

Abstract
Idiopathic pulmonary fibrosis (IPF) is a fibroproliferative lung disease, and fibroblast-myofibroblast differentiation (FMD) is thought to be a key event in the pathogenesis of IPF. Histone deacetylase-8 (HDAC8) has been shown to associate with α-smooth muscle actin (α-SMA; a marker of FMD) and regulates cell contractility in vascular smooth muscle cells. However, the role of HDAC8 in FMD or pulmonary fibrosis has never been reported. This study investigated the role of HDAC8 in pulmonary fibrosis with a focus on FMD. We observed that HDAC8 expression was increased in IPF lung tissue as well as transforming growth factor (TGF)β1-treated normal human lung fibroblasts (NHLFs). Immunoprecipitation experiments revealed that HDAC8 was associated with α-SMA in TGFβ1-treated NHLFs. HDAC8 inhibition with NCC170 (HDAC8-selective inhibitor) repressed TGFβ1-induced fibroblast contraction and α-SMA protein expression in NHLFs cultured in collagen gels. HDAC8 inhibition with HDAC8 siRNA also repressed TGFβ1-induced expression of profibrotic molecules such as fibronectin and increased expression of antifibrotic molecules such as peroxisome proliferator-activated receptor-γ (PPARγ). Chromatin immunoprecipitation quantitative PCR using an antibody against H3K27ac (histone H3 acetylated at lysine 27; a known HDAC8 substrate and a marker for active enhancers) suggested that HDAC8 inhibition with NCC170 ameliorated TGFβ1-induced loss of H3K27ac at the PPARγ gene enhancer. Furthermore, NCC170 treatment significantly decreased fibrosis measured by Ashcroft score as well as expression of type 1 collagen and fibronectin in bleomycin-treated mouse lungs. These data suggest that HDAC8 contributes to pulmonary fibrosis and that there is a therapeutic potential for HDAC8 inhibitors to treat IPF as well as other fibrotic lung diseases.
Keywords: α-SMA, bleomycin, fibronectin, FMD, HDAC8, IPF, PPARγ, TGFβ1, type 1 collagen
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and fatal disease of unclear etiology, with increasing incidence and mortality worldwide (19, 37). Current therapies have only partial efficacy, and thus, additional medications are needed (38). A prominent pathological feature of IPF is the formation of fibroblast foci, which consist of myofibroblasts and the extracellular matrix that they produce. Myofibroblasts are the principal effector cells synthesizing profibrotic proteins such as α-smooth muscle actin (α-SMA), type 1 collagen, and fibronectin. Although multiple types of cells can differentiate into myofibroblasts, fibroblast to myofibroblast differentiation (FMD) is the major source for myofibroblast accumulation (16). Among many fibrogenic cytokines implicated in the pathogenesis of pulmonary fibrosis, transforming growth factor (TGF)β1 has been shown to play a crucial role (52).
Histone deacetylases (HDACs) are enzymes that catalyze the removal of acetyl functional groups from the lysine residues of both histone and nonhistone proteins. Removal of histone acetyl epigenetic modification by HDACs regulates chromatin structure and transcription, whereas deacetylation of nonhistone proteins controls diverse cellular processes. HDAC inhibitors are potential anticancer agents and show promise for the treatment of many diseases including fibrotic diseases (15, 34, 42).
It has been reported that IPF lungs exhibit distinct expression patterns of HDACs, including HDAC8 (21). Immunohistochemical staining of IPF lungs demonstrates HDAC8 expression in myofibroblasts of fibroblastic foci as well as vascular smooth muscle cells and bronchiolar epithelial cells. Furthermore, immunoblots reveal significantly elevated protein expression of HDAC8 in IPF fibroblasts compared with control fibroblasts.
Although a few studies have investigated the effects of nonselective pan-HDAC inhibitors as well as class I HDAC inhibitors on pulmonary fibrosis (2, 3, 21, 41, 47), the effects of selective inhibition of HDAC8 on pulmonary fibrosis have never been reported. Therefore, we set out to investigate the role of HDAC8 in pulmonary fibrosis by use of cells and tissue from humans and mice.
We observed that HDAC8 expression is increased in IPF lungs. We also found that inhibition of HDAC8 at least partially represses TGFβ1-induced FMD and increases peroxisome proliferator-activated receptor-γ (PPARγ) mRNA transcription via an epigenetic mechanism. The data that an HDAC8 inhibitor ameliorates bleomycin-induced pulmonary fibrosis in mice suggest a therapeutic potential for HDAC8 inhibitors to treat IPF and possibly other fibrotic lung diseases.
METHODS
Reagents and antibodies.
NCC170, an HDAC8-selective inhibitor, was synthesized as described previously (43) and prepared in DMSO at a stock concentration of 10 mM. Human recombinant TGFβ1 was purchased from R&D Systems (Minneapolis, MN). Bleomycin was purchased from TEVA Pharmaceutical Industries (Petach Tikva, Israel). The following primary antibodies were purchased from the following companies: Santa Cruz Biotechnology (Dallas, TX; HDAC8), Abcam (Cambridge, MA; type 1 collagen), Sigma-Aldrich (St. Louis, MO; α-SMA), Cell Signaling Technology (Danvers, MA); β-actin, Smad2, phosphorylated Smad2, Smad3, phosphorylated Smad3, Akt, phosphorylated Akt, PPARγ, structural maintenance of chromosomes protein-3 (SMC-3), acetylated histone 3 K27 (H3K27ac). Anti-acetylated SMC3 antibody was a generous gift from Dr. Katsuhiko Shirahige (University of Tokyo) (32). Secondary antibodies used were anti-mouse or anti-rabbit IgG horseradish peroxidase-linked antibodies (Cell Signaling Technology).
Human lung tissue samples.
Frozen lung tissue samples of patients with IPF (n = 20) and normal lung controls (n = 10) were obtained from the Lung Tissue Research Consortium (LTRC), a program sponsored by the National Heart, Lung and Blood Institute. The clinical data and specimens had been deidentified by the LTRC. Lung tissue was homogenized, and proteins were extracted in RIPA buffer (Cell Signaling Technology), according to the manufacturer’s protocols (51).
Cell culture of human lung fibroblasts.
Normal human lung fibroblasts (NHLFs) were purchased from Lonza (Allendale, NJ) and maintained in fibroblast growth medium 2 (FGM-2, Lonza) for experiments up to passage 6 per the provider’s instruction. Before the treatment, when the cells reached 80% confluence, they were serum starved in fibroblast basal medium 2 (FBM-2, Lonza) with 0.2% bovine serum albumin (BSA) overnight (27).
RNA interference.
RNA interference was carried out with HDAC8 siRNA, PPARγ siRNA (Gene Solution FlexiTube), and negative control siRNA (All Star Negative), which were purchased from Qiagen (Hilden, Germany). For interference experiments, 100 pmol siRNA oligo was transfected into NHLFs by using Lipofectamine RNAiMAX reagent (Invitrogen; Waltham, MA) according to the manufacturer’s protocol.
Immunoblots.
Cells were harvested using 1× RIPA buffer with protease inhibitors (Complete Mini, EDTA-free; Roche, Basel, Switzerland), PMSF (Roche), and phosphatase inhibitors (Phosphatase Inhibitor Cocktail 2 and 3, Sigma-Aldrich). Twenty micrograms of protein per sample was loaded onto NuPAGE Novex Bis-Tris 4–12% protein gels (Invitrogen) for electrophoresis and then transferred onto polyvinylidene difluoride membranes (0.45 μm; Millipore, Darmstadt, Germany). Membranes were then blocked in 5% nonfat dry milk (Bio-Rad, Hercules, CA) for 1 h at room temperature and then incubated with appropriate primary antibodies overnight at 4°C. Secondary antibodies and an ECL kit from GE Healthcare Life Sciences (Pittsburgh, PA) were applied for generating chemiluminescent signals. All immunoblot data represent triplicate repeats. Densitometry analysis was performed using National Institutes of Health (NIH) ImageJ software (27).
Immunoprecipitation.
Immunoprecipitation was performed using the Pierce IP kit (magnetic beads) according to the manufacturer’s instruction and as described previously (40).
Immunofluorescent staining.
Immunofluorescent staining was conducted as described previously (11). Briefly, NHLFs were seeded onto eight-well slides at 2 × 104 cells/well. When the cells were 80% confluent, they were serum starved overnight and then treated with or without TGFβ1 for 48 h. Cells were fixed in 4% paraformaldehyde and stained using the specified primary antibodies.
Type I collagen gel contraction assay.
The contraction assay was conducted as described previously (12). Twelve-well cell culture plates were precoated with 5% BSA-PBS coating solution overnight. On the next day, rat tail type I collagen (BD Biosciences, Bedford, MA) was prepared and mixed with NHLFs according to the provider's instructions. Briefly, NHLFs in FBM-2 with 0.2% BSA were added to type I collagen at a final concentration of 2 × 105 cells/ml. FBM-2 with 0.2% BSA was added to the collagen mixture to make a final concentration of 2 mg/ml collagen. Then, NaOH was added to the collagen-FBM-2 mixture (0.023 ml of 1 N NaOH/1 ml of the collagen/FBM-2 mixture). The 5% BSA-PBS coating solution was aspirated, and the plates were washed twice with PBS. Eight-hundred microliters of the cell-gel mixture was added to each well, and the plates were kept in a 37°C incubator for 30 min before treatment with TGFβ1, and DMSO or NCC170. Gel contraction was assessed as the ratio of the gel surface area to the area of the well.
Quantitative real-time PCR.
Quantitative real-time PCR (qRT-PCR) was performed using the iCycler (Bio-Rad), and SYBR Green supermix (Bio-Rad) was employed according to the manufacturer’s instructions, along with gene-specific primers. mRNA expression was corrected to expression of the 36B4 housekeeping gene. The specific gene’s cycle threshold (CT) values were normalized to the housekeeping gene 36B4 and compared with the control group that was assigned a value of 1 to calculate the relative fold change in expression as previously described (12). The results represent at least three independent experiments.
Chromatin immunoprecipitation-quantitative PCR.
Chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR) was performed using the SimpleChIP Enzymatic Chromatin IP Kit (Magnetic Beads; Cell Signaling Technology no. 9003) and antibodies against H3K27ac or rabbit IgG (as a control), according to the manufacturer’s protocol. Cistrome DB (http://cistrome.org/db/#/) was used to identify PPARγ gene enhancer regions (marked by H3K27ac) and design primers for those regions.
Mouse experiments with bleomycin and NCC170.
Six- to ten-week-old C57BL/6 male mice (Charles River, Wilmington, MA) were given bleomycin (2 U/kg, dissolved in 50 μl PBS) or the same volume of PBS by oropharyngeal aspiration on day 0, which was followed by daily intraperitoneal injection of NCC170 (40 mg·kg−1·day−1) or the same amount of vehicle (DMSO) on day 7 through day 13 (n = 6–7/group). Mice were euthanized on day 14. Right lungs were harvested and frozen in liquid nitrogen for subsequent qRT-PCR and immunoblot analysis. Total RNA and proteins were obtained using TRIzol reagent (Invitrogen) and RIPA buffer, respectively, according to the manufacturers’ instructions. Left lungs were perfused with 10% formalin and paraffin embedded, and then tissue slides (5 μm thickness) were prepared for Masson’s trichrome staining. The stained whole lung tissue section on a single slide was scanned with an Aperio Digital Pathology Whole Slide Scanner (Leica Biosystems, Buffalo Grove, IL). The extent of lung fibrosis was quantified according to the modified Ashcroft scale (18). Investigators conducting the scoring (M. E. Bateman and A. L. Alkhatib) were blinded to the treatment groups. Collagen content was also assessed using an internal program of the Aperio Slide Scanner, as described previously (27).
Statistical analysis.
Data are expressed as means ± SE. Unless otherwise indicated, statistical analyses for multiple group comparison were conducted using one-way analysis of variance followed by Bonferroni’s multiple comparison test, or Kruskal-Wallis test followed by Dunn’s multiple comparison test (GraphPad Prism, v. 5, La Jolla, CA). P < 0.05 was considered significant.
RESULTS
HDAC8 expression is increased in IPF lungs.
To compare the HDAC8 expression pattern in IPF and control lungs, we first examined the level of HDAC8 expression in lung tissue homogenates from individuals with IPF and individuals without IPF. Patient demographics are shown in Table 1. In our IPF lung tissue specimens, HDAC8 expression was mildly increased (Fig. 1A), which was consistent with a published report (21). This increase was statistically significant, based on densitometry analysis (Fig. 1B). Furthermore, HDAC8 expression appeared to correlate with disease severity as measured by forced vital capacity (FVC), a physiological parameter of lung function (Fig. 1B).
Table 1.
Patient demographics: donors of IPF lungs and control lungs
| Diagnosis | Sex | Age | Cigarette Smoking | Pack Years | Years Not Smoking Cigarettes | Race | FVC Pre-Bronchodilator (%Predicted) | DLCO (%Predicted) |
|---|---|---|---|---|---|---|---|---|
| Control | Woman | 88 | Unknown | White (Caucasian) | 104 | 66 | ||
| Control | Man | 44 | Ever (>100) | 15 | 9 | White (Caucasian) | 97 | 116 |
| Control | Man | 79 | Unknown | White (Caucasian) | 90 | 110 | ||
| Control | Woman | 61 | Ever (>100) | 50 | 0 | White (Caucasian) | 80 | 34 |
| Control | Woman | 81 | Never | White (Caucasian) | 100 | |||
| Control | Man | 82 | Never | White (Caucasian) | 77 | 75 | ||
| Control | Man | 69 | Unknown | White (Caucasian) | 96 | |||
| Control | Man | 77 | Unknown | White (Caucasian) | 88 | 83 | ||
| Control | Woman | 81 | Ever (>100) | 25 | 39 | White (Caucasian) | 107 | 66 |
| Control | Man | 79 | Ever (>100) | 23 | 28 | White (Caucasian) | 103 | 108 |
| IPF | Woman | 61 | Ever (>100) | 4 | 29 | White (Caucasian) | 60 | 41 |
| IPF | Man | 55 | Never | White (Caucasian) | 51 | |||
| IPF | Man | 66 | Unknown | White (Caucasian) | 51 | |||
| IPF | Woman | 65 | Never | White (Caucasian) | 50 | 27 | ||
| IPF | Man | 55 | Ever (>100) | 18 | 18 | White (Caucasian) | 67 | 25 |
| IPF | Man | 59 | Ever (>100) | 18 | 22 | White (Caucasian) | 66 | 52 |
| IPF | Man | 53 | Ever (>100) | 8 | 13 | White (Caucasian) | 64 | |
| IPF | Man | 60 | Unknown | White (Caucasian) | 53 | 20 | ||
| IPF | Man | 62 | Ever (>100) | 2 | 0 | African American | 69 | 28 |
| IPF | Man | 53 | Ever (>100) | 33 | 6 | White (Caucasian) | 53 | 22 |
| IPF | Woman | 62 | Never | White (Caucasian) | 43 | 30 | ||
| IPF | Man | 55 | Ever (>100) | 25 | 13 | White (Caucasian) | 44 | 16 |
| IPF | Man | 54 | Never | White (Caucasian) | 39 | 25 | ||
| IPF | Man | 37 | Unknown | White (Caucasian) | 26 | 23 | ||
| IPF | Woman | 50 | Ever (>100) | 5 | 33 | White (Caucasian) | 44 | 35 |
| IPF | Man | 61 | Never | White (Caucasian) | 24 | 18 | ||
| IPF | Man | 42 | Never | African American | 32 | |||
| IPF | Man | 62 | Never | Hispanic | 45 | 23 | ||
| IPF | Man | 51 | Ever (>100) | 20 | 0 | White (Caucasian) | 34 | 29 |
| IPF | Woman | 50 | Never | African American | 40 | 9 |
IPF, idiopathic pulmonary fibrosis; FVC, forced vital capacity; DLCO, diffusing capacity of lungs for carbon monoxide.
Fig. 1.

Histone deacetylase-8 (HDAC8) expression is increased in idiopathic pulmonary fibrosis (IPF) lungs. A: immunoblots of lung homogenates. β-Actin was used as a loading control. Control: forced vital capacity (FVC) ≥ 80% predicted; “Mild” IPF: FVC 50–79% predicted; “Severe” IPF: FVC < 50% predicted. FVC is a physiological parameter of lung function and a marker for disease severity in IPF. B: densitometry analyses of the immunoblots. HDAC8 protein expression is significantly increased in IPF lungs compared with control lungs. Left: HDAC8 protein expression is significantly increased in severe IPF lungs compared with controls (**P < 0.01, control vs. severe IPF). Although the comparison between control vs. mild IPF or mild IPF vs. severe IPF did not reach statistical significance, HDAC8 expression appeared to correlate with disease severity. Right: HDAC8 protein expression is significantly increased in IPF lungs compared with controls, when mild IPF and severe IPF were combined for analysis [**P < 0.01, control vs. IPF (mild + severe)].
HDAC8 is increased and associated with α-SMA in TGFβ1-treated NHLFs.
We next investigated the effect of TGFβ1 on HDAC8 expression and deacetylase activity in NHLFs. Acetylated SMC3 was measured as a surrogate marker for HDAC8 deacetylase activity, since SMC3 is deacetylated by HDAC8 but not by other HDACs (4). TGFβ1 mildly increased HDAC8 protein expression and decreased SMC3 acetylation (Fig. 2). This is consistent with the previous finding that fibroblasts isolated from IPF lungs showed increased HDAC8 expression compared with their controls (21). Immunoblots confirmed that the cells responded to TGFβ1 treatment with strong induction of α-SMA expression (Fig. 2).
Fig. 2.
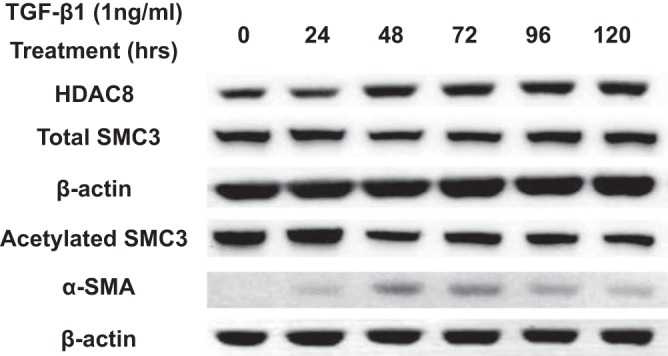
Transforming growth factor-β1 (TGFβ1) mildly increases histone deacetylase-8 (HDAC8) protein expression and decreases structural maintenance of chromosomes protein-3 (SMC3) acetylation. Subconfluent normal human lung fibroblasts (NHLFs) were serum starved overnight and then treated with 1 ng/ml TGFβ1 for the designated durations. Immunoblots of HDAC8 and β-actin were performed on lysates prepared from NHLFs. TGFβ1 treatment mildly increased HDAC8 protein expression and decreased acetylation of SMC3 (a known HDAC8 substrate). Immunoblots of the extracts for α-smooth muscle actin (α-SMA) supported the occurrence of fibroblast-myofibroblast differentiation (FMD).
We also investigated whether HDAC8 associates with α-SMA in TGFβ1-treated NHLFs, because HDAC8 has been shown to associate with α-SMA in smooth muscle cells (45). Immunoprecipitation with an antibody to HDAC8 showed that HDAC8 associated with α-SMA in TGFβ1-treated NHLFs (Fig. 3A). Immunofluorescent staining also suggested colocalization of HDAC8 and α-SMA within stress fibers in TGFβ1-treated NHLFs (Fig. 3B). Taken together, HDAC8 is slightly increased and associated with α-SMA in TGFβ1-treated NHLFs.
Fig. 3.

Histone deacetylase-8 (HDAC8) associates with α-smooth muscle actin (α-SMA) in transforming growth factor-β1 (TGFβ1)-treated normal human lung fibroblasts (NHLFs). Subconfluent NHLFs were serum starved overnight and then treated with TGFβ1 (1ng/ml) for 48 h. A: lysates from untreated and TGFβ1-treated cells were immunoprecipitated (IP) with an anti-HDAC8 antibody, anti-α-SMA antibody, or a nonspecific IgG control. Immunoprecipitated protein was immunoblotted for HDAC8 and α-SMA. The 2 right lanes (“Input”) represent an immunoblot of the lysates without IP. IP using anti-HDAC8 antibody suggested association between HDAC8 and α-SMA. Reverse IP using anti-α-SMA antibody also suggested association between HDAC8 and α-SMA, although HDAC8 was still weakly detected in IP using nonspecific IgG. This weak band was still detected even without nonspecific IgG (data not shown), which suggests that this weak band was due to nonspecific binding of α-SMA to beads. B: immunofluorescent staining revealed colocalization of HDAC8 and α-SMA within stress fibers in TGFβ1-treated NHLFs.
HDAC8 inhibition represses TGFβ1-induced contraction and α-SMA protein expression in NHLFs in a three-dimensional collagen gel.
We next investigated whether HDAC8 inhibition could reduce TGFβ1-induced contraction in NHLFs, because HDAC8 inhibition has been shown to reduce smooth muscle cell contractility (13) and because we found that a pan-HDAC inhibitor, trichostatin A (TSA), reduces TGFβ1-induced contraction in NHLFs (12). NHLFs cultured in a three-dimensional (3D) collagen gel were untreated or treated with TGFβ1 in the presence of an HDAC8-selective inhibitor (NCC170) or vehicle (DMSO). Untreated cells exposed to vehicle reduced the gel size by ~60% at 96 h (Fig. 4A). Addition of 1 ng/ml TGFβ1 increased the contractility of the cells as measured by reduction of the gel size by ~70% (Fig. 4A). Addition of NCC170 reversed the increased contraction associated with TGFβ1 treatment to that of untreated cells (○, Fig. 4A). These data agree with the concept that HDAC8 inhibition using NCC170 repressed TGFβ1-induced contraction of NHLFs (Fig. 4A). Furthermore, NCC170 prevented induction of α-SMA expression by TGFβ1. Repression of TGFβ1-mediated cell contractility by NCC170 correlated with repression of TGFβ1-dependent induction of α-SMA (Fig. 4B).
Fig. 4.
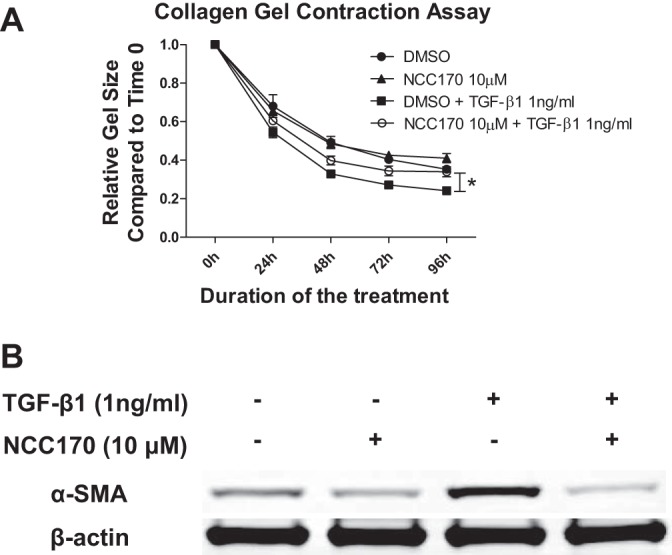
Histone deacetylase-8 (HDAC8) inhibitor NCC170 represses transforming growth factor-β1 (TGFβ1)-induced fibroblast contraction and α-smooth muscle actin (α-SMA) protein expression in collagen gels. normal human lung fibroblasts (NHLFs) were cultured in rat tail type I collagen gels (2 mg/ml) and treated with DMSO (vehicle) or NCC170, with or without TGFβ1 (1 ng/ml). A: NCC170 repressed TGFβ1-induced fibroblast contraction. The ratio of the area of the gel to culture well measured at 24 h intervals was used to assess gel contraction. Graph shows the area of the gel at indicated time points after treatment with DMSO (●), NCC170 (▲), DMSO+TGFβ1 (■), or NCC170+TGFβ1 (○). NCC170 ameliorated TGFβ1-induced collagen gel contraction (*P < 0.05; TGFβ1+DMSO vs. TGFβ1+NCC170). B: NCC-170 repressed TGFβ1-induced α-SMA protein expression. Gels were digested using type I collagenase (5 mg/ml), and cells were harvested in RIPA buffer at 48 h for detection of α-SMA by immunoblot. An immunoblot of β-actin was included as a loading control.
HDAC8 knockdown represses TGFβ1-induced expression of several profibrotic proteins while inducing antifibrotic proteins in NHLFs.
We next investigated the effects of HDAC8 inhibition on TGFβ1-induced FMD in NHLFs on six-well plates. Consistent with the HDAC8 inhibitor studies in the 3D gels, immunoblots showed that HDAC8 inhibition by HDAC8 siRNA repressed TGFβ1-induced α-SMA protein expression (Fig. 5A). HDAC8 knockdown also repressed TGFβ1-induced expression of other profibrotic proteins such as type 1 collagen, fibronectin, connective tissue growth factor (CTGF) (23), plasminogen activator inhibitor 1 (PAI-1) (33), and CCN1 (22) (Fig. 5A). On the other hand, HDAC8 knockdown ameliorated TGFβ1-induced repression of anti-fibrotic proteins such as PPARγ (1) and CCN3 (24) (Fig. 5A). Furthermore, HDAC8 inhibition repressed TGFβ1-induced phosphorylation of cofilin, an actin-binding protein essential for myofibroblast function (Fig. 5A) (20, 35, 49).
Fig. 5.

Histone deacetylase-8 (HDAC8) knockdown represses transforming growth factor-β1 (TGFβ1)-induced expression of several profibrotic proteins while inducing antifibrotic proteins in normal human lung fibroblasts (NHLFs). Subconfluent NHLFs were treated with negative control siRNA or HDAC8 siRNA for 72 h and then treated with TGFβ1 (1 or 2 ng/ml) for an additional 48 h before preparation of cell extracts in RIPA buffer (A) or an additional 24 h before preparation of total cellular RNA (B). A: immunoblots of the RIPA buffer extract revealed efficient knockdown of HDAC8 120 h posttransfection. HDAC8 knockdown repressed TGFβ1-induced expression of profibrotic proteins such as α-smooth muscle actin (α-SMA), type 1 collagen, fibronectin, connective tissue growth factor (CTGF), plasminogen activator inhibitor 1 (PAI-1), and CCN1, whereas HDAC8 knockdown increased expression of antifibrotic proteins such as peroxisome proliferator-activated receptor-γ (PPARγ) and CCN3. HDAC8 inhibition also repressed TGFβ1-induced phosphorylation of cofilin, an actin-binding protein essential for myofibroblast function. An immunoblot of β-actin is included as a loading control. B: quantitative RT-PCR analyses of total RNA isolated 96 h posttransfection of negative control siRNA or HDAC8 siRNA and 24 h after TGFβ1 treatment. Graphs show fold changes of the indicated mRNA after correction to the mRNA level of a housekeeping gene (36B4, a ribosomal protein) calculated by the 2−ΔΔCT method. Analyses confirmed efficient knockdown of HDAC8 mRNA (top left). HDAC8 knockdown repressed TGFβ1-induced mRNA expression of profibrotic genes such as α-SMA, fibronectin, CTGF, PAI-1, and lysyl oxidase (LOX), whereas HDAC8 knockdown increased mRNA expression of antifibrotic genes such as PPARγ and CCN3 (*P < 0.05, **P < 0.01, ***P < 0.001).
Our qRT-PCR results showed that HDAC8 inhibition significantly repressed TGFβ1-induced mRNA expression of Acta2 (= α-SMA), Fn1 (= fibronectin), Ctgf, Serpine1 (= PAI-1), but not Col1a1 (= type 1 collagen alpha 1) or Cyp61 (= CCN1). We also found that HDAC8 inhibition significantly repressed TGFβ1-induced expression of lysyl oxidase (LOX), an enzyme that posttranslationally modifies collagens and elastin to enable covalent cross-linking (2). On the other hand, HDAC8 knockdown ameliorated TGFβ1-induced repression of antifibrotic genes such as PPARγ and CCN3 at mRNA level (Fig. 5B). Taken together, these data suggest that HDAC8 inhibition represses several FMD markers in TGFβ1-treated NHLFs on two-dimensional six-well plates.
HDAC8 knockdown represses TGFβ1-induced expression of fibronectin, likely via PPARγ-dependent mechanism.
We next set out to explore potential mechanisms whereby HDAC8 inhibition represses TGFβ1-induced FMD. First, we investigated both the Smad and PI3K/Akt pathways, because these pathways are believed to be central to TGFβ1-induced FMD (6, 31, 52). However, HDAC8 inhibition did not appear to alter phosphorylation of Smad2, Smad3, or Akt (Fig. 6).
Fig. 6.
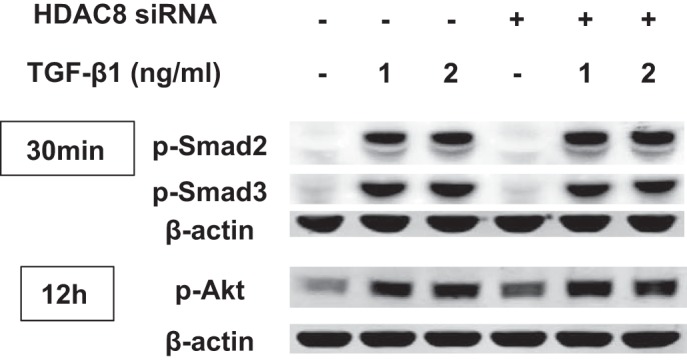
Histone deacetylase-8 (HDAC8) knockdown does not repress transforming growth factor-β1 (TGFβ1)-induced phosphorylation of Smad2/3 or Akt. Subconfluent normal human lung fibroblasts (NHLFs) were treated with negative control siRNA or HDAC8 siRNA for 72 h and then treated with TGFβ1 (1 ng/ml) for 30 min or 12 h. Immunoblots show that HDAC8 inhibition did not repress TGFβ1-induced phosphorylation of Smad2/3 or Akt.
We then investigated whether the repressive effects of HDAC8 knockdown on TGFβ1-induced FMD was due to upregulation of PPARγ. This was because PPARγ agonists inhibits FMD, although some of their inhibitory effects are PPARγ independent (1). PPARγ itself and PPARγ agonists have been shown to inhibit TGFβ-induced expression of fibronectin (10, 28, 46) and CTGF (7) in fibroblasts and smooth muscle cells.
Our immunoblots showed that HDAC8 knockdown failed to significantly repress TGFβ1-induced expression of fibronectin when PPARγ was knocked down. This suggests that the repressive effect of HDAC8 inhibition on TGFβ1-induced expression of fibronectin was mostly PPARγ dependent (Fig. 7). In contrast, HDAC8 knockdown did ameliorate TGFβ1-induced expression of CTGF, PAI-1, and α-SMA even when PPARγ was knocked down. This suggests that the repressive effect of HDAC8 inhibition on TGFβ1-induced expression of CTGF/PAI-1/α-SMA was likely PPARγ independent and was not due to upregulation of PPARγ (Fig. 7).
Fig. 7.
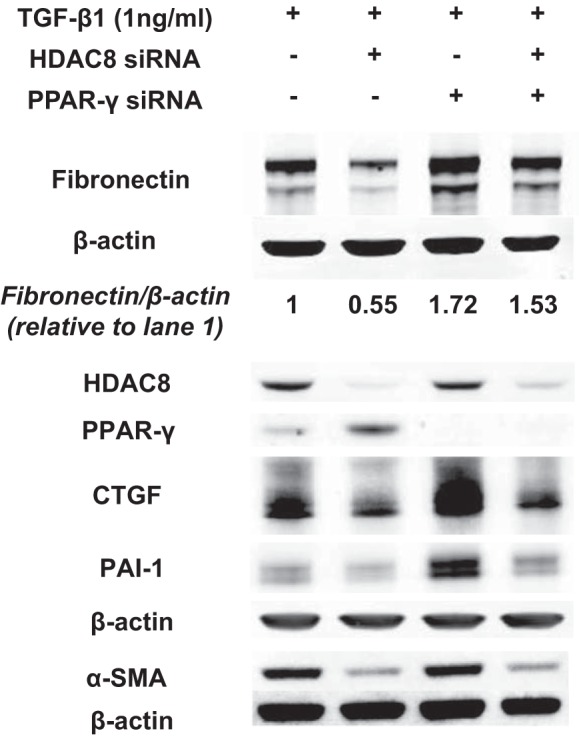
Histone deacetylase-8 (HDAC8) knockdown represses transforming growth factor-β1 (TGFβ1)-induced expression of fibronectin, likely via peroxisome proliferator-activated receptor-γ (PPARγ)-dependent mechanism. Subconfluent normal human lung fibroblasts (NHLFs) were treated with negative control siRNA or HDAC8 siRNA, or PPARγ siRNA, or HDAC8 siRNA + PPARγ siRNA for 72 h and then treated with TGFβ1 (1 ng/ml) for an additional 48 h. Immunoblots were performed to probe for HDAC8, PPARγ, fibronectin, connective tissue growth factor (CTGF), plasminogen activator inhibitor 1 (PAI-1), and α-smooth muscle actin (α-SMA). An immunoblot of β-actin was included as a loading control. HDAC8 knockdown failed to significantly repress TGFβ1-induced expression of fibronectin when PPARγ was knocked down. This suggests that the repressive effect of HDAC8 inhibition on TGFβ1-induced expression of fibronectin was mostly PPARγ dependent. In contrast, HDAC8 knockdown was still able to repress TGFβ1-induced expression of CTGF, PAI-1, and α-SMA when PPARγ was knocked down. This suggests that the repressive effect of HDAC8 inhibition on TGFβ1-induced expression of CTGF/PAI-1/α-SMA was likely PPARγ independent and not due to upregulation of PPARγ. Fibronectin/β-actin denotes the ratio of fibronectin to β-actin expression, calculated using densitometry analyses of the immunoblots.
HDAC8 inhibition increases H3K27 acetylation of PPARγ gene enhancer regions.
We next set out to explore potential mechanisms whereby HDAC8 inhibition increases PPARγ mRNA expression. To determine if the increased PPARγ mRNA levels after HDAC8 knockdown was due to increased mRNA stability, we assessed PPARγ mRNA levels in TGFβ1-treated NHLFs 8 h after inhibition of RNA synthesis with actinomycin D (10 μg/ml). HDAC8 inhibition had no effect upon PPARγ mRNA levels in NHLFs treated with TGFβ1 and actinomycin D (Fig. 8A). This suggests that HDAC8 inhibition does not affect PPARγ mRNA stability and that HDAC8 regulates PPARγ expression at the level of transcription. We then hypothesized that HDAC8 deacetylates H3K27 at PPARγ gene enhancer regions and reduces PPARγ mRNA transcription, because 1) the active enhancer mark H3K27ac (= acetylated H3K27) is highly induced on the PPARγ gene locus during adipogenesis and correlates with PPARγ gene expression (25); and 2) H3K27 is a known substrate of HDAC8 (13). Therefore, we performed ChIP-qPCR on NHLFs treated with or without TGFβ1 ± NCC170, using an antibody against H3K27ac and primers for PPARγ gene enhancer regions. The ChIP-qPCR experiments suggested that, in a subset of PPARγ gene enhancer regions, 1) HDAC8 inhibition significantly increased the H3K27ac level in the absence of TGFβ1; 2) TGFβ1 decreased the H3K27ac level; and 3) HDAC8 inhibition ameliorated TGFβ1-induced loss of H3K27ac (Fig. 8B). The most direct interpretation of these data is that HDAC8 inhibition activates PPARγ gene expression by increasing H3K27 acetylation in the PPARγ gene enhancer regions.
Fig. 8.

Histone deacetylase-8 (HDAC8) inhibition increases peroxisome proliferator-activated receptor-γ (PPARγ) expression, possibly by increasing H3K27 acetylation of the PPARγ gene enhancer. A: subconfluent normal human lung fibroblasts (NHLFs) were treated with negative control siRNA or HDAC8 siRNA for 72 h and then treated with transforming growth factor-β1 (TGFβ1, 1 ng/ml) for 24 h followed by addition of actinomycin D (Act D; 10 μg/ml) for 8 h to evaluate the effect of HDAC8 inhibition on PPARγ mRNA stability. Levels of PPARγ were determined by quantitative RT-PCR using 36B4 as an internal control. HDAC8 inhibition did not enhance PPARγ mRNA stability in NHLFs treated with TGFβ1 and actinomycin D. B: subconfluent NHLFs were pretreated with DMSO or NCC170 for 24 h and then treated with DMSO or NCC170 ± TGFβ1 for 24 h. Chromatin immunoprecipitation (ChIP)-PCR was performed using an antibody against acetylated histone 3 (H3)K27 (H3K27ac, a known HDAC8 substrate and marker for active enhancers) or IgG (as a control) and primers for the PPARγ gene enhancer region. For each treatment group, the ChIP signal was normalized to IgG control and expressed as “Fold Enrichment.” Averaged results of 3 independent experiments suggested that, in the enhancer region, 1) HDAC8 inhibition significantly increased the level of H3K27ac in the absence of TGFβ1; 2) TGFβ1 decreased the H3K27ac level; and 3) HDAC8 inhibition ameliorated TGFβ1-induced loss of H3K27ac.
HDAC8 inhibitor-treated mice are protected against bleomycin-induced pulmonary fibrosis.
We then investigated the effect of HDAC8 inhibition by NCC170 in a bleomycin-induced pulmonary fibrosis mouse model. Sections of formalin-fixed paraffin-embedded lung tissue from bleomycin-DMSO-treated mice displayed more fibrosis relative to negative control PBS-DMSO-treated mice. NCC170 treatment significantly reduced the histologic appearance of fibrogenesis in bleomycin-treated mice (Fig. 9A). Blinded histopathologic scoring of parenchymal changes using the modified Ashcroft scoring system revealed lower fibrosis scores among bleomycin-NCC170-treated mice compared with bleomycin-DMSO-treated mice (bleomycin-NCC170 = 3.31 ± 0.19 vs. bleomycin-DMSO = 4.05 ± 0.22, P < 0.05; Fig. 9B, top). This result was also supported by quantification of deposited collagen using automated image analysis of Masson trichrome staining. (Fig. 9B, bottom). We then assessed whether NCC170 altered expression of known fibrosis mediators and TGFβ targets in the bleomycin model. qRT-PCR showed that NCC170 significantly repressed bleomycin-induced mRNA expression of Col1a1 (type 1 collagen) and Fn1 (fibronectin). There was a trend for reduced mRNA expression of Lox and Ctgf, although it was not statistically significant (Fig. 9C). Immunoblots also showed that NCC170 repressed bleomycin-induced expression of type 1 collagen and fibronectin (Fig. 9D). These data indicate that HDAC8 inhibition diminishes bleomycin-induced pulmonary fibrosis in mice.
Fig. 9.

NCC170-treated mice are protected against bleomycin-induced pulmonary fibrosis. Mice were treated with PBS or bleomycin (2 U/kg, by oropharyngeal aspiration) on day 0, followed by daily intraperitoneal injection of DMSO or NCC170 (40 mg·kg−1·day−1) on day 8 through day 13 (n = 6–7/group) and harvest of the lungs on day 14. A and B: collagen expression was assessed using Masson’s trichrome staining of lung sections. The entire lung tissue section on a single slide was scanned with Aperio Digital Pathology Whole Slide Scanner. A: representative images (taken at ×40 magnification) are shown. Scale bars, 3 mm (left), 300 μm (right). B, top: extent of lung fibrosis was quantified according to an 8-tier, modified Ashcroft scale. NCC170 treatment significantly ameliorated bleomycin-induced pulmonary fibrosis. Bottom: amount of deposited collagen was quantified using automated image analysis of the Masson trichrome staining. NCC170 treatment significantly repressed collagen deposition in bleomycin-induced pulmonary fibrosis. *P < 0.05, **P < 0.01, ***P < 0.001. C: mRNA expression levels were assessed using quantitative RT-PCR of lung homogenates. NCC170 treatment significantly diminished bleomycin-induced expression of type 1 collagen (Col1a1) and fibronectin (Fn1) mRNA. Lox, lysyl oxidase; Ctgf, connective tissue growth factor; PAI-1, plasminogen activator inhibitor 1; α-SMA, α-smooth muscle actin; Pparg, peroxisome proliferator-activated receptor-γ. *P < 0.05, **P < 0.01, ***P < 0.001. D: protein expression levels were assessed using immunoblots of lung homogenates. NCC170 treatment significantly diminished bleomycin-induced expression of type 1 collagen and fibronectin proteins.
Fig. 9.

Continued
DISCUSSION
This study addressed the role of HDAC8 in lung fibrogenesis by examining HDAC8 expression in IPF lung homogenates and by examining the effect of HDAC8 inhibition on fibrogenesis in experimental models. First, we confirmed the previous finding that HDAC8 protein expression is slightly but significantly increased in IPF lungs (21). We next found that, in TGFβ1-treated NHLFs, HDAC8 associated with α-SMA and inhibition of HDAC8 repressed fibroblast contraction, as in smooth muscle cells (44, 45). This finding is relevant to the pathogenesis of and therapy for pulmonary fibrosis, because myofibroblast contraction induces profibrotic cascades extracellularly (e.g., activation of TGFβ in extracellular matrix) and intracellularly (e.g., integrin-mediated signal transduction) (17).
In NHLFs, we also found that HDAC8 inhibition repressed TGFβ1-induced expression of profibrotic proteins such as α-SMA, type 1 collagen, fibronectin, CTGF, PAI-1, and CCN1 (9, 23, 33, 36, 50). HDAC8 inhibition significantly repressed TGFβ1-induced expression of those genes at the mRNA level, except for Col1a1 and Cyr61 ( = CCN1). A mechanism how HDAC8 knockdown repressed type 1 collagen expression at the protein level but not the mRNA level may be that HDAC8 regulates collagen expression through posttranslational modification, such as oxidation by LOX (2). We found that HDAC8 inhibition repressed TGFβ1-induced expression of LOX. We suspect that the previously reported repressive effect of romidepsin (a class I HDAC inhibitor) on TGFβ1-induced LOX expression and FMD (2) was at least partially mediated by HDAC8 inhibition. A reason for no statistically significant reduction of CCN1 mRNA level by HDAC8 knockdown might simply be the small sample size, since there was a trend toward reduction. However, it is also possible that HDAC8 regulates CCN1 protein expression at the posttranslational level.
On the other hand, HDAC8 inhibition increased expression of antifibrotic genes such as PPARγ and CCN3,at both mRNA and protein levels.
Furthermore, we found that HDAC8 inhibition promoted dephosphorylation of cofilin in TGFβ1-treated fibroblasts. This is in line with the finding that HDAC8 inhibition repressed TGFβ1-induced fibroblast contraction, because dephosphorylation of cofilin promotes actin depolymerization and reduces cell contraction (30). A recent study in smooth muscle cells suggested that HDAC8 inhibition promotes dephosphorylation of cofilin by inducing acetylation of heat shock protein-20. Therefore, HDAC8 inhibition appears to reduce myofibroblast contractility through at least two mechanisms: reduction of α-SMA expression, and cofilin dephosphorylation (20, 26).
We examined the Smad and PI3K-Akt pathways in NHLFs because they have been shown to mediate TGFβ1-induced FMD (11, 29, 40, 52), and we observed that HDAC8 inhibition did not repress TGFβ1-induced phosphorylation of Smad2/3 or Akt. Since PPARγ has been shown to inhibit TGFβ1 signaling in fibroblasts by acting as a corepressor of Smad-mediated transcription (without preventing Smad2/3 activation) (8), we hypothesized that upregulation of PPARγ by HDAC8 inhibition is at least partially responsible for the repressive effects of HDAC8 inhibition on TGFβ1-induced FMD. We found that HDAC8 knockdown failed to repress TGFβ1-induced expression of fibronectin when PPARγ was also knocked down. This suggests that the repressive effect of HDAC8 inhibition on fibronectin expression was likely PPARγ dependent. In contrast, HDAC8 knockdown did repress TGFβ1-induced expression of CTGF, PAI-1, and α-SMA, even when PPARγ was knocked down. This suggests that the repressive effect of HDAC8 inhibition on CTGF/PAI-1/α-SMA expression was likely PPARγ independent. Therefore, HDAC8 inhibition appears to repress TGFβ1-induced FMD in both a PPARγ-dependent and -independent manner. Further research is needed to address the PPARγ-independent mechanisms.
With regard to the mechanism of how HDAC8 inhibition increases PPARγ expression, our qRT-PCR data suggested that HDAC8 inhibition increased PPARγ expression at the level of mRNA synthesis. Consistent with this view, our ChIP-qPCR experiments indicated that HDAC8 inhibition increases H3K27 acetylation at the PPARγ gene enhancer regions in both the presence and the absence of TGFβ1.
It has been shown that PPARγ levels are diminished in the lungs of patients with IPF or scleroderma (5, 48) and that TGFβ1 inhibits PPARγ expression in primary human lung fibroblasts, likely in part though regulation of Smad3 signaling (39). However, the detailed mechanism has been unclear. Our data suggest that 1) TGFβ1 decreases PPARγ mRNA transcription by decreasing H3K27ac at PPARγ enhancer regions, possibly by recruiting HDAC8 (and/or other HDACs) to those regions (via an unknown mechanism); and 2) HDAC8 inhibition increases PPARγ mRNA transcription by increasing H3K27ac at PPARγ enhancer regions. There may be additional mechanisms for how HDAC8 regulates expression of PPARγ. For example, HDAC8 may interact with (and deacetylate) other transcriptional factors or nonhistone proteins that regulate PPARγ expression. Our findings suggest an avenue for treating fibrotic diseases and, possibly other diseases (e.g., metabolic diseases and cardiovascular diseases) by promoting PPARγ signaling through HDAC8 inhibition.
Finally, we showed that NCC170 treatment significantly decreased fibrogenesis in bleomycin-treated mouse lungs. We demonstrated that NCC170 significantly repressed bleomycin-induced expression of type 1 collagen and fibronectin. NCC170 also appeared to repress bleomycin-induced protein expression of LOX and CTGF, but the reduction of their mRNA expression levels did not reach statistical significance. We did not observe significant differences in mRNA levels of PAI-1, α-SMA, or PPARγ, either. Possible reasons why mRNA levels of those genes were not significantly different are 1) the sample size was too small, 2) their expression levels were measured in whole lung homogenate and any significant changes seen in certain types of cells (e.g., fibroblasts) were diluted by other types of cells in the lungs, and 3) the time point we chose (14 days after bleomycin injection) was too late. We believe that the antifibrotic effect of NCC170 was mediated by HDAC8 inhibition, since NCC170 is an HDAC8-selective inhibitor; however, we cannot exclude the possibility that NCC170 inhibits pulmonary fibrosis independently of HDAC8. To address these points, further experiments using HDAC8 conditional knockout mice are under way in our laboratory. (We are generating conditional knockout mice because global deletion of HDAC8 in mice leads to perinatal lethality due to skull instability (14).)
In summary, this work demonstrates that HDAC8 expression is increased during lung fibrogenesis. More importantly, HDAC8 inhibition represses TGFβ1-induced FMD, at least partially, by increasing PPARγ gene transcription via restoration of H3K27 acetylation at enhancer region. The data that an HDAC8 inhibitor ameliorates bleomycin-induced pulmonary fibrosis in mice suggest a therapeutic potential for HDAC8 inhibitors to treat IPF and possibly other fibrotic diseases.
GRANTS
This work was upported by Wetmore Foundation Grant 555007 Task: M1 Award: 555007G1 and National Institute of General Medical Sciences of the National Institutes of Health (which funds the Louisiana Clinical and Translational Science Center) Grant 1-U54-GM-104940.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.S., T.S., Y.O., G.F.M., and J.A.L. conceived and designed research; S.S. and Y.Z. performed experiments; S.S., Y.Z., T.S., Y.O., M.E.B., A.L.A., G.F.M., and J.A.L. analyzed data; S.S., Y.Z., T.S., Y.O., M.E.B., A.L.A., G.F.M., and J.A.L. interpreted results of experiments; S.S. and Y.Z. prepared figures; S.S. drafted manuscript; S.S., G.F.M., and J.A.L. edited and revised manuscript; S.S., Y.Z., T.S., Y.O., M.E.B., A.L.A., G.F.M., and J.A.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Katsuhiko Shirahige (University of Tokyo) for supplying anti-acetylated SMC3 antibody, Dr. Philip Daroca (Tulane University) for assistance with Ashcroft scoring, and Dina Gaupp (Tulane University) for assistance with immunohistochemistry staining.
REFERENCES
- 1.Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM, Redonnet M, Phipps RP, Sime PJ. PPARγ agonists inhibit TGF-β-induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol 288: L1146–L1153, 2005. doi: 10.1152/ajplung.00383.2004. [DOI] [PubMed] [Google Scholar]
- 2.Conforti F, Davies ER, Calderwood CJ, Thatcher TH, Jones MG, Smart DE, Mahajan S, Alzetani A, Havelock T, Maher TM, Molyneaux PL, Thorley AJ, Tetley TD, Warner JA, Packham G, Ganesan A, Skipp PJ, Marshall BJ, Richeldi L, Sime PJ, O’Reilly KMA, Davies DE. The histone deacetylase inhibitor, romidepsin, as a potential treatment for pulmonary fibrosis. Oncotarget 8: 48737–48754, 2017. doi: 10.18632/oncotarget.17114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies ER, Haitchi HM, Thatcher TH, Sime PJ, Kottmann RM, Ganesan A, Packham G, O’Reilly KM, Davies DE. Spiruchostatin A inhibits proliferation and differentiation of fibroblasts from patients with pulmonary fibrosis. Am J Respir Cell Mol Biol 46: 687–694, 2012. doi: 10.1165/rcmb.2011-0040OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489: 313–317, 2012. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El Agha E, Moiseenko A, Kheirollahi V, De Langhe S, Crnkovic S, Kwapiszewska G, Szibor M, Kosanovic D, Schwind F, Schermuly RT, Henneke I, MacKenzie B, Quantius J, Herold S, Ntokou A, Ahlbrecht K, Braun T, Morty RE, Günther A, Seeger W, Bellusci S. Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell 20: 261–273, .e3, 2017. doi: 10.1016/j.stem.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez IE, Eickelberg O. The impact of TGFβ on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc 9: 111–116, 2012. doi: 10.1513/pats.201203-023AW. [DOI] [PubMed] [Google Scholar]
- 7.Fu M, Zhang J, Zhu X, Myles DE, Willson TM, Liu X, Chen YE. Peroxisome proliferator-activated receptor gamma inhibits transforming growth factor beta-induced connective tissue growth factor expression in human aortic smooth muscle cells by interfering with Smad3. J Biol Chem 276: 45888–45894, 2001. doi: 10.1074/jbc.M105490200. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh AK, Bhattacharyya S, Wei J, Kim S, Barak Y, Mori Y, Varga J. Peroxisome proliferator-activated receptor-gamma abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J 23: 2968–2977, 2009. doi: 10.1096/fj.08-128736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol 227: 493–507, 2012. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo B, Koya D, Isono M, Sugimoto T, Kashiwagi A, Haneda M. Peroxisome proliferator-activated receptor-gamma ligands inhibit TGF-beta 1-induced fibronectin expression in glomerular mesangial cells. Diabetes 53: 200–208, 2004. doi: 10.2337/diabetes.53.1.200. [DOI] [PubMed] [Google Scholar]
- 11.Guo W, Saito S, Sanchez CG, Zhuang Y, Gongora Rosero RE, Shan B, Luo F, Lasky JA. TGFβ1 stimulates HDAC4 nucleus-to-cytoplasm translocation and NADPH oxidase 4-derived reactive oxygen species in normal human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 312: L936–L944, 2017. doi: 10.1152/ajplung.00256.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo W, Shan B, Klingsberg RC, Qin X, Lasky JA. Abrogation of TGF-β1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am J Physiol Lung Cell Mol Physiol 297: L864–L870, 2009. doi: 10.1152/ajplung.00128.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ha SD, Han CY, Reid C, Kim SO. HDAC8-mediated epigenetic reprogramming plays a key role in resistance to anthrax lethal toxin-induced pyroptosis in macrophages. J Immunol 193: 1333–1343, 2014. doi: 10.4049/jimmunol.1400420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haberland M, Mokalled MH, Montgomery RL, Olson EN. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev 23: 1625–1630, 2009. doi: 10.1101/gad.1809209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10: 32–42, 2009. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 180: 1340–1355, 2012. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang X, Gai Y, Yang N, Lu B, Samuel CS, Thannickal VJ, Zhou Y. Relaxin regulates myofibroblast contractility and protects against lung fibrosis. Am J Pathol 179: 2751–2765, 2011. doi: 10.1016/j.ajpath.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hübner RH, Gitter W, El Mokhtari NE, Mathiak M, Both M, Bolte H, Freitag-Wolf S, Bewig B. Standardized quantification of pulmonary fibrosis in histological samples. Biotechniques 44: 507–517, 2008. doi: 10.2144/000112729. [DOI] [PubMed] [Google Scholar]
- 19.Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 46: 795–806, 2015. doi: 10.1183/09031936.00185114. [DOI] [PubMed] [Google Scholar]
- 20.Karolczak-Bayatti M, Sweeney M, Cheng J, Edey L, Robson SC, Ulrich SM, Treumann A, Taggart MJ, Europe-Finner GN. Acetylation of heat shock protein 20 (Hsp20) regulates human myometrial activity. J Biol Chem 286: 34346–34355, 2011. doi: 10.1074/jbc.M111.278549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korfei M, Skwarna S, Henneke I, MacKenzie B, Klymenko O, Saito S, Ruppert C, von der Beck D, Mahavadi P, Klepetko W, Bellusci S, Crestani B, Pullamsetti SS, Fink L, Seeger W, Krämer OH, Guenther A. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 70: 1022–1032, 2015. doi: 10.1136/thoraxjnl-2014-206411. [DOI] [PubMed] [Google Scholar]
- 22.Kurundkar AR, Kurundkar D, Rangarajan S, Locy ML, Zhou Y, Liu RM, Zmijewski J, Thannickal VJ. The matricellular protein CCN1 enhances TGFβ1/SMAD3-dependent profibrotic signaling in fibroblasts and contributes to fibrogenic responses to lung injury. FASEB J 30: 2135–2150, 2016. doi: 10.1096/fj.201500173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lasky JA, Ortiz LA, Tonthat B, Hoyle GW, Corti M, Athas G, Lungarella G, Brody A, Friedman M. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am J Physiol 275: L365–L371, 1998. [DOI] [PubMed] [Google Scholar]
- 24.Leask A. Yin and Yang revisited: CCN3 as an anti-fibrotic therapeutic? J Cell Commun Signal 9: 97–98, 2015. doi: 10.1007/s12079-015-0281-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JE, Ge K. Transcriptional and epigenetic regulation of PPARγ expression during adipogenesis. Cell Biosci 4: 29, 2014. doi: 10.1186/2045-3701-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Chen S, Cleary RA, Wang R, Gannon OJ, Seto E, Tang DD. Histone deacetylase 8 regulates cortactin deacetylation and contraction in smooth muscle tissues. Am J Physiol Cell Physiol 307: C288–C295, 2014. doi: 10.1152/ajpcell.00102.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo F, Zhuang Y, Sides MD, Sanchez CG, Shan B, White ES, Lasky JA. Arsenic trioxide inhibits transforming growth factor-β1-induced fibroblast to myofibroblast differentiation in vitro and bleomycin induced lung fibrosis in vivo. Respir Res 15: 51, 2014. doi: 10.1186/1465-9921-15-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maeda A, Horikoshi S, Gohda T, Tsuge T, Maeda K, Tomino Y. Pioglitazone attenuates TGF-beta(1)-induction of fibronectin synthesis and its splicing variant in human mesangial cells via activation of peroxisome proliferator-activated receptor (PPAR)gamma. Cell Biol Int 29: 422–428, 2005. doi: 10.1016/j.cellbi.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 29.Mercer PF, Woodcock HV, Eley JD, Platé M, Sulikowski MG, Durrenberger PF, Franklin L, Nanthakumar CB, Man Y, Genovese F, McAnulty RJ, Yang S, Maher TM, Nicholson AG, Blanchard AD, Marshall RP, Lukey PT, Chambers RC. Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax 71: 701–711, 2016. doi: 10.1136/thoraxjnl-2015-207429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal 25: 457–469, 2013. doi: 10.1016/j.cellsig.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Moore B, Lawson WE, Oury TD, Sisson TH, Raghavendran K, Hogaboam CM. Animal models of fibrotic lung disease. Am J Respir Cell Mol Biol 49: 167–179, 2013. doi: 10.1165/rcmb.2013-0094TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishiyama T, Ladurner R, Schmitz J, Kreidl E, Schleiffer A, Bhaskara V, Bando M, Shirahige K, Hyman AA, Mechtler K, Peters JM. Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell 143: 737–749, 2010. doi: 10.1016/j.cell.2010.10.031. [DOI] [PubMed] [Google Scholar]
- 33.Omori K, Hattori N, Senoo T, Takayama Y, Masuda T, Nakashima T, Iwamoto H, Fujitaka K, Hamada H, Kohno N. Inhibition of plasminogen activator inhibitor-1 attenuates transforming growth factor-β-dependent epithelial mesenchymal transition and differentiation of fibroblasts to myofibroblasts. PLoS One 11: e0148969, 2016. doi: 10.1371/journal.pone.0148969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pang M, Zhuang S. Histone deacetylase: a potential therapeutic target for fibrotic disorders. J Pharmacol Exp Ther 335: 266–272, 2010. doi: 10.1124/jpet.110.168385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pho M, Lee W, Watt DR, Laschinger C, Simmons CA, McCulloch CA. Cofilin is a marker of myofibroblast differentiation in cells from porcine aortic cardiac valves. Am J Physiol Heart Circ Physiol 294: H1767–H1778, 2008. doi: 10.1152/ajpheart.01305.2007. [DOI] [PubMed] [Google Scholar]
- 36.Ponticos M, Holmes AM, Shi-wen X, Leoni P, Khan K, Rajkumar VS, Hoyles RK, Bou-Gharios G, Black CM, Denton CP, Abraham DJ, Leask A, Lindahl GE. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum 60: 2142–2155, 2009. doi: 10.1002/art.24620. [DOI] [PubMed] [Google Scholar]
- 37.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis . An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183: 788–824, 2011. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, Brozek JL, Collard HR, Cunningham W, Homma S, Johkoh T, Martinez FJ, Myers J, Protzko SL, Richeldi L, Rind D, Selman M, Theodore A, Wells AU, Hoogsteden H, Schünemann HJ; American Thoracic Society; European Respiratory Society; Japanese Respiratory Society; Latin American Thoracic Association . An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 192: e3–e19, 2015. doi: 10.1164/rccm.201506-1063ST. [DOI] [PubMed] [Google Scholar]
- 39.Ramirez A, Ballard EN, Roman J. TGFβ1 controls PPARγ expression, transcriptional potential, and activity, in part, through Smad3 signaling in murine lung fibroblasts. PPAR Res 2012: 375876, 2012. doi: 10.1155/2012/375876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saito S, Zhuang Y, Shan B, Danchuk S, Luo F, Korfei M, Guenther A, Lasky JA. Tubastatin ameliorates pulmonary fibrosis by targeting the TGFβ-PI3K-Akt pathway. PLoS One 12: e0186615, 2017. doi: 10.1371/journal.pone.0186615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanders YY, Hagood JS, Liu H, Zhang W, Ambalavanan N, Thannickal VJ. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur Respir J 43: 1448–1458, 2014. doi: 10.1183/09031936.00095113. [DOI] [PubMed] [Google Scholar]
- 42.Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 6: a018713, 2014. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki T, Muto N, Bando M, Itoh Y, Masaki A, Ri M, Ota Y, Nakagawa H, Iida S, Shirahige K, Miyata N. Design, synthesis, and biological activity of NCC149 derivatives as histone deacetylase 8-selective inhibitors. ChemMedChem 9: 657–664, 2014. doi: 10.1002/cmdc.201300414. [DOI] [PubMed] [Google Scholar]
- 44.Waltregny D, De Leval L, Glénisson W, Ly Tran S, North BJ, Bellahcène A, Weidle U, Verdin E, Castronovo V. Expression of histone deacetylase 8, a class I histone deacetylase, is restricted to cells showing smooth muscle differentiation in normal human tissues. Am J Pathol 165: 553–564, 2004. doi: 10.1016/S0002-9440(10)63320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waltregny D, Glénisson W, Tran SL, North BJ, Verdin E, Colige A, Castronovo V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J 19: 966–968, 2005. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- 46.Wang W, Liu F, Chen N. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) agonists attenuate the profibrotic response induced by TGF-beta1 in renal interstitial fibroblasts. Mediators Inflamm 2007: 62641, 2007. doi: 10.1155/2007/62641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Chen C, Finger SN, Kwajah S, Jung M, Schwarz H, Swanson N, Lareu FF, Raghunath M. Suberoylanilide hydroxamic acid: a potential epigenetic therapeutic agent for lung fibrosis? Eur Respir J 34: 145–155, 2009. doi: 10.1183/09031936.00084808. [DOI] [PubMed] [Google Scholar]
- 48.Wei J, Ghosh AK, Sargent JL, Komura K, Wu M, Huang QQ, Jain M, Whitfield ML, Feghali-Bostwick C, Varga J. PPARγ downregulation by TGFß in fibroblast and impaired expression and function in systemic sclerosis: a novel mechanism for progressive fibrogenesis. PLoS One 5: e13778, 2010. doi: 10.1371/journal.pone.0013778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolfson NA, Pitcairn CA, Fierke CA. HDAC8 substrates: Histones and beyond. Biopolymers 99: 112–126, 2013. doi: 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang J, Velikoff M, Canalis E, Horowitz JC, Kim KK. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am J Physiol Lung Cell Mol Physiol 306: L786–L796, 2014. doi: 10.1152/ajplung.00243.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yue X, Li X, Nguyen HT, Chin DR, Sullivan DE, Lasky JA. Transforming growth factor-beta1 induces heparan sulfate 6-O-endosulfatase 1 expression in vitro and in vivo. J Biol Chem 283: 20397–20407, 2008. doi: 10.1074/jbc.M802850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yue X, Shan B, Lasky JA. TGFβ: titan of lung fibrogenesis. Curr Enzym Inhib 6: 6, 2010. doi: 10.2174/157340810791233033. [DOI] [PMC free article] [PubMed] [Google Scholar]