FITC-inulin, p-aminohippuric acid, and kallikrein measurements.
Urine fluorescence from fluorescein isothiocyanate-inulin (Inulin-FITC; Sigma-Aldrich) was measured in triplicate in a black 96-well microplate according to the manufacturer’s instructions. Urine PAH was measured with a commercial assay kit according to the manufacturer’s protocol (MAK101; Sigma-Aldrich). Kallikrein activity was measured via fluorometric assay using 5 µl of urine with 0.5 mM H-D-Val-Leu-Arg-AFC (sc-391026; Santa Cruz Biotechnology). Enzymatic activity was expressed as relative fluorescence units.
Renal fibrosis measurement.
Sprague-Dawley rats were infused for 4 wk with vehicle (DMSO), the POP inhibitor KYP-2047 (1 mg·kg−1·day−1), or the POP inhibitor and Ac-SDKP (1.6 mg·kg−1·day−1) using subcutaneously implanted osmotic minipumps (ALZET 2004). Renal Ac-SDKP urinary excretion was measured to evaluate the effect of the POP inhibitor at 2 wk of treatment. Systolic blood pressure was measured in conscious rats with a noninvasive, computerized tail-cuff system (CODA; Kent Scientific, Torrington, CT), as described previously (26). At the end of the protocol, kidneys were harvested for analysis. Picrosirius red staining was used to quantify the renal interstitial collagen deposition, a marker of fibrosis, as previously described. For the renal interstitial collagen fraction, ≥20 images were taken with a ×100 objective using a microscope with a digital camera attached. The interstitial collagen content was quantified by computerized image analysis software (MicroSuite Biological imaging software; Olympus) and was expressed as the ratio of the collagen-positive area to the entire area of the captured field.
Statistics.
t-Tests and the nonparametric two-sample Wilcoxon test were used to compare the differences of interest. Significance was determined using Hochberg method to adjust for multiple testing. The adjustment was applied to groups of similar tests. In the stop-flow technique, the average sample contents from fractions 2–8, 16–22, and 23–24 were compared with ANOVA.
All statistical calculations were performed with SAS version 9.3. A P value <0.05 was considered significant.
RESULTS
Ac-SDKP in the kidney.
To understand the renal handling of Ac-SDKP, we evaluated the presence of Ac-SDKP in kidney parenchyma, Ac-SDKP clearance, and the difference of Ac-SDKP concentrations between the artery and renal vein (n = 6). We found that Ac-SDKP is present in kidney tissue, with a concentration of 29.9 ± 1.5 ng/mg protein. Urinary Ac-SDKP excretion was 1.9 ± 0.7 µg/day. Renal Ac-SDKP clearance was 0.10 ml·min−1·100 g body wt (BW)−1, and inulin clearance was 1.27 ± 0.2 ml·min−1·100 g BW−1. Therefore, the fractional excretion of Ac-SDKP was 8.1 ± 3%. Despite the urinary excretion, the Ac-SDKP concentration in the renal vein did not differ from the arterial concentration (Fig. 1), and the mean arterial-to-vein difference was 0.48 ± 1.1 ng/ml (P = 0.75).
Fig. 1.

A: inulin and N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) renal plasma clearance in Sprague-Dawley rats. Fractional excretion of Ac-SDKP was 8.1 ± 3%. B: quantitative analysis of Ac-SDKP concentrations in the artery and the renal vein. Individual values are plotted. No differences were observed between Ac-SDKP concentrations entering the kidney and Ac-SDKP exiting in the renal vein. n = 7. P = 0.67. BW, body wt.
Nephron expression of meprin-α and prolyl oligopeptidase.
To explore the renal presence of the Ac-SDKP precursor and the releasing enzyme in Sprague-Dawley rats, we evaluated the mRNA and protein expression and the enzymatic activity of the Tβ4-Ac-SDKP axis. Figure 2 shows the expression of the Tβ4 gene (referred to as Tmsb4x), the meprin-α gene (referred to as Mep1a), and the POP gene (referred to as Prep) in 14 tubular microdissected nephron subsegments, determined in a subanalysis using NephronRNAseq database (20). Tβ4 mRNA was expressed in all nephron subsegments. The meprin gene (Mep1a) was expressed only in the 2nd (S2) and 3rd (S3) subsegments of the proximal tubule. The presence of meprin-α was confirmed by immunofluorescence (Figs. 3 and 4B). Meprin-α was present in the brush border of the proximal tubule in the cortex (S2 segment) and in the outer medulla (S3 segment; n = 3). We confirmed meprin-α expression by measuring enzyme activity in the cortex and the medulla (n = 8; Fig. 4). The POP gene (Prep) was expressed in the S2 and S3 segments in the proximal tubule, in the medullary region of the loop of Henle, and in the distal nephron (Fig. 2). The protein expression of the POP enzyme was evaluated by immunohistochemistry (n = 3). Immunostaining for the POP enzyme was observed in the distal nephron, with higher expression being found in the kidney medulla (Fig. 5). The POP enzyme colocalized with the distal nephron marker ENaC (Fig. 5). The POP enzyme was also observed in ENaC-negative tubules, in agreement with the mRNA expression results. Consistent with the protein expression results, POP enzyme activity was 2.16-fold higher in the medulla than in the cortex (n = 8; Fig. 5).
Fig. 2.
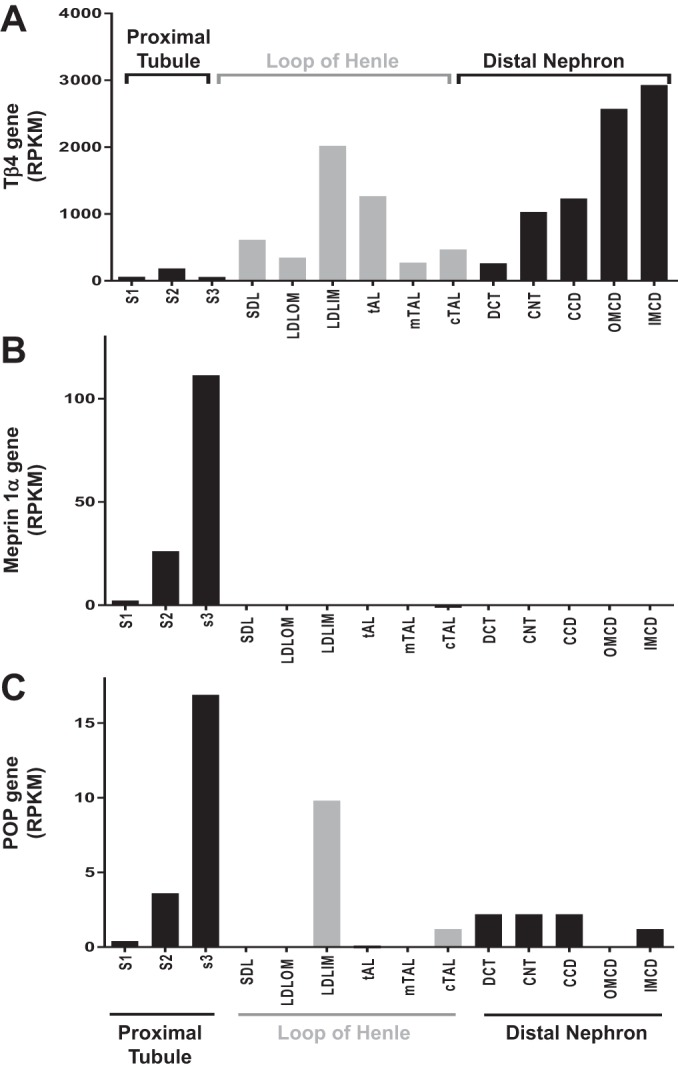
Thymosin-β4 (Tβ4), meprin-α, and prolyl oligopeptidase (POP) gene expression along the renal tubule (n = 7–11). Thymosin-β4 (Tmsb4x gene; A) is expressed along the nephron. Meprin-α (Mep1a; B) is expressed in the proximal tubules. Prolyl oligopeptidase (Prep; C) is expressed in the proximal tubule, the long descending limb in the inner medulla, and in the distal nephron. Results are plotted as the median reads per kilobase of exon model per million mapped reads (RPKM). CCD, cortical collecting duct; CNT, connecting tubule; cTAL, cortical thick ascending limb of the loop of Henle; DCT, distal convoluted tubule; IMCD, inner medullary collecting duct; LDLIM, long descending limb of the loop of Henle in the inner medulla; LDLOM, long descending limb of the loop of Henle in the outer medulla; mTAL, medullary thick ascending limb of the loop of Henle; OMCD, outer medullary collecting duct; S1, 1st segment of the proximal tubule; S2, 2nd segment of the proximal tubule; S3, 3rd segment of the proximal tubule; SDL, short descending limb of the loop of Henle; tAL, thin ascending limb of the loop of Henle.
Fig. 3.

Renal expression of meprin-α. Images show meprin expression (green), nuclear staining with 4′,6′-diamidino-2-phenylindole (DAPI; blue), and merged images of the 2 in the cortical areas (top) and the medullary region (bottom). Arrows indicate structures that are compatible with the glomerulus in the cortex (scale bar, 200 µm).
Fig. 4.

Meprin-α activity in the kidney. Meprin-α activity was measured in homogenates from the kidney cortex, medulla, and papilla (negative control; n = 8). A shows no differences in meprin-α activity between the cortex and medulla. No meprin-α activity was detected in the kidney papilla. B illustrates the meprin-α distribution at low magnification (×20) in the kidney. Meprin was expressed extensively in the cortex, which is compatible with the 2nd segment of the proximal tubule, and in outer medulla region (3rd segment). No signal was detected in the kidney papilla. AFU, arbitrary fluorescence units; prot, protein.
Fig. 5.

Prolyl oligopeptidase (POP) in the kidney. Brown color shows the peroxidase activity related to the presence of the POP enzyme. A: in the cortex, a strong signal is observed in the tubules, which is compatible with the distal convoluted tubule, the cortical collecting ducts (asterisk), and the thick ascending limbs of the loop of Henle (arrow; scale bar, 50 µm). B: POP expression at low magnification in the cortex and the medulla (scale bar, 200 µm). C: coimmunostaining of the POP enzyme (red) and the distal tubule marker epithelial sodium channel (green). POP enzyme is expressed in epithelial sodium channel-positive cells (distal nephron) as well as in other tubule segments (scale bar, 50 µm). D: POP enzymatic activity was significantly higher in the medullary region than in cortical areas (n = 8).
Ac-SDKP is produced in the distal nephron.
To examine Ac-SDKP formation, secretion, and degradation in different nephron segments, the stop-flow technique (12, 44) was applied to normal rats and rats that were administered ACE and POP enzyme inhibitors (n = 7). The stop-flow technique, developed by Vander et al. (43), allows the renal handling of several substances to be examined in different nephron segments by stopping the urine flow and exposing the tubular fluid to a specific modification in each nephron segment. The early urine fractions (2–8) corresponding to the highest activity of the distal tubule marker kallikrein exhibited the highest Ac-SDKP content (Figs. 6 and 7). The lowest amounts of Ac-SDKP were found in the later fractions (16–22) corresponding to the proximal tubule marker p-aminohippurate (PAH). Therefore, the Ac-SDKP content in the distal nephron was 2.75-fold higher than in the proximal tubule. Treatment with a POP inhibitor directly in the renal parenchyma decreased the amount of Ac-SDKP along the nephron (Figs. 6 and 7). Under local infusion of the POP inhibitor KYP-2047 into the kidney, no effects were observed in systemic Ac-SDKP plasma levels (vehicle 3.1 ± 0.5 nM vs. POP inhibitor 2.2 ± 0.2 nM; P = 0.054), minimizing the potential effects of a large drop in Ac-SDKP filtration into the tubules. On the other hand, ACEi increased the Ac-SDKP content in all nephron segments, but the highest content was observed in the distal nephron (Fig. 7).
Fig. 6.

Quantitative analysis of N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) in different nephron fractions through the stop-flow technique. A: PAH concentrations in consecutive urine fractions were analyzed to identify the fractions representing the proximal part of the nephron. B: kallikrein activity in consecutive urine fractions was analyzed to identify the fractions representing the distal part of the nephron. C: Ac-SDKP adjusted by water reabsorption along the nephron. Greatest amounts of Ac-SDKP are observed in the distal portion of the nephron, and smallest amounts are found in the proximal part of the nephron. Bottom curve (open squares) represents the Ac-SDKP-to-inulin ratio under infusion of the prolyl oligopeptidase enzyme inhibitor (POP inh) KYP-2047. Last 2 fractions in all panels represent the amounts of free flow (n = 7). *P < 0.01, distal nephron vs. proximal nephron. #P < 0.05, vehicle vs. POP inhibitor.
Fig. 7.

Effects of prolyl oligopeptidase (POP) and angiotensin-converting enzyme inhibitors on N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) amounts in the nephron. Amounts of Ac-SDKP in the distal nephron and the proximal nephron and free-flow levels were adjusted by inulin content in rats treated with vehicle, the POP inhibitor (inh) KYP-2047, or captopril. Distal nephron was expressed as the mean of fractions 2–8, proximal nephron as the mean of fractions 16–22, and free-flow levels as the mean of fractions 23 and 24 in the stop-flow experiments. Inhibition of the POP enzyme decreased the amount of Ac-SDKP in all nephron segments, whereas captopril treatment (angiotensin-converting enzyme inhibitor) significantly increased Ac-SDKP in all nephron regions (n = 7). *P < 0.001.
Ac-SDKP prevents medullary interstitial fibrosis in the kidney.
To evaluate the potential role of Ac-SDKP under physiological conditions, subcutaneous infusion of the POP inhibitor KYP-2047 was performed for 4 wk in normal rats (n = 7). The POP inhibitor decreased Ac-SDKP excretion in the urine, which was reversed by the coinfusion of Ac-SDKP (Fig. 8). No differences in blood pressure were found between the groups (vehicle: 132.4 ± 19.2 mmHg; POP inhibitor: 134.7 ± 16.4 mmHg; POP inhibitor and Ac-SDKP: 123.4 ± 14.5 mmHg; P = 0.29). After 4 wk of POP inhibition, interstitial fibrosis in the kidney medulla had increased compared with that in vehicle-infused rats (Fig. 9). No such increase was observed in the renal cortex. The simultaneous infusion of the POP inhibitor and Ac-SDKP prevented the increase in collagen content observed in the kidney medulla (Fig. 9). Histological examination of the kidneys did not show any significant immune cell infiltrates in animals exposed to the POP inhibitor.
Fig. 8.
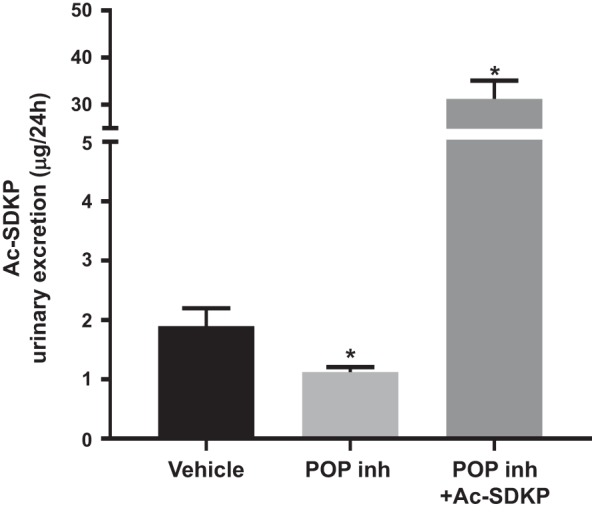
Twenty-four-hour urinary excretion of N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP). During prolyl oligopeptidase inhibition (POP inh), Ac-SDKP excretion was significantly reduced, which was reversed via the simultaneous infusion of Ac-SDKP (n = 7). *P < 0.05 vs. vehicle.
Fig. 9.

Effects of the prolyl oligopeptidase inhibitor (POP inh) and N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) on kidney fibrosis. Four-week infusion of the POP inhibitor increased interstitial fibrosis in the kidney medulla compared with vehicle-infused rats. This increase was not observed in the renal cortex. Simultaneous infusion of the POP inhibitor and Ac-SDKP prevented the increase in collagen content observed in the kidney medulla. Representative images and quantitative analysis of picrosirius red staining (n = 7) are shown. Scale bar, 200 µm. n.s., Not significant.
DISCUSSION
In this study, we demonstrate the renal production of Ac-SDKP. We detected Ac-SDKP along the nephron, with the highest level being found in the distal nephron. The enzymes that participate in Ac-SDKP release were also explored in the kidney. Meprin-α was observed in the proximal nephron, and the POP enzyme was found in more distal areas, suggesting sequential action inside of the tubule. Tβ4 expression was detected along the nephron; however, Tβ4 is a small peptide that is also freely filtered in the glomerulus (28). Furthermore, local inhibition of the POP enzyme in the kidney decreased the amount of Ac-SDKP in the nephron. These data indicate that all of the components necessary for the release of Ac-SDKP are present in the kidney and that the Ac-SDKP peptide is released in vivo in the nephron. In contrast, inhibition of ACE, the main Ac-SDKP hydrolyzing enzyme, dramatically increased the amount of Ac-SDKP in all nephron segments, including the distal nephron.
Since meprin-α is only expressed in the proximal tubule and the soluble form of meprin-α is inactive, the first step in Ac-SDKP release must occur in the proximal tubule. Therefore, we speculate that the main source of Tβ4 for Ac-SDKP release in the kidney is glomerular filtration. Previous reports are consistent with this possibility, showing that exogenous injection of Tβ4 increases both Tβ4 and Ac-SDKP in the urine within minutes (28). Therefore, we propose a peptidergic model in which Tβ4 is filtered in the glomerulus and meprin-α cleaves Tβ4 in the proximal tubules and releases the intermediary peptides that will ultimately release Ac-SDKP through POP activity in the more distal part of the nephron. This two-step successive enzymatic metabolism model has been reported for other peptides, such as B-type natriuretic peptide, and for intratubular angiotensin II formation (31, 33). Meprin-α is also the first-step enzyme in B-type natriuretic peptide metabolism, and the second enzyme is neprilysin. The presence of meprin (2, 16) and POP (11, 38) enzymes in the kidney has been confirmed in other reports. The highest POP mRNA expression is found in the S3 segment of the proximal tubule, which may generate controversy, considering the high protein expression observed in the distal nephron. However, RNA expression does not necessarily represent the amount of protein expression, since several posttranslational modifications can alter the amount and function of a protein. The protein expression and activity measured in our work confirmed that the majority of POP enzyme expression occurs in the most distal part of the nephron, including the thick ascending limb, distal convoluted tubule, connecting tubule, and collecting ducts. Renal release of Ac-SDKP in vivo has not been demonstrated previously. Furthermore, the kidney has been proposed as the sole Ac-SDKP clearance organ in humans and in rodents (1). In humans, the excretion fraction of Ac-SDKP is <10% of filtered Ac-SDKP, but in the presence of ACEi, this proportion increases to 80% (1). In our study, the renal clearance of Ac-SDKP in rats was similar to that in humans under normal conditions, with fractional Ac-SDKP excretion of 8%. Classically, renal fractional excretion <1 (<100%) indicates that the substance is degraded or reabsorbed in the nephron, but if the substance is produced or secreted in the kidney, fractional excretion is >1. This concept may not apply to substances with more complex behavior, where the same substance is degraded/reabsorbed and secreted or released at the same time, such as Ac-SDKP. Surprisingly, under normal conditions, we did not find any difference between the arterial and venous Ac-SDKP concentrations in the kidney, providing the first indication that Ac-SDKP could be produced in the kidney. Supporting this interpretation, a previous report showed that a neutralizing antibody against Ac-SDKP that prevented Ac-SDKP filtration did not decrease urinary Ac-SDKP levels (34), indicating that most of the Ac-SDKP in urine must be produced inside of the tubule. Similarly, decreased POP enzyme activity in the kidneys of Munich Wistar Frömter rats is associated with lower Ac-SDKP levels in the urine (25). Altogether, these observations indicate that at least in rodents, Ac-SDKP formation occurs in the nephron.
To explore the localization of Ac-SDKP within the nephron, we used the renal stop-flow technique. This technique has been employed to examine the formation, secretion, and degradation of various substances in different nephron segments (12). Our results indicate that in rats, the distal nephron releases Ac-SDKP into the urine. Renal POP inhibition attenuated Ac-SDKP release, indicating that Ac-SDKP is produced in the nephron. Supporting this interpretation and consistent with previous reports (11, 25, 29), we showed that POP gene (Prep) and corresponding enzyme expression mainly occur in the distal nephron. Moreover, we found that POP activity was higher in the kidney medulla than in the cortex. Treatment of animals with ACEi caused the amount of Ac-SDKP to increase in all nephron segments, with higher levels being observed in the distal part. This pattern could be due to reduced Ac-SDKP degradation by ACE, even in the distal tubule. However, it has been reported that ACEi increases the expression of the POP enzyme in the kidney (25). This observation could explain the greater Ac-SDKP increase detected in the distal tubule under ACEi treatment. The stop-flow technique also allowed us to examine Ac-SDKP handling in the more-proximal region of the nephron. Consequently, we observed that after glomerular filtration, Ac-SDKP was decreased by at least 25–30% in the proximal nephron compared with the free-flow values of the vehicle-treated groups. Small peptides are subjected to strong hydrolysis and reabsorption in the proximal tubules (3). The decrease in Ac-SDKP in the proximal tubule was partially reversed by ACEi. This suggests that at least a portion of the filtered Ac-SDKP is degraded by ACE in the proximal tubule, where ACE is highly expressed in the brush borders of proximal tubule cells (5). The absence of differences in the arterial-to-vein Ac-SDKP concentration indicates that Ac-SDKP must be produced in the kidney; however, we cannot at this point confirm or rule out the possibility that Ac-SDKP is reabsorbed in either the distal nephron or in more-proximal regions after glomerular filtration. Additionally, we cannot discard the possibility that Ac-SDKP release occurs simultaneously on the basolateral side of epithelial cells.
As a preliminary approach for examining the physiological function of Ac-SDKP in the distal portion of the nephron, we performed subcutaneous (systemic) infusion of the POP inhibitor with and without concomitant Ac-SDKP infusion. We were forced to perform systemic infusion, since local (intrakidney) delivery of the inhibitor and or Ac-SDKP was not achievable in the long-term experiments due to technical difficulties with the catheter and the system did not deliver the drugs. We found that the use of a POP inhibitor increases renomedullary fibrosis without any changes in the cortex. No significant cell infiltrates were found in the histological examination. The increased collagen content was prevented when Ac-SDKP was simultaneously infused. No blood pressure changes were observed between the groups. Ac-SDKP has antifibrotic and anti-inflammatory properties in several tissues and also shows antiproliferative effects in epithelial and mesenchymal cells (14). Cavasin et al. (7) demonstrated that the POP inhibitor S17092 induces collagen deposition in several organs, including the kidney, when animals are subjected to angiotensin II treatment. In an aging mouse model, the inhibition of ACE dramatically decreased renomedullary fibrosis, which was independent of changes in blood pressure (27), in agreement with our findings. This prevention of medullary fibrosis could be a consequence of an increased Ac-SDKP concentration mediated by ACE inhibition. At this point, we do not know the mechanism driving medullary fibrosis. The kidney medulla presents an unfavorable internal environment, due to a low oxygen concentration and high osmolarity, which may require protective mechanisms. Therefore, we believe that in association with other mechanisms, the Tβ4-Ac-SDKP axis is a peptidergic system that participates in the prevention of fibrosis in the renal medullary region under normal conditions. The system can potentially be damaged or be insufficient in some pathological conditions promoting renal fibrosis. Subjecting renal epithelial cells to the influence of a stressor such as hypoxia or high osmolarity can lead to cytokine release to the interstitium, particularly the release of transforming growth factor-β (TGF-β), a key factor that promotes interstitial fibrosis and epithelial-to mesenchymal transition (10, 41). Previous studies by our group (35) have shown that Ac-SDKP decreases TGF-β levels and downstream signaling. Thus we may speculate that Ac-SDKP acting in the epithelial cells in the tubular lumen can have an antifibrotic effect through decreasing TGF-β signaling and preventing epithelial-to mesenchymal transition.
The lack of knowledge regarding the exact localization of Ac-SDKP release is a limitation of our work. The stop-flow technique is not a very precise method for localization of the subsegments of the nephron in rats. With the use of tubule markers, we can roughly separate the proximal and distal nephron segments. However, the expression of the POP enzyme suggests that some Ac-SDKP is released from the medullary part of the loop of Henle, from the distal convoluted tubule, and along the connecting and collecting ducts. The stop-flow technique is a method that allows the evaluation of each nephron segment through simply increasing the time that the fluid is in contact with the segment, thus exaggerating the process of absorption, release, or secretion. Then, when the flow is resumed, the very high flow induced by mannitol would prevent or at least minimize conversion because of the time necessary for the process to occur. However, it may be possible for minor conversion to occur, which is a limitation of the technique.
As we noted above, the use of chronic subcutaneous systemic infusion is another limitation of this work. Currently, we cannot discard any potential systemic effects of the POP inhibition as a mediator of the observed renal fibrotic changes, and future development of a kidney-specific POP knockout animal model will help to clarify these possibilities. The POP enzyme participates in the hydrolysis of several peptides, mainly in the central nervous system (9). Although we did not perform systematic behavioral measurements, we did not notice any specific behavioral changes in the animals receiving the POP inhibitor. Nevertheless, through the coinfusion of Ac-SDKP and the POP inhibitor, we tried to reverse the effects related to the lack of Ac-SDKP while the other potential POP inhibitory effects were still present.
Finally, we believe that the renal Tβ4-Ac-SDKP peptidergic system may play an important role in renal diseases. Future work should focus on renal diseases in which hypoxia and hyperosmolarity are involved, such as acute kidney injury, diabetic nephropathy, and progressive chronic kidney disease. The role of ACE inhibitors in the Tβ4-Ac-SDKP peptidergic system observed in this study could explain some of the beneficial effects of these drugs on kidney diseases, both in animals (13, 18, 22, 25, 32, 45, 46) and in humans (8, 40). We conclude that Ac-SDKP is released in the rat nephron, predominantly in the distal part, and that Ac-SDKP is part of an antifibrotic peptidergic system in the kidney.
GRANTS
This work was funded by the National Heart, Lung, and Blood Institute Grant 5-P01-HL-028982.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.A.R. and O.A.C. conceived and designed research; C.A.R., N.K., M.E.W., and T.-D.L. performed experiments; C.A.R. and E.L.P. analyzed data; C.A.R., P.N., T.-D.L., and O.A.C. interpreted results of experiments; C.A.R. prepared figures; C.A.R. drafted manuscript; C.A.R., N.K., P.N., M.E.W., T.-D.L., and O.A.C. edited and revised manuscript; C.A.R., N.K., P.N., M.E.W., T.-D.L., and O.A.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Gülser Gürocak for technical assistance in the Ac-SDKP measurements.
REFERENCES
- 1.Azizi M, Ezan E, Reny JL, Wdzieczak-Bakala J, Gerineau V, Ménard J. Renal and metabolic clearance of N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP) during angiotensin-converting enzyme inhibition in humans. Hypertension 33: 879–886, 1999. doi: 10.1161/01.HYP.33.3.879. [DOI] [PubMed] [Google Scholar]
- 2.Bylander J, Li Q, Ramesh G, Zhang B, Reeves WB, Bond JS. Targeted disruption of the meprin metalloproteinase β gene protects against renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 294: F480–F490, 2008. doi: 10.1152/ajprenal.00214.2007. [DOI] [PubMed] [Google Scholar]
- 3.Carone FA, Peterson DR, Oparil S, Pullman TN. Renal tubular transport and catabolism of proteins and peptides. Kidney Int 16: 271–278, 1979. doi: 10.1038/ki.1979.129. [DOI] [PubMed] [Google Scholar]
- 4.Carretero OA, Scicli AG. Renal kallikrein: its localization and possible role in renal function. Fed Proc 35: 194–198, 1976. [PubMed] [Google Scholar]
- 5.Casarini DE, Boim MA, Stella RC, Krieger-Azzolini MH, Krieger JE, Schor N. Angiotensin I-converting enzyme activity in tubular fluid along the rat nephron. Am J Physiol Renal Physiol 272: F405–F409, 1997. doi: 10.1152/ajprenal.1997.272.3.F405. [DOI] [PubMed] [Google Scholar]
- 6.Cavasin MA, Liao TD, Yang XP, Yang JJ, Carretero OA. Decreased endogenous levels of Ac-SDKP promote organ fibrosis. Hypertension 50: 130–136, 2007. doi: 10.1161/HYPERTENSIONAHA.106.084103. [DOI] [PubMed] [Google Scholar]
- 7.Cavasin MA, Rhaleb NE, Yang XP, Carretero OA. Prolyl oligopeptidase is involved in release of the antifibrotic peptide Ac-SDKP. Hypertension 43: 1140–1145, 2004. doi: 10.1161/01.HYP.0000126172.01673.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiurchiu C, Remuzzi G, Ruggenenti P. Angiotensin-converting enzyme inhibition and renal protection in nondiabetic patients: the data of the meta-analyses. J Am Soc Nephrol 16, Suppl 1: S58–S63, 2005. doi: 10.1681/ASN.2004110968. [DOI] [PubMed] [Google Scholar]
- 9.García-Horsman JA, Männistö PT, Venäläinen JI. On the role of prolyl oligopeptidase in health and disease. Neuropeptides 41: 1–24, 2007. doi: 10.1016/j.npep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Gewin L, Zent R. How does TGF-β mediate tubulointerstitial fibrosis? Semin Nephrol 32: 228–235, 2012. doi: 10.1016/j.semnephrol.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goossens F, De Meester I, Vanhoof G, Scharpé S. Distribution of prolyl oligopeptidase in human peripheral tissues and body fluids. Eur J Clin Chem Clin Biochem 34: 17–22, 1996. [DOI] [PubMed] [Google Scholar]
- 12.Harvey AM, Malvin RL. The effect of androgenic hormones on creatinine secretion in the rat. J Physiol 184: 883–888, 1966. doi: 10.1113/jphysiol.1966.sp007954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inserra F, Romano LA, de Cavanagh EM, Ercole L, Ferder LF, Gomez RA. Renal interstitial sclerosis in aging: effects of enalapril and nifedipine. J Am Soc Nephrol 7: 676–680, 1996. [DOI] [PubMed] [Google Scholar]
- 14.Iwamoto N, Xano HJ, Yoshioka T, Shiraga H, Nitta K, Muraki T, Ito K. Acetyl-seryl-aspartyl-lysyl-proline is a novel natural cell cycle regulator of renal cells. Life Sci 66: 221–226, 2000. doi: 10.1016/S0024-3205(00)00460-4. [DOI] [PubMed] [Google Scholar]
- 15.Jalkanen AJ, Hakkarainen JJ, Lehtonen M, Venäläinen T, Kääriäinen TM, Jarho E, Suhonen M, Forsberg MM. Brain pharmacokinetics of two prolyl oligopeptidase inhibitors, JTP-4819 and KYP-2047, in the rat. Basic Clin Pharmacol Toxicol 109: 443–451, 2011. doi: 10.1111/j.1742-7843.2011.00747.x. [DOI] [PubMed] [Google Scholar]
- 16.Jiang W, Sadler PM, Jenkins NA, Gilbert DJ, Copeland NG, Bond JS. Tissue-specific expression and chromosomal localization of the alpha subunit of mouse meprin A. J Biol Chem 268: 10380–10385, 1993. [PubMed] [Google Scholar]
- 17.Junot C, Nicolet L, Ezan E, Gonzales MF, Menard J, Azizi M. Effect of angiotensin-converting enzyme inhibition on plasma, urine, and tissue concentrations of hemoregulatory peptide acetyl-Ser-Asp-Lys-Pro in rats. J Pharmacol Exp Ther 291: 982–987, 1999. [PubMed] [Google Scholar]
- 18.Knowles JW, Reddick RL, Jennette JC, Shesely EG, Smithies O, Maeda N. Enhanced atherosclerosis and kidney dysfunction in eNOS−/−Apoe−/− mice are ameliorated by enalapril treatment. J Clin Invest 105: 451–458, 2000. doi: 10.1172/JCI8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar N, Nakagawa P, Janic B, Romero CA, Worou ME, Monu SR, Peterson EL, Shaw J, Valeriote F, Ongeri EM, Niyitegeka JM, Rhaleb NE, Carretero OA. The anti-inflammatory peptide Ac-SDKP is released from thymosin-β4 by renal meprin-α and prolyl oligopeptidase. Am J Physiol Renal Physiol 310: F1026–F1034, 2016. doi: 10.1152/ajprenal.00562.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lenfant M, Wdzieczak-Bakala J, Guittet E, Prome JC, Sotty D, Frindel E. Inhibitor of hematopoietic pluripotent stem cell proliferation: purification and determination of its structure. Proc Natl Acad Sci USA 86: 779–782, 1989. doi: 10.1073/pnas.86.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao TD, Nakagawa P, Janic B, D’Ambrosio M, Worou ME, Peterson EL, Rhaleb NE, Yang XP, Carretero OA. N-Acetyl-Seryl-Aspartyl-Lysyl-Proline: mechanisms of renal protection in mouse model of systemic lupus erythematosus. Am J Physiol Renal Physiol 308: F1146–F1154, 2015. doi: 10.1152/ajprenal.00039.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao TD, Yang XP, D’Ambrosio M, Zhang Y, Rhaleb NE, Carretero OA. N-acetyl-seryl-aspartyl-lysyl-proline attenuates renal injury and dysfunction in hypertensive rats with reduced renal mass: council for high blood pressure research. Hypertension 55: 459–467, 2010. doi: 10.1161/HYPERTENSIONAHA.109.144568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lombard MN, Sotty D, Wdzieczak-Bakala J, Lenfant M. In vivo effect of the tetrapeptide, N-acetyl-Ser-Asp-Lys-Pro, on the G1-S transition of rat hepatocytes. Cell Tissue Kinet 23: 99–103, 1990. [DOI] [PubMed] [Google Scholar]
- 25.Macconi D, Tomasoni S, Romagnani P, Trionfini P, Sangalli F, Mazzinghi B, Rizzo P, Lazzeri E, Abbate M, Remuzzi G, Benigni A. MicroRNA-324-3p promotes renal fibrosis and is a target of ACE inhibition. J Am Soc Nephrol 23: 1496–1505, 2012. doi: 10.1681/ASN.2011121144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maheshwari M, Romero CA, Monu SR, Kumar N, Liao TD, Peterson EL, Carretero OA. Renal protective effects of N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) in obese rats on a high-salt diet. Am J Hypertens 31: 902–909, 2018. doi: 10.1093/ajh/hpy052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol 77: 1–10, 1999. doi: 10.1046/j.1440-1711.1999.00795.x. [DOI] [PubMed] [Google Scholar]
- 28.Mora CA, Baumann CA, Paino JE, Goldstein AL, Badamchian M. Biodistribution of synthetic thymosin β4 in the serum, urine, and major organs of mice. Int J Immunopharmacol 19: 1–8, 1997. doi: 10.1016/S0192-0561(97)00005-2. [DOI] [PubMed] [Google Scholar]
- 29.Myöhänen TT, Venäläinen JI, García-Horsman JA, Piltonen M, Männistö PT. Distribution of prolyl oligopeptidase in the mouse whole-body sections and peripheral tissues. Histochem Cell Biol 130: 993–1003, 2008. doi: 10.1007/s00418-008-0468-x. [DOI] [PubMed] [Google Scholar]
- 30.Nakagawa P, Liu Y, Liao TD, Chen X, González GE, Bobbitt KR, Smolarek D, Peterson EL, Kedl R, Yang XP, Rhaleb NE, Carretero OA. Treatment with N-acetyl-seryl-aspartyl-lysyl-proline prevents experimental autoimmune myocarditis in rats. Am J Physiol Heart Circ Physiol 303: H1114–H1127, 2012. doi: 10.1152/ajpheart.00300.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navar LG, Imig JD, Zou L, Wang CT. Intrarenal production of angiotensin II. Semin Nephrol 17: 412–422, 1997. [PubMed] [Google Scholar]
- 32.Omata M, Taniguchi H, Koya D, Kanasaki K, Sho R, Kato Y, Kojima R, Haneda M, Inomata N. N-acetyl-seryl-aspartyl-lysyl-proline ameliorates the progression of renal dysfunction and fibrosis in WKY rats with established anti-glomerular basement membrane nephritis. J Am Soc Nephrol 17: 674–685, 2006. doi: 10.1681/ASN.2005040385. [DOI] [PubMed] [Google Scholar]
- 33.Pankow K, Wang Y, Gembardt F, Krause E, Sun X, Krause G, Schultheiss HP, Siems WE, Walther T. Successive action of meprin A and neprilysin catabolizes B-type natriuretic peptide. Circ Res 101: 875–882, 2007. doi: 10.1161/CIRCRESAHA.107.153585. [DOI] [PubMed] [Google Scholar]
- 34.Peng H, Carretero OA, Liao TD, Peterson EL, Rhaleb NE. Role of N-acetyl-seryl-aspartyl-lysyl-proline in the antifibrotic and anti-inflammatory effects of the angiotensin-converting enzyme inhibitor captopril in hypertension. Hypertension 49: 695–703, 2007. doi: 10.1161/01.HYP.0000258406.66954.4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng H, Carretero OA, Peterson EL, Rhaleb NE. Ac-SDKP inhibits transforming growth factor-β1-induced differentiation of human cardiac fibroblasts into myofibroblasts. Am J Physiol Heart Circ Physiol 298: H1357–H1364, 2010. doi: 10.1152/ajpheart.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peng H, Carretero OA, Vuljaj N, Liao TD, Motivala A, Peterson EL, Rhaleb NE. Angiotensin-converting enzyme inhibitors: a new mechanism of action. Circulation 112: 2436–2445, 2005. doi: 10.1161/CIRCULATIONAHA.104.528695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pradelles P, Frobert Y, Créminon C, Ivonine H, Frindel E. Distribution of a negative regulator of haematopoietic stem cell proliferation (AcSDKP) and thymosin β4 in mouse tissues. FEBS Lett 289: 171–175, 1991. doi: 10.1016/0014-5793(91)81062-D. [DOI] [PubMed] [Google Scholar]
- 38.Quinto BM, Juliano MA, Hirata I, Carmona AK, Juliano L, Casarini DE. Characterization of a prolyl endopeptidase (kininase) from human urine using fluorogenic quenched substrates. Int J Biochem Cell Biol 32: 1161–1172, 2000. doi: 10.1016/S1357-2725(00)00060-1. [DOI] [PubMed] [Google Scholar]
- 39.Rasoul S, Carretero OA, Peng H, Cavasin MA, Zhuo J, Sanchez-Mendoza A, Brigstock DR, Rhaleb NE. Antifibrotic effect of Ac-SDKP and angiotensin-converting enzyme inhibition in hypertension. J Hypertens 22: 593–603, 2004. doi: 10.1097/00004872-200403000-00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Romero CA, Orias M, Weir MR. Novel RAAS agonists and antagonists: clinical applications and controversies. Nat Rev Endocrinol 11: 242–252, 2015. doi: 10.1038/nrendo.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romero CA, Remor A, Latini A, De Paul AL, Torres AI, Mukdsi JH. Uric acid activates NRLP3 inflammasome in an in-vivo model of epithelial to mesenchymal transition in the kidney. J Mol Histol 48: 209–218, 2017. doi: 10.1007/s10735-017-9720-9. [DOI] [PubMed] [Google Scholar]
- 42.Scicli AG, Gandolfi R, Carretero OA. Site of formation of kinins in the dog nephron. Am J Physiol Renal Physiol 234: F36–F40, 1978. doi: 10.1152/ajprenal.1978.234.1.F36. [DOI] [PubMed] [Google Scholar]
- 43.Vander AJ, Malvin RL, Wilde WS, Sullivan LP. Localization of the site of action of mercurial diuretics by stop flow analysis. Am J Physiol 195: 558–562, 1958. doi: 10.1152/ajplegacy.1958.195.3.558. [DOI] [PubMed] [Google Scholar]
- 44.Wilde WS, Malvin RL. Graphical placement of transport segments along the nephron from urine concentration pattern developed with stop flow technique. Am J Physiol 195: 153–160, 1958. doi: 10.1152/ajplegacy.1958.195.1.153. [DOI] [PubMed] [Google Scholar]
- 45.Worou ME, Liao TD, D’Ambrosio M, Nakagawa P, Janic B, Peterson EL, Rhaleb NE, Carretero OA. Renal protective effect of N-acetyl-seryl-aspartyl-lysyl-proline in Dahl salt-sensitive rats. Hypertension 66: 816–822, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuo Y, Chun B, Potthoff SA, Kazi N, Brolin TJ, Orhan D, Yang HC, Ma LJ, Kon V, Myöhänen T, Rhaleb NE, Carretero OA, Fogo AB. Thymosin β4 and its degradation product, Ac-SDKP, are novel reparative factors in renal fibrosis. Kidney Int 84: 1166–1175, 2013. doi: 10.1038/ki.2013.209. [DOI] [PMC free article] [PubMed] [Google Scholar]