

Abstract
Cleft lip and palate are common craniofacial deformities. The etiology underlying these deformities is complex and multifactorial and they can occur as part of one of many chromosomal syndromes, Mendelian single gene disorders, teratogenic effects, and as yet uncharacterized syndromes. Our paper will provide an overview of the multiple genes and molecular pathways that have been implicated in palatal fusion. We believe that understanding the molecular mechanisms of cleft formation can help clinicians anticipate which patients may have difficulties healing and in the future allow them to make surgical and medical treatment decisions based on genetic information.
Keywords: Cleft lip, cleft palate, genetics, molecular pathways, embryology
INTRODUCTION
Cleft lip and cleft palate are common congenital deformities resulting from failure of the facial processes to grow or fuse appropriately during early embryologic development (fourth through 12th weeks of gestation). The incidence of cleft lip and palate (CLP) varies based on race, with a review article analyzing incidence rate of CLP within livebirths, stillbirths, and abortions finding the following data: Asian and American Indian populations have the highest incidence with 0.79 to 4.04 per 1000, Caucasian populations have an intermediate incidence with 0.91 to 2.69 per 1000, and African populations have the lowest incidence at 0.18 to 1.67 per 1000.1 The etiology of CLP is multifactorial and complex and includes both genetic and environmental factors. Orofacial clefting can be classified as nonsyndromic, or found as an isolated defect, which occurs in about 85% of cleft lip with or without cleft palate and about 45% of cleft palate alone.2 Syndromic clefting can be further subdivided as occurring in over 150 chromosomal syndromes, such as van der Woude syndrome, and velocardiofacial syndrome3, 4; over 300 Mendelian single gene disorders; effects of teratogens, such as alcohol, tobacco smoke, antiepileptic drugs, and organic solvents5; and as yet uncategorized syndromes.
CLP are classified by laterality and completeness, with this classification scheme based on embryologic development. Cleft lips can be unilateral or bilateral; complete, in which the cleft involves the entire thickness of the upper lip and in which the alveolar ridge is often cleft as well; or incomplete, in which there is a variable continuous segment across the cleft. A Simonart band is a band of lip tissue bridging the cleft region (Fig. 1). Cleft palates are also described as being unilateral or bilateral and as complete or incomplete (Fig. 2). However, they are also further classified based on the location of the cleft with respect to the incisive foramen, with clefts of the primary palate occurring anterior to the incisive foramen and those of the secondary palate occurring posterior. Cleft of the soft palate alone may also occur.
Figure 1.
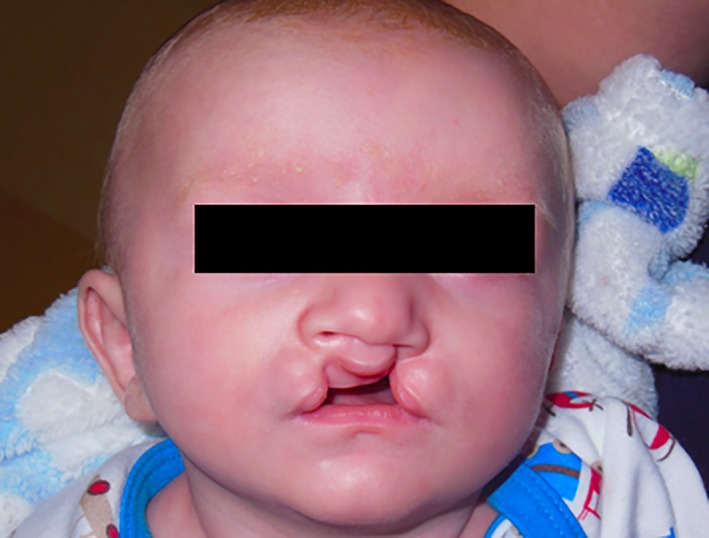
A child with bilateral cleft lip.
Figure 2.
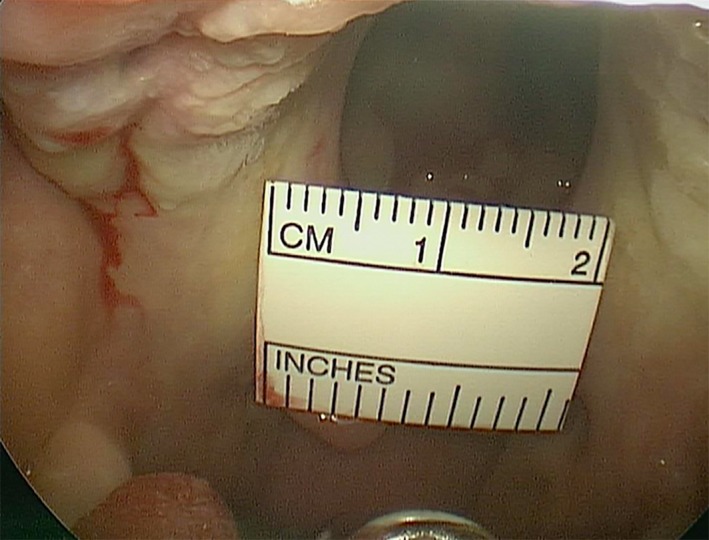
Intraoral view of cleft palate
FACIAL DEVELOPMENT
The development of the human face takes place between the fourth and 10th weeks of gestation by rapid delamination and migration of the cranial neural crest cells (CNC) from the neural placode. Migration and proliferation of the CNC leads to growth and fusion of paired maxillary prominences growing from the lateral sides towards the midline to join the frontonasal prominence, which descends in the midline from the forebrain, and concomitant development of paired mandibular prominences. Helms et al. have published a thorough review of the molecular mechanisms involved in neural crest migration.6 A pair of thickened ectodermal nasal placodes develop on either side of the frontonasal prominence. When these invaginate to form nasal pits during the fifth week of development, the frontonasal prominence becomes divided into the medial and lateral nasal processes. The medial nasal processes fuse to create the philtrum, medial upper lip, nasal tip, columella, and primary palate, and the lateral nasal processes fuse to create the lateral aspect of the nose. The maxillary processes give rise to the cheeks and lateral upper lip. Palatogenesis occurs during weeks five through 12 of development. The primary palate, which includes the central maxillary alveolar arch with the four incisor teeth and the hard palate anterior to the incisive foramen, develops first from the rapid expansion of the frontonasal prominence and fusion of the medial nasal prominences. The secondary palate then develops from the fusion of the palatine shelves which elongate adjacent to the tongue, and as the mandible grows, the tongue descends in the oral cavity allowing the palate shelves to elongate and elevate above the tongue (Fig. 3).
Figure 3.
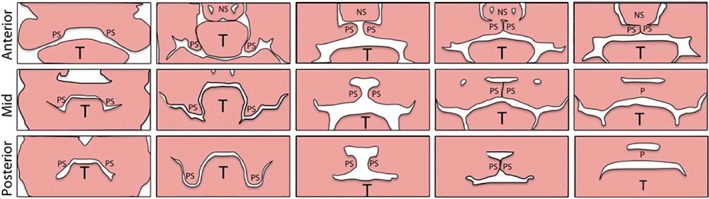
Palatal growth and fusion. Palatal shelves (PS), grow down the sides of the tongue (T), elevate, contact each other and the nasal septum (NS) and eventually fuse. (From Lane J, Kaartinen V. Signaling networks in palate development. Wiley Interdiscip Rev Syst Biol Med 2014;6:11.)
Any interruption of mandibular elongation (as occurs in Pierre Robin sequence, Stickler syndrome, and Treacher Collins syndrome) can lead to profound tongue‐based airway obstruction and cleft palate. The palatine shelves fuse with the primary palate and the primary and secondary palates fuse with the nasal septum. Fusion of the palate occurs from an anterior to posterior direction beginning at the incisive foramen and occurs from weeks eight through 12 of gestation, concluding with uvular fusion. The point during development during which fusion is interrupted determines the degree of clefting noted clinically.7, 8
MOLECULAR GENETICS
More than 300 genes have been implicated in palatal fusion in human and experimental animal models. Single gene mutations can lead to clefting. Examples including Stickler syndrome, which is associated with pathogenic variants in one of six genes (COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, or COL9A3) 9 or Treacher Collins syndrome, which is associated with a mutation in one of three genes TCOF1 (which encodes the protein Treacle, responsible for ribosome production10), POLR1C, or POLR1D.11 However, the etiology of clefting is still poorly known with multiple molecular pathways associated with cleft lip and palate. The most commonly studied molecular pathways include those involved with extracellular signaling factors, transcription factors, and cell adhesion molecules12 (Fig. 4). Though there are many molecules with known or suspected association with CLP, we will aim to provide a review of those pathways most commonly implicated (Table 1).
Figure 4.
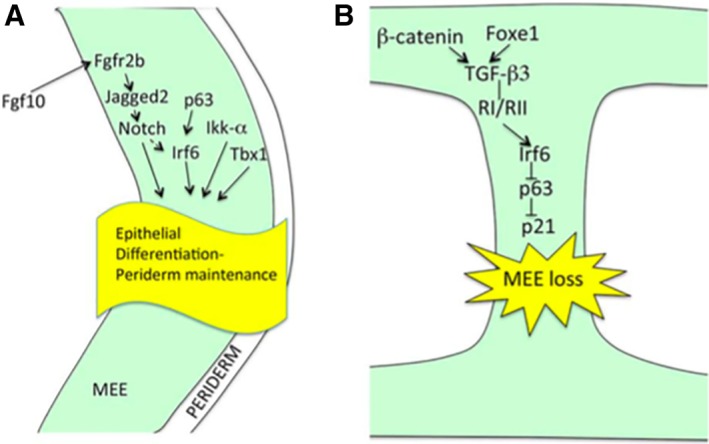
A. Signalling pathways involved in palatal epithelial differentiation include Fgf10, Irf6, and Tbx1 pathways. Mutations of genes involved in these pathways can lead to clefting. B. Signalling cascade involved in loss of the medial edge epithelium. (From Lane J, Kaartinen V. Signaling networks in palate development. Wiley Interdiscip Rev Syst Biol Med 2014;6:13.)
Table 1.
Common Clefting Syndromes With Their Associated Genetic Mutations and Clinical Features
| Syndrome | Associated Gene or Genetic Pathway | Clinical features |
|---|---|---|
| Loeys‐Dietz Syndrome | TGFRβ1, TGFβR2, TGF‐β2 | Craniosyostosis, cleft palate, and hypertelorism |
| Pierre Robin Sequence | BMPR1B | Micrognathia, glossoptosis, and cleft palate |
| Apert Syndrome (FGF2 mutation) | FGF2 | Flat forehead, retracted midface, cleft palate, and hypertelorism, learning disability, poor joint mobility, and severe symmetric syndactyly of fingers and toes |
| Crouzon Syndrome | FGF2, FGF3 | Flat forehead, midface hypoplasia, and cleft palate, normal intelligence, normal hands and feet |
| Van der Woude Syndrome | IRF6 | Cleft lip and palate with lip pits |
| DiGeorge/Velocardiofacial Syndrome | TBX1 | Cleft palate, congenital cardiac and renal abnormalities, neonatal hypocalcemia, micrognathia, low set ears, telecanthus |
| Stickler Syndrome | COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, or COL9A3 | Cleft palate, bifid uvula, flat midface, hearing loss, retinal detachments, cataracts |
| Treacher Collins Syndrome | TCOF1, POLR1C or POLR1D | Cleft palate, conductive hearing loss, coloboma, micrognathia, microtia |
Online Mendelian Inheritance in Man, OMIM (https://omim.org/).
TGF‐β and BMP Pathways
The Transforming Growth Factor β (TGFβ) superfamily is comprised of the transforming growth factor beta family, as well as other families, including bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs), activins (ACTs), and inhibins (INHs). The TGFβ family is comprised of three isoforms, TGF‐β1, TGF‐β2, and TGF‐β3, which are signaling molecules important for the mediation of many events in normal growth and development.13, 14 These growth factors are secreted by cells and the factors are bound by adjacent receptors, that then signal intracellularly through cytoplasmic proteins belonging to the Smad family.15 Receptors TgfβR1 and TgfβR2 phosphorylate SMADs 2/3.16 This complex is then transported to the nucleus, where transcriptional changes are induced.
TgfβR3 binds all three Tgfβ ligands as well as BMP2. The loss of TgfβR3 results in arrested palatal shelf elevation and elongation and interfered and also led to atypical expression of both TGFβ and BMP signaling pathways within the palatal shelves.17 Loeys‐Dietz syndrome, which includes craniosyostosis, cleft palate, and hypertelorism is associated with mutations in genes encoding TGF‐β receptors 1and 2 (TGFBR1 and TGFBR2), as well as TGF‐β2.18 As TGFβ1 and 2 have been shown to have an effect on inducing chondrogensis and osteogenesis, knowledge of these genetic mutations in a patient with a cleft may aid in surgical planning and in anticipation of possible complications following repair.19
Tgfβ3, which is expressed in medial edge epithelial cells, is involved in multiple cellular processes including epithelial‐to‐mesenchymal transformation as well as programmed cell death of the palate medial edge epithelium. Mice lacking Tgfβ3 have been shown to exhibit lung agenesis and cleft palate due to impaired adhesion of medial edge epithelia and elimination of midline epithelial seam.20 Conditional deletion of Tgfβ3 from the medial edge epithelium alone was associated with cleft palate formation, and correction of this using in vitro techniques reversed the cleft palate phenotype.
SHH, FGF10, BMP, AND MSX1
The Sonic hedgehog (Shh) gene is part of the Hedgehog gene family, which encode proteins important for cell–cell interaction and has is required for palate and frontonasal development.21 Shh is a downstream target of Fibroblast Growth Factor 10 (Fgf10) and its receptor Fgfr2b, signaling during palate development. Shh has been found to be predominantly expressed at sites of epithelia‐mesenchymal interactions and is critically important for the induction of facial primordia, particularly the frontonasal process. Loss of Shh is associated with midline facial defects including holoprosencephaly as well as cleft palate formation, and it is also expressed in the medial edge epithelium.22
Epithelially expressed Shh has been shown to directly signal to the mesenchyme to regular palate outgrowth, with Shh signaling regulating Bmp2, Bmp4, Msx1, and Fgf10 expression in adjacent palatal mesenchyme. Conversely, inactivation of smoothened (Smo), a protein involved in the hedgehog signaling pathway, in palatal mesenchyme affected palatal epithelial cell proliferation.23 Bone morphogenic protein 4 (Bmp4) is expressed within the palatal shelf mesenchyme and epithelium, in similar or neighboring areas in which Shh is expressed.22 BMP mutations have also been associated with cleft palate; for example, BMPR1B mutation causes Pierre Robin sequence.24
Fibroblast growth factor receptors have been associated with multiple forms of syndromic craniosynostosis which include cleft palate as a feature, including Apert syndrome (FGF2) and Crouzon syndrome (FGF2, FGF3).25, 26, 27 Apert syndrome is characterized by craniofacial malformations, including flat forehead, retracted midface, cleft palate, and hypertelorism, learning disability, poor joint mobility, and severe symmetric syndactyly of fingers and toes, which is the distinguishing feature of Apert syndrome from the other FGFR2‐related syndromes. Crouzon syndrome also presents with flat forehead, midface hypoplasia, and cleft palate, but is associated with less severe facial deformity when compared to Apert syndrome and normal intelligence, hands, and feet.28, 29
The Msx1 homeobox gene is also expressed in the facial promorida and is required for the expression of Bmp2 and Bmp4 in palatal mesenchyme and Shh in medial edge epithelium.30 Msx1 regulates outgrowth of the maxillary prominances.31 Msx1‐deficicient mice demonstrate complete cleft of the secondary palate along with abnormal maxillary development and absent alveolar process.32 In addition, Msx1 has been found to regulate angiogenesis in the developing face; abnormal angiogenesis has been hypothesized to lead to foreshortened maxillary prominances in Msx1 null mice as a result of decreased oxygen and nutrient delivery.31
IRF6
Interferon regulatory factor 6 (IRF6) belongs to a family of nine transcription factors that bind to specific DNA sequences and regulate gene expression. Van der Woude syndrome (VWS) is an autosomal dominant syndrome that is the most common syndromic form of cleft lip or palate. A set of monozygotic twins who were disconcordant for VWS led to the identification of a nonsense mutation in IRF6. IRF6 mutations were then identified in 45 additional unrelated families affected with VWS. Expression analysis then identified high levels of Irf6 mRNA along the medial edge of the fusing palate. Mice lacking Irf6 had multiple anomalies in epithelial development with sygnathia and fused esophageal mucosa, highlighting the importance of transcriptional regulation of the epithlium during cleft lip and palate formation. Similar to TGF‐β3, Irf6 is also expressed along the medial edge of the palate, and IRF6 may be involved in the TGF‐β signaling pathway.33 Patients with VWS have been shown to be at increased risk for wound complications following cleft repair, suggesting that suggesting that IRF6 plays a role in wound healing.34
PDGF
Platelet‐derived growth factor (PDGF) and its receptors PDGFRα and PDGFRβ have been shown to be crucial for cell proliferation, survival, and migration.35 Pdgfc and Pdgfa, which encode PDGF ligands, are expressed in palatal shelf epithelium. PDGFRα, which encodes the receptor, is expressed in mesenchyme.36 PDGFRα mutant mice demonstrated defects in neural crest derivatives contributing to the frontonasal process and subsequently leading to cleft palate.37 Both Pdgfc and Pdgfa mutant mice developed clefting, subepidermal blistering, spina bifida, and other skeletal and vascular defects. Pdgfc null mice displayed failure of palatal shelf elevation, with hypoplastic palatal shelves and improper fusion. Pdgfa null mice displayed a severe cleft palate phenotype.
TBX1
The T‐box transcription factor TBX1 is a mediator of developmental abnormalities and has been shown to be essential for normal palatal elongation and elevation.38 Mutations have been associated with DiGeorge and Velocardiofacial Syndromes phenotypes in mice.39, 40 TBX1 null mouse embryos all exhibited cleft palate, with shortened palatal shelves that were unable to elongate and elevate, as well as increased tongue musculature height. Fifty percent of TBX1 null mice demonstrated palatal‐oral fusions, which tethered the posterior palate. This suggests that TBX1 may also be responsible for maintenance of epithelial integrity in the posterior palate.38
VEGFA
Vascular endothelial growth factor A, a growth factor involved in angiogenesis and ossification has also been shown to be essential for palatal development. Deletion of VEGFa in cranial neural crest cells caused cleft palate in mice, associated with reduced palatal shelf width and with palatal shelves failing to elongate and elevate above the tongue. These mice also displayed abnormal vascular development and less ossification of the maxillary and palatine bones.41
CONCLUSION
Cleft lip and palate are common craniofacial deformities resulting from improper growth or fusion of the facial processes during embryologic development. Their etiology is complex and multifactorial. They can occur as part of one of many chromosomal syndromes, Mendelian single gene disorders, teratogenic effects, and as yet uncharacterized syndromes. Multiple genes and molecular pathways have been implicated in palatal fusion in human and animal studies and additional research to further elucidate the etiology of clefting is ongoing. Understanding the molecular mechanisms of cleft formation will help the clinician anticipate which patients may have difficulties healing, such as those with mutations in Irf6,42, 43 BMP4,44 or the TGF‐β family,45 and in the future hopefully make surgical and medical treatment decisions based on genetic information.
FINANCIAL DISCLOSURES
None
Conflict of Interest: None
BIBLIOGRAPHY
- 1. Vanderas AP. Incidence of cleft lip, cleft palate, and cleft lip and palate among races: a review. Cleft Palate J 1987;24:216–225. [PubMed] [Google Scholar]
- 2. Jones MC. Etiology of facial clefts: prospective evaluation of 428 patients. Cleft Palate J 1988;25:16–20. [PubMed] [Google Scholar]
- 3. Cohen MM Jr. Syndromes with cleft lip and cleft palate. Cleft Palate J 1978;15:306–328. [PubMed] [Google Scholar]
- 4. Venkatesh R. Syndromes and anomalies associated with cleft. Indian J Plast Surg 2009;42 Suppl:S51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wyszynski DF, Beaty TH. Review of the role of potential teratogens in the origin of human nonsyndromic oral clefts. Teratology 1996;53:309–317. [DOI] [PubMed] [Google Scholar]
- 6. Cordero DR, Brugmann S, Chu Y, Bajpai R, Jame M, Helms JA. Cranial neural crest cells on the move: their roles in craniofacial development. Am J Med Genet A 2011;155:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schoenwolf GCB, SB , Brauer PR, Francis‐West PH. Development of the pharyngeal apparatus and face In: Larsen's Human Embryology. 5th ed. Philadelphia, PA: Elsevier, Inc.; 2015:429–472. [Google Scholar]
- 8. Sadler TW. Head and neck embryology In: Sadler T, ed. Langman's Medical Embryology. 6th ed. Baltimore, MD: Williams & Wilkins; 1990:313–320. [Google Scholar]
- 9. Robin NH, Moran RT, Ala‐Kokko L. Stickler syndrome In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews(R). Seattle, WA: University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 10. Dixon J, Jones NC, Sandell LL, et al. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A 2006;103(36):13403–13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Katsanis SH, Jabs EW. Treacher Collins syndrome In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews(R). Seattle, WA: University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 12. Levi B, Brugman S, Wong VW, Grova M, Longaker MT, Wan DC. Palatogenesis: engineering, pathways and pathologies. Organogenesis 2011;7:242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kingsley DM. The TGF‐beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev 1994;8:133–146. [DOI] [PubMed] [Google Scholar]
- 14. Poniatowski ŁA, Wojdasiewicz P, Gasik R, Szukiewicz D. Transforming growth factor beta family: insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediators Inflamm 2015;2015:137823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Inman GJ. Linking Smads and transcriptional activation. Biochem J 2005;386(Pt 1):e1–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Inman GJ, Nicolas FJ, Hill CS. Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF‐beta receptor activity. Mol Cell. 2002;10:283–294. [DOI] [PubMed] [Google Scholar]
- 17. Hill CR, Jacobs BH, Brown CB, Barnett JV, Goudy SL. The type III transforming growth factor beta receptor regulates vascular and osteoblast development during palatogenesis. Dev Dyn 2015;244:122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loeys BL, Dietz HC. Loeys‐Dietz syndrome In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews(R). Seattle, WA: University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 19. Joyce ME, Roberts AB, Sporn MB, Bolander ME. Transforming growth factor‐beta and the initiation of chondrogenesis and osteogenesis in the rat femur. J Cell Biol 1990;110:2195–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Proetzel G, Pawlowski SA, Wiles MV, et al. Transforming growth factor‐β3 is required for secondary palate fusion. Nat Genet 1995;11:1295–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tapadia MD, Cordero DR, Helms JA. It's all in your head: new insights into craniofacial development and deformation. J Anat 2005;207:461–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bitgood MJ, McMahon AP. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell–cell interaction in the mouse embryo. Dev Biol 1995;172:126–138. [DOI] [PubMed] [Google Scholar]
- 23. Lan Y, Jiang R. Sonic hedgehog signaling regulates reciprocal epithelial‐mesenchymal interactions controlling palatal outgrowth. Development 2009;136:1387–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang Y, Yuan J, Yao X, et al. BMPR1B mutation causes Pierre Robin sequence. Oncotarget 2017;8:25864–25871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilkie AOM, Slaney SF, Oldridge M, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet 1995;9:165–172. [DOI] [PubMed] [Google Scholar]
- 26. Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet 1994;8:98–103. [DOI] [PubMed] [Google Scholar]
- 27. Meyers GA, Orlow SJ, Munro IR, Przylepa KA, Jabs EW. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat Genet 1995;11:462–464. [DOI] [PubMed] [Google Scholar]
- 28. Ko JM. Genetic syndromes associated with craniosynostosis. J Korean Neurosurg Soc 2016;59:187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kreiborg S, Cohen MM, Jr . Is craniofacial morphology in Apert and Crouzon syndromes the same? Acta Odontol Scand 1998;56:339–341. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in Msx1‐deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 2002;129:4135–4146. [DOI] [PubMed] [Google Scholar]
- 31. Medio M, Yeh E, Popelut A, Babajko S, Berdal A, Helms JA. Wnt/β‐catenin signaling and Msx1 promote outgrowth of the maxillary prominences. Front Physiol 2012;3:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 1994;6:348–356. [DOI] [PubMed] [Google Scholar]
- 33. Kondo S, Schutte BC, Richardson RJ, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet 2002;32:285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jones JL, Canady JW, Brookes JT, et al. Wound complications after cleft repair in children with Van der Woude syndrome. J Craniofac Surg 2010;21:1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoch RV, Soriano P. Roles of PDGF in animal development. Development 2003;130:4769–4784. [DOI] [PubMed] [Google Scholar]
- 36. Ding H, Wu X, Bostrom H, et al. A specific requirement for PDGF‐C in palate formation and PDGFR‐alpha signaling. Nat Genet 2004;36:1111–1116. [DOI] [PubMed] [Google Scholar]
- 37. Tallquist MD, Soriano P. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development 2003;130:507–518. [DOI] [PubMed] [Google Scholar]
- 38. Goudy S, Law A, Sanchez G, Baldwin HS, Brown C. Tbx1 is necessary for palatal elongation and elevation. Mech Dev 2010;127:292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T‐box gene, Tbx1. Nat Genet 2001;27:286–291. [DOI] [PubMed] [Google Scholar]
- 40. Merscher S, Funke B, Epstein JA, et al. TBX1 is responsible for cardiovascular defects in velo‐cardio‐facial/DiGeorge syndrome. Cell 2001;104:619–629. [DOI] [PubMed] [Google Scholar]
- 41. Hill C, Jacobs B, Kennedy L, et al. Cranial neural crest deletion of VEGFa causes cleft palate with aberrant vascular and bone development. Cell Tissue Res 2015;361:711–722. [DOI] [PubMed] [Google Scholar]
- 42. Ingraham CR, Kinoshita A, Kondo S, et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat Genet 2006;38:1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Richardson RJ, Dixon J, Malhotra S, et al. Irf6 is a key determinant of the keratinocyte proliferation‐differentiation switch. Nat Genet 2006;38:1329–1334. [DOI] [PubMed] [Google Scholar]
- 44. Suzuki S, Marazita ML, Cooper ME, et al. Mutations in BMP4 are associated with subepithelial, microform, and overt cleft lip. Am J Hum Genet 2009;84:406–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ferguson MW, O'Kane S. Scar‐free healing: from embryonic mechanisms to adult therapeutic intervention. Philos Trans R Soc Lond B Biol Sci 2004;359:839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]