

Abstract
Objectives
To describe the clinical presentation of lymphatic malformations (LM) and genotypically associated disorders and to summarize the recent literature regarding the genetic etiology of LM and provide a biologic correlation to medical and surgical management.
Results
LM are congenital lesions derived from a developmental abnormality of the lymphatic vessels. The severity of disease varies widely and complications can occur with higher staged disease and those associated with a known constellation of symptoms. Somatic mutations of the PIK3CA gene have been found to be an etiologic factor in the development of LM and associated overgrowth syndromes. Sirolimus is a mammalian target of rapamycin (mTOR) inhibitor that inhibits the pathway downstream of PIK3CA. Preliminary studies in select groups of patients suggest that sirolimus has a role in the medical management of certain aspects of this disease.
Conclusions
Discovery of LM molecular genetics has led to the possibility of targeted therapies and highlights the importance of precision medicine in rare diseases. Identifying genetic mutations in larger cohorts of patients with LM will lead to additional insights. Knowledge of the genetic basis for disease can then lead to discovery of directed medical therapy. A specific molecular diagnosis can also help families understand better why their child is different and provide accurate counseling for subsequent pregnancies.
Level of Evidence
6
Keywords: Lymphatic malformation, genetics, sirolimus, overgrowth syndrome, molecular genetics, bone, somatic mutation, activating mutation, lymphocytopenia
CLINICAL CHARACTERISTICS OF LYMPHATIC MALFORMATIONS
Vascular anomalies are grouped into “vascular tumors” and “vascular malformations.” Vascular tumors, which include infantile and congenital hemangiomas, are hyperplastic, proliferative lesions. To identify literature pertinent to this report a search was conducted in Ovid MEDLINE and EMBASE, 1995 to June 2018, using a combination of controlled vocabulary and key words for the concepts of all therapeutic interventions for vascular anomalies. Information from this search regarding lymphatic malformations and overgrowth conditions was used to create this report. Vascular malformations are lesions that demonstrate abnormal vascular development and structure.1 Isolated lymphatic malformations (LM) are congenital, arising from disordered lymphatic vessel growth, structure and function.2 The incidence is 1 in 2000 to 4000 live births and 75% occur in the head and neck region.3, 4, 5 LM can be divided radiographically into microcystic lesions (cysts <2 cm) or macrocystic lesions (cysts ≥2 cm).6 Clinically, head and neck LM (HNLM) are staged based on whether or not the HNLM is unilateral or bilateral and the location of the lesion in relationship to the hyoid bone. Higher staged (4 or 5) HNLM represent 15% of all malformations and have a higher rate of surgical complications and a poorer prognosis.7, 8
LYMPHATIC MALFORMATION RELATED TO SYNDROMES
Staging HNLM allows lesion categorization based on disease severity which has been correlated with treatment needs and outcomes.8 Though the majority of HNLM occur spontaneously, outside of the head and neck, there are various syndromes and conditions associated with abnormal lymphatic tissue and a spectrum of other phenotypic characteristics, which in some cases have been shown to be genotypically related to HNLM (Table 1). Many of these syndromes have associated locoregional limb or organ overgrowth, similar to facial skeletal overgrowth that occurs in some HNLM. For example, Klippel‐Trenaunay syndrome (KTS) includes capillary malformation, aberrant veins, limb overgrowth, and LM. Patients with KTS can also experience chronic pain, depression, and thrombosis which can require more extensive treatment.9 CLOVES syndrome is associated with congenital/lipomatous overgrowth, lymphatic/capillary/venous vascular malformations, epidermal nevi, skeletal, and spinal anomalies. The lipomatous overgrowth is often severely disfiguring and can cause scoliosis and restrictive lung disease. Hydrocephalus, Chiari malformations, and epilepsy can also accompany CLOVES.10 Fibroadipose hyperplasia is described as fibrofatty overgrowth in anatomic regions with or without LM.10 The overgrowth causes disfigurement and functional compromise. Some less defined congenital conditions are associated with progressive bone loss that can also be associated with HNLM.8, 11 Gorham‐Stout syndrome or lymphangiomatosis is a rare, lymphatic disorder that is associated with osteolysis. Also called “vanishing bone disease,” this syndrome can lead to pathologic fractures, pain and swelling of the involved areas, and can involve any bone in the head and neck.12
Table 1.
International Society for the Study of Vascular Anomalies 2014 Classification Scheme and Associated Genetic Basis.
| LM | |
| Primary lymphedema | ‐ |
| Nonne‐Milroy syndrome | FLT4/VEGRFR3 |
| Primary hereditary lymphedema | VEGFC |
| Primary hereditary lymphedema | GJC2/CX47 |
| Lymphedema‐distichiasis | FOXC2 |
| Hypotrichosis‐lymphedema‐telangiectasia | SOX18 |
| Primary lymphedema with myelodysplasia | GATA2 |
| Primary generalized lymphatic anomaly | CCBE1 |
| Microcephaly with/without chorioretinopathy | KIF11 |
| Lymphedema or mental retardation syndrome | ‐ |
| Lymphedema‐choanal atresia | PTEN14 |
| Vascular Malformations associated with other anomalies | |
| Klippel‐Trenaunay syndrome | ‐ |
| Parkes Weber syndrome | RASA1/EPHB4 |
| Servelle‐Martorell syndrome | ‐ |
| Sturge‐Weber syndrome | GNAQ |
| Limb capillary malformation + congenital nonprogressive limb overgrowth | ‐ |
| Maffucci syndrome | ‐ |
| Megalencephaly with Capillary Malformations | PIK3CA |
| Microcephaly with Capillary Malformations | STAMBP |
| CLOVES syndrome | PIK3CA |
| Proteus syndrome | AKT1 |
| Bannayan‐Riley‐Ruvalcaba syndrome/Cowden Syndrome | PTEN |
UNEXPLAINED FEATURES OF HEAD AND NECK LYMPHATIC MALFORMATIONS
HNLM can be associated with chronic pain, disfigurement, ulcerations, infection, coagulopathies, bone overgrowth, progressive osteolysis, and even death.13 Large, high‐stage HNLM cause significant morbidity and mortality via airway obstruction or airway bleeding, facial skeletal overgrowth and recurrent infections. Lesion persistence and disfigurement following large HNLM treatment extends beyond the mass itself as there is also adjacent tissue and bony overgrowth.14 Due to this soft and bony tissue overgrowth, excision or ablative treatment of the lesion will not completely rid the patient of the residual asymmetry or overgrowth, making current therapy inadequate. The factors causing association of some HNLM with progressive bone loss, chronic pain, and hematologic abnormalities, such as lymphocytopenia, are currently unknown.
GENETIC ETIOLOGY OF HEAD AND NECK LYMPHATIC MALFORMATION
Within the past decade, the genetic etiology of HNLM and other rare overgrowth conditions has been explored and have led to a better understanding of the formation of these lesions, why treatment has not always been effective, and possible new treatment options.15 Genetic mutations can be divided into somatic or germline mutations.15 Germline mutations are alterations in the genetic sequence of the gamete, involve all cells in the organism and can be passed on to future generations. Somatic mutations are post‐zygotic DNA variations occurring after fertilization that are more difficult to detect without the use of advanced DNA sequencing technologies.15 These mutations are passed on to the progeny of the surviving affected cell; however, the passing of the mutation to the offspring of the individual depends on whether or not the gonads receive the mutation.16
The most commonly identified mutations found in vascular anomalies involve the tyrosine kinase receptor signaling pathway (Fig. 1). Through this signaling, the RAS or phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K) pathway can be activated. The most commonly associated mutation in HNLM is a postzygotic somatic mutation in the phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit alpha (PIK3CA) gene.17 Three locations or “hotspots” within PIK3CA are frequently involved in sporadic human cancers and are thought to enhance tissue overgrowth.18, 19, 20 Samples of affected tissue from patients with isolated HNLM, CLOVES, KTS, or fibroadipose hyperplasia are required to detect PIK3CA hotspot mutations.21 The majority of the tissue samples revealed activating hotspot mutations in PIK3CA.21 Interestingly, these exact same mutations are some of the most commonly identified mutations in a large number of human cancers. It is unknown why PIK3CA mutations that occur during early embryonic development can lead to the mosaic, vascular overgrowth phenotypes, and yet the exact same mutations occurring in adult tissues can promote tumorigenesis. Clearly, there is much to discover regarding the molecular pathophysiology of these entities.22 Future work is necessary to understand the molecular pathophysiology of PIK3CA and how it contributes to a variety of rare conditions that occur throughout the body, not just the head and neck.
Figure 1.
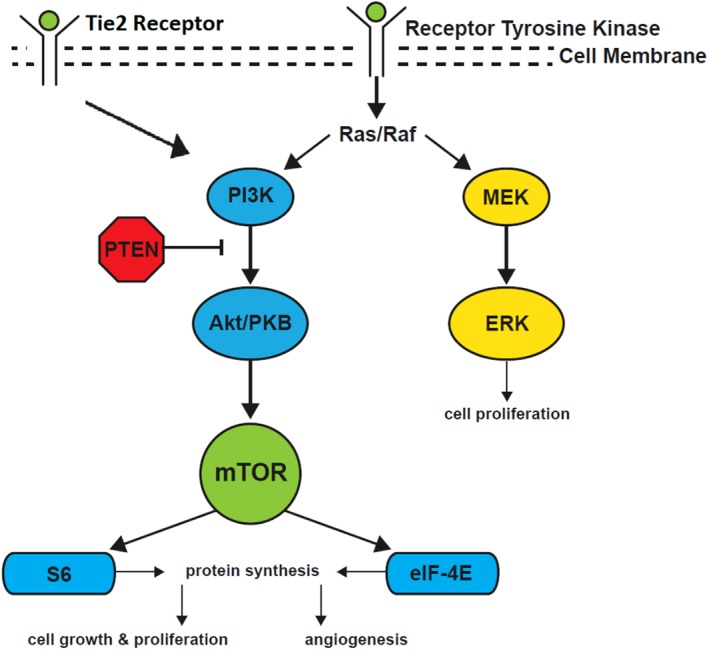
PI3K pathway leading to cell growth and proliferation.
THERAPY FOR HEAD AND NECK LYMPHATIC MALFORMATIONS BASED ON MALFORMATION GENOTYPE
The treatment options for HNLM have included surgical resection, laser therapy, and sclerotherapy ablation. With the application of molecular genetics to HNLM, the possibility of targeted primary or adjunctive medical therapy and/or surgery has become real. As previously stated, LM tissue and associated overgrowth can recur after initial treatment and may be difficult to completely eradicate.8, 23 Targeted medical therapy to cells with activating somatic mutations, as determined with directed genetic testing, could theoretically aid in regression or prevention of disease recurrence.17 Rapamycin, also known as sirolimus, is a macrolide produced by Streptomyces hygroscopicus. This drug inhibits a downstream component of the PI3K pathway, mTOR, an acronym for the “mammalian target of rapamycin” (Fig. 1).24 The mTOR enzyme is the master switch for cellular growth or destruction.24 Inhibition of mTOR is effective in halting disease processes related to cellular proliferation and has been widely used to inhibit B‐ and T‐cell activation to avoid posttransplant rejection.25 Although many studies have focused on the role of PI3K activation and inhibition in vascular endothelial cells, it is important to keep in mind that sirolimus is an immunosuppressant, and it may have greater than one mechanism of action in LM. Clinical response of HNLM or LM associated syndromes/disorders such as Gorham‐Stout disease, kaposiform hemangioendothelioma, combined lymphatico‐venous malformation, pulmonary lymphangectasia, generalized lymphangiomatosis, and LM of the tongue to sirolimus has varied.26, 27, 28, 29, 30, 31, 32, 33 Literature to date does not yet distinguish which patients are the best candidates for sirolimus and what true benefits it has. A phase 2 trial analyzed the outcomes after empiric use of sirolimus in patients with complicated LM throughout the body. This study showed a decreased incidence of cellulitis and a decrease in hospitalized days and frequency of infections. Metabolic toxicity (3%), gastrointestinal disturbance (3%), and blood/bone marrow abnormalities (27%) were the reported adverse effects of the medication. Patients were included in the trial based on their clinical presentation; genetic testing was not performed to confirm a PIK3CA mutation. Another study described 19 high‐stage HNLM treated with sirolimus and showed qualitative reduction of malformation size. Additionally, oral mucosal bleeding and presence of mucosal vesicles improved in some cases.34
Currently, there is not enough evidence to identify which patients would benefit most from sirolimus. It is also unknown at what age and dose it is appropriate to begin therapy and how to use this medication as an adjunct to surgical excision.35 Directed genetic testing for PIK3CA mutations in lesion tissue provide clues to which patients will benefit from mTOR inhibition. Better understanding the pharmacology of sirolimus in the setting of specific PIK3CA mutations in HNLM tissue is the only way to predict the efficacy of sirolimus. It may be that inhibition of other components of the PI3K pathway are more effective for treatment of HNLM.36
Another consideration is genetic testing to direct HNLM therapy with selective use of surgery to remove all cells with mutations in smaller HNLM. There are many phenotypic presentations of HNLM and some may not need combined medical and surgical therapy; instead, these lesions could be managed with surgical excision alone, potentially avoiding any biologic agents and their associated adverse effects. For large lesions that defy complete removal, a combination of medical and invasive therapy could allow for optimal treatment outcomes. Further research is necessary to answer these questions.
CONCLUSIONS
The advent of sophisticated genetic testing has furthered our understanding of HNLM and has introduced directed treatment modalities. An objective molecular etiology can also provide families and individuals with HNLMs knowledge that can assist with coping and acceptance of this disease. Creating standardized outcome measures to systematically collect data for medical trial studies in various LM populations is needed.37 Use of sirolimus as a medical treatment modality for HNLM has made the need for standardized reporting all the more urgent. Precision medicine bases treatment decisions on specific molecular, genetic and other biologic factors unique to an individual patient's condition. HNLM research is making precision based treatment a possibility for patients with HNLM and other rare conditions with LM.
Funding: Seattle Children's Guild Association Funding Focus Award, NIH RO1 NS092772 and NIHMS 905693.
Conflicts of Interest: None
BIBLIOGRAPHY
- 1. Enjolras O, Mulliken JB. Vascular tumors and vascular malformations (new issues). Adv Dermatol 1997;13:375–423. [PubMed] [Google Scholar]
- 2. Boscolo E, Coma S, Luks VL, et al. AKT hyper‐phosphorylation associated with PI3K mutations in lymphatic endothelial cells from a patient with lymphatic malformation. Angiogenesis 2015;18:151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kennedy TL, Whitaker M, Pellitteri P, Wood WE. Cystic hygroma/lymphangioma: a rational approach to management. Laryngoscope 2001;111:1929–1937. [DOI] [PubMed] [Google Scholar]
- 4. Churchill P, Otal D, Pemberton J, Ali A, Flageole H, Walton JM. Sclerotherapy for lymphatic malformations in children: a scoping review. J Pediatr Surg 2011;46:912–922. [DOI] [PubMed] [Google Scholar]
- 5. Schoinohoriti OK, Theologie‐Lygidakis N, Tzerbos F, Iatrou I. Lymphatic malformations in children and adolescents. J Craniofac Surg 2012;23:1744–1747. [DOI] [PubMed] [Google Scholar]
- 6. Malic CC, Guilfoyle R, Courtemanche RJM, Arneja JS, Heran MKS, Courtemanche DJ. Lymphatic malformation architecture: implications for treatment with OK‐432. J Craniofac Surg 2017;28:1721–1724. [DOI] [PubMed] [Google Scholar]
- 7. de Serres LM, Sie KC, Richardson MA. Lymphatic malformations of the head and neck. A proposal for staging. Arch Otolaryngol Head Neck Surg 1995;121:577–582. [DOI] [PubMed] [Google Scholar]
- 8. Balakrishnan K, Menezes MD, Chen BS, Magit AE, Perkins JA. Primary surgery vs primary sclerotherapy for head and neck lymphatic malformations. JAMA Otolaryngol Head Neck Surg 2014;140:41–45. [DOI] [PubMed] [Google Scholar]
- 9. Harvey JA, Nguyen H, Anderson KR, et al. Pain, psychiatric comorbidities, and psychosocial stressors associated with Klippel‐Trenaunay Syndrome. J Am Acad Dermatol 2018;79(5):899–903. [DOI] [PubMed] [Google Scholar]
- 10. Mirzaa G, Conway R, Graham JM Jr, Dobyns WB. PIK3CA‐Related Segmental Overgrowth In: Adam MP, Ardinger HH, Pagon RA, et al. eds. GeneReviews((R)). Seattle (WA): University of Washington, Seattle; 1993. –2018. [Google Scholar]
- 11. Lee JH, Huynh M, Silhavy JL, et al. De novo somatic mutations in components of the PI3K‐AKT3‐mTOR pathway cause hemimegalencephaly. Nat Genet 2012;44:941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nikolaou VS, Chytas D, Korres D, Efstathopoulos N. Vanishing bone disease (Gorham‐Stout syndrome): a review of a rare entity. World J Orthop 2014;5:694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adams DM, Trenor CC, 3rd , Hammill AM, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics 2016;137:e20153257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gilony D, Schwartz M, Shpitzer T, Feinmesser R, Kornreich L, Raveh E. Treatment of lymphatic malformations: a more conservative approach. J Pediatr Surg 2012;47:1837–1842. [DOI] [PubMed] [Google Scholar]
- 15. Shendure J, Balasubramanian S, Church GM, et al. DNA sequencing at 40: past, present and future. Nature 2017;550:345–353. [DOI] [PubMed] [Google Scholar]
- 16. Karki R, Pandya D, Elston RC, Ferlini C. Defining "mutation" and "polymorphism" in the era of personal genomics. BMC Med Genomics 2015;8:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Greene AK, Goss JA. Vascular anomalies: from a clinicohistologic to a genetic framework. Plast Reconstr Surg 2018;141:709e–717e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kinross KM, Montgomery KG, Kleinschmidt M, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest 2012;122:553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304:554. [DOI] [PubMed] [Google Scholar]
- 20. Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol 2006;18:77–82. [DOI] [PubMed] [Google Scholar]
- 21. Luks VL, Kamitaki N, Vivero MP, et al. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr 2015;166:1048–1054 e1041–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keppler‐Noreuil KM, Rios JJ, Parker VE, et al. PIK3CA‐related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A 2015;167A:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Odeyinde SO, Kangesu L, Badran M. Sclerotherapy for vascular malformations: complications and a review of techniques to avoid them. J Plast Reconstr Aesthet Surg 2013;66:215–223. [DOI] [PubMed] [Google Scholar]
- 24. Vignot S, Faivre S, Aguirre D, Raymond E. mTOR‐targeted therapy of cancer with rapamycin derivatives. Ann Oncol 2005;16:525–537. [DOI] [PubMed] [Google Scholar]
- 25. Sasongko TH, Ismail NF, Zabidi‐Hussin Z. Rapamycin and rapalogs for tuberous sclerosis complex. Cochrane Database Syst Rev 2016;7:CD011272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alemi AS, Rosbe KW, Chan DK, Meyer AK. Airway response to sirolimus therapy for the treatment of complex pediatric lymphatic malformations. Int J Pediatr Otorhinolaryngol 2015;79:2466–2469. [DOI] [PubMed] [Google Scholar]
- 27. Cramer SL, Wei S, Merrow AC, Pressey JG. Gorham‐Stout disease successfully treated with sirolimus and zoledronic acid therapy. J Pediatr Hematol Oncol 2016;38:e129–132. [DOI] [PubMed] [Google Scholar]
- 28. Dvorakova V, Rea D, O'Regan GM, Irvine AD. Generalized lymphatic anomaly successfully treated with long‐term, low‐dose sirolimus. Pediatr Dermatol 2018;35(4):533–534. [DOI] [PubMed] [Google Scholar]
- 29. Lackner H, Karastaneva A, Schwinger W, et al. Sirolimus for the treatment of children with various complicated vascular anomalies. Eur J Pediatr 2015;174:1579–1584. [DOI] [PubMed] [Google Scholar]
- 30. Laforgia N, Schettini F, De Mattia D, Martinelli D, Ladisa G, Favia V. Lymphatic malformation in newborns as the first sign of diffuse lymphangiomatosis: successful treatment with sirolimus. Neonatology 2016;109:52–55. [DOI] [PubMed] [Google Scholar]
- 31. Lagreze WA, Joachimsen L, Gross N, Taschner C, Rossler J. Sirolimus‐induced regression of a large orbital lymphangioma. Orbit 2018:1–2. [DOI] [PubMed] [Google Scholar]
- 32. Yesil S, Tanyildiz HG, Bozkurt C, Cakmakci E, Sahin G. Single‐center experience with sirolimus therapy for vascular malformations. Pediatr Hematol Oncol 2016;33:219–225. [DOI] [PubMed] [Google Scholar]
- 33. Yesil S, Bozkurt C, Tanyildiz HG, et al. Successful treatment of macroglossia due to lymphatic malformation with sirolimus. Ann Otol Rhinol Laryngol 2015;124:820–823. [DOI] [PubMed] [Google Scholar]
- 34. Strychowsky JE, Rahbar R, O'Hare MJ, Irace AL, Padua H, Trenor CC 3rd. Sirolimus as treatment for 19 patients with refractory cervicofacial lymphatic malformation. Laryngoscope 2018;128:269–276. [DOI] [PubMed] [Google Scholar]
- 35. Czechowicz JA, Long‐Boyle JR, Rosbe KW, Mathes EF, Frieden IJ, Shimano KA. Sirolimus for management of complex vascular anomalies ‐ A proposed dosing regimen for very young infants. Int J Pediatr Otorhinolaryngol 2018;105:48–51. [DOI] [PubMed] [Google Scholar]
- 36. Venot Q, Blanc T, Rabia SH, et al. Targeted therapy in patients with PIK3CA‐related overgrowth syndrome. Nature 2018;558:540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Balakrishnan K, Bauman N, Chun RH, et al. Standardized outcome and reporting measures in pediatric head and neck lymphatic malformations. Otolaryngol Head Neck Surg 2015;152:948–953. [DOI] [PubMed] [Google Scholar]