

Abstract
The nuclear receptor retinoic acid-related orphan receptor-α (RORα) regulates numerous critical biological processes, including central nervous system development, lymphocyte differentiation, and lipid metabolism. RORα has been recently identified in the heart, but very little is known about its role in cardiac physiology. We sought to determine whether RORα regulates myocardial hypertrophy and cardiomyocyte survival in the context of angiotensin II (ANG II) stimulation. For in vivo characterization of the function of RORα in the context of pathological cardiac hypertrophy and heart failure, we used the “staggerer” (RORαsg/sg) mouse, which harbors a germline mutation encoding a truncated and globally nonfunctional RORα. RORαsg/sg and wild-type littermate mice were infused with ANG II or vehicle for 14 days. For in vitro experiments, we overexpressed or silenced RORα in neonatal rat ventricular myocytes (NRVMs) and human cardiac fibroblasts exposed to ANG II. RORαsg/sg mice developed exaggerated myocardial hypertrophy and contractile dysfunction after ANG II treatment. In vitro gain- and loss-of-function experiments were consistent with the discovery that RORα inhibits ANG II-induced pathological hypertrophy and cardiomyocyte death in vivo. RORα directly repressed IL-6 transcription. Loss of RORα function led to enhanced IL-6 expression, proinflammatory STAT3 activation (phopho-STAT3 Tyr705), and decreased mitochondrial number and function, oxidative stress, hypertrophy, and death of cardiomyocytes upon ANG II exposure. RORα was less abundant in failing compared with nonfailing human heart tissue. In conclusion, RORα protects against ANG II-mediated pathological hypertrophy and heart failure by suppressing the IL-6-STAT3 pathway and enhancing mitochondrial function.
NEW & NOTEWORTHY Mice lacking retinoic acid-related orphan receptor-α (RORα) develop exaggerated cardiac hypertrophy after angiotensin II infusion. Loss of RORα leads to enhanced IL-6 expression and NF-κB nuclear translocation. RORα maintains mitochondrial function and reduces oxidative stress after angiotensin II. The abundance of RORα is reduced in failing mouse and human hearts.
Keywords: angiotensin II; fibroblasts; group F, member 1; hypertrophy; left ventricular; mitochondria; nuclear; nuclear receptor subfamily 1; retinoic acid-related orphan receptor-α; signal transducer and activator of transcription 3
INTRODUCTION
The contributions of the nuclear receptor superfamily to regulating cardiac physiology through ligand-dependent transcriptional regulation have increasingly been recognized (17, 27). Although cardiomyocyte nuclear receptors are best known for regulating metabolism, they also participate in numerous other cellular functions (42), including the transcriptional regulation of sarcomeric and Ca2+-handling genes (8). The retinoic acid-related orphan receptor (ROR) subfamily of nuclear receptors consists of RORα, RORβ, and RORγ (NR1F1–NR1F 3) (6, 19). RORs regulate transcription by binding to ROR response elements (ROREs) consisting of the consensus sequence AGGTCA preceded by a 6-bp AT-rich region in the promoter region of target genes (11). Endogenous ligands have been identified, including several cholesterol and vitamin D metabolites, and a number of recently developed synthetic ligands function either as RORα agonists or inverse agonists (21, 39, 41).
RORα is expressed abundantly in cerebellar Purkinje cells and exhibits a critical role in cerebellar development (12). RORα also plays a key role in the regulation of several metabolic pathways in the liver (33) and skeletal muscle (25) and controls differentiation of T helper cells, including the generation of innate lymphoid 2 cells (18). RORα has been recently identified in the heart (15). It protects against myocardial ischemia-reperfusion injury (15), but its other myocardial functions and its operative pathways remain unknown.
In the present study, we investigated whether RORα regulates angiotensin II (ANG II)-induced pathological cardiomyocyte hypertrophy and heart failure using the “staggerer” (RORαsg/sg) mouse model, which contains a natural deletion mutation in the Rora locus (14), and complementary in vitro approaches. We found that RORαsg/sg mice undergo enhanced pathological ventricular remodeling after ANG II infusion, which is characterized by exaggerated cardiomyocyte hypertrophy, fibrosis, cell death, and mitochondrial depletion. Loss of RORα facilitates the activation of critical proinflammatory IL-6 and NF-κB pathways, leading to phosphorylation of proinflammatory STAT3 Tyr705 rather than adaptive mitochondrial STAT3 Ser727. These findings expand our limited understanding of the role of RORα in the heart and suggest a protective role for RORα in the pathobiology of heart failure.
MATERIALS AND METHODS
Experimental animals.
Heterozygous RORαsg/sg mice on a C57BL/6J background were purchased from The Jackson Laboratory and maintained as previously described (22). Homozygous mice RORαsg/sg mice, the products of heterozygous breeding, and wild-type (WT) littermates were used in all experiments at 12–16 wk of age. All animal experiments followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and animal protocols were approved by the National Institute of Environmental Health Sciences Animal Care and Use Committee.
Mouse echocardiography.
To evaluate cardiac structure and function in vivo, transthoracic echocardiography was performed on conscious, loosely restrained mice at the National Institute of Environmental Health Sciences using a VisualSonics Vevo 770 ultrasound system (VisualSonics, Toronto, ON, Canada). Two-dimensional and M-mode echocardiography were performed in the parasternal long-axis view at the level of the papillary muscles. Left ventricular (LV) systolic function was assessed by fractional shortening (FS; in %) calculated as follows: FS = [(LVEDD − LVESD)/LVEDD] × 100, where LVEDD is LV end-diastolic dimension and LVESD is LV end-systolic dimension. Reported FS values are the average of at least five cardiac cycles per mouse. Sonographers and investigators were blinded to mouse treatment conditions during image acquisition and analysis.
Histology and immunofluorescence microscopy.
To fix heart tissue for immunohistochemistry, mice were heparinized, and the heart was perfused with 10 ml of PBS followed by 20 ml of 4% paraformaldehyde (PFA)-PBS through a 23-gauge butterfly needle and then excised and placed in 4% PFA-PBS for 24 h before transfer to 70% ethanol. Hearts were sectioned and stained using standard methods in the University of North Carolina Histology Research Core. Slides were scanned using an Aperio ScanScope (Aperio Technologies, Vista, CA) and analyzed in Aperio ImageScope software. At least three sections from three or more mice within each group were examined for automated quantitation of fibrosis by Masson trichrome and cardiomyocyte cross-sectional area by wheat germ agglutinin (WGA).
For Masson trichrome, the algorithm positive pixel count version 9 was used to measure collagen staining by Masson trichrome using hue value (0.66) and hue width (0.1) The number positive/number total value was used to determine weighted average fibrosis (in %) for each section. For immunohistochemistry, hearts were fixed overnight in 4% PFA-PBS, incubated in 30% sucrose-PBS, and then frozen in OTC medium (Tissue-Tek, Hatfield, PA). Frozen sections (10 μm) obtained with a Leica cryostat (Leica, Buffalo Grove, IL) were placed on glass slides, dried at room temperature, and then incubated with primary antibodies against WGA. After being washed, sections were incubated for 3 h at room temperature with anti-mouse, anti-rabbit, anti-goat, or anti-rat Alexa Fluor 488- or 594-conjugated secondary antibodies (1:1,000, Life Technologies, Grand Island, NY). Fluorescence was observed with a Zeiss LSM710 confocal microscope.
Quantitative real-time RT-PCR.
Total RNA from WT and RORαsg/sg mouse hearts was isolated with a RNeasy mini kit (Qiagen, Valencia, CA) or RNAqueous Micro RNA isolation kit (Ambion, Austin, TX) following the manufacturers’ instructions. RNA was reverse transcribed using a High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). Quantitative real-time RT-PCRs were carried out in triplicate in a LightCycler 480 System (Roche Diagnostics, Indianapolis, IN) using either probe/primer sets or SYBR Green I. Relative quantitation of PCR products used the ΔΔCT method (where CT is threshold cycle) relative to two validated reference genes [TATA box-binding protein (Tbp) and RNA polymerase II subunit A (Polr2a)]. All probes and primers were from Roche Diagnostics or ThermoFisher.
Reference genes were as follows: mouse Tbp, forward 5′-GGCGGTTTGGCTAGGTTT-3′ and reverse 5′-GGGTTATCTTCACACACCATGA-3′; rat Tbp, forward 5′-GGGGAGCTGTGATGTGAAGT-3′ and reverse 5′-CCAGGAAATAATTCTGGCTCATA-3′; mouse Polr2a, forward 5′-AATCCGCATCATGAACAGTG-3′ and reverse 5′-TCATCATCCATTTTATCCACCA-3′; and rat Polr2a, forward 5′-TTCGGCTCAGTGGAGAGG-3′ and reverse 5′-GCTCCCACCATTTCTCCAG-3′. Target genes were as follows: mouse atrial natriuretic factor (ANF), forward 5′-CACAGATCTGATGGATTTCAAGA-3′ and reverse 5′-CCTCATCTTCTACCGGCATC-3′; rat ANF, forward 5′-CACAGATCTGATGGATTTCAAGA-3′ and reverse 5′-CCTCATCTTCTACCGGCATC-3′; mouse myosin heavy chain (MHC)-β (Myh7), forward 5′-CTGCAGGACCTGGTGGAC-3′ and reverse 5′-GGAACTTGGACAGGTTGGTG-3′; rat MHC-β (Myh7), forward 5′-CTCCACGCACCCTCACTT-3′ and reverse 5′-CATGACCAGGGGGTTGTC-3′; mouse RORα, forward 5′-GAGGTATCTCAGTCACGAAG-3′ and reverse 5′-AACAGTTCTTCTGACGAGGACAGG-3′; rat RORα, forward 5′-ACCAGCATCTGGCTTCTTCCCCT-3′ and reverse 5′-GTGAAGGTGTTCTAGTTCCGCCA-3′; mouse MHC-α (Myh6), forward 5′-CCAAGACTGTCCGGAATGA-3′ and reverse 5′-TCCAAAGTGGATCCTGATGA-3′; rat MHC-α (Myh6), forward 5′-TGCAGAAGAAACTGAAGGAAAA-3′ and reverse 5′-GCTCGGCCTCTAGCTCCT-3′; mouse IL-6, forward 5′-ACCTGTCTATACCACTTC-3′ and reverse 5′-GCATCATCGTTGTTCATA-3′; rat IL-6, 5′-TAGTGTGCTATGCCTAAG-3′ and reverse 5′-TATTGCCAGTTCTTCGTA-3′; mouse TNF-α, forward 5′-TTCTGTCTACTGAACTTC-3′ and reverse 5′-CCATAGAACTGATGAGAG-3′; rat TNF-α, forward 5′-CCAATCTGTGTCCTTCTAA-3′ and reverse 5′-TTCTGAGCATCGTAGTTG-3′; mouse STAT3, 5′-CAAGATTGACCTAGAGAC-3′ and reverse 5′-TTATACCACAGGATTGATG-3′; mouse glycoprotein 130 (gp130), forward 5′-GCTCCTTGCTGTTGGTAT-3′ and reverse 5′-TTATACCACAGGATTGATG-3′.
Osmotic minipumps.
For continuous in vivo infusion, micro-osmotic pumps (model 1002, Alzet) releasing ANG II (1.4 μg·kg−1·min−1 in 0.9% NaCl-0.01N acetic acid, Sigma) or vehicle were implanted subcutaneously after anesthesia with ketamine (30 mg/kg ip)-xylazine (10 mg/kg ip).
Immunoblotting.
Whole tissue or cell lysates were produced in RIPA buffer supplemented with PhosSTOP (Roche Diagnostics, Indianapolis, IN) and protease inhibitor cocktail (Roche Diagnostics). Subsequently, samples were incubated in 4× LDS sample buffer, including 2% β-mercaptoethanol, for 10 min at 70°C. SDS-PAGE and immunoblot analysis were performed using the 4–12% Nupage gel system (Life Technology, Foster City, CA). Membranes were blocked in 5% milk-Tris-buffered saline-Tween 20 and incubated in primary antibody overnight at 4°C and then secondary horseradish peroxidase (HRP)-conjugated antibodies for 1 h at room temperature. Images were generated using Amersham ECL Select Western Blotting Detection Reagent (GE Healthcare Life Sciences, Marlborough, MA) and the MultiDoc-It Imaging System (UVP Gel Image System, UVP, Upland, CA).
Antibodies.
The following antibodies were used: RORα (N2N3, 1:1,000, Genetex, Irvine, CA); GAPDH (MAB374 clone 6C5, 1:10,000, Millipore); ANP (ab14348, 1:1,000, Abcam, Cambridge, MA); pan collagen (Maine Medical Center Research Institute, Scarborough, ME); STAT3 (no. 9139, 1:1,000), histone H3 (no. 4499, 1:1,000), cofilin (no. 5175, 1:1,000), IκBα (no. 4812, 1:500), p-IκBα (no. 2859, 1:500), cytochrome c (no. 12963, 1:2,000), poly(ADP-ribose) polymerase (PARP; no. 9532, 1:1,000), cleaved PARP (no. 5625, 1:1,000), and phosphorylated (p-)STAT3/Tyr705 (no. 9145, 1:1000) (all from Cell Signaling Technology, Danvers, MA); p-STAT3 Ser727 (no. 44-384G, 1:1,000, ThermoFisher Scientific); NF-κB (sc-8008, 1:1,000), IL-6 (sc-1265, 1:500), and translocase of outer membrane 20 (TOM20; sc-17764, 1:1000) (all from Santa Cruz Biotechnology, Dallas, TX); an α-actinin (A7811, 1:1,000), Myh7 (M8421), polyclonal goat anti-rabbit IgG/HRP (A9169, 1:5,000), polyclonal rabbit anti-mouse IgG/HRP (A9044, 1:5,000), and polyclonal rabbit anti-goat IgG-HRP (A5420, 1:5,000) (all from Sigma-Aldrich, St. Louis, MO).
Neonatal rat ventricular myocyte cultures, immunocytochemistry, and lentiviral infections.
Female Sprague-Dawley rats and newborn litters were from Charles River. Neonatal rat ventricular myocyte (NRVMs) were isolated as previously described (37). Experiments were carried out after 36–96 h of serum starvation in the presence of insulin, transferrin, and BrdU. To visualize NRVM size and sarcomeric organization, cells were stained for α-actinin (1:500, Sigma Aldrich) and F-actin with phalloidin Alexa 594 (1:1,000, Life Technologies, Grand Island, NY). Nuclear staining was performed with ProLong Diamond Antifade Mounting Medium (Life Technologies) with DAPI. NRVM hypertrophy was induced by stimulation for 24 h with ANG II (200 nM) after infection with empty lentivirus or inducible RORα-lentivirus (pINDUCER21-RORA, Addgene plasmid no. 51303, a gift from George Daley). Lentiviral shRNAs (shControl and shRORα) were designed specifically for the rat (iO51217 or iV051217, ABM, Richmond, BC, Canada) or human (TRCN0000022154, Sigma-Aldrich).
Rat target sequences were as follows: 5′-TGTCATTACGTGTGAAGGCTGCAAGGGCT-3′, 5′-ACCTACAACATCTCAGCCAATGGGCTGAC-3′, 5′-GGACTGGACATCAATGGGATCAAACCCGA-3′, and 5′-AGAGGTGATGTGGCAGTTGTGTGCTATCA-3′. The human target sequence was as follows: 5′-CCGGCCAGACATTGTGCGACTTCATCTCGAGATGAAGTCGCACAATGTCTGGTTTTT-3′.
Human cardiac fibroblast cultures.
Human primary cardiac fibroblasts (CFBs) from a 63-yr-old white man were provided kindly by Dr. Li Qian (University of North Carolina McAllister Heart Institute). Human CFBs were cultured in human cardiac fibroblast growth medium (Cell Applications, San Diego, CA).
Isolation of mitochondria.
Mitochondria from frozen heart tissue were isolated using a mitochondria isolation kit (ab110168, Abcam). Tissue was briefly washed in isolation buffer, dried with Whatman filter paper, weighed, and then placed in glass beaker, thoroughly minced, and homogenized with Dounce homogenizer. The homogenate was centrifuged at 1,000 g for 10 min at 4°C. The supernatant was centrifuged at 12,000 g for 15 min at 4°C and then saved as crude cytosolic and nuclear fractions for further purification. The homogenate pellets were resuspended in isolation buffer with protease and phosphatase inhibitor cocktails (Roche Diagnostics). The protein concentration was measured by a BCA protein assay kit (Thermo Scientific, Waltham, MA).
RORE and IL-6 reporter assays.
For RORE reporter assays, H9c2 rat myoblasts were transfected with four copies of the RORE sequence (6-bp AT-rich sequence preceding the GGTCA core motif, 5′-CGCGTGGTAAGTAGGTCACTCTC-3′) inserted in CAT reporter vector. For IL-6 reporter assays, H9c2 cells were transfected with plasmids, including the promoter region of mouse IL-6 [−1277, a gift from Gail Bishop (Addgene plasmid nos. 61286 and 61293)]. H9c2 cells were cotransfected with IL-6 reporter plasmid, pCMV-β-Gal, pCMV10-3xFlag-RORα, or pCMV10-3xFlag-Empty using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and varying doses of pGL4-RORα plasmid (0, 0.1, and 0.2 μg). After 24 h of incubation, luciferase and β-galactosidase activities were measured by Luciferase Assay Substrate (Promega, Madison, WI) and a Luminescent β-galactosidase Detection Kit II (Clontech, Mountain View, CA). The transfection efficiency in each sample was normalized by β-galactosidase activity. All transfections were performed in triplicate and repeated at least twice.
IL-6 promoter constructs and analysis of promoter activities by luciferase assay.
To quantify the role of RORα in IL-6 promoter activity, a DNA fragment of the mouse IL-6 gene promoter, including −1,044 bp from the transcription start site, was synthesized by GenScript (Piscataway, NJ). This fragment was inserted between the KpnI and HindIII sites in front of the luciferase reporter gene in the pGL 4.10 Luc vector (Promega, Madison, WI) and used as the IL-6 WT promoter pGL4.10. The generated construct was confirmed by sequencing. A reporter plasmid with a mutation (as underlined) in the putative ROREs (RORα‐binding sites) at −935/−928 (5′‐TGACCTG‐3′) or at −555/−548 (5′-CAGGTCA-3′), and including the KpnI and HindIII sites in the IL-6 promoter, was generated by GenScript. The fragment was inserted between the KpnI and HindIII sites in the IL-6 WT promoter pGL4.10 plasmid construct and used as IL6-mut#1 (mutated at −935/−928) or IL6-mut#2 (mutated at −555/−548). To examine RORα transcriptional activity on the IL-6 promoter, pGL4.10_IL6 WT, _IL6-mut#1 (mutated at −858/−852), or _IL6-mut#2 (mutated at −478/−472) was cotransfected with p3xFLAG-CMV or p3xFLAG-CMV-RORα and pCMV-β-Gal in H9c2 myocytes using Lipofectamine 2000 per the manufacturer’s protocol. Cells were harvested and lysed in 1× passive lysis buffer (Promega) at 24 h posttransfection. Luciferase and β-galactosidase activities were measured by Luciferase Assay Substrate (Promega) and the Luminescent β-galactosidase Detection Kit II (Clontech), respectiviely. Luciferase activity was normalized by β-galactosidase activity. All transfections were performed in triplicate and repeated at least twice.
Chromatin immunoprecipitation-PCR.
To demonstrate direct transcriptional regulation of IL-6 by RORα, H9c2 cells (3 × 106 cells) treated with vehicle or ANG II (200 nM) for 24 h were washed in PBS containing protease and phosphatase inhibitor cocktail (PIC; Thermo Scientific) and then fixed in 1% formaldehyde at 25°C for 8 min for cross-linking and followed by quenching by 0.125 M glycine at 25°C for 8 min. After a wash in PBS and lysis in IP lysis buffer (catalog no. 87788, ThermoFisher Scientific) containing PIC, cross-linked chromatin was sonicated for 20 min (S220 focused ultrasonicator, Covaris) and then incubated overnight with RORα antibody generated against amino acids 121–213 of mouse RORα4 or control IgG antibody. After an incubation with protein G agarose beads for 2 h, DNA-protein complexes were eluted. The cross-links were reversed by an overnight incubation at 65°C with 25 mM NaCl, RNase A, and proteinase K, and then chromatin immunoprecipitated (ChIPed)-DNA was purified. The amount of ChIPed-DNA relative to each input DNA was determined by quantitative PCR in triplicate with primers amplifying RORE or nontargeting Polr2a) in the IL6 promoter.
Chromatin immunoprecipitation (ChIP)-PCR primers (rat) were as follows: Il6-RORE, forward 5′-CAACGAGGTCCTTCTTTGCT-3′ and reverse 5′-TAGTGCTGATCCCACTGCTG-3′; Il6-non-RORE, forward 5′-CAAGCTCTGGGGTTGTCAAG-3′ and reverse 5′-CCCCGAAGCCTTCTGTTAAG-3′; IL6-3.2k, forward 5′-GGACACAGTCAGCCCCATAC-3′ and reverse 5′-AAAAATTCGGGGACTTCAGC-3′; and IL6-2.9k, forward 5′-CAAATGGGAAACCTGTCAAGA-3′ and reverse 5′-TGGCATTTGTTTATGGGAGA-3′.
ATP bioluminescence assay.
ATP content was determined using a luciferin-luciferase ATP assay (A22066, ThermoFisher Scientific) according to the manufacturer’s protocol. All reagents were placed on ice before the experiments were carried out. The ATP solution (100 nM) was prepared by mixing 50 μl of a stock solution (5 mM) with 950 μl deionized water and buffer in a microcentrifuge tube. The mixture was then transferred to a 96-well microplate for measurement of ATP bioluminescence. In brief, 100 μl of the ATP solution was mixed with 100 μl of the luciferin-luciferase reagent (10 mg/ml, reconstituted with deionized water) in each well of the microplate. ATP bioluminescence was measured immediately using a microplate luminometer, with the integration time set at 1 s with normal gain.
mtDNA copy number by quantitative PCR.
Total DNA was extracted from whole heart homogenates using the DNeasy Tissue Kit (Qiagen) or from NRVM cells using PureLink genomic DNA mini kit (ThermoFisher Scientific) to quantify mtDNA copy number. mtDNA was quantified by amplifying mitochondrial cytochrome c oxidase I (mtCox I), mitochondrial D-Loop (D-Loop), and mitochondrial NADH dehydrogenase 2 (mtND2), and nuclear amplicons were generated by amplification of the Tbp gene (nuDNA) segment. mtDNA copy number was expressed as the mean mtDNA copy number relative to the nuclear genome.
mtDNA PCR primers (rat) were as follows: Tbp, forward 5′-GAGAAACGGGTCTCAAGATGAA-3′ and reverse 5′-CATTATCCACCTGGGCCTAAA-3′; D-Loop, forward 5′-CGGATGCCTTCCTCAACATA-3′ and reverse 5′-AGTCTTTCGAGCTTTGTCTATGA-3′; Cox1, forward 5′-TGAGCAGGAATAGTAGGGACAG-3′ and reverse 5′-GGGCTGTGACGATGACATTATAG-3′; and ND2, forward 5′-CCACCATTCTCGCAATTTCATC-3′ and reverse 5′-TCATCCTATGTGGGCAATTGAT-3′.
Dichlorofluorescein staining.
Dichlorofluorescein (DCF) staining was used to detect ROS in fixed mouse heart sections and H9c2 rat ventricular myoblasts. For heart sections, unfixed hearts were embedded in OCT compound and sectioned into 5-μm slices. Slices were incubated with carboxy-H2DCF-DA (C-400, Molecular Probes, 100 μmol/l) at 37°C for 30 min followed by three 5-min washes in PBS. Images were obtained with a confocal microscope (LSM 710, Zeiss). Fluorescence was quantified in ImageJ. H9c2 cells were cultured in 24-well plates, infected with doxycycline (Dox)-inducible RORα lentivirus, and then treated with ANG II (200 nM for 24 h) in the presence or absence of Dox. Cells were then incubated with 100 μM carboxy-H2DCF-DA in loading medium at 37°C for 30 min and then washed with HBSS. DCF fluorescence was measured with a plate reader (CLARIOstar, BMG Labtech, Cary, NC).
Fluorometric intracellular ROS assay.
Isolated NRVMs were seeded into a 96-well, flat-bottom plate at 100 μl/well with 0.5 × 105 cells. NRVMs were infected with RORα-expressing lentivirus for 48 h and then treated with ANG II (200 nM) for 24 h in serum-free medium. Intracellular ROS were measured using a Fluorometric Intracellular ROS kit (MAK142, Sigma-Aldrich). The reaction mix (containing 20 μl of ROS detection reagent stock solution and 10 ml of assay buffer) was added into each well and incubated (5% CO2, 37°C) for 1 h. The fluorescence intensity reading at an excitation wavelength of 490 nm and emission wavelenght of 525 nm was measured by a 96-well plate reader.
MitoTracker assay.
To visualize mitochondria, cells were incubated with MitoTracker Red CMXRos (100 nM) for 15 min.
Human heart samples.
Ventricular myocardium was obtained from nonfailing and failing human hearts through the Duke Human Heart Repository, as previously described (Table 1) (35). Briefly, failing human myocardium was acquired from the left ventricular free wall of explanted hearts after cardiac transplantation. Nonfailing LV tissue was acquired from donors whose hearts were not used for transplant with permission from Carolina Donor Services. No Health Insurance Portability and Accountability Act information was provided with any of the samples used in this study.
Table 1.
Patient characteristics
| Mean Age (Range) | Sex (Women/Men), % | Mean Ejection Fraction (Range), % | |
|---|---|---|---|
| Nonfailing (n = 5) | 49 (35–62) | 40/60 | 62 (55–70) |
| Failing (n = 7) | 46 (30–55) | 28/72 | 16 (15–20) |
Transverse aortic constriction.
Transverse aortic constriction (TAC) and sham surgeries were done at the University of North Carolina McAllister Heart Institute Mouse Models core, as previously described (46). Briefly, mice were anesthetized with isoflurane. The chest was opened sterilely, and a semiocclusive ligature was tied around a 27-gauge needle in the transverse aorta between the right and left carotid arteries. The resultant pressure gradient was confirmed by carotid Doppler ultrasound.
Statistics.
All results are presented as means ± SE. Comparisons were made in GraphPad Prism (San Diego, CA) using an unpaired t-test (two groups) or two-way ANOVA (type III sum of squares) with Tukey’s post hoc analysis (four groups).
RESULTS
RORα protects against ANG II-induced pathological hypertrophy in vivo.
Heart failure is characterized by persistent activation of the renin-angiotensin-aldosterone system. ANG II induces pathological cardiac hypertrophy directly by activating angiotensin receptors on cardiomyocytes and cardiac fibroblasts.
To understand the in vivo function of RORα in ANG II-induced pathological hypertrophy and heart failure, we treated RORαsg/sg mice and WT littermates with ANG II (2 mg·kg−1·day−1 by osmotic minipump) or vehicle for 14 days. ANG II infusion was associated with an increase in cardiomyocyte surface area. RORαsg/sg myocytes increased roughly twofold in cross-sectional area compared with a 50% increase in WT myocytes (P = 0.002; Fig. 1A). Heart weight indexed to tibia length increased modestly in WT mice (13%, P = 0.04), whereas heart weight indexed to tibia length increased 30% in RORαsg/sg mice (P < 0.0001; Fig. 1B and Table 2). There was no significant difference in baseline fibrosis of WT and RORαsg/sg hearts, as measured by Masson trichrome staining. ANG II infusion increased fibrosis in both groups, although the response was more robust in RORαsg/sg mice (Fig. 1C). Collectively, these results suggest that the loss of RORα facilitates a more pronounced hypertrophic response to ANG II.
Fig. 1.

Retinoic acid-related orphan receptor-α (RORα)sg/sg mice display exaggerated pathological hypertrophy after 14-day angiotensin II (ANG II) infusion. Wild-type (WT) and RORαsg/sg mice were treated with ANG II (2 mg·kg−1·day−1) or vehicle control (VC) by osmotic minipump for 14 days. A: heart sections were stained with wheat germ agglutinin (WGA; green), isolectin B4 (red), and DAPI (blue). Cardiomyocyte cross-sectional area (CSA) was analyzed with ImageJ software (≥4 random areas in ≥3 mice/group). The ANG II-treated CSA was indexed to the vehicle-treated CSA. B: heart weight (mg) was indexed to tibia length (mm). C: heart sections were stained with Masson trichrome. The percentage of fibrosis (trichrome blue) was determined algorithmically using Aperio ImageScope (3 sections in ≥3 mice/group). D: echocardiography compared fractional shortening in WT and RORαsg/sg mice treated with VC or ANG II. E and F: quantitative RT-PCR (n = 3–4 per group; E) and immunoblot analysis (F) comparing the abundance of RORα in ANG II- and VC-treated mouse hearts. G: immunoblot analysis for RORα in primary neonatal rat ventricular myocytes (NRVMs) and rat cardiac fibroblast (rCFBs). *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001. Four-group comparisons used two-way ANOVA; two-group comparisons used an unpaired t-test.
Table 2.
Mouse morphometrics
| Treatment | Body Weight, g | Tibia Length, mm | Heart Weight, mg | Heart Weight/Body Weight, % | Heart Weight/Tibia Length, mg/mm |
|---|---|---|---|---|---|
| Vehicle | |||||
| Wild type (n = 8) | 30.6 ± 1.5 | 17.7 ± 0.0 | 128 ± 5 | 0.42 ± 0.01 | 7.3 ± 0.2 |
| RORαsg/sg (n = 10) | 24.0 ± 0.8* | 17.6 ± 0.2 | 108 ± 3* | 0.45 ± 0.01 | 6.2 ± 0.2* |
| Angiotensin II | |||||
| Wild type (n = 9) | 27.0 ± 0.7† | 17.8 ± 0.3 | 142 ± 3 | 0.53 ± 0.02† | 8.4 ± 0.2† |
| RORαsg/sg (n = 10) | 23.9 ± 0.8 | 17.4 ± 0.3 | 135 ± 5† | 0.57 ± 0.01† | 8.0 ± 0.4† |
All values are means ± SE. RORα, retinoic acid-related orphan receptor-α.
P < 0.05 vs. wild-type mice;
P < 0.05 vs. vehicle by two-way ANOVA.
To assess the functional consequences of this exaggerated pathological hypertrophy, we performed conscious echocardiography on RORαsg/sg mice and WT littermates infused with ANG II for 14 days. ANG II increased septal and posterior wall thickness in both WT and RORαsg/sg mice (Table 3) and significantly increased estimated LV mass in RORαsg/sg but not WT mice. FS, a measure of contractile function, was lower in ANG II-infused RORαsg/sg mice than in ANG II-infused WT littermates (44 ± 2 vs. 50 ± 1%, P = 0.01; Fig. 1D).
Table 3.
Echocardiographic parameters at baseline and after angiotensin II treatment (14 days)
| Heart Rate, beats/min | Left Ventricular Internal Diameter at Diastole, mm | Left Ventricular Internal Diameter at Systole, mm | Fractional Shortening, % | Left Ventricular Diastolic Volume, µl | Left Ventricular Systolic Volume, µl | Interventricular Septal Thickness at Diastole, mm | Posterior Wall Thickness at Diastole, mm | Left Ventricular Mass (Calculated), mg | |
|---|---|---|---|---|---|---|---|---|---|
| Baseline | |||||||||
| Wild type (n = 6) | 631 ± 24 | 3.08 ± 0.12* | 1.23 ± 0.09 | 57.1 ± 2.1 | 34.0 ± 4.6 | 3.9 ± 0.7 | 0.99 ± 0.03 | 0.94 ± 0.04 | 95 ± 9 |
| RORαsg/sg (n = 6) | 650 ± 9 | 2.53 ± 0.08 | 1.33 ± 0.06 | 47.3 ± 0.8* | 23.2 ± 1.9 | 4.6 ± 0.5 | 0.93 ± 0.03 | 0.90 ± 0.02 | 70 ± 2* |
| Angiotensin II | |||||||||
| Wild type (n = 9) | 686 ± 9 | 3.13 ± 0.19 | 1.61 ± 0.10 | 50.4 ± 1.2† | 41.0 ± 5.6 | 8.2 ± 1.6 | 1.23 ± 0.05† | 1.12 ± 0.01† | 115 ± 11 |
| RORαsg/sg (n = 10) | 641 ± 15 | 2.88 ± 0.17 | 1.62 ± 0.12 | 43.7 ± 1.7*† | 33.4 ± 4.8 | 7.6 ± 0.6 | 1.28 ± 0.03† | 1.12 ± 0.05† | 110 ± 10† |
All values are means ± SE. Echocardiography was performed on nonanesthetized mice. RORα, retinoic acid-related orphan receptor-α.
P < 0.05 vs. wild-type mice;
P < 0.05 for angiotensin II vs. baseline (comparing only mice for which both baseline and posttreatment values were available) by two-way ANOVA.
Cardiac RORα mRNA (Fig. 1E) and protein (Fig. 1F) abundance were decreased by ANG II. Given that both cardiomyocytes and fibroblasts participate in the cardiac response to ANG II, we assayed RORα abundance in NRVMs and neonatal rat CFBs. Immunoblot analysis indicated that the abundance of RORα was markedly higher in NRVMs than in rat CFBs (Fig. 1G), suggesting that the in vivo differences between WT and RORαsg/sg hearts after ANG II treatment may be attributed largely to RORα-mediated events in cardiomyocytes, although fibroblasts could play a role as well.
Taken together, these data indicate that the loss of RORα enhances adverse ventricular remodeling induced by chronic ANG II exposure.
RORα prevents ANG II-induced cardiomyocyte hypertrophy in vitro.
To further elucidate the role of RORα in regulating ANG II-induced cardiomyocyte hypertrophy, we infected NRVMs with a lentivirus containing a Dox-inducible RORα, an in vitro gain-of-function approach to complement our in vivo loss-of-function experiments. NRVMs were treated with ANG II (200 nM) for 24 h, and myocyte cell surface area was measured after immunofluorescence staining with F-actin (red) and DAPI (blue) (Fig. 2A). Mean cell surface area was increased after ANG II in lentivirus-infected NRVMs without Dox induction (“no Dox”), but myocyte hypertrophy was abrogated by Dox-induced overexpression of RORα (Fig. 2B).
Fig. 2.

Retinoic acid-related orphan receptor-α (RORα) regulates angiotensin II (ANG II)-induced neonatal rat ventricular myocyte hypertrophy. Neonatal rat ventricular myocytes (NRVMs) were infected with doxycycline (Dox)-inducible lentiviral constructs containing RORα. A and B: lentivirus-infected NRVMs with (RORα) or without (no Dox) induction of RORα were treated with vehicle control (VC) or ANG II (200 nM) for 24 h. Cell surface area was analyzed in NRVMs stained for F-actin (red) and DAPI (blue) using ImageJ. Whiskers are 5th to 95th percentile. C: quantitative RT-PCR comparing myosin heavy chain (MHC)-β and atrial natriuretic peptide (ANP) mRNA expression (n = 3 independent experiments). D: representative immunoblot and summary densitometry of NRVM lysates with and without Dox induction of enhanced green fluorescent protein (eGFP)-tagged RORα. E: RORα was silenced in NRVMs using shRNAs; scrambled RNA sequence was used as a control (shCtrl). F: myocyte cross-sectional area was measured in NRVMs. G: representative immunoblot and summary densitometry of NRVMs infected with shCtrl or shRORα. H: quantitative RT-PCR comparing the abundance of hypertrophic markers in NRVMs infected with shCtrl or shRORα (n = 3 for shRNA experiments). *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001 by two-way ANOVA.
To further evaluate our in vitro findings, we assayed for typical molecular markers of hypertrophy. Expression of MHC-β and ANP mRNA (Fig. 2C) and protein (Fig. 2D) was lower in ANG II-treated NRVMs with Dox-induced overexpression of RORα. Conversely, silencing RORα expression with shRNA (Fig. 2E) yielded a robust increase in ANG II-induced NRVM hypertrophy compared with control shRNA (shCtrl; Fig. 2F). Silencing RORα with shRNA also increased abundance of ANP, BNP, and MHC-β in NRVMs in the presence of ANG II (Fig. 2, G and H).
Collectively, these gain- and loss-of-function data support our in vivo findings using RORαsg/sg mice and are consistent with the concept that RORα inhibits maladaptive cardiac hypertrophy induced by ANG II.
RORα loss-of-function promotes activation of the IL-6/STAT3 pathway and NF-κB.
IL-6 and NF-κB are recognized mediators of ANG II-induced cardiomyocyte hypertrophy (9, 34). RORα negatively regulates both IL-6 and NF-κB in other tissues (7, 20), leading us to explore whether the antihypertrophic effects of RORα are mediated by repression of IL-6 and NF-κB.
We found that ANG II infusion increased IL-6 protein abundance to a greater extent in RORαsg/sg compared with WT hearts (Fig. 3A). ANG II increased IL-6 mRNA in both groups, although the abundance was nearly twice as high in RORαsg/sg hearts than in WT hearts (Fig. 3B). Expression of the IL-6 signal transducer gp130 was lower in vehicle-treated but higher in ANG II-treated RORαsg/sg hearts compared with WT hearts. ANG II increased TNF-α in both WT and RORαsg/sg hearts, although there was no statistically significant difference between the two genotypes (P = 0.09). Taken together, these results suggest that the absence of RORα augments expression of cardiac inflammatory mediators after ANG II exposure, consistent with a previous report (7) indicating that RORα represses inflammatory responses in smooth muscle cells.
Fig. 3.

The IL-6/STAT3 pathway is upregulated by angiotensin II (Ang II) treatment in retinoic acid-related orphan receptor-α (RORα)sg/sg mouse hearts. Wild-type (WT) and RORαsg/sg mice were treated with angiotensin II (Ang II) or vehicle control (VC) by osmotic minipump for 14 days. A: immunoblot analysis compared the abundance of IL-6 with the loading control GAPDH. B: quantitative RT-PCR compared the abundance of mRNAs associated with the IL-6 pathway (n = 3–5/group). C: immunoblot analysis for phosphorylated STAT3 (Tyr705) and total STAT3 with the loading control GAPDH. D: immunoblot analysis comparing the abundance of IκBα, phospho-IκBα, and NF-κB with the loading control GAPDH. E: immunoblot analysis of fractionated heart lysates comparing the nuclear abundance of NF-κB with the loading control histone H3 (HH3). Comparative densitometry (n = 3/group) used ImageJ. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001 by two-way ANOVA. gp130, glycoprotein 130 (IL-6 signal transducer).
The STAT3 pathway is the primary operative signaling pathway downstream of IL-6 in ANG II-induced cardiac hypertrophy (32), leading us to ask whether the loss of RORα affected STAT3 expression and function. Basal STAT3 expression in RORαsg/sg hearts was low, but ANG II significantly derepressed STAT3 in the absence of RORα (Fig. 3, B and C). STAT3 activation through phosphorylation of the Tyr705 residue (p-STAT3 Tyr705) was also greater in RORαsg/sg mice than in WT mice, consistent with elevated IL-6 levels (Fig. 3C) and the general increase in proinflammatory signaling.
The proinflammatory transcription factor NF-κB was more abundant in RORαsg/sg mice at baseline and after ANG II infusion (Fig. 3D). This increase of NF-κB was coupled with an increase in IκBα in RORαsg/sg mice treated with ANG II (Fig. 3D). IκBα binds and inhibits NF-κB by sequestering it in the cytoplasm. When IκBα is phosphorylated, it dissociates from the complex, allowing translocation of NF-κB to the nucleus. We found that ANG II treatment of RORαsg/sg mice led to increased p-IκBα (Fig. 3D) and nuclear NF-κB (Fig. 3E), suggesting a significant increase in NF-κB activity in RORαsg/sg mice treated with ANG II. These findings are consistent with the known regulation of NF-κB by RORα through direct transcriptional upregulation of IκBα in smooth muscle cells (7). Nuclear localization of NF-κB increases IL-6 abundance (4), and, hence, RORα may regulate IL-6 through both direct and indirect transcriptional regulation.
Collectively, these findings indicate that the exaggerated pathological cardiac hypertrophy in ANG II-treated mice lacking RORα is associated with increased abundance and activity of the key proinflammatory mediators IL-6 and NF-κB in the heart.
RORα controls IL-6 expression in cultured cardiomyocytes through direct transcriptional regulation.
Our in vivo experiments indicated that loss of RORα increased activity and abundance of both IL-6 and NF-κB in the setting of ANG II infusion. The relationship between IL-6 and NF-κB is complex; IL-6 activates NF-κB, and NF-κB in turn increases transcription of IL6 (29).
To elucidate the role of RORα in regulating IL-6 expression, we transfected H9c2 rat ventricular myoblasts with plasmids containing the native IL6 promoter or an IL6 promoter containing point mutations within a predicted RORE (Fig. 4A). H9c2 cells were cotransfected with varying doses of RORα-containing plasmids that had been previously validated by dose-response activation of a luciferase-tagged RORE. Coexpression of RORα with the native IL6 promoter construct led to a dose-dependent decrease in IL6 promoter activity, but this effect was abrogated by mutating the RORE 935 bp upstream of the IL6 transcriptional start site (Fig. 4A).
Fig. 4.

Retinoic acid-related orphan receptor-α (RORα) represses IL-6 expression in cardiomyocytes and cardiac fibroblasts through direct transcriptional regulation. A: H9c2 rat ventricular myoblasts were cotransfected with IL6 promoter constructs containing the native IL6 promoter or the IL6 promoter mutated at one of two ROR response elements (ROREs) and varying amounts of RORα-containing plasmids. IL6 promoter activity was assayed by luciferase activity. B: H9c2 ventricular myocytes were treated with vehicle control (VC) or angiotensin II (ANG II; 200 nM) for 24 h and then incubated with anti-RORα or anti-IgG (control) antibodies. Chromatin was cross linked and used in chromatin immunoprecipitation (ChIP)-PCR with primers amplifying the RORE in the IL6 promoter, a mutated IL6 promoter lacking the RORE (IL-6 non-RORE), or the negative control RNA polymerase II subunit A (Polr2a). C: representative immunoblot and summary densitometry (n = 3 independent experiments) of IL-6 in the absence and presence of doxycycline (Dox) induction of lentiviral RORα. D: ELISA assaying IL-6 levels in conditioned medium (48 h) from lentivirus-infected neonatal rat ventricular myocytes (NRVMs; n = 3 independent experiments). E: human cardiac fibroblasts (hCFBs) were infected with either scrambled control (shCtrl) or RORα shRNA for 24 h and then treated with ANG II (200 nM) for 24 h. Lysates were immunoblotted (n = 3). F: hCFBs were treated with shCtrl or shRORα and cultured for 48 h. IL-6 in hCFB-conditioned medium was assayed by ELISA (n = 2 independent experiments). G: hCFBs were treated with control (mCherry) or RORα-expressing lentivirus for 48 h. IL-6 in hCFB-conditioned medium was assayed by ELISA (n = 2 independent experiments). Four-group comparisons used two-way ANOVA; two-group comparisons used an unpaired t-test. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001.
To confirm that RORα directly binds to the IL6 promoter, we performed ChIP-PCR with H9c2 ventricular myoblasts. Enrichment of RORα binding to the proximal promoter region of IL6, which encompasses the RORE, was observed in samples immunoprecipitated with anti-RORα antibody, but the enrichment was not observed in the −2.8 kbp upstream of the IL6 promoter. Interestingly, RORα binding to the IL6 promoter was significantly increased by ANG II exposure (200 nM for 24 h) compared with vehicle. Abundance of the control DNA Polr2a was low and equal in all samples (Fig. 4B).
To further evaluate RORα regulation of IL-6 in the context of ANG II exposure, we infected NRVMs with Dox-inducible RORα-containing lentivirus (as in Fig. 2B). Treatment with ANG II (200 nM for 24 h) increased IL-6 mRNA in NRVMs in the absence of Dox induction, but RORα overexpression abrogated this effect. Overexpression of RORα suppressed IL-6 protein levels in untreated NRVMs, potently inhibited IL-6 upregulation after ANG II treatment (Fig. 4C), and reduced NRVM IL-6 secretion into culture medium (Fig. 4D). These in vitro gain-of-function findings are consistent with our in vivo loss-of-function findings indicating that RORα represses IL-6 transcription.
RORα blunts the human cardiac fibroblast response to ANG II.
Cardiac hypertrophy due to ANG II occurs partially through direct cardiomyocyte stimulation and partially through paracrine stimulation of cardiomyocytes by substances released from CFBs, most notably IL-6 (38). RORα abundance is substantially greater in cardiomyocytes than in CFBs (Fig. 1F), and RORα mediates myocyte-autonomous responses to ANG II (Fig. 2, A–F). However, we sought to test whether RORα might also influence the CFB response to ANG II using primary human CFBs (courtesy of Dr. Li Qian, University of North Carolina McAllister Heart Institute) for gain- and loss-of-function experiments.
RORα expression was downregulated in human CFBs using shRNAs; scrambled RNA sequence was used as a control (shCtrl). Silencing RORα (shRORα) in human CFBs increased STAT3 phosphorylation (p-STAT3 Tyr705) in both the presence and absence of ANG II (Fig. 4E). We also found that silencing RORα modestly increased and overexpressing RORα significantly decreased IL-6 content in human CFB-conditioned medium using an IL-6 ELISA (Fig. 4, F and G). To test whether altering RORα activity in CFBs affected cardiomyocytes, we cultured NRVMs in medium conditioned (48 h) by human CFBs treated with either shRORα or shCtrl. There was no detectable difference in the surface area of NRVMs cultured with medium conditioned by human CFB-shRORα compared with human CFB-shCtrl (not shown).
RORα deletion impairs STAT3 Ser727 phosphorylation and mitochondrial function in vivo.
STAT3 is well recognized as a transcriptional regulator of multiple important processes in the heart (2). More recently, STAT3 was shown to translocate to the mitochondria, where it activates the electron transport chain to promote oxidative phosphorylation (45). STAT3-mediated transcriptional regulation downstream of IL-6 is activated largely by phosphorylation at the Tyr705 residue, whereas phosphorylation of the Ser727 residue is associated with increased mitochondrial STAT3 trafficking and enhanced mitochondrial respiration (13, 30).
In light of our finding that the loss of RORα enhances STAT3 phosphorylation at Tyr705 (Fig. 3C), we examined whether mitochondrial p-STAT3 Ser727 was affected by RORα in vivo using fractionated lysates from the hearts of WT and RORαsg/sg mice infused with ANG II for 2 wk. We found that the ratio of mitochondrial p-STAT3 Ser727 to total STAT3 was lower in vehicle-treated RORαsg/sg mice (50% of WT mice), an effect that was exaggerated in ANG II-treated RORαsg/sg mice (32% of ANG II-treated WT mice; Fig. 5A). The purity of these respective fractions was confirmed by blotting for the cytoplasmic marker GAPDH and the mitochondrial marker TOM20 (Fig. 5B).
Fig. 5.

Loss of retinoic acid-related orphan receptor-α (RORα) limits mitochondrial phosphorylated (p-)STAT3 Ser727 and compromises mitochondrial function in vivo. Wild-type (WT) and RORαsg/sg mice were treated with angiotensin II (ANG II; 2 mg·kg−1·day−1) or vehicle control (VC) by osmotic minipump for 14 days. A: heart tissue lysates were separated into mitochondrial and cytoplasmic/nuclear fractions. Immunoblot analysis compared the abundance of p-STAT3 Ser727 and total STAT3. The mitochondria-specific loading control was translocase of outer membrane 20 (TOM20). Comparative densitometry used ImageJ (n = 3/group). B: purity of mitochondrial and cytoplasmic fractions was confirmed by immunoblot analysis for TOM20 (mitochondrial) and GAPDH (cytoplasmic). C: immunoblot analysis comparing the abundance of cytochrome c (CytC) in whole heart lysates (n = 3/group). D: ATP bioluminescence assay quantifing ATP content in the mitochondrial fraction (n = 2–4/group). E: mouse heart sections were stained with dichlorofluorescin (DCF) as an index of oxidative stress (n = 2–4/group). F: heart lysates were immunoblotted for poly ADP-ribose polymerase (PARP) and cleaved PARP as a marker of apoptosis (n = 3/group). G: TUNEL staining detecting apoptotic cells. *P < 0.05, ** P < 0.01, ***P < 0.005, and ****P < 0.001 by two-way ANOVA.
To explore the potential effects of the loss of mitochondrial STAT3, we next asked whether RORα deletion affects mitochondrial abundance and function in vivo. Vehicle- and ANG II-treated RORαsg/sg mice had lower levels of cytochrome c by immunoblot analysis (Fig. 5C), which is potentially consistent with mitochondrial depletion. To test the functional consequences of this finding, we measured ATP content in the mitochondrial fraction of ANG II-treated mouse hearts and assayed for oxidative stress in mouse heart sections. We found that RORαsg/sg mice had markedly lower mitochondrial ATP than WT littermates in both vehicle- and ANG II-treated groups (Fig. 5D). Staining with DCF (28) revealed significantly greater oxidative stress in RORαsg/sg mice treated with either vehicle or ANG II (Fig. 5E), consistent with mitochondrial dysfunction. To understand whether these defects in mitochondrial function affected cell survival, we immunoblotted for the cleavage of PARP, a marker of apoptosis. We found that the abundance of cleaved PARP was modestly higher at baseline and markedly enhanced after 2 wk of ANG II infusion in RORαsg/sg mice compared with WT littermates (Fig. 5F). TUNEL staining showed greater apoptosis in RORαsg/sg hearts compared with WT hearts. ANG II increased TUNEL staining to a greater extent in RORαsg/sg hearts than in WT hearts (Fig. 5G).
Coupled with our data demonstrating increased p-STAT3 Tyr705 and NF-κB activation (Fig. 3), these findings suggest that the loss of RORα biases signaling toward detrimental, proinflammatory pathways and away from cytoprotective pathways potentially enhancing cell death.
RORα preserves mitochondrial function and p-STAT3 Ser727 in vitro.
Our loss-of-function in vivo experiments suggest that RORα mediates STAT3 Ser727 phosphorylation and mitochondrial translocation. This regulatory pathway may have favorable effects on both mitochondrial function (Fig. 5, D and E) and cell survival (Fig. 5F). To expand on these in vivo findings, we treated NRVMs with ANG II (200 nM for 24 h) after infection with lentivirus containing either scrambled shRNA (shCtrl) or shRORα. Immunoblot analysis demonstrated decreased mitochondrial p-STAT3 Ser727 in lysates of vehicle- and ANG II-treated NRVMs with RORα silenced (Fig. 6A). Interestingly, silencing RORα also led to increased abundance of activated NF-κB in the nucleus, consistent with our in vivo findings (Fig. 6A). We then infected NRVMs with Dox-inducible RORα lentivirus and treated them with ANG II in the absence or presence of Dox. RORα overexpression increased STAT3 Ser727 phosphorylation more than twofold after treatment with either vehicle or ANG II (Fig. 6B).
Fig. 6.

Retinoic acid-related orphan receptor-α (RORα) promotes mitochondrial phosphorylated (p-)STAT3 Ser727 translocation and mitochondrial function in vitro. A: neonatal rat ventricular myocytes (NRVMs) were infected with lentivirus containing scrambled shRNA (shRNA) or shRNA against RORα (shRORα) and then treated with angiotensin II (ANG II; 200 nM for 24 h). Cell lysates were fractionated and immunoblotted (n = 3 independent experiments). B: NRVMs were infected with doxycycline (Dox)-inducible RORα lentivirus and then treated with ANG II (200 nM) or vehicle for 24 h in the absence (−) or presence (+) of Dox. Representative immunoblot analysis and summary quantitative densitometry (ImageJ) are shown (n = 3 independent experiments). C: after infection and treatment, NRVM genomic DNA was used in quantitative PCR for mitochondrial genes normalized to the nuclear reference gene TATA box-binding protein (Tbp). D: NRVM mitochondria were labeled with MitoTracker red CMXRos. E: H9c2 rat ventricular myoblasts were infected with inducible RORα lentivirus and treated with ANG II (200 nM for 24 h) in the presence or absence of Dox and then stained with dichlorofluorescin (DCF) to assay oxidative stress. F: NRVMs were infected with inducible RORα lentivirus or RORα shRNA. Intracellular ROS was measured using a fluorometric assay. Four-group comparisons used two-way ANOVA; two-group comparisons used an unpaired t-test. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001.
In vivo, loss of RORα was associated with decreased mitochondrial p-STAT3 Ser727 as well as impaired mitochondrial function. We found that shRNA-mediated knockdown of RORα in vitro decreases mitochondrial DNA abundance (Fig. 6C) and reduces MitoTracker red staining in NRVMs (Fig. 6D). Taken together, these readouts suggest that silencing RORα may reduce the number of functional mitochondria in NRVMs, consistent with our in vivo findings (Fig. 5). Overexpression of RORα in H9c2 ventricular myoblasts did not affect baseline redox status but decreased ANG II-induced oxidative stress, as measured by DCF fluorescence (Fig. 6E) and fluorometric intracellular ROS assay (Fig. 6F), whereas silencing RORα with shRNA increased oxidative stress in both vehicle and ANG II-treated NRVMs (Fig. 6F).
Collectively, our in vivo and in vitro findings indicate that RORα preserves mitochondrial abundance and function, possibly through enhanced mitochondrial p-STAT3 Ser727.
RORα deficiency is associated with hypocontractility and heart failure in mouse and human hearts.
To expand upon our finding that the loss of RORα is associated with maladaptive hypertrophy after ANG II infusion, we assayed the abundance of RORα in another mouse model of heart failure. WT C57BL/6 mice underwent TAC or sham surgery. Eight weeks after surgery, they underwent echocardiography and were then euthanized. Immunoblot analysis indicated that TAC mice had significantly lower expression of RORα than sham-operated mice (Fig. 7A), and conscious echocardiography confirmed that contractile function was impaired in TAC mice (Fig. 7B). Coupled with our finding from the ANG II infusion model (Fig. 1, E and F), we now show that RORα abundance is decreased in two mouse models of hypertrophy and heart failure.
Fig. 7.

Loss of retinoic acid-related orphan receptor-α (RORα) is associated with impaired contractility in mouse and human hearts. A and B: wild-type (WT) male C57BL6 mice underwent transverse aortic constriction (TAC) or sham surgery. Eight weeks after surgery, mice underwent conscious echocardiography and then euthanasia. A: RORα abundance was measured using immunoblot analysis of heart lysates. B: fractional shortening, a measure of contractile function, was derived from M-mode echocardiography. Results were compared in WT (n = 6) and RORαsg/sg mice (n = 6) using a Student’s t-test. C: nonfailing and failing human heart tissue were obtained from the Duke Human Heart Repository, and immunoblot analysis was used to compare the abundance of RORα. **P < 0.01 using an unpaired t-test.
We then sought to determine whether RORα was present in the human heart and how its abundance was affected by heart failure. We found that RORα was robustly expressed in nonfailing human ventricular myocardium but that its abundance was decreased more than twofold by heart failure (Fig. 7C). To our knowledge, ours is the first identification of RORα in the human heart.
DISCUSSION
RORα has been recently identified in the mouse heart (15), but its contributions to cardiac physiology remain poorly understood. Using complementary in vivo and in vitro approaches, we present evidence that RORα inhibits pathological cardiac hypertrophy, reduces fibrosis and cell death, and preserves mitochondrial function after chronic ANG II infusion (shown in Fig. 8), novel functions for this nuclear receptor. Collectively, these processes define pathological ventricular remodeling, suggesting that RORα may play a protective role in heart failure.
Fig. 8.
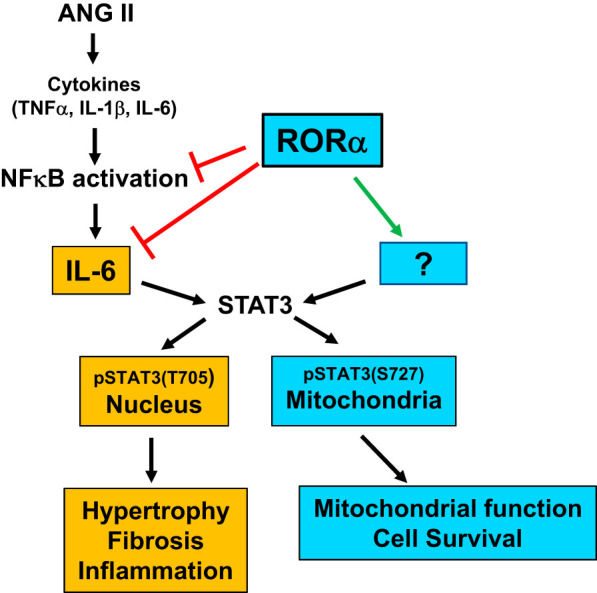
Retinoic acid-related orphan receptor-α (RORα) mitigates angiotensin (ANG II)-induced cardiac hypertrophy through regulation of NF-κB, IL-6, and STAT3.
The primary objective of the present study was to identify a role for RORα in the cardiac response to the pathological stimulus of ANG II. Mice lacking functional bx;1RORα (RORαsg/sg) mice exposed to chronic ANG II infusion developed exaggerated cardiomyocyte hypertrophy, cardiac fibrosis, and enhanced cell death. In considering molecular mechanism, we focused initially on inflammatory pathways, particularly the IL-6-STAT3 pathway. IL-6 is one of the central mediators of cardiomyocyte hypertrophy induced by ANG II (34) and pressure overload (47) and is regulated by RORα in other tissues (7, 20). IL-6 abundance is elevated at baseline in the absence of RORα and robustly upregulated in RORαsg/sg hearts after ANG II treatment such that its abundance exceeds levels in WT hearts.
We show here that RORα directly regulates IL-6 abundance in cardiomyocytes by binding to a RORE in the IL6 promoter. It is possible that RORα may also indirectly affect IL-6 abundance through its regulation of NF-κB, as we found that the absence of RORα promotes NF-κB translocation to the nucleus. In RORαsg/sg mice, ANG II robustly upregulates NF-κB and IκBα abundance, facilitating NF-κB nuclear translocation and increasing the expression of NF-κB transcriptional targets, including IL6 (29), the IL-6 receptor, gp130 (9), and TNF-α (5). Collectively, these targets serve as canonical upstream activators of STAT3 Tyr705 phosphorylation, one of the central events in the induction of ANG II-induced pathological cardiomyocyte hypertrophy (34) and cardiac fibrosis (16, 38).
We also found that the abundance of RORα was decreased in human heart failure as well as two mouse models of heart failure (ANG II infusion and TAC). Downregulation of RORα in the setting of ischemic injury has been previously reported (15). Although the regulation of RORα activity by agonist binding and posttranslational modifications has been previously described (18), little is known about the regulation of RORA transcription. Using the online tool ConTra V3 (http://bioit2.irc.ugent.be/contra/v3/#/step/1) to identify predicted transcription factor-binding sites within 1,000 bp of the RORA promoter, we found putative binding sites for two transcription factors that are known to be regulated by ANG II: SP1 in cardiac fibroblasts (36) and activator protein-2 activity in cultured vascular smooth muscle cells (43). Future studies may elucidate whether either of these transcription factors regulates RORα abundance in the injured or failing heart.
Interestingly, we found that loss of RORα has opposite effects on ANG II-induced phosphorylation of the two recognized STAT3 phosphorylation sites; p-STAT3 Tyr705 increases but mitochondrial p-STAT3 Ser727 decreases. Whereas p-STAT3 Tyr705 contributes to hypertrophy and fibrosis, p-STAT3 Ser727 enhances mitochondrial oxidative phosphorylation by activation of complexes I, II, and III (3, 13, 45) and protects against ANG II-induced hypertrophy (49). Our data suggest that augmented STAT3 Tyr705 phosphorylation likely occurs as a direct result of transcriptional derepression of IL6 in the absence of RORα. However, we also show that loss of RORα dramatically decreases and overexpression of RORα increases mitochondrial p-STAT3 Ser727. The mechanisms that regulate STAT3 Ser727 phosphorylation are less well characterized in general, and it appears likely that RORα affects p-STAT3 Ser727 through a yet-to-be-defined nontranscriptional mechanism.
We also find that RORα deletion leads to impaired ATP production and increased oxidative stress, hallmarks of mitochondrial dysfunction. We propose that these adverse effects may arise in part from a lack of cardioprotective RORα-mediated STAT3 Ser727 phosphorylation and mitochondrial translocation. Collectively, our findings position RORα as a novel regulator of the cardiac response to ANG II, promoting mitochondrial function and cell survival and mitigating inflammation and oxidative stress.
We show that RORαsg/sg mice have small hearts at baseline when heart weight is indexed to tibia length, although that difference was not evident when heart weight was indexed to body weight. RORαsg/sg mice have low body weight due to multiple metabolic abnormalities (for a review, see Ref. 6), including lower adipose mass than and improved insulin sensitivity compared with WT littermates (23, 24, 26). Although RORα plays a role in bone metabolism, tibial length of RORαsg/sg mice is normal (31), leading us to normalize heart weight to tibia length. The small heart of uninjured RORαsg/sg mice suggests that RORα may also participate in the regulation of either developmental (physiological) cardiac hypertrophy or atrophy, processes that are molecularly distinct from pathological hypertrophy, as induced by ANG II. We are actively exploring the transcriptional pathways that may contribute to this phenotype.
Agonist-induced pathological cardiac hypertrophy can be induced directly through receptors on myocardial cells or indirectly through increased load due to activation of vascular receptors. Given that RORαsg/sg mice globally lack functional RORα, we cannot entirely exclude a contribution of extracardiac effects on the pronounced ventricular remodeling. However, RORαsg/sg mice have blunted pressor responses to vasoconstrictors (1), and, hence, the pronounced hypertrophy that we observed was unlikely to have resulted from increased blood pressure. Furthermore, our in vitro experiments using NRVMs strongly support the concept that the RORαsg/sg phenotype arises from cardiomyocyte-autonomous effects of ANG II. Nevertheless, we are in the process of generating cardiomyocyte-specific RORα knockout mice to further define the roles of RORα in the heart. Future studies could also explore whether in vivo gain-of-RORα function is protective against ANG II-induced cardiac injury using mice with cardiomyocyte-specific RORα overexpression (15) or an RORα agonist currently in development (44).
Very little is known about the role of RORα in the cardiac response to injury. One group found that RORαsg/sg mice sustained larger infarcts after ischemia-reperfusion injury than WT littermates (15). The absence of RORα also was associated with greater oxidative stress and enhanced susceptibility to apoptosis, similar to our findings in the context of ANG II exposure. Another group showed that melatonin protected against cardiomyopathy in a mouse model of sepsis by mitigating NF-κB activation. This effect was blunted in RORαsg/sg mice, suggesting that RORα contributed to the cardioprotective effects of melatonin (10). Interpretation of these results is complicated somewhat by persistent uncertainty as to whether melatonin is a bona fide RORα ligand (40). Nevertheless, these studies, coupled with our present findings, suggest that RORα may exert cardioprotective effects in multiple contexts.
Collectively, our experiments indicate that RORα protects against ANG II-induced cardiac remodeling, joining other nuclear receptors known to inhibit cardiomyocyte hypertrophy (15, 48). We also found that RORα abundance is decreased in two mouse models of heart failure as well as in failing human heart tissue, suggesting that RORα depletion could contribute to the pathobiology of heart failure. Coupled with s recent report showing that RORα may protect against ischemia-reperfusion injury (15), RORα is emerging as a novel cardioprotective nuclear receptor.
GRANTS
B. C. Jensen was supported by American Heart Association Grant-in-Aid 17GRNT33710008 and National Institutes of Health (NIH) Grant R01-HL-140067 as well as the Hugh A. McAllister Research Foundation. A. M. Jetten was supported by the NIH Grant Z01-ES-101586).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.Y.B., H.S.K., and B.J. conception and design of research; J.Y.B., H.S.K., W.H., P.H.M., D.E.B., and A.M.J. performed experiments; J.Y.B., H.S.K., W.H., P.H.M., D.E.B., and B.J. analyzed data; J.Y.B., H.S.K., W.H., A.M.J., and B.J. interpreted results of experiments; J.Y.B. and B.J. prepared figures; J.Y.B., H.S.K., and B.J. drafted manuscript; J.Y.B., H.S.K., W.H., D.E.B., A.M.J., and B.J. edited and revised manuscript; J.Y.B., H.S.K., W.H., A.M.J., and B.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Laura M. DeGraff (National Institute of Environmental Health Sciences) for the minipump implantation surgery and Dr. Min Shi (National Institute of Environmental Health Sciences) for the expert statistical consultation.
REFERENCES
- 1.Besnard S, Bakouche J, Lemaigre-Dubreuil Y, Mariani J, Tedgui A, Henrion D. Smooth muscle dysfunction in resistance arteries of the staggerer mouse, a mutant of the nuclear receptor RORalpha. Circ Res 90: 820–825, 2002. doi: 10.1161/01.RES.0000014489.24705.71. [DOI] [PubMed] [Google Scholar]
- 2.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther 120: 172–185, 2008. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 105: 771–785, 2010. doi: 10.1007/s00395-010-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brasier AR. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res 86: 211–218, 2010. doi: 10.1093/cvr/cvq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol 10: 1498–1506, 1990. doi: 10.1128/MCB.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cook DN, Kang HS, Jetten AM. Retinoic acid-related orphan receptors (RORs): regulatory functions in immunity, development, circadian rhythm, and metabolism. Nucl Receptor Res 2: 2, 2015. doi: 10.11131/2015/101185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delerive P, Monté D, Dubois G, Trottein F, Fruchart-Najib J, Mariani J, Fruchart JC, Staels B. The orphan nuclear receptor ROR alpha is a negative regulator of the inflammatory response. EMBO Rep 2: 42–48, 2001. doi: 10.1093/embo-reports/kve007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguère V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab 5: 345–356, 2007. doi: 10.1016/j.cmet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, El-Jamali A, Dietz R, Scheidereit C, Bergmann MW. Requirement of nuclear factor-kappaB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation 111: 2319–2325, 2005. doi: 10.1161/01.CIR.0000164237.58200.5A. [DOI] [PubMed] [Google Scholar]
- 10.García JA, Volt H, Venegas C, Doerrier C, Escames G, López LC, Acuña-Castroviejo D. Disruption of the NF-κB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-α and blocks the septic response in mice. FASEB J 29: 3863–3875, 2015. doi: 10.1096/fj.15-273656. [DOI] [PubMed] [Google Scholar]
- 11.Giguère V, Tini M, Flock G, Ong E, Evans RM, Otulakowski G. Isoform-specific amino-terminal domains dictate DNA-binding properties of ROR alpha, a novel family of orphan hormone nuclear receptors. Genes Dev 8: 538–553, 1994. doi: 10.1101/gad.8.5.538. [DOI] [PubMed] [Google Scholar]
- 12.Gold DA, Gent PM, Hamilton BA. ROR alpha in genetic control of cerebellum development: 50 staggering years. Brain Res 1140: 19–25, 2007. doi: 10.1016/j.brainres.2005.11.080. [DOI] [PubMed] [Google Scholar]
- 13.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324: 1713–1716, 2009. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamilton BA, Frankel WN, Kerrebrock AW, Hawkins TL, FitzHugh W, Kusumi K, Russell LB, Mueller KL, van Berkel V, Birren BW, Kruglyak L, Lander ES. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature 379: 736–739, 1996. [Erratum in Nature 381: 346, 1996. 10.1038/381346b0] doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- 15.He B, Zhao Y, Xu L, Gao L, Su Y, Lin N, Pu J. The nuclear melatonin receptor RORα is a novel endogenous defender against myocardial ischemia/reperfusion injury. J Pineal Res 60: 313–326, 2016. doi: 10.1111/jpi.12312. [DOI] [PubMed] [Google Scholar]
- 16.Hilfiker-Kleiner D, Shukla P, Klein G, Schaefer A, Stapel B, Hoch M, Müller W, Scherr M, Theilmeier G, Ernst M, Hilfiker A, Drexler H. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation 122: 145–155, 2010. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- 17.Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res 95: 568–578, 2004. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- 18.Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal 7: e003, 2009. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jetten AM, Kurebayashi S, Ueda E. The ROR nuclear orphan receptor subfamily: critical regulators of multiple biological processes. Prog Nucleic Acid Res Mol Biol 69: 205–247, 2001. doi: 10.1016/S0079-6603(01)69048-2. [DOI] [PubMed] [Google Scholar]
- 20.Journiac N, Jolly S, Jarvis C, Gautheron V, Rogard M, Trembleau A, Blondeau JP, Mariani J, Vernet-der Garabedian B. The nuclear receptor ROR(alpha) exerts a bi-directional regulation of IL-6 in resting and reactive astrocytes. Proc Natl Acad Sci USA 106: 21365–21370, 2009. doi: 10.1073/pnas.0911782106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. Crystal structure of the human RORalpha Ligand binding domain in complex with cholesterol sulfate at 2.2 A. J Biol Chem 279: 14033–14038, 2004. doi: 10.1074/jbc.M400302200. [DOI] [PubMed] [Google Scholar]
- 22.Kang HS, Angers M, Beak JY, Wu X, Gimble JM, Wada T, Xie W, Collins JB, Grissom SF, Jetten AM. Gene expression profiling reveals a regulatory role for ROR alpha and ROR gamma in phase I and phase II metabolism. Physiol Genomics 31: 281–294, 2007. doi: 10.1152/physiolgenomics.00098.2007. [DOI] [PubMed] [Google Scholar]
- 23.Kang HS, Okamoto K, Takeda Y, Beak JY, Gerrish K, Bortner CD, DeGraff LM, Wada T, Xie W, Jetten AM. Transcriptional profiling reveals a role for RORalpha in regulating gene expression in obesity-associated inflammation and hepatic steatosis. Physiol Genomics 43: 818–828, 2011. doi: 10.1152/physiolgenomics.00206.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lau P, Fitzsimmons RL, Pearen MA, Watt MJ, Muscat GE. Homozygous staggerer (sg/sg) mice display improved insulin sensitivity and enhanced glucose uptake in skeletal muscle. Diabetologia 54: 1169–1180, 2011. doi: 10.1007/s00125-011-2046-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lau P, Nixon SJ, Parton RG, Muscat GE. RORα regulates the expression of genes involved in lipid homeostasis in skeletal muscle cells: caveolin-3 and CPT-1 are direct targets of ROR. J Biol Chem 279: 36828–36840, 2004. doi: 10.1074/jbc.M404927200. [DOI] [PubMed] [Google Scholar]
- 26.Lau P, Tuong ZK, Wang SC, Fitzsimmons RL, Goode JM, Thomas GP, Cowin GJ, Pearen MA, Mardon K, Stow JL, Muscat GE. Rorα deficiency and decreased adiposity are associated with induction of thermogenic gene expression in subcutaneous white adipose and brown adipose tissue. Am J Physiol Endocrinol Metab 308: E159–E171, 2015. doi: 10.1152/ajpendo.00056.2014. [DOI] [PubMed] [Google Scholar]
- 27.Lazar MA. Maturing of the nuclear receptor family. J Clin Invest 127: 1123–1125, 2017. doi: 10.1172/JCI92949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5: 227–231, 1992. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- 29.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol 10: 2327–2334, 1990. doi: 10.1128/MCB.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meier JA, Larner AC. Toward a new STATe: the role of STATs in mitochondrial function. Semin Immunol 26: 20–28, 2014. doi: 10.1016/j.smim.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer T, Kneissel M, Mariani J, Fournier B. In vitro and in vivo evidence for orphan nuclear receptor RORalpha function in bone metabolism. Proc Natl Acad Sci USA 97: 9197–9202, 2000. doi: 10.1073/pnas.150246097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mir SA, Chatterjee A, Mitra A, Pathak K, Mahata SK, Sarkar S. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J Biol Chem 287: 2666–2677, 2012. doi: 10.1074/jbc.M111.246173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pathak P, Li T, Chiang JY. Retinoic acid-related orphan receptor α regulates diurnal rhythm and fasting induction of sterol 12α-hydroxylase in bile acid synthesis. J Biol Chem 288: 37154–37165, 2013. doi: 10.1074/jbc.M113.485987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sano M, Fukuda K, Kodama H, Pan J, Saito M, Matsuzaki J, Takahashi T, Makino S, Kato T, Ogawa S. Interleukin-6 family of cytokines mediate angiotensin II-induced cardiac hypertrophy in rodent cardiomyocytes. J Biol Chem 275: 29717–29723, 2000. doi: 10.1074/jbc.M003128200. [DOI] [PubMed] [Google Scholar]
- 35.Schechter MA, Hsieh MK, Njoroge LW, Thompson JW, Soderblom EJ, Feger BJ, Troupes CD, Hershberger KA, Ilkayeva OR, Nagel WL, Landinez GP, Shah KM, Burns VA, Santacruz L, Hirschey MD, Foster MW, Milano CA, Moseley MA, Piacentino V III, Bowles DE. Phosphoproteomic profiling of human myocardial tissues distinguishes ischemic from non-ischemic end stage heart failure. PLoS One 9: e104157, 2014. doi: 10.1371/journal.pone.0104157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siddesha JM, Valente AJ, Sakamuri SS, Yoshida T, Gardner JD, Somanna N, Takahashi C, Noda M, Chandrasekar B. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J Mol Cell Cardiol 65: 9–18, 2013. doi: 10.1016/j.yjmcc.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpson P. Norepinephrine-stimulated hypertrophy of cultured rat myocardial cells is an alpha 1 adrenergic response. J Clin Invest 72: 732–738, 1983. doi: 10.1172/JCI111023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skoumal R, Tóth M, Serpi R, Rysä J, Leskinen H, Ulvila J, Saiho T, Aro J, Ruskoaho H, Szokodi I, Kerkelä R. Parthenolide inhibits STAT3 signaling and attenuates angiotensin II-induced left ventricular hypertrophy via modulation of fibroblast activity. J Mol Cell Cardiol 50: 634–641, 2011. doi: 10.1016/j.yjmcc.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 39.Slominski AT, Kim TK, Takeda Y, Janjetovic Z, Brozyna AA, Skobowiat C, Wang J, Postlethwaite A, Li W, Tuckey RC, Jetten AM. RORα and ROR γ are expressed in human skin and serve as receptors for endogenously produced noncalcemic 20-hydroxy- and 20,23-dihydroxyvitamin D. FASEB J 28: 2775–2789, 2014. doi: 10.1096/fj.13-242040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slominski AT, Zmijewski MA, Jetten AM. RORα is not a receptor for melatonin (response to DOI 10.1002/bies.201600018). BioEssays 38: 1193–1194, 2016. doi: 10.1002/bies.201600204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, Liu J, Istrate MA, Kamenecka TM, Roush WR, Vidović D, Schürer SC, Xu J, Wagoner G, Drew PD, Griffin PR, Burris TP. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 472: 491–494, 2011. doi: 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vega RB, Kelly DP. Cardiac nuclear receptors: architects of mitochondrial structure and function. J Clin Invest 127: 1155–1164, 2017. doi: 10.1172/JCI88888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang S, Zhang C, Zhang M, Liang B, Zhu H, Lee J, Viollet B, Xia L, Zhang Y, Zou MH. Activation of AMP-activated protein kinase α2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med 18: 902–910, 2012. doi: 10.1038/nm.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Billon C, Walker JK, Burris TP. Therapeutic effect of a synthetic RORα/γ agonist in an animal model of autism. ACS Chem Neurosci 7: 143–148, 2016. doi: 10.1021/acschemneuro.5b00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. Function of mitochondrial Stat3 in cellular respiration. Science 323: 793–797, 2009. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willis MS, Schisler JC, Li L, Rodríguez JE, Hilliard EG, Charles PC, Patterson C. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ Res 105: 80–88, 2009. doi: 10.1161/CIRCRESAHA.109.194928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao L, Cheng G, Jin R, Afzal MR, Samanta A, Xuan YT, Girgis M, Elias HK, Zhu Y, Davani A, Yang Y, Chen X, Ye S, Wang OL, Chen L, Hauptman J, Vincent RJ, Dawn B. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ Res 118: 1918–1929, 2016. doi: 10.1161/CIRCRESAHA.116.308688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao YC, Xu LW, Ding S, Ji QQ, Lin N, He Q, Gao LC, Su YY, Pu J, He B. Nuclear receptor retinoid-related orphan receptor α deficiency exacerbates high-fat diet-induced cardiac dysfunction despite improving metabolic abnormality. Biochim Biophys Acta Mol Basis Dis 1863: 1991–2000, 2017. doi: 10.1016/j.bbadis.2016.10.029. [DOI] [PubMed] [Google Scholar]
- 49.Zouein FA, Zgheib C, Hamza S, Fuseler JW, Hall JE, Soljancic A, Lopez-Ruiz A, Kurdi M, Booz GW. Role of STAT3 in angiotensin II-induced hypertension and cardiac remodeling revealed by mice lacking STAT3 serine 727 phosphorylation. Hypertens Res 36: 496–503, 2013. doi: 10.1038/hr.2012.223. [DOI] [PMC free article] [PubMed] [Google Scholar]