

Abstract
During aerobic exercise (>65% of maximum oxygen consumption), the primary source of acetyl-CoA to fuel oxidative ATP synthesis in muscle is the pyruvate dehydrogenase (PDH) reaction. This study investigated how regulation of PDH activity affects muscle energetics by determining whether activation of PDH with dichloroacetate (DCA) alters the dynamics of the phosphate potential of rat gastrocnemius muscle during contraction. Twitch contractions were induced in vivo over a broad range of intensities to sample submaximal and maximal aerobic workloads. Muscle phosphorus metabolites were measured in vivo before and after DCA treatment by phosphorus nuclear magnetic resonance spectroscopy. At rest, DCA increased PDH activation compared with control (90 ± 12% vs. 23 ± 3%, P < 0.05), with parallel decreases in inorganic phosphate (Pi) of 17% (1.4 ± 0.2 vs. 1.7 ± 0.1 mM, P < 0.05) and an increase in the free energy of ATP hydrolysis (ΔGATP) (−66.2 ± 0.3 vs. −65.6 ± 0.2 kJ/mol, P < 0.05). During stimulation DCA increased steady-state phosphocreatine (PCr) and the magnitude of ΔGATP, with concomitant reduction in Pi and ADP concentrations. These effects were not due to kinetic alterations in PCr hydrolysis, resynthesis, or glycolytic ATP production and altered the flow-force relationship between mitochondrial ATP synthesis rate and ΔGATP. DCA had no significant effect at 1.0- to 2.0-Hz stimulation because physiological mechanisms at these high stimulation levels cause maximal activation of PDH. These data support a role of PDH activation in the regulation of the energetic steady state by altering the phosphate potential (ΔGATP) at rest and during contraction.
Keywords: magnetic resonance, mitochondria, phosphate potential, pyruvate dehydrogenase, substrate disposal
INTRODUCTION
Skeletal muscle utilizes glucose and fatty acids as substrates for mitochondrial ATP production at rest and during contraction (37, 51). The rate of mitochondrial ATP synthesis and magnitude of substrate oxidation are dependent upon the contractile intensity and thus ATP demand of the contractile ATPases [actinomyosin and sarco(endo)plasmic reticulum Ca2+-ATPase]. In the transition from rest to submaximal work, ATP demand from contractile processes reaches a steady state with mitochondrial ATP synthesis. This response to a step change in cytosolic ATP demand results in a new steady state whereby the phosphate potential (i.e., free energy of ATP hydrolysis, ΔGATP) is maintained by the proton motive force (PMF), which is supported through increased rates of substrate oxidation and delivery of reducing equivalents (NADH, FADH2) to the electron transport chain (ETC). At steady state, the rate of mitochondrial ATP synthesis and ETC flux is determined by cytosolic ATPase activity. The magnitudes of the steady-state phosphate potential and the PMF are controlled primarily by feedback via the concentrations of the products of ATP hydrolysis, ADP and inorganic phosphate (Pi) (62). However, additional regulatory mechanisms have been proposed through which mitochondrial matrix redox status (NADH/NAD+) and consequently the steady-state phosphate potential may be modified independently of feedback regulation by ATP hydrolysis (3, 7, 32).
Stimulation of mitochondrial matrix dehydrogenase activity via intracellular Ca2+ concentration ([Ca2+]i) is one mechanism by which redox regulation of the phosphate potential is proposed to occur, not only through the direct link to matrix redox status via NADH production but also through regulation of mitochondrial substrate selection and subsequent reducing equivalent generation by tricarboxylic acid (TCA) cycle flux (3, 7). Among those enzymes sensitive to [Ca2+]i, only pyruvate dehydrogenase (PDH) has the capacity to increase acetyl-CoA (A-CoA) delivery to the TCA cycle through regulation of substrate oxidation (40). PDH catalyzes the irreversible decarboxylation of pyruvate to A-CoA for use in the TCA cycle and subsequent production of reducing equivalents (NADH) for maintenance of oxidative ATP synthesis (22, 45). Production of A-CoA by PDH represents the entry point for carbohydrates into the TCA cycle and thus potentially plays a role in governing the cell’s preference for oxidation of carbohydrates versus fatty acids (28). PDH activity is regulated via reversible phosphorylation catalyzed by pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP) (22, 45). In the phosphorylated state, PDH is inactive and restricts pyruvate flux at times when ATP demand is low, as in resting skeletal muscle. However, during muscle contraction increases in [Ca2+]i and pyruvate as well as a decrease in the intramitochondrial ratios ATP/ADP, NADH/NAD+, and A-CoA/CoA activate PDH through coordinated allosteric stimulation of PDP activity and inhibition of PDK activity resulting in net PDH dephosphorylation (22, 45). As a result, PDH activation during contraction is proportional to exercise intensity and serves to increase pyruvate flux to provide substrate (A-CoA, NADH) in support oxidative ATP synthesis (27).
Treatment with dichloroacetate (DCA), a potent inhibitor of PDK activity, results in dephosphorylation and complete activation of PDH in resting or contracting skeletal muscle (26, 44, 52, 53, 60) concomitant with increased rates of glucose oxidation (8, 10, 12, 16, 39). PDH activation through DCA treatment is believed to increase the matrix redox status (NADH/NAD+) as evidenced by significant increases in skeletal muscle content of both A-CoA and acetylcarnitine (14, 26, 50, 56). Acetylcarnitine formation occurs through carnitine acetyltransferase (CAT), the mitochondrial matrix enzyme that catalyzes the conversion of A-CoA and carnitine to acetylcarnitine and free CoA. CAT flux is stimulated when there is excess A-CoA production not matched to TCA cycle utilization (13). PDH production of A-CoA occurs with NADH at a 1-to-1 ratio, and the millimolar increases in acetylcarnitine of DCA-treated skeletal muscle likely result in an increase in the matrix redox state. However, it remains unclear whether alteration in the activation status of skeletal muscle PDH can exert any regulation over mitochondrial oxidative phosphorylation or muscle energetics at rest and/or during exercise. Therefore, the purpose of this study was to quantify the effect of pharmacological activation of PDH at rest and during contraction on rat gastrocnemius skeletal muscle energetics and function. Here DCA treatment was used to fully activate PDH irrespective of the gastrocnemius contractile state. PDH activation via DCA treatment resulted in a significant increase in the magnitude of the steady-state phosphate potential (ΔGATP) at rest and during muscle twitch contractile intensities below the reported aerobic threshold (25). This effect occurred without alteration to the contractile metabolic demand, glycolytic ATP contribution, or the kinetics of PCr hydrolysis or resynthesis. At twitch contractile intensities above the expected aerobic threshold (1.0–2.0 Hz), PDH activation with DCA had no significant effect on the net bioenergetic response. Taken together, these data suggest that PDH activity does regulate steady-state energetics during muscle contraction below the maximal aerobic capacity and that this effect is likely mediated through alteration in the steady-state mitochondrial redox potential.
MATERIALS AND METHODS
Animal care and feeding.
Male Wistar rats (250–350 g; Charles River) were housed three per cage in a temperature (22°C)- and humidity (50%)-controlled room on a 12:12-h light-dark cycle. Rats were provided Purina Rat Chow and tap water ad libitum except when noted below. All procedures were approved by the Michigan State University Institutional Animal Care and Use Committee and complied with The American Physiological Society’s “Guiding Principles in the Care and Use of Animals.”
Muscle stimulation and sampling for PDH activity.
The average body weight of all treatment groups was 240 ± 9 g. Animals were fasted 12 h overnight before isoflurane anesthesia induction at 5% for 3 min. Anesthesia was maintained at 1.5–2.5% during the surgical procedure, and body temperature was monitored via rectal thermocouple (Omega HH11) and maintained at 36.5–37.5°C. For control resting (n = 5) and DCA (n = 4) treatment groups no stimulation was performed. The control gastrocnemius-plantaris muscle group was excised immediately after anesthesia induction, whereas DCA-treated muscles were excised 1 h after tail vein injection of DCA (150 mg/kg at 220 mg/ml) dissolved in isotonic saline. Muscles were immediately freeze-clamped after excision in liquid N2-precooled Wollenberger tongs and stored at −80°C for analysis. For 0.5 Hz (n = 4) and 1.0 Hz (n = 4) stimulation groups the sciatic nerve of one hindlimb was exposed as described previously (33). In brief, a 1.5-cm-wide incision was made on the lateral aspect of the hip, and blunt dissection was performed through the gluteus adjacent to the sciatic nerve. The nerve was then insulated from surrounding tissues with a small strip of Parafilm, and a bipolar platinum electrode was placed around the nerve and the wound closed with cyanoacrylate glue. A length of suture (20-lb test braided nylon) was fixed to the patellar tendon, which was fixed in place on a custom Perspex animal holder. The lower limb was dissected free of overlying skin, and a length of suture was secured to the Achilles tendon. The calcaneus was cut to remove anterior muscles from interfering with the force recording from the gastrocnemius-plantaris-soleus muscles. The animal was positioned prone in the Perspex animal holder with the knee fixed in place and the Achilles tendon attached to a strain gauge (Grass FT03C). Maximum force output was then achieved by determining the supramaximal stimulation voltage and the optimal resting length with the length-tension relation. After initial maximal tension was measured, muscles were stimulated at either 0.5 Hz or 1.0 Hz for 5 min. Immediately after this period, the gastrocnemius-plantaris muscle group was freeze-clamped in liquid N2-precooled Wollenberger tongs while the muscles were still contracting to maintain the phosphorylation status of PDH. Samples were then stored at −80°C for later biochemical analyses.
[2-14C]pyruvate tracer solution for PDH activity assay.
A standard solution of 35 mM pyruvate was made in water and its concentration verified. Briefly, 1 µl of 35 mM pyruvate was added to 0.99 ml of 303 mM triethanolamine (TEA)-3.03 mM EDTA and 10 µl of 25 mM NADH, pH 7.5 at room temperature (RT). Pyruvate concentration was assayed in triplicate with a spectrophotometric lactate dehydrogenase (LDH) assay after consumption of NADH at 340 nm at 25°C. Baseline absorbance was measured, and excess LDH (5–10 units) was added and incubated for 5 min or until no absorbance change was present. The pyruvate concentration was then calculated from the change in absorbance from baseline with the extinction coefficient of NADH (ε340 = 6,220 M−1·cm−1) assuming that the reaction of NADH is 1:1 with pyruvate. Five milliliters of standardized 35 mM pyruvate solution were then added to 250 µCi of [2-14C]pyruvate sodium salt (specific activity 7.3 mCi/mmol; Perkin Elmer no. NEC256), and the final concentration was verified by spectrophotometric LDH assay as described above. Specific activity of the [2-14C]pyruvate tracer solution was then determined via reaction with glutamate with glutamate pyruvate transaminase to form [2-14C]alanine. This step was performed to determine the fraction of the 14C label that is specific to pyruvate in the tracer solution. In brief, 1 µl of [2-14C]pyruvate tracer solution was added to 500 µl of 50 mM TEA, 0.5 mM EDTA, and 250 mM glutamate, pH 7.5 at RT. Assays were carried out in duplicate with two negative controls. Glutamate pyruvate transaminase (~2 units) was then added to initiate the reaction but omitted from negative controls. Reactions were carried out at RT, and after 1 h 400 µl aliquots were removed and loaded into gravity flow columns containing a 1-ml settled volume of anion exchange resin (Bio-Rad AG1x8 resin, 200–400 mesh, acetate form) contained in a 3-ml syringe with a Luer lock stopcock. This resin traps the negatively charged [2-14C]pyruvate while the positively charged [2-14C]alanine passes through. Resin was then washed three times with 500 µl of water, and column eluate was collected into 25 ml glass scintillation vials containing 20 ml of Safety-Solve scintillant and counted with a Packard Tri-Carb liquid scintillation analyzer. Background counts from negative controls were subtracted from sample values to obtain the specific activity (cpm/µmol) of the [2-14C]pyruvate tracer solution. Aliquots (100 µl) of the [2-14C]pyruvate tracer solution were stored at −80°C until needed.
Homogenization and assay buffers for PDH activity assay.
Homogenization buffer A, for measurement of PDH active fraction (PDHa), contained (in M) 0.05 Tris·HCl, 0.005 EGTA, 0.005 MgCl2·6H2O, 0.05 KCl, 0.05 NaF, 0.005 DCA, and 0.001 DTT, with 0.1% Triton X-100, pH 7.8 on ice. Homogenization buffer B, for measurement of total PDH activity (PDHt), contained (in M) 0.05 Tris·HCl, 0.024 CaCl2·2H2O, 0.005 MgCl2·6H2O, 0.05 KCl, and 0.005 DCA, with 0.1% Triton X-100 and with 0.01 M glucose, 0.001 M DTT, and 5 U/ml hexokinase added fresh before use, pH 7.8 on ice. Assay buffer contained 0.12 M Tris·HCl, 0.577 mM Na2EDTA, 0.001 M MgCl2·6H2O, and 0.001 M DCA, with 3.86 mM NAD+, 5.77 mM CoA, 0.001 M thiamine pyrophosphate, 0.012 M l-carnitine-HCl, and 0.8 U/ml CAT added fresh before use, pH 7.8 at 37°C. For assay of PDHt, 2.88 mM NaF was also added to assay buffer.
PDH activity assay.
PDH activity was determined by a method developed previously by Sterk et al. (2003) with modifications outlined below (55). In brief, PDH activity in muscle homogenates was determined by a coupled enzyme assay after the conversion of the radiolabeled substrate ([2-14C]pyruvate) to radiolabeled product ([1-14C]acetylcarnitine):
Reaction time points were collected and then passaged through gravity flow columns containing anion exchange resin allowing for the separation of 14C-labeled compounds by trapping the unreacted negatively charged substrate, [2-14C]pyruvate, while allowing the passage of the positively charged product, [1-14C]acetylcarnitine, to quantify the reaction rate. Overall reaction rate was limited by the amount of PDH activity, as CAT and substrates of the reaction were added in excess. Thus the rate of [1-14C]acetylcarnitine production was assumed to be 1:1 with [1-14C]A-CoA production and used to quantify PDH activity in muscle samples. Reactions were carried out in homogenates produced from freeze-clamped muscle samples of the gastrocnemius-plantaris group. Samples were first powdered under liquid nitrogen by mortar and pestle, followed by two separate assays for determination of PDHa and PDHt. Approximately 30–50 mg of muscle powder was added to 250 µl of ice-cold homogenization buffer A (PDHa) and immediately mixed with vigorous shaking. Buffer volume was then adjusted by adding buffer to a final 10% homogenate concentration. Homogenates were then sonicated on ice with a Branson sonicator at 20 kc three times for 15 s with 15-s breaks in between. Each homogenate was then immediately assayed by adding 15 µl of homogenate to 85 µl of assay buffer prewarmed to 37°C and vortexed to mix. Reactions were carried out in triplicate at time points of 30 s, 1 min, and 1.5 min and initiated by the addition of [2-14C]pyruvate tracer solution to a final concentration of 1 mM. [2-14C]pyruvate tracer was added as a drop on the side of the reaction Eppendorf tube, and time points were started by vortexing, mixing the [2-14C]pyruvate tracer into the assay solution and initiating the reaction. Reaction temperature was maintained at 37°C for the entire duration by means of a heated water bath. Reactions were then quenched at appropriate time points via the addition of 500 µl of ice-cold methanol followed by vortexing. Each time point was run separately, and quenched samples were stored on ice until the reaction series of that sample was complete. Once the reaction series was completed for PDHa, the entire process was repeated by producing another muscle homogenate with homogenization buffer B for determination of PDHt activity. Quenched reaction time points were then loaded into gravity flow columns containing a 1-ml settled volume of anion exchange resin (Bio-Rad AG1x8 resin, 200–400 mesh, acetate form) contained in a 3 ml syringe with a Luer lock stopcock. Columns were then washed three times with 500 µl of water, with column eluate collected into 25-ml glass scintillation vials containing 20 ml of Safety-Solve liquid scintillation cocktail. The radioactivity in each vial was then counted with a Packard Tri-Carb liquid scintillation analyzer, and, using the specific activity of the tracer solution, the reaction rate was determined for quantification of PDH activity.
Assay for muscle ATP content.
Muscle samples were assayed for ATP content by the method of Lowry et al. (1972) (38). In brief, the superficial gastrocnemius was sampled under isoflurane anesthesia and immediately freeze-clamped. Muscles were powdered under liquid nitrogen and stored at −80°C until analysis. Metabolites were extracted from powdered muscle samples by adding ice-cold perchloric acid solution (2 N HClO4, 5 mM EDTA) at a final dilution of 1:10 as previously described (61). Extracts were mixed at 4°C for 20 min and subsequently centrifuged at 20,000 g for 20 min. The supernatant was neutralized by the addition of an equivalent volume of potassium hydroxide buffer (2 N KOH, 150 mM TES, 300 mM KCl), and a second centrifugation (3,000 g, 10 min) step removed precipitated perchlorate salts. Neutralized extracts were assayed for ATP content at RT with a coupled spectrophotometric enzyme assay (38).
NMR spectroscopy during stimulation.
In vivo rat muscle preparations were prepared as previously described for benchtop experiments, but for these experiments the skin and calcaneus process were left intact. An intraperitoneal (IP) catheter (PE 20) was inserted for DCA administration and sealed in place with cyanoacrylate glue. The animal was secured in a custom 74-mm-diameter phosphorus nuclear magnetic resonance (31P NMR) probe with the knee secured via a tungsten pin fixed through the femur and attached to brass supports. The Achilles tendon was tied to a custom-built isometric force transducer at the top of the probe. This arrangement positioned the center of the superficial gastrocnemius muscle directly over a 1.7-cm-diameter circular surface coil. Isoflurane anesthesia was maintained at 1.5–2.5% for the duration of the experiment through a nose cone built into the body of the NMR probe, and temperature was monitored via rectal thermistor (YSI model 73A) and maintained at 36.5–37.5°C with thermostated air. In each experiment the muscle length and supramaximal stimulation voltage (5–15 V) were adjusted to yield peak isometric tension development, and the probe was inserted into a Bruker AM400 spectrometer (9.4 T, 7.4-cm vertical bore magnet). Magnetic field homogeneity was optimized by shimming on the available proton signal. 31P NMR spectra were acquired at 161.8 MHz (4096 complex points, 8,012-Hz sweep width). The summed free induction decays (FIDs) were apodized with a 30-Hz exponential filter before the Fourier transform. The pulse width (15 µs) was chosen to yield maximum signal-to-noise ratio at a repetition time (TR) of 3.0 s. The NMR signal was corrected for partial saturation by acquiring 128 FIDs under the experimental conditions and a second at 5*T1 for PCr at 9.4 T (TR = 15 s). For each stimulation series, spectra (TR = 3 s, 8 scans) were acquired at rest (6 spectra), during stimulation (12 spectra), and during recovery after stimulation cessation (16 spectra). Muscles were stimulated at twitch contractile intensities of 0.35, 0.5, 0.75, 1.0, and 2.0 Hz, but no more than four stimulations were used in any experiment and frequencies were randomized. There was no effect of stimulation order on the resulting PCr transients. After the first series of stimulations, DCA was administered IP (150 mg/kg at 220 mg/ml in isotonic saline), followed by a 1 h incubation before an identical stimulation series was performed in the exact order. The triceps surae muscle group was then excised and weighed for normalization of force recordings.
High-resolution NMR spectroscopy for resting metabolites.
To obtain quantitative metabolite values in resting muscle, animals were mounted in the NMR probe and a series of spectra (20) were acquired (2,400 FIDs, 4,096 complex points, 8,012-Hz sweep, 3-s recycle delay, 30-Hz exponential filter) under the control conditions. A second series was acquired 1 h after IP administration of DCA (150 mg/kg at 220 mg/ml in isotonic saline) with the same acquisition settings.
Spectral analysis.
The FIDs of the stimulation series were analyzed with jMRUI software (version 3.0), and relative peak areas were integrated with AMARES (54). Areas of Pi, PCr, γ-ATP, α-ATP, and β-ATP peaks were normalized to the total phosphorus integral of each individual spectrum. PCr and Pi were quantified from their ratio to γ-ATP and adjusted to absolute chemical content using the enzymatically determined ATP content of the rat superficial gastrocnemius muscle determined from perchloric acid extracts described previously (34). Creatine content was estimated assuming that PCr content is 82% of total creatine (34). Intracellular pH (pHi) was estimated from the chemical shift of Pi relative to PCr (42):
| (1) |
The PCr content during steady-state contractions was calculated as a percentage of the initial value by using the summed data from the six resting spectra and the final six spectra during the contractile phase. The dynamics of PCr changes were fit to a monoexponential function by an iterative least squares algorithm to obtain the time constants of both PCr hydrolysis and resynthesis. Glycolytic ATP synthesis rate (JGLY) was estimated as follows:
| (2) |
where 1.5 is the stoichiometric coefficient for ATP/H+, β is the buffering capacity (1), and d(pHi)/dt is the rate of acidification during contraction (29). Cytosolic ATPase rate (JATPase) was calculated from the initial rate of PCr hydrolysis at the start of stimulation plus the basal ATPase rate of 0.012 mM/s (21, 29). Mitochondrial ATP synthesis rate (JMITO) was calculated by subtracting JGLY from JATPase, assuming that the steady-state phosphate potential is maintained via the matching of cytosolic ATPase activity (JATPase) to JMITO after JGLY is accounted for (29). ΔGATP was estimated from Pi, PCr, and Cr with the creatine kinase equilibrium equation:
| (3) |
and substituting for [ATP]/[ADP] with KeqCK = 1.66 × 109 M−1 (36):
| (4) |
Analysis of high-resolution, nonstimulation spectra was done with NUTS analysis software version 6.1 and manual Lorentzian line fitting. Individual peaks were normalized to the total phosphorus integral and quantified as described previously.
Statistics.
All data are reported as means ± SE. Comparisons of group means were by paired two-tailed Student’s t-test at the P < 0.05 level of significance.
RESULTS
PDH activity at rest, during contraction, or during DCA treatment.
The percent active fraction of the total PDH activity (PDHa/PDHt) present in rat gastrocnemius-plantaris muscle samples collected at rest, during twitch stimulation, or during DCA treatment is presented in Fig. 1. The active fraction of PDH in resting muscle was 23 ± 3%, consistent with published values in the rat (11, 20, 24). Muscle twitch stimulation resulted in significant increases in the active fraction of PDH compared with resting values, resulting in 53 ± 7% and 75 ± 13% active fractions for 0.5 Hz and 1.0 Hz, respectively (Fig. 1). Treatment of resting muscle with DCA resulted in complete activation of PDH, consistent with previous reports in both rat and human skeletal muscle (26, 44, 52, 53, 60). PDH activity with DCA treatment was significantly increased compared with control values at rest (90 ± 12% vs. 23 ± 3%, P < 0.05) and during 0.5-Hz stimulation (90 ± 12% vs. 53 ± 7%, P < 0.05) but was not significantly different from the activity measured during stimulation at 1.0 Hz (90 ± 12% vs. 75 ± 13%, not significant).
Fig. 1.

Pyruvate dehydrogenase activity (PDHa) expressed as a % of total PDH activity (PDHt) in rat gastrocnemius-plantaris muscle at rest, during 0.5 Hz or 1.0 Hz stimulation, and after dichloroacetate (DCA) treatment at rest. Freeze-clamped muscle samples were collected from anesthetized Wistar rats and assayed in vitro for PDH activity via radioisotopic assay described in materials and methods. Values are means ± SE for resting (n = 5) and 0.5 Hz (n = 4), 1.0 Hz (n = 4), and DCA (n = 4) treatment groups. *Significantly different from resting (P < 0.05); †significantly different from 0.5 Hz (P < 0.05). Significance determined by 2-tailed Student’s t-test.
ATP free energy in resting muscle.
In vivo metabolite levels measured by 31P NMR spectroscopy in control and DCA treated superficial gastrocnemius muscles are presented in Fig. 2. Activation of PDH with DCA resulted in a 17% decrease in resting Pi compared with control values (P < 0.05) within the same animal (Table 1). However, this was not met with a measurable stoichiometric increase in PCr or any alteration in pHi. This decrease in intracellular Pi concentration through PDH activation with DCA treatment resulted in a measurable increase in the magnitude of ΔGATP from −65.7 ± 0.22 to −66.4 ± 0.30 kJ/mol (P < 0.05) in control and DCA-treated animals, respectively. (Table 1).
Fig. 2.

Representative high-resolution 31P nuclear magnetic resonance spectrum of control (A) and dichloroacetate (DCA)-treated (B) resting rat gastrocnemius muscle within the same animal. Acquisition parameters: 2,400 free induction decays, 4,096 complex points, 8,012-Hz sweep, 3-s repetition time, and 30 Hz exponential filter. EC, extracellular; IC, intracellular; PCr, phosphocreatine; Pi, inorganic phosphate; PME, phosphomonoester.
Table 1.
Resting metabolite, pH, and ΔGATP before and after DCA treatment
| Control (n = 6) | DCA (n = 6) | |
|---|---|---|
| Pi, mmol/LCW | 1.61 ± 0.11 | 1.34 ± 0.16* |
| PCr, mmol/LCW | 38.0 ± 1.21 | 37.2 ± 1.63 |
| ATP, mmol/LCW | 9.24 ± 0.7 | |
| PCr/ATP | 4.01 ± 0.13 | 3.92 ± 0.18 |
| pH | 7.01 ± 0.02 | 6.98 ± 0.01 |
| ΔGATP, kJ/mol | −65.7 ± 0.22 | −66.4 ± 0.30* |
Values are means ± SE; n = 6 per group. Resting values were calculated from high-resolution spectra as seen in Fig. 2. Peak areas were quantified with NUTS line-fitting algorithm and normalized to total 31P peak area for each spectrum. Spectra were quantified with γ-ATP peak area at ATP concentration of 9.24 mM determined enzymatically in control white gastrocnemius muscle. pH was calculated via chemical shift of inorganic phosphate (Pi) relative to phosphocreatine (PCr) as shown in Eq. 1. Free energy of ATP hydrolysis (ΔGATP) was calculated as described in Eq 3. DCA, dichloroacetate; LCW, liter cell water.
Significant difference between control and DCA conditions (P < 0.05) by 2-tailed paired Student’s t-test.
Effect of PDH activation on PCr dynamics during contractile activity.
Dynamic changes in PCr and reciprocal stoichiometric changes in Pi occur at the onset and cessation of contractile activity and deviate further from their resting levels as contraction intensity increases. Figure 3 shows a representative stack plot of individual 31P NMR spectra serially acquired over the time course of one such experiment in control and DCA-treated conditions. Resting 31P NMR spectra were acquired for the first 2 min 24 s (spectra 1–6), during the next 4 min 48 s of 0.5-Hz stimulation (spectra 7–18), and during the subsequent 7 min 12 s of recovery (spectra 19–36) to quantify the steady-state and kinetic response of PCr hydrolysis and resynthesis in control and DCA-treated animals (Fig. 3). Force recordings from the triceps surae group were measured concurrently, and muscle force generation was quantified as the total tension time integral (TTI) for each muscle twitch and averaged between groups. No difference in force production between control and DCA treatment conditions was found at 0.35 Hz (67.1 ± 8.1 vs. 58.2 ± 5.9 g·s, n = 6), 0.5 Hz (62.4 ± 13.0 vs. 59.3 ± 13.1 g·s, n = 7), 1.0 Hz (45.5 ± 3.0 vs. 39.2 ± 1.9 g·s, n = 7), and 2.0 Hz (47.0 ± 4.7 vs. 43.6 ± 5.1 g·s, n = 6), with the exception of 0.75 Hz (58.0 ± 10.9 vs. 49.4 ± 10.5 g·s, n = 6, P < 0.05).
Fig. 3.
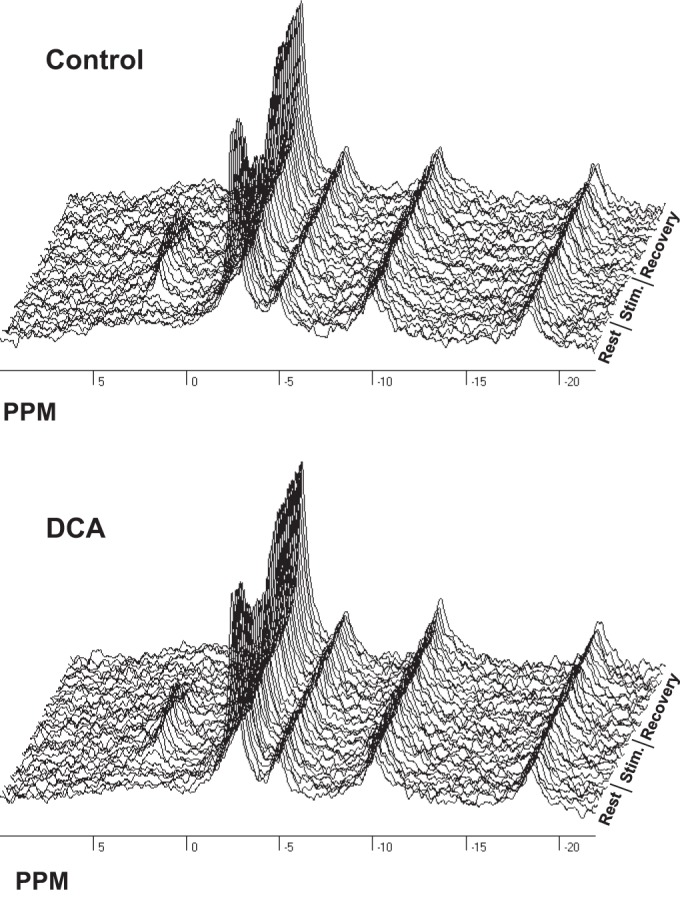
Sample 31P nuclear magnetic resonance spectra acquired in the rat gastrocnemius muscle before (Rest), during (Stim), and after (Recovery) 0.5-Hz stimulation within 1 animal in control and dichloroacetate (DCA) treatment conditions. Acquisition parameters: total of 36 spectra composed of 8 free induction decays, 4,096 complex points, 8,012-Hz sweep, 3-s repetition time, and 30-Hz exponential filter.
Representative PCr transients during stimulation and recovery before and after DCA treatment are depicted in Fig. 4, A and B, respectively. During PCr consumption at the onset of stimulation, ATP hydrolysis rates (JATPase) are derived from linear fitting to the initial rate of PCr hydrolysis (29). PCr transients quantified from both control and DCA-treated rats showed that there was no difference in ATP use irrespective of treatment and that a linear relation exists between increases in ATP use and increased stimulation frequency (see Table 3). In addition, PDH activation with DCA treatment did not alter the time constant for PCr resynthesis depicted as the rising monoexponential functions in Fig. 4, A and B and further quantified in Table 2, indicating that ATP production is also unaffected by DCA treatment (Table 2). During steady-state contractions, pHi values decreased with increasing stimulation frequency as previously shown in other studies (41). DCA treatment did not result in significant deviation in pHi from control conditions, with the exception of 0.35-Hz stimulation intensity (Table 2). A representative summed spectrum from one animal during steady-state contraction at 0.5-Hz intensity (spectra 13–18) is shown in Fig. 4, C and D, for control and DCA conditions, respectively. Figure 4E depicts a difference spectrum computed by subtracting the control spectrum (Fig. 4C) from that of the DCA treated condition (Fig. 4D), showing the differences in phosphorus metabolite peak areas. This resulted in residual areas for PCr and Pi indicating elevated PCr and reduced Pi with DCA compared with control stimulation within the same animal at the same intensity.
Fig. 4.

Phosphocreatine (PCr) content expressed as % of initial resting values and steady-state 31P nuclear magnetic resonance spectrum during 0.5-Hz stimulation before and after dichloroacetate (DCA) treatment in 1 animal. A: control PCr content during 0.5-Hz stimulation and recovery. B: PCr content during 0.5-Hz stimulation and recovery after DCA administration. Dashed line indicates control steady-state PCr content at the end of the stimulation, taken as the average PCr content in the last 6 spectra during stimulation. Linear regression fits of the initial rate of PCr hydrolysis were used to determine the ATP hydrolysis rate (JATPase) during muscle stimulation. Time constant of PCr recovery (τPCr) was calculated by fitting normalized PCr spectral area to a monoexponential function by an iterative least squares algorithm. C: summed steady-state 31P nuclear magnetic resonance (NMR) spectrum of the last 6 spectra acquired during 0.5-Hz stimulation in the control condition. D: summed steady-state 31P NMR spectrum of the last 6 spectra acquired during 0.5-Hz stimulation after DCA treatment. E: difference spectrum: DCA − control spectrum. Intensity adjusted to 4 times that of C and D. Pi, inorganic phosphate.
Table 3.
Metabolic flux and steady-state ΔGATP in control and DCA treatment conditions
| 0.35 Hz (n = 6) |
0.5 Hz (n = 7) |
0.75 Hz (n = 6) |
1.0 Hz (n = 7) |
2.0 Hz (n = 6) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Control | DCA | Control | DCA | Control | DCA | Control | DCA | Control | DCA | |
| JATPase, mM/s | 0.09 ± 0.04 | 0.19 ± 0.05 | 0.16 ± 0.02 | 0.21 ± 0.01 | 0.23 ± 0.01 | 0.32 ± 0.04 | 0.34 ± 0.04 | 0.40 ± 0.05 | 0.87 ± 0.14 | 0.68 ± 0.11* |
| JGLY, mM/s | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.02 ± 0.00 | 0.02 ± 0.01 | 0.06 ± 0.02 | 0.03 ± 0.01 | 0.10 ± 0.02 | 0.07 ± 0.01 | 0.43 ± 0.08 | 0.40 ± 0.07 |
| JMITO. mM/s | 0.09 ± 0.03 | 0.12 ± 0.01 | 0.14 ± 0.02 | 0.18 ± 0.02 | 0.16 ± 0.02 | 0.27 ± 0.04 | 0.22 ± 0.02 | 0.32 ± 0.05 | 0.40 ± 0.16 | 0.40 ± 0.10 |
| ΔGATP, kJ/mol | −58.2 ± 0.3 | −59.9 ± 0.4* | −56.8 ± 0.4 | −57.7 ± 0.6* | −55.6 ± 0.3 | −56.0 ± 0.4* | −55.4 ± 0.3 | −55.6 ± 0.4 | −54.6 ± 0.6 | −54.5 ± 0.3 |
Values are means ± SE, n = 6 or 7 per stimulation. Glycolytic ATP synthesis rate (JGLY), cytosolic ATP hydrolysis rate (JATPase), mitochondrial ATP synthesis rate (JMITO), and free energy of ATP hydrolysis (ΔGATP) were calculated as described in materials and methods. DCA, dichloroacetate.
Significant difference between control and DCA (P < 0.05) by 2-tailed paired Student’s t-test.
Table 2.
PCr kinetics and end of stimulation pH in control and DCA treatment conditions
| 0.35 Hz (n = 6) |
0.5 Hz (n = 7) |
0.75 Hz (n = 6) |
1.0 Hz (n = 7) |
2.0 Hz (n = 6) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Control | DCA | Control | DCA | Control | DCA | Control | DCA | Control | DCA | |
| Time constant of PCr hydrolysis, s | 75.3 ± 12.7 | 47.1 ± 20.7 | 71.1 ± 5.3 | 72.5 ± 7.2 | 56.8 ± 4.8 | 66.4 ± 5.5 | 45.4 ± 7.6 | 61.1 ± 13.9 | 25.2 ± 4.5 | 33.3 ± 9.1 |
| Time constant of PCr resynthesis, s | 46.7 ± 5.8 | 43.5 ± 12.62 | 61.1 ± 6.1 | 58.6 ± 4.6 | 59.8 ± 6.6 | 47.2 ± 6.1 | 65.0 ± 6.2 | 60.0 ± 6.6 | 72.9 ± 5.6 | 68.1 ± 4.2 |
| pH | 6.96 ± 0.02 | 6.93 ± 0.02* | 6.94 ± 0.02 | 6.90 ± 0.01 | 6.79 ± 0.09 | 6.84 ± 0.03 | 6.75 ± 0.06 | 6.78 ± 0.04 | 6.65 ± 0.12 | 6.65 ± 0.08 |
Values are means ± SE; n = 6 or 7 per stimulation. Time constants of phosphocreatine (PCr) hydrolysis and resynthesis were calculated by fitting normalized PCr spectral area to a monoexponential function by an iterative least squares algorithm. pH at the end of stimulation was calculated with Eq. 1 from the average chemical shift of the inorganic phosphate (Pi) peak relative to PCr during the last 6 spectra during stimulation. DCA, dichloroacetate.
Significant difference between control and DCA conditions (P < 0.05) by 2-tailed paired Student’s t-test.
At intensities below the reported aerobic threshold for rat hindlimb muscles (25) (0.35 Hz, 0.5 Hz, 0.75 Hz) PDH activation with DCA resulted in significantly higher steady-state PCr levels relative to the comparable control condition within the same animal (Fig. 5). This effect was lost with stimulation at intensities of 1.0 Hz and 2.0 Hz, with no discernible differences in the observed steady-state energetics. Increased PCr levels at each stimulation frequency were associated with equivalent decreases in Pi levels as illustrated in Fig. 6A. The plot of Pi against PCr shows a linear relationship with a slope of −1.0 Pi/PCr (R2 = 0.97), indicating a 1:1 stoichiometric balance between PCr reduction and Pi increase, validating the in vivo quantification of these metabolites. Figure 6, B and C, shows Pi and calculated free ADP levels against JATPase showing distinct reductions in Pi and ADP in DCA-treated contraction frequencies compared with controls.
Fig. 5.
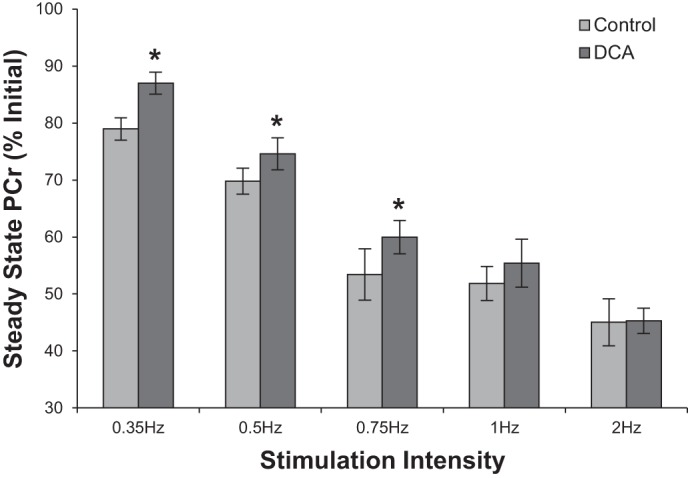
Steady-state phosphocreatine (PCr) expressed as % of initial resting values before and after dichloroacetate (DCA) treatment. PCr peak areas of the last 6 spectra during gastrocnemius muscle stimulation were averaged and normalized to the average PCr peak area of the first 6 spectra acquired in the gastrocnemius muscle at rest. Values are means ± SE. *Significantly different from control (P < 0.05) of the same stimulation intensity by 2-tailed paired Student’s t-test.
Fig. 6.

Steady-state phosphorus metabolites during contraction at 0.35 Hz, 0.5 Hz, 0.75 Hz, and 1.0 Hz before and after dichloroacetate (DCA) treatment. A: reciprocal relationship between steady-state inorganic phosphate (Pi) and phosphocreatine (PCr) during contraction. Linear regression was fit to group data, as there is no significant difference in slope between conditions. B: steady-state Pi content as a function of cytosolic ATP hydrolysis rate (JATPase). C: steady-state ADP content calculated from the creatine kinase equilibrium reaction as a function of JATPase. Linear regressions fit to initial linear portion of the data that include the first 3 points from 0.35 Hz, 0.5 Hz, and 0.75 Hz data.
Effect of PDH activation on mitochondrial flow-force relationship.
At stimulation intensities below the reported aerobic threshold (25), PDH activation with DCA resulted in a significant increase in the magnitude of steady-state ΔGATP. At stimulation rates above 1.0 Hz, this effect was lost (Table 3). As a result of the changes in concentration of phosphate metabolites associated with DCA treatment, PDH activation resulted in a leftward shift in the flow-force relationship between the steady-state ΔGATP maintained for a given JMITO (Fig. 7). The shift in the relationship between JMITO and ΔGATP stems from the significant elevation in the magnitude of steady-state ΔGATP at intensities below the aerobic threshold, as JMITO was not affected by PDH activation (Table 3). Figure 7, inset, shows the linear relationship between JMITO and ΔGATP for stimulation intensities below (0.35–0.75 Hz) as well as at (1.0 Hz) the reported aerobic threshold of the rat hindlimb (25). PDH activation with DCA treatment resulted in a leftward shift in the flow-force relationship between JMITO and ΔGATP without significant alteration in slope (P = 0.7). These data support a shift in the thermodynamic set point between the free energy transduction from substrate oxidation to ΔGATP and not an alteration in the kinetic response of oxidative phosphorylation to muscle contraction.
Fig. 7.
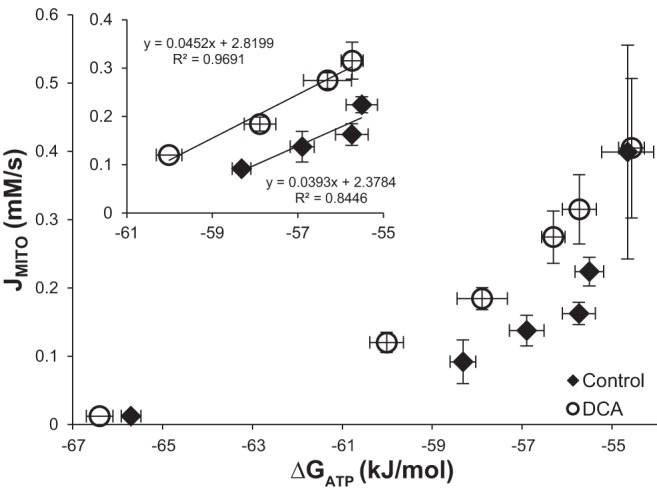
Steady-state free energy of ATP hydrolysis (ΔGATP) as a function of steady-state mitochondrial ATP synthesis rate (JMITO) at rest and during contraction at 0.35 Hz, 0.5 Hz, 0.75 Hz, 1.0 Hz, and 2.0 Hz in the rat gastrocnemius muscle before (Control) and after dichloroacetate (DCA) treatment. Inset: steady-state ΔGATP vs. JMITO during contraction at 0.35 Hz, 0.5 Hz, 0.75 Hz, and 1.0 Hz. Data were fit to linear regression with no significant difference in slope between control and DCA treatment.
DISCUSSION
Pharmacological activation of PDH resulted in an increase in the magnitude of steady-state ΔGATP in rat gastrocnemius muscle at rest and during contraction at intensities below the reported aerobic threshold. At rest, treatment with DCA resulted in complete conversion of PDH into the active state concurrent with a significant decrease in the resting Pi levels of 17% and increase in the magnitude of ΔGATP (Fig. 1, Table 1). During muscle stimulation at contractile intensities below the reported aerobic threshold of 1.0 Hz (25), PDH activation resulted in significant elevation of steady-state PCr (lowered free ADP) with a concomitant reduction in Pi content, resulting in an increase in the magnitude of ΔGATP at any contractile frequency (Fig. 5, Fig. 6, Table 3). This resulted in a leftward shift in the flow-force relationship between JMITO and the steady-state ΔGATP that was maintained (Fig. 7). The increase in free energy with PDH activation occurred without alteration of the estimated glycolytic contribution to ATP production (Table 3), changes in muscle force generation (TTI) and the corresponding cytosolic ATPase load (Table 3), or the time constants of PCr hydrolysis and resynthesis (Table 2). At or above the reported aerobic threshold for rat hindlimb (1.0–2.0 Hz), PDH activation with DCA had no significant effect on steady-state energetics. Taken together these data suggest that at exercise intensities below maximum JMITO fluxes, the free energy at which ATP is synthesized to meet ATP hydrolysis demand is affected by PDH activity.
The magnitude of PDH activation in skeletal muscle determines the capacity for oxidative glucose disposal. In the present study, resting rat gastrocnemius-plantaris muscle PDH activity was 23% of the total activity, consistent with previous findings in both rat (14–25%) (11, 20, 24) and human (17–25%) (14, 26, 44) skeletal muscle at rest. This agrees with the relatively low whole body respiratory exchange ratio in the fasted rat of 0.77 corresponding to 23.9% and 76.1% energy expenditure (kcal/l O2) from glucose and free fatty acid oxidation, respectively (6, 46), and is consistent with respiratory quotients from human leg and forearm skeletal muscle arterial-venous differences (respiratory quotient = 0.74–0.77) (2, 5, 15, 30, 31). Low fractional PDH activity in the resting fasted state restricts skeletal muscle glucose oxidation at times when glucose availability is limited and contractile ATP demand is low. This occurs via preferential covalent phosphorylation and inhibition of the enzyme complex through allosteric stimulation of PDK activity (47, 63). DCA is a potent inhibitor of PDK, resulting in PDH complex dephosphorylation via endogenous PDP activity even where the energetic demand (0.01 mM ATP/s) (21) and Ca2+ concentration (~40–50 nM) (4, 23, 59) are low. DCA treatment at rest caused a significant increase in PDH activity toward maximum activation of the enzyme complex (90 ± 12%) similar to that found in both rat (53, 60) and human (26, 44, 52) skeletal muscle. Furthermore, PDH activation with DCA treatment at rest resulted in a significant increase in the magnitude of steady-state ΔGATP (Table 1). This effect was due to a significant reduction in intracellular Pi of 17% as quantified through 31P NMR before and after DCA treatment within the same animal. This reduction in cytosolic Pi content would result in a stoichiometric increase in PCr; however, the 0.3 mM reduction in Pi found represents a 0.8% change in PCr content that was below detection limits of the 31P NMR method used because of signal to noise considerations.
Since ATP consumption is balanced by oxidative ATP synthesis under steady-state conditions, the increase in the magnitude of ΔGATP associated with DCA administration is necessarily associated with increases in the free energy at which ATP is synthesized and transported out of the mitochondrial matrix to the cytosol. This increase is likely explained by a shift in mitochondrial matrix redox potential (NADH/NAD+) to a more reduced steady state, in turn resulting in an increase in mitochondrial PMF that drives the ATP synthesis machinery (F1FO-ATPase and adenine nucleotide translocase) to achieve a new steady-state relation with ΔGATP. This has been shown in isolated liver mitochondria in vitro, where markedly different redox states can be obtained under identical ATPase loads by switching the carbon fuel source utilized to support oxidative phosphorylation (32). Furthermore, the magnitude of the extramitochondrial ΔGATP was substrate dependent and highest with substrates that resulted in a more reduced matrix for a given ATPase stimulation (32). DCA treatment results in significant increases in skeletal muscle glucose utilization, shifting mitochondrial substrate oxidation from primarily fatty acid to glucose in the resting state. In fasting rat epitrochlearis muscle in vitro, DCA treatment resulted in a threefold increase in glucose oxidation (12). This effect was also observed in the isolated fasted rat soleus muscle, where DCA treatment increased glucose oxidation by 45%, with concomitant reduction in oleate oxidation (53). Furthermore, in isolated rat diaphragm, treatment with DCA resulted in 32% and 54% increases in glucose oxidation (16, 39). To our knowledge, the effect of DCA solely on human skeletal muscle substrate oxidation has not been determined; however, DCA treatment in humans resulted in 14% and 34% increases in whole body glucose oxidation as well (8, 10). This effect is attributed to a significant increase in oxidative pyruvate flux, as blood concentrations of both lactate and alanine are significantly reduced with DCA treatment, shifting anaplerotic pyruvate utilization toward oxidation via PDH activation (12, 18, 53).
If enhancing pyruvate oxidation yields an increase in steady-state redox potential, it would be expected that shifting substrate utilization toward glucose or pyruvate would increase the cytosolic phosphate potential (ΔGATP). In fact, this effect has been demonstrated in both skeletal muscle and cardiac tissue preparations. The addition of 20 mM pyruvate to isolated mouse soleus muscles resulted in a 17% reduction and an 8% increase in resting Pi and PCr, respectively, compared with 11 mM glucose alone when measured by 31P NMR spectroscopy (48). Furthermore, removal of pyruvate in the superfusate reversed this effect within 30 min (48). The effect of increased pyruvate oxidation on tissue bioenergetics, particularly Pi and PCr, was also observed in both working isolated guinea pig heart ex vivo and canine heart in vivo. Incubation of the isolated guinea pig heart with 5 mM pyruvate, glucose, and lactate nearly doubled both the PCr-to-Pi ratio (4.93 ± 0.18 vs. 2.63 ± 0.16) and cytosolic phosphate potential [ATP]/([ADP] + [Pi]) (7.3 vs. 3.5), yet there was no significant alteration in either rate pressure product or oxygen consumption compared with glucose and lactate alone (9). In separate experiments using isolated guinea pig heart, switching the perfusate medium containing 16.7 mM glucose to one containing 10 mM pyruvate resulted in a significant increase in intracellular [PCr] (18.3 ± 0.3 vs. 15.2 ± 0.2 mM) and PCr/Pi (30.4 ± 2.2 vs. 10.3 ± 0.09) (64). This effect was also observed in the paced canine heart in vivo, where perfusion with 5.26 mM pyruvate resulted in a 23% increase in PCr/ATP and 25% reduction in calculated [ADP] with no change in heart rate, ventricular pressure, or oxygen consumption compared with preperfusion values with blood alone (35). Together these data suggest not only that PDH activation via pharmacological treatment with DCA results in increases in pyruvate oxidation but also that this switch in substrate utilization would be expected to increase the phosphate potential at similar rates of oxygen consumption or ETC flux.
Our results indicate that activation of PDH results in an increase in the magnitude of ΔGATP not only at rest but also during submaximal exercise conditions, with the magnitude of the effect of DCA on ΔGATP dependent on the stimulation level. When DCA treatment was used at stimulation intensities (0.35–0.75 Hz) below the expected aerobic maximum of 1.0 Hz of the rat hindlimb, a significant increase in the magnitude of steady-state ΔGATP was found relative to the matched control condition (Table 3). This was evidenced by a significant increase in the steady-state PCr (Fig. 5) through a reduction in ADP and Pi levels (Fig. 6). This effect was not attributed to any significant alteration in the kinetics of PCr hydrolysis or resynthesis, estimated glycolytic ATP production, muscle force generation (TTI), or cytosolic ATPase load (Tables 2 and 3). However, the relationship between PDH activation and steady-state ΔGATP is not linear even for stimulations below the aerobic threshold. For example, the magnitude of the difference in ΔGATP between control and DCA treatment conditions is much more pronounced during stimulation at 0.35 Hz compared with rest (1.7 vs. 0.6 kJ/mol). If an increase in the mitochondrial redox status is the mechanism responsible for the increase in the magnitude of ΔGATP with PDH activation, these data suggest that at rest mitochondria are in a highly reduced state and therefore PDH activation has less ability to alter the redox status than during exercise states. It may also be the case that any increase in the redox status, and therefore PMF, may not be sufficient to stimulate rephosphorylation of ADP to ATP, as the ADP concentration at rest is sufficiently low (~10 µM) and potentially limiting to ATP synthesis. However, during stimulation mitochondria become more oxidized as the concentration of ADP increases (32). This may allow PDH activation to exert a greater influence over cytosolic ΔGATP and account for the large differences observed in ΔGATP between control and DCA conditions at rest and during stimulation. The effect of PDH activation on steady-state ΔGATP was largest at the lowest stimulation intensity tested and progressively became smaller as the stimulation intensity increased to 0.75 Hz. This result could be expected, as the activation state of PDH increases proportional to stimulation intensity until complete activation is reached at maximal workloads (27, 43). Therefore, compared with the control condition, activation of PDH with DCA would result in the greatest difference in fractional PDH activity at lower stimulation intensity and result in the smallest difference in activity as muscle stimulation approaches the aerobic threshold. It is also possible that as stimulation intensity approaches the reported aerobic capacity, limitations to ETC capacity become more important than substrate selection in modifying the flow-force relationship between ATPase activity and cytosolic ΔGATP.
Prior studies utilizing DCA have also observed increased steady-state PCr in skeletal muscle during contraction but have attributed this effect to relief of an “acetyl-group deficit” that mitigates “metabolic inertia” (19, 56, 57). These studies proposed that the increased acetylcarnitine present in resting skeletal muscle after DCA treatment relieves this deficit, reducing the amount of PCr hydrolyzed during muscle contraction. However, muscle acetylcarnitine has been altered before contraction without significant changes in PDH activity and had no effect on phosphorus metabolites during moderate-intensity exercise (17, 49, 58). Therefore, the effect of DCA treatment on muscle energetics is not likely due to changes in acetylcarnitine content but rather to the activation state of PDH. Furthermore, the present data do not support the concept that activation of PDH relieves “metabolic inertia,” as the time constants of both PCr hydrolysis and resynthesis were unaffected by DCA treatment. This suggests that both the “on”-kinetics of oxidative phosphorylation and the mitochondrial oxidative capacity were not affected by PDH activation. Instead, the present data support an increase in matrix redox state driven by PDH activation and increases in glucose oxidation that are transduced through the PMF and ATP synthesis machinery resulting in increased magnitude of steady-state ΔGATP.
Perspectives and Significance
In summary, PDH activation through DCA treatment resulted in a significant increase in the magnitude of steady-state ΔGATP at rest and during stimulation below the reported aerobic threshold of 1.0 Hz. This increase in energetic status was achieved without a significant change in the glycolytic contribution to ATP production during stimulation, muscle force generation (excluding 0.75 Hz), cytosolic ATPase load, or the time constants of PCr hydrolysis or resynthesis. From the present data we proposed that the most likely mechanism for the increase in the magnitude of ΔGATP with PDH activation is an increase in the mitochondrial redox status (NADH/NAD+) contributing to an increase in mitochondrial PMF and driving the ATP synthesis machinery (F1FO-ATPase, adenine nucleotide translocase) to a new steady state supporting the elevated ΔGATP. Furthermore, at exercise intensities above this threshold PDH activation had no significant effect on steady-state energetics, as PDH activity was similar between conditions and the maximal rate of ATP synthesis was likely limiting. Taken together, these data do not support a role of PDH activation in relieving “metabolic inertia” but rather support a shift in the thermodynamic set point that is likely attributable to an increase in the steady-state redox potential through altering mitochondrial substrate oxidation in favor of pyruvate utilization.
GRANTS
This work was funded by National Institute of Diabetes and Digestive and Kidney Diseases Grant RO1 DK-095210.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.D.K., R.A.M., and R.W.W. conceived and designed research; J.D.K., R.A.M., and R.W.W. performed experiments; J.D.K. and R.W.W. analyzed data; J.D.K., R.A.M., D.A.B., and R.W.W. interpreted results of experiments; J.D.K. and R.W.W. prepared figures; J.D.K. and R.W.W. drafted manuscript; J.D.K., R.A.M., D.A.B., and R.W.W. edited and revised manuscript; R.A.M., D.A.B., and R.W.W. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the work of Dr. Anne Tonson for help with quantification of 31P NMR data and Dr. Jason Bazil and Matthew Lewis for critical reading of this manuscript.
REFERENCES
- 1.Adams GR, Foley JM, Meyer RA. Muscle buffer capacity estimated from pH changes during rest-to-work transitions. J Appl Physiol (1985) 69: 968–972, 1990. doi: 10.1152/jappl.1990.69.3.968. [DOI] [PubMed] [Google Scholar]
- 2.Andres R, Cader G, Zierler KL. The quantitatively minor role of carbohydrate in oxidative metabolism by skeletal muscle in intact man in the basal state; measurements of oxygen and glucose uptake and carbon dioxide and lactate production in the forearm. J Clin Invest 35: 671–682, 1956. doi: 10.1172/JCI103324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balaban RS. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta 1787: 1334–1341, 2009. doi: 10.1016/j.bbabio.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balnave CD, Allen DG. Evidence for Na+/Ca2+ exchange in intact single skeletal muscle fibers from the mouse. Am J Physiol Cell Physiol 274: C940–C946, 1998. doi: 10.1152/ajpcell.1998.274.4.C940. [DOI] [PubMed] [Google Scholar]
- 5.Baltzan MA, Andres R, Cader G, Zierler KL. Heterogeneity of forearm metabolism with special reference to free fatty acids. J Clin Invest 41: 116–125, 1962. doi: 10.1172/JCI104453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brooks GA, Donovan CM. Effect of endurance training on glucose kinetics during exercise. Am J Physiol Endocrinol Metab 244: E505–E512, 1983. doi: 10.1152/ajpendo.1983.244.5.E505. [DOI] [PubMed] [Google Scholar]
- 7.Brown GC. Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem J 284: 1–13, 1992. doi: 10.1042/bj2840001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown JA, Gore DC. In vivo metabolic response of glucose to dichloroacetate in humans. J Surg Res 61: 391–394, 1996. doi: 10.1006/jsre.1996.0135. [DOI] [PubMed] [Google Scholar]
- 9.Bünger R, Mallet RT, Hartman DA. Pyruvate-enhanced phosphorylation potential and inotropism in normoxic and postischemic isolated working heart. Near-complete prevention of reperfusion contractile failure. Eur J Biochem 180: 221–233, 1989. doi: 10.1111/j.1432-1033.1989.tb14637.x. [DOI] [PubMed] [Google Scholar]
- 10.Carraro F, Klein S, Rosenblatt JI, Wolfe RR. Effect of dichloroacetate on lactate concentration in exercising humans. J Appl Physiol (1985) 66: 591–597, 1989. doi: 10.1152/jappl.1989.66.2.591. [DOI] [PubMed] [Google Scholar]
- 11.Caterson ID, Fuller SJ, Randle PJ. Effect of the fatty acid oxidation inhibitor 2-tetradecylglycidic acid on pyruvate dehydrogenase complex activity in starved and alloxan-diabetic rats. Biochem J 208: 53–60, 1982. doi: 10.1042/bj2080053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark AS, Mitch WE, Goodman MN, Fagan JM, Goheer MA, Curnow RT. Dichloroacetate inhibits glycolysis and augments insulin-stimulated glycogen synthesis in rat muscle. J Clin Invest 79: 588–594, 1987. doi: 10.1172/JCI112851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Constantin-Teodosiu D, Carlin JI, Cederblad G, Harris RC, Hultman E. Acetyl group accumulation and pyruvate dehydrogenase activity in human muscle during incremental exercise. Acta Physiol Scand 143: 367–372, 1991. doi: 10.1111/j.1748-1716.1991.tb09247.x. [DOI] [PubMed] [Google Scholar]
- 14.Constantin-Teodosiu D, Simpson EJ, Greenhaff PL. The importance of pyruvate availability to PDC activation and anaplerosis in human skeletal muscle. Am J Physiol Endocrinol Metab 276: E472–E478, 1999. doi: 10.1152/ajpendo.1999.276.3.E472. [DOI] [PubMed] [Google Scholar]
- 15.Dagenais GR, Tancredi RG, Zierler KL. Free fatty acid oxidation by forearm muscle at rest, and evidence for an intramuscular lipid pool in the human forearm. J Clin Invest 58: 421–431, 1976. doi: 10.1172/JCI108486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.deBoisblanc BP, Meszaros K, Burns A, Bagby GJ, Nelson S, Summer WR. Effect of dichloroacetate on mechanical performance and metabolism of compromised diaphragm muscle. J Appl Physiol (1985) 72: 1149–1155, 1992. doi: 10.1152/jappl.1992.72.3.1149. [DOI] [PubMed] [Google Scholar]
- 17.Evans MK, Savasi I, Heigenhauser GJ, Spriet LL. Effects of acetate infusion and hyperoxia on muscle substrate phosphorylation after onset of moderate exercise. Am J Physiol Endocrinol Metab 281: E1144–E1150, 2001. doi: 10.1152/ajpendo.2001.281.6.E1144. [DOI] [PubMed] [Google Scholar]
- 18.Goodman MN, Ruderman NB, Aoki TT. Glucose and amino acid metabolism in perfused skeletal muscle. Effect of dichloroacetate. Diabetes 27: 1065–1074, 1978. doi: 10.2337/diab.27.11.1065. [DOI] [PubMed] [Google Scholar]
- 19.Greenhaff PL, Campbell-O’Sullivan SP, Constantin-Teodosiu D, Poucher SM, Roberts PA, Timmons JA. An acetyl group deficit limits mitochondrial ATP production at the onset of exercise. Biochem Soc Trans 30: 275–280, 2002. doi: 10.1042/bst0300275. [DOI] [PubMed] [Google Scholar]
- 20.Hagg SA, Taylor SI, Ruderman NB. Glucose metabolism in perfused skeletal muscle. Pyruvate dehydrogenase activity in starvation, diabetes and exercise. Biochem J 158: 203–210, 1976. doi: 10.1042/bj1580203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harkema SJ, Meyer RA. Effect of acidosis on control of respiration in skeletal muscle. Am J Physiol Cell Physiol 272: C491–C500, 1997. doi: 10.1152/ajpcell.1997.272.2.C491. [DOI] [PubMed] [Google Scholar]
- 22.Harris RA, Bowker-Kinley MM, Huang B, Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul 42: 249–259, 2002. doi: 10.1016/S0065-2571(01)00061-9. [DOI] [PubMed] [Google Scholar]
- 23.Head SI. Membrane potential, resting calcium and calcium transients in isolated muscle fibres from normal and dystrophic mice. J Physiol 469: 11–19, 1993. doi: 10.1113/jphysiol.1993.sp019801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hennig G, Löffler G, Wieland OH. Active and inactive forms of pyruvatedehydrogenase in skeletal muscle as related to the metabolic and functional state of the muscle cell. FEBS Lett 59: 142–145, 1975. doi: 10.1016/0014-5793(75)80361-9. [DOI] [PubMed] [Google Scholar]
- 25.Hood DA, Gorski J, Terjung RL. Oxygen cost of twitch and tetanic isometric contractions of rat skeletal muscle. Am J Physiol Endocrinol Metab 250: E449–E456, 1986. doi: 10.1152/ajpendo.1986.250.4.E449. [DOI] [PubMed] [Google Scholar]
- 26.Howlett RA, Heigenhauser GJ, Hultman E, Hollidge-Horvat MG, Spriet LL. Effects of dichloroacetate infusion on human skeletal muscle metabolism at the onset of exercise. Am J Physiol Endocrinol Metab 277: E18–E25, 1999. doi: 10.1152/ajpendo.1999.277.1.E18. [DOI] [PubMed] [Google Scholar]
- 27.Howlett RA, Parolin ML, Dyck DJ, Hultman E, Jones NL, Heigenhauser GJ, Spriet LL. Regulation of skeletal muscle glycogen phosphorylase and PDH at varying exercise power outputs. Am J Physiol Regul Integr Comp Physiol 275: R418–R425, 1998. doi: 10.1152/ajpregu.1998.275.2.R418. [DOI] [PubMed] [Google Scholar]
- 28.Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297: E578–E591, 2009. doi: 10.1152/ajpendo.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeneson JA, Wiseman RW, Kushmerick MJ. Non-invasive quantitative 31P MRS assay of mitochondrial function in skeletal muscle in situ. Mol Cell Biochem 174: 17–22, 1997. doi: 10.1023/A:1006891420109. [DOI] [PubMed] [Google Scholar]
- 30.Kelley DE, Reilly JP, Veneman T, Mandarino LJ. Effects of insulin on skeletal muscle glucose storage, oxidation, and glycolysis in humans. Am J Physiol Endocrinol Metab 258: E923–E929, 1990. doi: 10.1152/ajpendo.1990.258.6.E923. [DOI] [PubMed] [Google Scholar]
- 31.Kelley DE, Simoneau JA. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J Clin Invest 94: 2349–2356, 1994. doi: 10.1172/JCI117600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koretsky AP, Balaban RS. Changes in pyridine nucleotide levels alter oxygen consumption and extra-mitochondrial phosphates in isolated mitochondria: a 31P-NMR and NAD(P)H fluorescence study. Biochim Biophys Acta 893: 398–408, 1987. doi: 10.1016/0005-2728(87)90092-2. [DOI] [PubMed] [Google Scholar]
- 33.Kushmerick MJ, Meyer RA. Chemical changes in rat leg muscle by phosphorus nuclear magnetic resonance. Am J Physiol Cell Physiol 248: C542–C549, 1985. doi: 10.1152/ajpcell.1985.248.5.C542. [DOI] [PubMed] [Google Scholar]
- 34.Kushmerick MJ, Moerland TS, Wiseman RW. Mammalian skeletal muscle fibers distinguished by contents of phosphocreatine, ATP, and Pi. Proc Natl Acad Sci USA 89: 7521–7525, 1992. doi: 10.1073/pnas.89.16.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laughlin MR, Taylor J, Chesnick AS, DeGroot M, Balaban RS. Pyruvate and lactate metabolism in the in vivo dog heart. Am J Physiol Heart Circ Physiol 264: H2068–H2079, 1993. doi: 10.1152/ajpheart.1993.264.6.H2068. [DOI] [PubMed] [Google Scholar]
- 36.Lawson JW, Veech RL. Effects of pH and free Mg2+ on the Keq of the creatine kinase reaction and other phosphate hydrolyses and phosphate transfer reactions. J Biol Chem 254: 6528–6537, 1979. [PubMed] [Google Scholar]
- 37.van Loon LJ, Greenhaff PL, Constantin-Teodosiu D, Saris WH, Wagenmakers AJ. The effects of increasing exercise intensity on muscle fuel utilisation in humans. J Physiol 536: 295–304, 2001. doi: 10.1111/j.1469-7793.2001.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowry OH, Passonneau JV. A Flexible System of Enzymatic Analysis. New York: Academic, 1972. [Google Scholar]
- 39.McAllister A, Allison SP, Randle PJ. Effects of dichloroacetate on the metabolism of glucose, pyruvate, acetate, 3-hydroxybutyrate and palmitate in rat diaphragm and heart muscle in vitro and on extraction of glucose, lactate, pyruvate and free fatty acids by dog heart in vivo. Biochem J 134: 1067–1081, 1973. doi: 10.1042/bj1341067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCormack JG, Denton RM. Role of calcium ions in the regulation of intramitochondrial metabolism. Properties of the Ca2+-sensitive dehydrogenases within intact uncoupled mitochondria from the white and brown adipose tissue of the rat. Biochem J 190: 95–105, 1980. doi: 10.1042/bj1900095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer RA. Linear dependence of muscle phosphocreatine kinetics on total creatine content. Am J Physiol Cell Physiol 257: C1149–C1157, 1989. doi: 10.1152/ajpcell.1989.257.6.C1149. [DOI] [PubMed] [Google Scholar]
- 42.Meyer RA, Brown TR, Kushmerick MJ. Phosphorus nuclear magnetic resonance of fast- and slow-twitch muscle. Am J Physiol Cell Physiol 248: C279–C287, 1985. doi: 10.1152/ajpcell.1985.248.3.C279. [DOI] [PubMed] [Google Scholar]
- 43.Parolin ML, Chesley A, Matsos MP, Spriet LL, Jones NL, Heigenhauser GJ. Regulation of skeletal muscle glycogen phosphorylase and PDH during maximal intermittent exercise. Am J Physiol Endocrinol Metab 277: E890–E900, 1999 10.1152/ajpendo.1999.277.5.E890. [DOI] [PubMed] [Google Scholar]
- 44.Parolin ML, Spriet LL, Hultman E, Matsos MP, Hollidge-Horvat MG, Jones NL, Heigenhauser GJ. Effects of PDH activation by dichloroacetate in human skeletal muscle during exercise in hypoxia. Am J Physiol Endocrinol Metab 279: E752–E761, 2000. doi: 10.1152/ajpendo.2000.279.4.E752. [DOI] [PubMed] [Google Scholar]
- 45.Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans 34: 217–222, 2006. doi: 10.1042/BST0340217. [DOI] [PubMed] [Google Scholar]
- 46.Péronnet F, Massicotte D. Table of nonprotein respiratory quotient: an update. Can J Sport Sci 16: 23–29, 1991. [PubMed] [Google Scholar]
- 47.Peters SJ, St Amand TA, Howlett RA, Heigenhauser GJ, Spriet LL. Human skeletal muscle pyruvate dehydrogenase kinase activity increases after a low-carbohydrate diet. Am J Physiol Endocrinol Metab 275: E980–E986, 1998. doi: 10.1152/ajpendo.1998.275.6.E980. [DOI] [PubMed] [Google Scholar]
- 48.Phillips SK, Wiseman RW, Woledge RC, Kushmerick MJ. The effect of metabolic fuel on force production and resting inorganic phosphate levels in mouse skeletal muscle. J Physiol 462: 135–146, 1993. doi: 10.1113/jphysiol.1993.sp019547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Putman CT, Spriet LL, Hultman E, Dyck DJ, Heigenhauser GJ. Skeletal muscle pyruvate dehydrogenase activity during acetate infusion in humans. Am J Physiol Endocrinol Metab 268: E1007–E1017, 1995. doi: 10.1152/ajpendo.1995.268.5.E1007. [DOI] [PubMed] [Google Scholar]
- 50.Roberts PA, Loxham SJ, Poucher SM, Constantin-Teodosiu D, Greenhaff PL. The acetyl group deficit at the onset of contraction in ischaemic canine skeletal muscle. J Physiol 544: 591–602, 2002. doi: 10.1113/jphysiol.2002.021097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E, Wolfe RR. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol Endocrinol Metab 265: E380–E391, 1993. doi: 10.1152/ajpendo.1993.265.3.E380. [DOI] [PubMed] [Google Scholar]
- 52.Savasi I, Evans MK, Heigenhauser GJ, Spriet LL. Skeletal muscle metabolism is unaffected by DCA infusion and hyperoxia after onset of intense aerobic exercise. Am J Physiol Endocrinol Metab 283: E108–E115, 2002. doi: 10.1152/ajpendo.00337.2001. [DOI] [PubMed] [Google Scholar]
- 53.Small L, Brandon AE, Quek LE, Krycer JR, James DE, Turner N, Cooney GJ. Acute activation of pyruvate dehydrogenase increases glucose oxidation in muscle without changing glucose uptake. Am J Physiol Endocrinol Metab 315: E258–E266, 2018. doi: 10.1152/ajpendo.00386.2017. [DOI] [PubMed] [Google Scholar]
- 54.Stefan D, Cesare FD, Andrasescu A, Popa E, Lazariev A, Vescovo E, Strbak O, Williams S, Starcuk Z, Cabanas M, van Ormondt D, Graveron-Demilly D. Quantitation of magnetic resonance spectroscopy signals: the jMRUI software package. Meas Sci Technol 20: 104035, 2009. doi: 10.1088/0957-0233/20/10/104035. [DOI] [Google Scholar]
- 55.Sterk JP, Stanley WC, Hoppel CL, Kerner J. A radiochemical pyruvate dehydrogenase assay: activity in heart. Anal Biochem 313: 179–182, 2003. doi: 10.1016/S0003-2697(02)00538-9. [DOI] [PubMed] [Google Scholar]
- 56.Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Greenhaff PL. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. Am J Physiol Endocrinol Metab 274: E377–E380, 1998. doi: 10.1152/ajpendo.1998.274.2.E377. [DOI] [PubMed] [Google Scholar]
- 57.Timmons JA, Poucher SM, Constantin-Teodosiu D, Worrall V, Macdonald IA, Greenhaff PL. Increased acetyl group availability enhances contractile function of canine skeletal muscle during ischemia. J Clin Invest 97: 879–883, 1996. doi: 10.1172/JCI118490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watt MJ, Heigenhauser GJ, Stellingwerff T, Hargreaves M, Spriet LL. Carbohydrate ingestion reduces skeletal muscle acetylcarnitine availability but has no effect on substrate phosphorylation at the onset of exercise in man. J Physiol 544: 949–956, 2002. doi: 10.1113/jphysiol.2002.026757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Westerblad H, Allen DG. Changes of myoplasmic calcium concentration during fatigue in single mouse muscle fibers. J Gen Physiol 98: 615–635, 1991. doi: 10.1085/jgp.98.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J 141: 761–774, 1974. doi: 10.1042/bj1410761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wiseman RW, Moerland TS, Chase PB, Stuppard R, Kushmerick MJ. High-performance liquid chromatographic assays for free and phosphorylated derivatives of the creatine analogues beta-guanidopropionic acid and 1-carboxy-methyl-2-iminoimidazolidine (cyclocreatine). Anal Biochem 204: 383–389, 1992. doi: 10.1016/0003-2697(92)90255-6. [DOI] [PubMed] [Google Scholar]
- 62.Wu F, Jeneson JA, Beard DA. Oxidative ATP synthesis in skeletal muscle is controlled by substrate feedback. Am J Physiol Cell Physiol 292: C115–C124, 2007. doi: 10.1152/ajpcell.00237.2006. [DOI] [PubMed] [Google Scholar]
- 63.Wu P, Inskeep K, Bowker-Kinley MM, Popov KM, Harris RA. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 48: 1593–1599, 1999. doi: 10.2337/diabetes.48.8.1593. [DOI] [PubMed] [Google Scholar]
- 64.Zweier JL, Jacobus WE. Substrate-induced alterations of high energy phosphate metabolism and contractile function in the perfused heart. J Biol Chem 262: 8015–8021, 1987. [PubMed] [Google Scholar]