

Abstract
TGFβ activation during newborn lung injury decreases the expression of pulmonary artery smooth muscle cell (PASMC)-soluble guanylate cyclase (sGC), a critical mediator of nitric oxide signaling. Using a rat PASMC line (CS54 cells), we determined how TGFβ downregulates sGC expression. We found that TGFβ decreases sGC expression through stimulating its type I receptor; TGFβ type I receptor (TGFβR1) inhibitors prevented TGFβ-1-mediated decrease in sGCα1 subunit mRNA levels in the cells. However, TGFβR1-Smad mechanisms do not regulate sGC; effective knockdown of Smad2 and Smad3 expression and function did not protect sGCα1 mRNA levels during TGFβ-1 exposure. A targeted small-molecule kinase inhibitor screen suggested that MEK signaling regulates sGC expression in TGFβ-stimulated PASMC. TGFβ activates PASMC MEK/ERK signaling; CS54 cell treatment with TGFβ-1 increased MEK and ERK phosphorylation in a biphasic, time- and dose-dependent manner. Moreover, MEK/ERK activity appears to be required for TGFβ-mediated sGC expression inhibition in PASMC; MEK and ERK inhibitors protected sGCα1 mRNA expression in TGFβ-1-treated CS54 cells. Nuclear ERK activity is sufficient for sGC regulation; heterologous expression of a nucleus-retained, constitutively active ERK2-MEK1 fusion protein decreased CS54 cell sGCα1 mRNA levels. The in vivo relevance of this TGFβ-MEK/ERK-sGC downregulation pathway is suggested by the detection of ERK activation and sGCα1 protein expression downregulation in TGFβ-associated mouse pup hyperoxic lung injury, and the determination that ERK decreases sGCα1 protein expression in TGFβ-1-treated primary PASMC obtained from mouse pups. These studies identify MEK/ERK signaling as an important pathway by which TGFβ regulates sGC expression in PASMC.
Keywords: pulmonary vascular smooth muscle cells, soluble guanylate cyclase, transforming growth factor-β signaling
INTRODUCTION
Cyclic guanosine monophosphate (cGMP) plays an important role in regulating pulmonary vascular tone and lung development. cGMP is synthesized by nitric oxide (NO)-stimulated soluble guanylate cyclase (sGC) (reviewed in Ref. 22). sGC is a heterodimeric protein consisting of two homologous subunits, sGCα and sGCβ, each of which are expressed as two isoforms. The sGCα1 and sGCβ1 heterodimer is the most abundant one in the vasculature (30) and in the lung (66). The COOH-terminal portions of both subunits constitute the catalytic domain of sGC. Accordingly, both of the sGC subunits must heterodimerize for cGMP to be synthesized by the enzyme. cGMP has three intracellular targets: cGMP-dependent protein kinase I (PKGI), phosphodiesterases, and cyclic nucleotide-gated ion channels. In vascular smooth muscle cells (SMC), cGMP-stimulated PKGI phosphorylates several cytosolic proteins that regulate intracellular Ca2+ levels and the cytoskeleton, thereby controlling vascular tone. Moreover, PKGI has an important role in regulating cell phenotype (17, 51). Upon cGMP stimulation, PKGI can localize to the nucleus and phosphorylate transcription regulators (13, 31, 32). In SMC, cGMP stimulates the proteolysis of PKGI, which is caused in part by proprotein convertases residing within the endomembrane system, releasing a COOH-terminal portion of the molecule (PKGIγ) (41, 42, 86). This constitutively active PKGIγ fragment migrates into the nucleus via mechanisms requiring importins, transactivates gene expression, and regulates cell phenotype (15).
Pulmonary sGC expression and activity are developmentally regulated. In the fetal rat, scant sGC expression is detected during early lung development (11), when the conducting airway structures form. However, the sGC expression level greatly increases later, commencing during the saccular and early alveolar phases of pulmonary development. This burst of sGC expression is followed by a precipitous decrease of sGC expression in the adult rat lung. A similar developmental regulation of sGC expression has been detected in pigs (63). sGC is also differentially expressed within structures of the developing lung. In the perinatal rat lung, sGCα1 and sGCβ1 mRNA are localized within SMC of blood vessels and cells in the parenchyma (11). During the alveolar phase of fetal lamb lung development, sGC immunoreactivity accumulates within SMC of pulmonary arteries and veins and in parenchyma cells (19). sGC appears to have a role in regulating the later stages of lung development. Because sGC is not detected before the saccular phase of lung development (11), sGC has a limited role in regulating conducting airway structures development. However, sGC likely aids in pulmonary microvascular and alveolar development. This is because reduced pulmonary sGC activity in newborn sGCα1-deficient mice is associated with a decrease in alveolar structure development (5). sGC also has a role in regulating pulmonary blood flow in the newborn lung. Inhibition of sGC stimulation by NO in the fetal lamb decreases the normal surge in lung blood flow that occurs at the time of birth (2, 26). Also, decreased NO-mediated sGC stimulation in newborn lambs breathing hypoxic gas mixtures causes pulmonary hypertension (75).
Pulmonary sGC expression is decreased in many models of newborn lung injury. For example, in prematurely born lambs with O2- and ventilator-induced lung injury, sGC protein expression is diminished in the intrapulmonary arterial SMC, and this inhibits alveolar development and NO-dependent pulmonary vasodilation (10). Moreover, fetal lambs with pulmonary vascular injury, caused by prenatal ligation of the ductus arteriosus, also exhibit decreased sGC expression and activity in pulmonary artery smooth muscle cells (PASMC) and disrupt pulmonary vascular development (8, 89). Mouse pups exposed to chronic hyperoxic lung injury have decreased sGC expression in PASMC and lung interstitial cells (6). Moreover, they exhibit dysregulated pulmonary microvascular and alveolar formation (6). In contrast, it is interesting to note that in fetal lambs with pulmonary hypertension induced by an aorto-pulmonary vascular graft, lung sGC expression and cGMP levels are increased (9). Moreover, sGC expression can be increased in some adult lung injury models (52).
Decreased sGC expression during newborn lung injury plays a role in the pathogenesis of pulmonary disease. Reduced sGC activity in the injured newborn lung causes pulmonary hypertension. The diminished sGC expression also limits the effectiveness of inhaled NO and phosphodiesterase inhibitors in ameliorating pulmonary hypertension in newborns with lung injury. Recent studies have also suggested that decreased sGC activity during newborn lung injury disrupts pulmonary development (5). This is because sGCα1-deficient mouse pups exhibit markedly disrupted pulmonary microvascular and alveolarization, in comparison with wild-type mouse pups, when exposed to mild lung injury. Studies suggest also that stimulation of residual sGC activity in the injured newborn lung can partially protect pulmonary development. For example, inhaled NO improves pulmonary vascular development, inhibiting PASMC hyperplasia, in the injured rat pup lung through mechanisms that are independent of its vasodilatory properties (76, 77). Moreover, inhaled NO improved pulmonary vascular function, alveolarization, and extracellular matrix organization in the oxygen-injured premature baboon lung (60). sGC activators also prevented pulmonary vascular remodeling in hypoxic newborn rats (23). However, in these cases the improvement in pulmonary vascular tone and development was incomplete with sGC stimulation. It is desired to protect sGC expression during lung injury to potentiate the protective effects of cGMP in the developing lung.
The mechanisms that regulate sGC expression during newborn lung injury are poorly understood. TGFβ is activated during some forms of newborn lung injury (3, 4, 21, 62, 67) and can directly inhibit pulmonary development (4, 48, 67). Moreover, TGFβ has been shown to decrease sGC expression in vivo and in PASMC and aortic SMC (6). Here, we identify intracellular mechanisms by which TGFβ downregulates sGC expression in PASMC.
MATERIALS AND METHODS
Antibodies and reagents.
For the immunoblotting studies, Smad protein expression was detected using anti-Smad2 (No. 3103,1:500) and anti-Smad3 (No. 9523, 1:500) antibodies, and MEK and ERK isoform expression and activation were determined using anti-MEK1/2 (No. 8727, 1:1,000), anti-phospho (p)-MEK1/2 (p-Ser217/221 No. 9154, 1:1,000), anti-ERK1/2 (No. 9102,1:2,000), and anti-p-ERK1/2 (p-Thr202/Tyr204 No. 9101,1:1,000) antibodies, which were purchased from Cell Signaling Technology. The expression of the ERK2-MEK1 fusion proteins, which harbor a Myc-tag, was determined using an anti-Myc antibody (No. 2278; Cell Signaling Technology). sGCα1 subunit protein expression was detected using an anti-sGCα1 antibody (G4280, 1:10,000; Sigma-Aldrich), and GAPH expression was determined using an anti-GAPDH antibody (G8795, 1:2,000; Sigma-Aldrich). Enzyme-conjugated secondary antibodies were purchased from Jackson ImmunoResearch. For detection of protein expression in tissue by immunohistochemistry (IHC) and in cells using immunofluorescence (IF), the following antibodies were used: anti-p-ERK1/2 (No. 4370, IHC 1:400; Cell Signaling Technology), anti-sGCα1 (IHC 1:20,000 and IF, 1:200), and anti-smoothelin antibody (sc-28562, IHC 1:300 and IF 1:400; Santa Cruz Biotechnology). Isotype antibodies were obtained from a commercial source (Abcam). Alexa Fluor 488-tagged secondary antibodies were obtained from Thermo Fisher Scientific. The esiRNA targeting enhanced green fluorescent protein (eGFP; EHUEGFP), mSmad2 (EMU022831), and mSmad3 (EMU014271) were obtained from Sigma-Aldrich. Recombinant human TGFβ-1 (No. 240-B; R & D Systems) was reconstituted using 4 mM HCl in 1 mg/ml BSA. The kinase inhibitor dorsomorphin dihydrochloride (ab144821) was obtained from Abcam, GSK1120212 (CT-GSK212) and SCH772984 (CT-SCH772) were obtained from Chemietek, BAY11-7082 (BML-EI278), LL-Z1640-2 (ALX-380–267) was obtained from Enzo. AZD6244 (S1008), JNK-IN-8 (S4901), SB203580 (S1076), LY-294002 (S1105), and MK2206 (S1078) were obtained from Selleck Chemicals, and SD208 (S7071), NG25 (SML1332), and SB505124 (S4696) were obtained from Sigma. NG25 was dissolved in water, whereas the other inhibitors were dissolved in DMSO according to manufacturer’s instructions. In the control studies, the cells were treated with an equivalent volume of the inhibitor diluent. The sGC simulator BAY 41-8543 (No. 10011131) was obtained from Cayman, and it was dissolved in DMSO until use.
Plasmid constructs.
p3TP-lux, which expresses Photinus pyralis luciferase under the control of three tandem TGFβ-response elements (TRE) and the PAI-1 promoter (97), was obtained from Addgene (Plasmid 11767). pCMV-RL, which encodes Renilla remiformis luciferase driven by a CMV promoter, was purchased from Promega (E2261). pCMV.myc·ERK2-MEK1 and pCMV.myc·ERK2-L4A-MEK1 (78) (plasmids 39194 and 39197, respectively) and pcDNA3.1Green cGull (59) (plasmid 86867) were also obtained from Addgene. pAcGFP1-Nuc, which encodes GFP, was purchased from Takara (No. 632431).
Cell culture and transfection.
CS54 cells, a spontaneous rat PASMC line, was generated by Rothman et al. (see Ref. 80; also known as PAC1 cells) and kindly provided by R. B. Pilz (University of California, San Diego, CA). Primary mouse pup (m)PASMC were obtained from postnatal day (P) 10 FVB/NCrl mouse pups (Charles River) and identified by their characteristic morphology and reactivity with an anti-smoothelin antibody (103). All cells were maintained in DMEM containing 4.5 g/l glucose (hDMEM, No. 11995; Life Technologies). Complete media was formulated with 10% (vol/vol) heat-inactivated FBS (SH300803; Hyclone), 0.29 mg/ml glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. The cells were maintained in a humidified 37°C incubator containing 5% CO2 and passaged using EDTA-trypsin before becoming confluent. The mPASMC were used before the third passage. When cells were 80% confluent, they were transiently transfected with plasmids using Xfect transfection reagent (No. 631317; Takara) and methods detailed by the manufacturer.
RNA isolation and quantification.
mRNA levels were determined using specific primers and quantitative real-time (q)PCR, with GAPDH mRNA as a reference gene. RNA was extracted from cell lysates using phenol and guanidine isothiocyanate reagent (TRIzol; Invitrogen), precipitated in the presence of glycogen, and dissolved in diethyl pyrocarbonate-treated water. After the RNA quality was verified using spectroscopy (NanoDrop), it was quantified using an RNA-binding fluoroprobe (RiboGreen; Invitrogen) and fluorescence spectroscopy. cDNA were synthesized using PrimeScript RT reagents (RR047A; Takara), and PCR was performed using primers for sGCα1 (forward: 5′-AAG CAT GCA TCT GGA GAA GG-3′; reverse: 5′-TCT AAA GCC AGG TGG CAA AT-3′) and GAPDH (forward: 5′-AGA ACA TCA TCC CTG CAT CCA-3′; reverse: 5′-GCC TGC TTC ACC ACC TTC TTG-3′), SYBR Premix EX TaqII (RR820; Takara), Quant-iT RiboGreen RNA reagents (R11490; Thermo Fisher Scientific), and a thermocycler instrument (QuantStudio 3; Applied Biosystems). The specificity of the PCR primers was validated empirically by examining the DNA melting profile. The relative sGCα1 mRNA expression level was determined using the ΔΔCT method by subtracting the ddCT of GAPDH from that of sGCα1. The relative sGCα1 mRNA expression level was normalized to the mean level detected in the samples obtained from control reagent-treated cells. Samples were run in triplicate, and the median values of the samples were utilized in the analysis.
RNA knockdown.
Cells were seeded onto 4-cm2 wells in complete medium. When the cells were 60% confluent, the medium was refreshed and esiRNA transfection was performed using 14 pmol of total esiRNA and 3 µl/well of RNAiMax reagent in Optimem medium (Thermo Fisher Scientific). For experiments requiring transfection of esiRNA and reporter plasmids, the promoter-reporter plasmid constructs were introduced into the cells 6 h after the esiRNA transfection, as described above.
Promoter activity measurement.
Promoter activity was determined by measuring luciferase activities in the cell lysates using the Dual-Luciferase Reporter Assay System (E1910; Promega) and a luminometer (FLUOstar Omega; BMG Labtech) according to the manufacturer’s instructions. The promoter activation was determined by dividing the TRE and PAI-1 promoter-regulated luciferase activity by the CMV promoter-driven luciferase activity.
Immunoblotting.
For cellular protein expression determination, cells were scraped into ice-cold lysis buffer containing 50 mM Tris·HCl (pH 7.4), 1 mM EDTA, 1 mM dithiothreitol, and protease and phosphatase inhibitors (Halt 78447; Thermo Fisher Scientific). The lysates were then triturated through a small-bore needle using a syringe, sonicated, and kept on ice. For pulmonary tissue protein expression analysis, mouse pups were euthanized using 200 mg/kg pentobarbital sodium intraperitoneal injection, and whole lung tissues were obtained by dissection and frozen in liquid N2. Subsequently, the tissue was pulverized, and proteins were solubilized using the ice-cold lysis buffer and inhibitors described above. Following centrifugation to remove insoluble materials, the protein concentrations in the lysates were determined using bicinchoninic acid protein assay reagent (23227; Thermo Fisher Scientific), and protein molecular weight standards (Bio-Rad) and equal amounts of lysate proteins were resolved using SDS-PAGE and then electroblotted onto polyvinylidene difluoride membranes. The membranes were blocked using 5% nonfat dry milk in TBS containing 0.05% Tween-20 and then exposed to primary antibodies. Afterward, immunocomplexes were detected using peroxidase-conjugated secondary antibodies and chemiluminescent substrates. Enhanced chemiluminescence signals were acquired using a cooled charge-coupled device (CCD) camera system (ChemiDoc XRS; Bio-Rad). Uncalibrated densitometry was performed using ImageJ (74). For the characterization of the anti-sGCα1 antibody, P10 wild-type or sGCα1 knockout mouse pups, which are detailed elsewhere (6), were used.
Cellular sGC activity measurement.
sGC activity was measured in cells treated with and without an sGC stimulator by measuring cGMP levels. Human embryonic kidney (HEK)-293 cells seeded on 1.7-cm2 chamber slides were transfected with 0.5 µg of pcDNA3.1Green cGull or pAcGFP1-Nuc. After 36 h, the cells were serum restricted for 1 h and then treated with 0 or 2 µM SCH772984 for 1 h before addition of 0 or 10 ng/ml TGFβ-1 to the media. After 6 h, the cells were transferred to the heated stage of an inverted microscope (TiE; Nikon), and 0 or 3 µM BAY 41-8543 was added to the media. Starting at 45 s, wide-field fluorescence images were acquired at 30-s intervals using a light-emitting diode illumination source (Sola light engine; Lumencor), a ×20 objective lens (Plan Apo, NA 0.75; Nikon), 440- to 520-nm excitation, a 505-nm dichroic mirror, and a 485- to 585-nm emission filter set (No. 96320; Nikon), CCD camera (DS-Ri1; Nikon), and image acquisition software (NIS Elements; Nikon). Image stacks were analyzed in a masked fashion using the following methods. For identification of the sGC-stimulation period leading to substrate-unlimited, linear fluorescence signal increase, the image stacks were adjusted using identical intensity lookup tables, region of interests were mapped on 10 cells in each treatment group expressing the fluorescent protein, and then the average signal intensity was determined for each of them at each time slice. For comparing the cGMP levels between treatment groups, 10 cells exhibiting green cGull fluorescence in each treatment group were identified, and the fluorescent signal was determined 9 min after the sGC stimulation.
Immunohistochemistry.
The Subcommittee for Research Animal Studies at the Massachusetts General Hospital approved the experiments described here. Protein expression was mapped in mouse pup lungs using specific antibodies, immunohistochemistry, and bright-field microscopy. Within 12 h of birth, FVB/NCrl mouse pups and dams commenced breathing either air or 85% O2, using methods described previously (67). We used this strain of mouse pups because others have shown that this oxygen exposure regimen disrupts alveolar development in them (94). On postnatal day 10, pups were euthanized with an intraperitoneal injection of 200 mg/kg pentobarbital sodium, and a thoracotomy was made to permit the lungs to collapse. The trachea was cannulated with a 0.6-mm outer diameter polyethylene tube (PE10; Harvard Apparatus), and the lungs were inflated with 3% formaldehyde in PBS at a distending pressure of 22 cm H2O pressure for 30 min. Subsequently, the airway was ligated while the lungs remained expanded, and then the pup was then submerged in the fixative overnight. The lungs were then dissected from the body and an ∼5-mm-thick transverse section of the left lung was obtained. After dehydration with graded EtOH solutions and equilibration in Clear Rite 3 (Richard Allen Scientific), the lung segments were embedded in paraffin. Subsequently, 6-µm-thick lung sections were obtained, cleared of paraffin, and rehydrated and then underwent antigen retrieval using 10 mM sodium citrate and 0.05% Tween-20, pH 6.0, under pressure for 15 min. After neutralizing the sections using PBS and quenching endogenous peroxide using 3% H2O2, the sections were permeabilized using 0.1% Triton X-100 in PBS, blocked with 5% goat serum in PBS containing 0.5% Tween-20, and then reacted with the primary antibodies overnight at 4°C. After washing, the sections were interacted with biotinylated secondary antibodies, avidin-biotin coupled peroxidase (Vector Laboratories), and metal enhanced DAB substrate (34065; Thermo Fisher Scientific) before being counterstained with Gill’s hematoxylin and dehydrated and having a coverslip mounted. Subsequently, 0.34-µm-thick z-axis stack images were acquired using a microscope with a motorized stage (Ti-E; Nikon) and integrated CCD camera system (DS-Ri1; Nikon), and then extended focus images were constructed (29).
Cellular sGCα1 protein expression measurement.
sGCα1 protein expression was quantified in WGA-labeled mPASMC using immunofluorescence and the following methods. mPASMC seeded on 1.7-cm2 chamber slides were treated with and without SCH772984 and TGFβ-1, as described for CS54 cells. Subsequently, they were washed with PBS, fixed with 4% formaldehyde in PBS, permeabilized with 0.1% Triton X-100, blocked with 5% goat serum in PBS, and incubated with the anti-sGCα1 antibody diluted in the blocking buffer. The next day, the unreacted antibody was washed off the cells using PBS, and they were then stained with Alexa Fluor 546-conjugated secondary and Alexa Fluor 488-conjugated WGA (5 µg/ml PBS) before being mounted with a coverslip. Subsequently, representative wide-field fluorescence images were obtained using an inverted microscope and integrated camera system (TiE; Nikon). In a masked fashion, regions of interest defined by the WGA reactivity were defined, and the integrated intensity representing the sGCα1 immunoreactivity was measured using an image analysis program (NIS Elements; Nikon).
Data analysis and statistical methods.
Unless otherwise indicated, the experiments were repeated at least three times, and representative data from one experiment are shown. For the inhibitor screen, the percent reduction of sGCα1 mRNA expression was determined by subtracting the ΔΔCT value determined using RNA from cells treated with TGFβ-1 and the inhibitor with the average ΔΔCT value measured in samples obtained from untreated control cells. This result was then normalized to the difference of the average ΔΔCT values for the TGFβ-1 and control cells and then multiplied by 100. To test whether or not the inhibitor prevented TGFβ-mediated decrease in sGCα1 mRNA levels, the ΔΔCT values determined using samples from the cells treated with TGFβ-1 and with the inhibitor were compared with those of the untreated control cells using a t-test. The resulting P values were then adjusted using the method of Benjamini and Yekutieli (7) to control for multiple testing and thereby diminish the false discovery rate during this analysis. A P value of >0.05 indicated that the inhibitor prevented the inhibition of sGCα1 mRNA expression by TGFβ treatment. The data were analyzed using R (73). For normally distributed data, when treatment-mediated variance was detected using a one-way model of ANOVA, a Student’s t-test was then used post hoc. For nonparametric data, treatment variance detected using a Kruskal-Wallis test was confirmed using a Mann-Whitney U-test. When three or more comparisons were made, a Bonferroni correction of the P value was used. Otherwise, P < 0.05 was considered to be significant.
RESULTS
TGFβ decreases sGCα1 mRNA expression in PASMC.
We studied the mechanisms by which TGFβ decreases sGC expression in PASMC because they express abundant sGC and exhibit decreased sGC levels in several newborn lung injury models (6, 10, 89). Previously, we showed that treatment of SMC with 10–20 ng/ml TGFβ-1 decreases sGCα1 mRNA levels by 3 h (6). However, as little as 2.5 ng/ml TGFβ-1 has been detected in the bronchoalveolar lavage of babies (47) and has been shown to be sufficient to increase intracellular PASMC signaling (103). To define the relationship between TGFβ and sGC expression in PASMC, we tested whether this lower TGFβ-1 level regulates sGCα1 expression in these cells. For these studies, TGFβ-1 was used because it is the archetypical TGFβ isoform, and it transduces intracellular signals through the same receptor-mediated mechanisms as TGFβ-2 and TGFβ-3. We detailed the expression of the sGCα1 isoform because its expression is abundant in PASMC, and this isoform plays a primary role in regulating cGMP production by nitric oxide (93). Moreover, we showed that reduced expression of the sGCα1 subunit alone is sufficient to decrease sGCβ1 protein levels and sGC enzyme activity in the lung (6). sGCα1 mRNA levels were evaluated because our previous work showed that TGFβ regulates sGCα1 gene expression at a transcriptional level (6). We used CS54 cells as a model PASMC line for this work because these cells express sGC despite passaging (6, 37).
Physiologically relevant TGFβ-1 levels were observed to rapidly decrease sGC subunit mRNA expression levels in PASMC. As shown in Fig. 1, 2.5 ng/ml TGFβ-1 decreased sGCα1 mRNA levels by ∼45% by 1 h and by nearly 90% after 6 h in the PASMC. Furthermore, the regulatory effect of TGFβ on sGC mRNA expression appeared to be saturable even at these low levels; at each time studied, the decrease in sGCa1 mRNA expression was similar over the range of TGFβ-1 treatments. sGCα1 mRNA levels decreased with the increasing duration of TGFβ-1 exposure. The data suggest also that as little as 2.5 ng/ml TGFβ-1 completely inhibits sGCα1 transcription in PASMC. This is because a similar reduction of sGCα1 mRNA expression was detected in vascular SMC with transcription inhibited by actinomycin D (6). As a result of these observations, and to be consistent with previous studies (6), in a balance of the studies detailed below we treated cells with 10 ng/ml TGFβ-1 for 6 h.
Fig. 1.
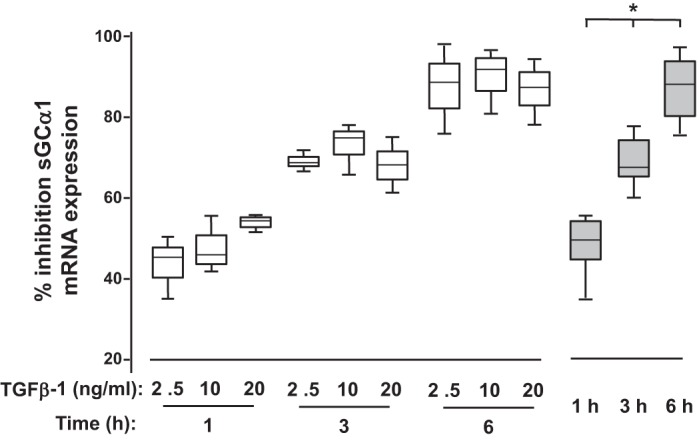
Transforming growth factor-β (TGFβ) decreases soluble guanylate cyclase-α1 (sGCα1) mRNA expression in pulmonary artery smooth muscle cells (PASMC). Serum-restricted CS54 cells were treated with either none or the indicated amounts of recombinant TGFβ-1 for the times shown. Subsequently, sGCα1 mRNA levels relative to that of GAPDH were determined in cell lysates using specific primers and quantitative PCR, and the %inhibition of sGCα1 mRNA expression was determined. Open bars, n = 3 in each group; gray bars, data combined from the cells treated with 2.5–20 ng/ml TGFβ-1 for the indicated times. *P < 0.015 vs. the other time intervals.
TGFβ decreases sGCα1 mRNA expression in PASMC by TGFβR1-dependent but TGFβR1/Smad-independent mechanisms.
TGFβ mediates intracellular signaling through canonical, TGFβR1/Smad-dependent, and TGFβR1/Smad-independent pathways (reviewed in Ref. 35). After extracellular activation, TGFβ binds to its type II receptor (TGFβR2), and this promotes the recruitment, phosphorylation, and activation of the type I TGFβ receptors (TGFβR1) activin-like kinase (ALK)4 and ALK5 in a heteromeric receptor complex. In some cases, accessory receptors such as betaglycan and endoglin assist in the TGFβ-receptor complex formation. In turn, the activated TGFβR1 in the complex phosphorylates COOH-terminal serine residues in Smad2 and Smad3. After Smad4 is recruited, the p-Smad2/3 proteins migrate as a complex into the nucleus, where they bind to TGFβ response elements in promoters and regulate the expression of specific genes. It is important to consider broadly the mechanisms by which physiological dosages of TGFβ might regulate PASMC sGC expression. This is because the manner in which TGFβ regulates gene expression is dependent on its dose level and duration, the cell type, and the tissue context.
To systematically define how TGFβ controls sGC expression in PASMC, we used small molecular kinase inhibitors and tested whether TGFβ-1 regulates sGCα1 mRNA levels in CS54 cells in a TGFβR1/Smad-dependent manner. Details about the inhibitory levels of the compounds used in the experiments and references describing their characterization are detailed in the Table 1. We determined that TGFβ mediates sGCα1 mRNA expression in a TGFβR1-dependent manner. As shown in Fig. 2A, treatment of the CS54 cells with SB-505124 and SD208, which are well-established TGFβR1 inhibitors, prevented a decrease in sGCα1 mRNA levels in the TGFβ-1-treated PASMC. However, decreasing Smad2 and Smad3 expression and activity in the PASMC using RNAi was determined to not protect sGCα1 mRNA expression in these TGFβ-1-exposed cells (Fig. 2, B and C). Together, these studies indicate that TGFβ decreases sGCα1 expression in PASMC via TGFβR1-dependent but not TGFβR1/Smad-dependent mechanisms.
Table 1.
TGFβ signaling kinase inhibitor compounds
| Target | Inhibitor | Dose, µM | Dose Ref. No. | IC50, nM | In Vitro or Cell Assay | IC50 Ref. No. |
|---|---|---|---|---|---|---|
| ALK1 | Dorsomorphin | 10.0 | 103 | 470* | Cell assay | 102 |
| TGFβR1 | SB-505124 | 1.0 | 103 | 47 | In vitro | 20 |
| TGFβR1 | SD208 | 1.0 | 92 | 48 | In vitro | 90 |
| TAK1 | LL-Z16402 | 1.0 | 68 | 8 | In vitro | 69 |
| TAK1 | NG25 | 2.0 | 24 | 149 | In vitro | 88 |
| P38 MAPK | SB-203580 | 1.0 | 18 | 600 | In vitro | 18 |
| JNK | JNK-IN-8 | 1.0 | 25 | 1–19 JNK isoforms | In vitro | 104 |
| IKKβ | BAY11–7082 | 1.0 | 101 | 7-fold range in the literature | Cell assay | 43 |
| PI3K | LY-294002 | 50.0 | 36 | 500–973 p110 isforms | In vitro | 14 |
| Akt | MK2206 | 10.0 | 71 | 5–65 Akt isoforms | In vitro | 99 |
| MEK1/2 | GSK1120212 | 1.0 | 64 | 1–2, MEK1 and 2 | In vitro | 98 |
| MEK1/2 | AZD6244 | 2.0 | 56 | 14 | In vitro | 100 |
| ERK1/2 | SCH772984 | 2.0 | 64 | 1–4 ERK1 and -2 | In vitro | 64 |
ALK1, activin-like kinase 1; JNK, c-Jun amino terminal kinase; PI3K, phosphatidylinositol 3-kinase; TAK1, transforming growth factor-β-activated kinase 1; TGFβR1, transforming growth factor-β receptor 1.
Based on BMP-induced Smad1/5/8 phosphorylation.
Fig. 2.
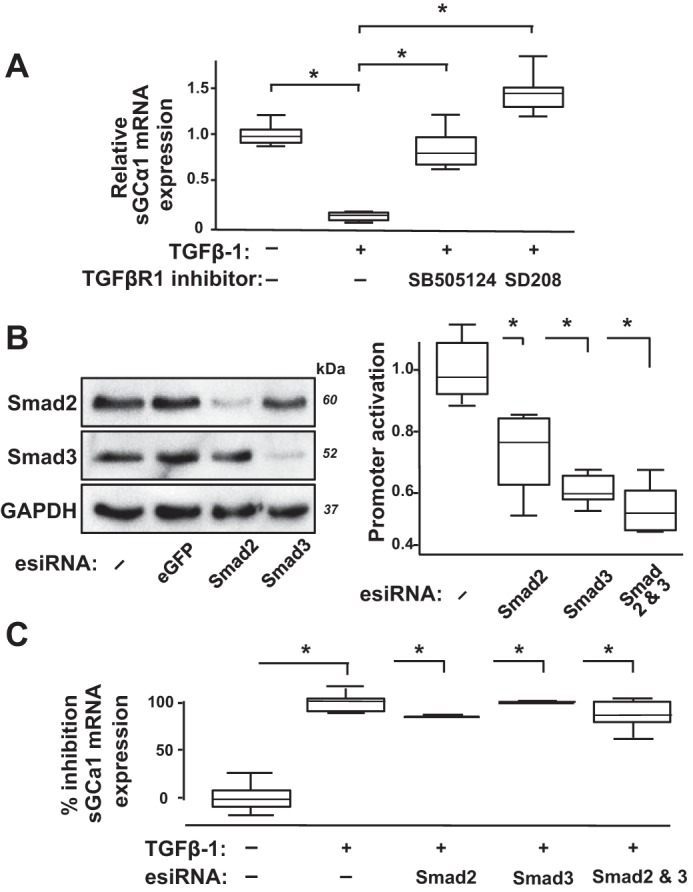
Transforming growth factor-β (TGFβ) decreases soluble guanylate cyclase-α1 (sGCα1) mRNA expression in pulmonary artery smooth muscle cells (PASMC) through TGFβ type I receptor-dependent but Smad2/3-independent mechanisms. A: TGFβ type I receptor (TGFβR1) kinase inhibitors SB-505124 and SD208 prevent TGFβ-mediated decrease in sGCα1 mRNA levels in PASMC. Serum-restricted CS54 cells were treated with 0 or 1 µM of the indicated inhibitors for 1 h before addition of 0 or 10 ng/ml TGFβ-1 to the media. The cells were lysed 6 h later, and the relative sGCα1 mRNA expression was determined; n = 6 in each group; *P < 0.015. B: esiRNA decreases Smad2/3 protein expression and function in PASMC. CS54 cells were transfected with either no esiRNA (−) or esiRNA targeting eGFP or the indicated Smad genes. The cells were lysed 48 h later, and protein expression was determined using immunoblotting. Additionally, CS54 cells were cotransfected with plasmids encoding Photinus pyralis luciferase driven by TGFβ-response elements and PAI-1 promoter and Renilla reniformis luciferase activated by a CMV promoter, and then 6 h later they were transfected with no esiRNA (−) or esiRNA targeting the indicated genes. After 36 h, the cells were serum restricted and treated with 10 ng/ml TGFβ-1 for 6 h, and the promoter activation was determined by measuring the luciferase activities in cell lysates and normalized to the average level detected in the cells without Smad knockdown; n = 6 in each group; *P < 0.015 vs. control. C: Smad1/2 knockdown did not prevent TGFβ-mediated decrease in sGCα1 mRNA expression in PASMC. CS54 cells were transfected without or with the indicated esiRNA, and after 48 h, the cells were serum restricted and treated with 0 or 10 ng/ml TGFβ-1 for 6 h, and the relative sGCα1 mRNA expression levels were determined and normalized to that of the TGFβ-1-treated cells; n = 3 in each group; *P < 0.012.
A small-molecule inhibitor screen identified possible noncanonical mechanisms by which TGFβ regulates sGC subunit mRNA expression.
TGFβ regulates gene expression through several mechanisms that are independent of Smad2/3. As illustrated in Fig. 3, left, TGFβR1 can stimulate ALK1. This leads to the activation of BMP R/Smads, Smad1/5, and downstream gene regulation in PASMC and fibroblasts in the developing lung (103). TGFβR1 can also activate TGFβ-activated kinase 1 (TAK1) (44) and thereby stimulate p38 mitogen-activated protein kinase (MAPK), c-Jun amino terminal kinase (JNK), and IkB kinase (IKKβ) activity. Moreover, through TAK1-independent mechanisms, TGFβR1 can stimulate phosphatidylinositol-3 kinase (PI3K)/Akt and mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling (50).
Fig. 3.

Small-molecule kinase inhibitor screen identified potential mechanisms by which Transforming growth factor-β (TGFβ) regulates soluble guanylate cyclase-α1 (sGCα1) mRNA expression in pulmonary artery smooth muscle cells (PASMC). Key noncanonical TGFβR1-regulated signaling systems (illustrated on the left and described in the text) were targeted using doses of small-molecule inhibitors shown by others to diminish the regulatory kinase activity. Serum-restricted CS54 cells were treated with or without the indicated inhibitor dose or diluent for 1 h, and then 0 or 10 ng/ml TGFβ-1 was added to the media. The cells were lysed 6 h later, the RNA was collected, and the relative sGCα1 mRNA expression was determined. The % reduction of sGCα1 mRNA expression caused by combined treatment with the inhibitor and TGFβ was calculated using methods defined in materials and methods; box plot data from 3 independent experiments are shown. The P values result from a comparison between the sGCα1 mRNA levels detected in cells treated with the TGFβ-1 and inhibitor and the levels determined in control, nontreated cells. ALK1, activin-like kinase 1; JNK, c-Jun amino terminal kinase; PI3K, phosphatidylinositol-3 kinase; TAK1, TGFβ-activated kinase 1.
To identify potential noncanonical pathways by which TGFβ regulates sGC expression in PASMC, we examined whether known inhibitory concentrations of small molecules that target TGFβR1-stimulated kinases protect sGCα1 expression in TGFβ-treated PASMC. We tested whether the compounds inhibited the percent reduction in sGCα1 mRNA levels detected in TGFβ-1-treated CS54 cells (Fig. 3). These studies determined that the TAK1 inhibitor LL-Z16402 increased and the MEK inhibitor GSK1120212 prevented a decrease in sGCα1 mRNA levels caused by TGFβ. Consistent with the data shown in the previous figure, the TGFβR1 inhibitor SB-505124 also prevented a decrease in sGCα1 expression by TGFβ. Inhibition of ALK1 using dorsomorphin did not protect sGCα1 mRNA levels in TGFβ-treated cells, suggesting that ALK1-stimulated Smad1/5-signaling does not regulate sGCα1 expression. Although treatment with LL-Z16402 implicated TAK1 as a mediator of sGC expression in TGFβ-treated cells, we found that exposure to inhibitors of TAK1 downstream signaling elements, e.g., p38 MAPK, JNK, and IKKβ, was not protective. This suggested that the effect of LL-Z16402 in TGFβ-treated cells might be mediated by an off-target effect of the inhibitor. Other investigators have reported that LL-Z16402 can regulate ERK signaling in hTERT human aortic SMC (70). Accordingly, we tested whether other TAK1 inhibition methods regulate TGFβ’s effect on sGCα1 mRNA expression in the cells. We determined that treatment with NG25, another TAK1 inhibitor, did not prevent decreased sGCα1 mRNA levels in TGFβ-treated CS54 cells. Additionally, we observed that effective TAK1 knockdown with targeting esiRNA did not inhibit TGFβ’s regulation of sGC expression in the cells (data not shown). These studies suggested that although TGFβR1-stimulated MEK signaling decreases sGC mRNA levels in TGFβ-treated CS54 cells, TAK1 does not play a role in this mechanism.
TGFβ activates MEK and ERK in PASMC.
Because the kinase inhibitor screen suggested that MEK mediates the regulation of sGC expression by TGFβ in PASMC, we next tested whether physiological dosages of TGFβ-1 stimulate MEK and ERK activation via phosphorylation in the CS54 cells. As shown in Fig. 4, although MEK and ERK exhibit basal phosphorylation in the CS54 cells, as little as 0.1 ng/ml TGFβ-1 increases MEK and ERK phosphorylation in these cells. Moreover, MEK and ERK phosphorylation were increased rapidly, within 5 min of the TGFβ-1 treatment. This was followed by a period of reduced MEK and ERK phosphorylation, despite continued TGFβ-1 exposure, and then phosphorylation of these mediators 3 h later. Growth factors have been shown to stimulate a similar biphasic pattern of MEK and ERK phosphorylation in some vascular SMC (83) and in fibroblasts (40, 61, 87). In fibroblasts, the rapid, initial burst of MEK and ERK phosphorylation following growth factor stimulation was found to be associated with the nuclear translocation of p-ERK; the later sustained phosphorylation of ERK in some cells was associated with an autocrine induction of growth fibroblast growth factor signaling (27).
Fig. 4.
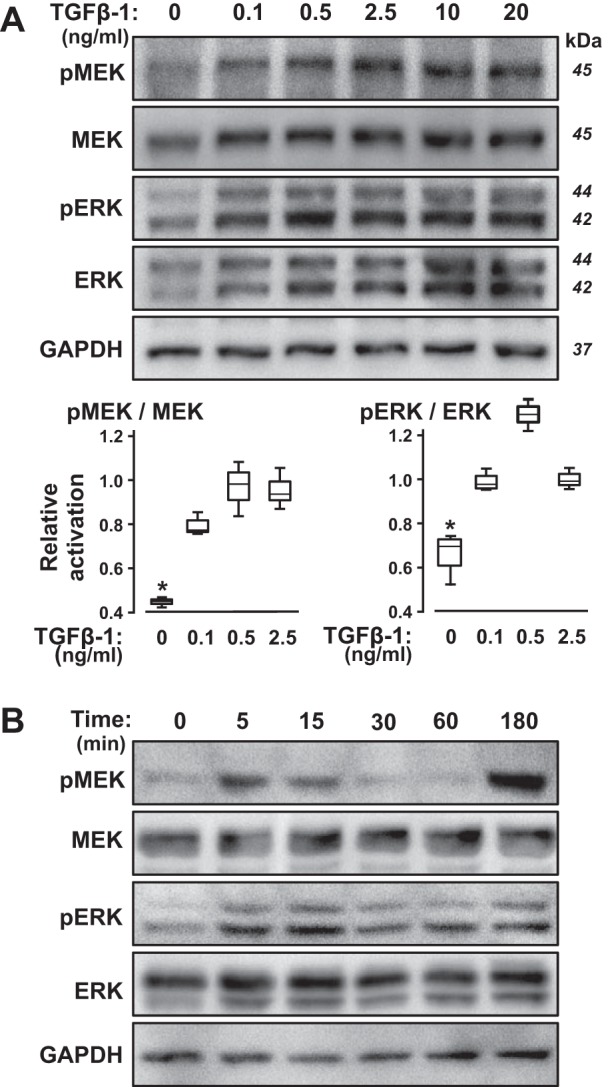
Transforming growth factor-β (TGFβ) induces MEK and ERK phosphorylation in pulmonary artery smooth muscle cells (PASMC) in a dose- and time-dependent manner in PASMC. Serum-restricted CS54 cells were treated with the indicated amounts of TGFβ-1 for 3 h (A) or with 10 ng/ml TGFβ-1 for the indicated times (B). Subsequently, the cells were lysed, and the level of the indicated phosphorylated (p) or total proteins was determined by immunoblotting. The molecular weights were determined using protein standards. The blot images are representative of 2 independent studies. The p-MEK/MEK and p-ERK/ERK levels were quantified in the lysates of CS54 cells treated with the indicated TGFβ-1 levels for 3 h using immunoblotting and densitometry in additional studies; n = 3; *P < 0.05 compared with the TGFβ-treated levels.
MEK and ERK inhibition protects sGCα1 expression and sGC enzyme activity.
To confirm the possible role of MEK and ERK signaling in regulating sGC expression in TGFβ-treated PASMC, we tested whether MEK and ERK inhibitors protect sGCα1 mRNA expression in cells treated with the cytokine. CS54 cells were treated without or with AZD6244 or SCH772984, MEK and ERK inhibitors, respectively, and TGFβ-1, and then sGCα1 mRNA levels were determined. As shown in Fig. 5A, these kinase inhibitors protected sGCα1 mRNA expression in the TGFβ-1-stimulated PASMC. A similar inhibition of TGFβ-mediated sGCα1 mRNA reduction by the MEK and ERK inhibitors suggests that the results were not due to an off-target effect of the agents.
Fig. 5.
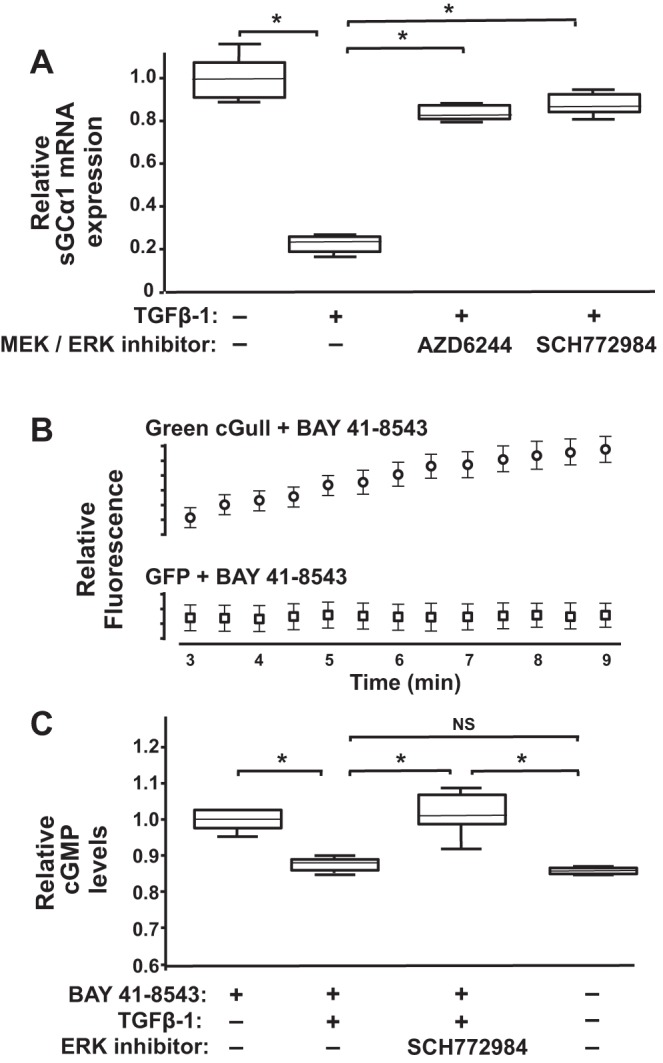
MEK and ERK control transforming growth factor-β (TGFβ)-mediated soluble guanylate cyclase-α1 (sGCα1) mRNA downregulation in pulmonary artery smooth muscle cells (PASMC). A: MEK and ERK kinase inhibitors protect sGCα1 mRNA levels in TGFβ-treated PASMC. Serum-restricted CS54 cells were treated with 0 or 2 µM of AZD6244 or SCH772984 for 1 h before addition of 0 or 10 ng/ml TGFβ-1 to the media. RNA was collected from lysed cells 6 h, later and then the relative sGCα1 mRNA expression levels were measured; n = 6/group; *P < 0.015. B: sGC stimulation increases green cGull but not green fluorescent protein (GFP) fluorescence in cells. Human embryonic kidney (HEK)-293 cells transfected with plasmids encoding the indicated fluorescent proteins were treated with 3 µM BAY 41-8543, and then fluorescence was measured at the indicated times; n = 10 cells/group; means ± SD. C: sGC activity in TGFβ-treated cells is protected by ERK inhibition. HEK-293 cells expressing green cGull were treated with the indicated reagents, and the mean cellular fluorescence was measured after 9 min. Results are representative of 2 independent experiments; n = 10 cells per group; *P < 0.012; NS, not significant.
We examined next whether ERK regulates sGC enzyme activity in TGFβ-treated cells. For this work, we used cells expressing green cGull, a newly developed, single-fluorescent protein, intracellular cGMP biosensor (59). Within this molecule, a mutated cGMP-binding domain from the mouse phosphodiesterase-5α was shown to bind to cGMP over a wide physiological range, increasing the sensor’s fluorescence in a dose-dependent manner. Moreover, we employed HEK-293 cells because they express sGC (34) and were shown previously to exhibit increased green cGull fluorescence in response to sGC stimulation (59). To identify the experimental conditions under which the cGMP detection exhibits substrate concentration independent, zero-order kinetics, first we determined the time period over which sGC stimulation causes a linear increase in the green cGull fluorescence signal in the cells. We observed that the sGC stimulator BAY 41-8543 caused a linear increase in green cGull fluorescence in the HEK-293 cells from 3 to 9 min after treatment (Fig. 5B). This rate and extent of fluorescence signal increase following BAY 41-8543 treatment was similar to that reported previously in these cells following sGC stimulation with a nitric oxide donor (59). In contrast, we found that although the initial fluorescence level of GFP-expressing HEK-293 cells was higher than that observed in cells harboring the cGMP biosensor, the GFP fluorescence was not modulated by BAY 41-8543 treatment. We determined that TGFβ-1 treatment decreased green cGull-detected cGMP levels in BAY 41-8543 treated HEK-293 cells to levels measured in cells without sGC stimulation. Importantly, ERK inhibition using SCH772984 prevented the decrease in sGC activity caused by the cytokine treatment in the cells (Fig. 5C). In additional control studies, the fluorescence level of green cGull expressing HEK-293 cells was not modulated in cells treated with the drug diluent (data not shown). Together these results suggest that TGFβ decreases sGC expression and activity in cells by MEK- and ERK-mediated mechanisms.
Nuclear ERK is sufficient to downregulate sGC mRNA expression in PASMC.
ERK regulates gene expression by phosphorylating transcription factors residing in differing cellular compartments (96). Upon MEK activation, ERK can phosphorylate transcription factors in the cytosol, such as RSK, that subsequently localize to the nucleus and regulate gene expression there. Activated ERK can also migrate into the nucleus and phosphorylate transcription factors that reside there, such as MSK, Elk-1, Myc, BRF1, and UBF.
To characterize how ERK regulates sGC, and to determine which cellular compartment mediates this effect in PASMC, we tested how heterologous expression of ERK-MEK fusion proteins with differing nuclear and cytoplasmic compartmentation might regulate sGC mRNA expression. Robinson et al. (78) generated a Myc-tagged ERK2-MEK1 fusion protein, which harbors ERK2 fused via a GLU-GLY linker to MEK1. Upon expression in mammalian cells, they demonstrated that the ERK2 portion of the protein becomes phosphorylated and constitutively active and that the fusion protein accumulates in the cytosol. Moreover, they demonstrated that when four leucines in the putative MEK nuclear export motif residing in the fusion protein are mutated to alanines (L4A), the encoded protein is retained also within the nuclear compartment and phosphorylates transcription factors residing there.
As shown in Fig. 6, A and B, transfecting CS54 cells with plasmids encoding these fusion proteins causes expression of proteins of the expected size that are retained within the expected cellular compartments, as described in other cell types (78). Whereas ERK2-L4A-MEK1 harboring a MEK nuclear export sequence mutation was immunolocalized to the nucleus and cytosol, ERK2-MEK1 was detected in the cytosol alone. Importantly, we determined that the nuclear but not cytosolic localization of the fusion protein was sufficient for sGCα1 mRNA expression downregulation. As shown in Fig. 6C, whereas expression of the nuclear and cytosolic ERK2-L4A-MEK1 protein decreased sGCα1 mRNA levels, expression of the fusion protein that accumulates within the cytosol alone did not. The decrease in sGCα1 mRNA expression associated with nuclear ERK2-L4A-MEK1 appears to be related to its ERK activity. This is because treatment of the cells with an ERK inhibitor (SCH772984) prevented the decrease in sGCα1 mRNA expression by the ERK2-L4A-MEK1 fusion protein. These data suggest that nuclear ERK activity plays a role in regulating sGCα1 mRNA expression in PASMC.
Fig. 6.
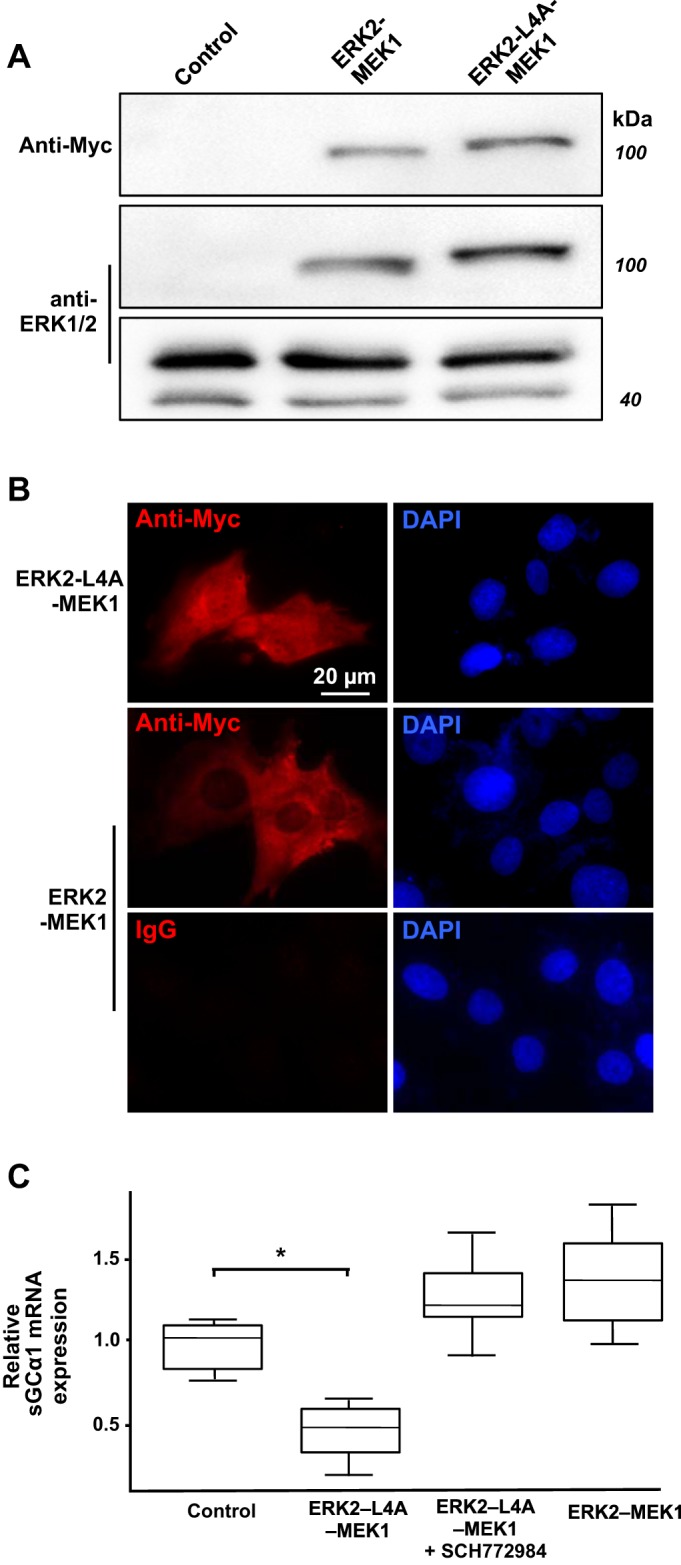
Nuclear ERK downregulates stimulated soluble guanylate cyclase (sGC) mRNA expression in pulmonary artery smooth muscle cells (PASMC). A: transient transfection induces ERK-MEK fusion protein expression in PASMC. CS54 cells were transfected with pcDNA3 (control) or plasmids that encode the indicated Myc-tagged ERK2-MEK1 fusion proteins. Cell lysates were obtained 1 day later, and fusion protein and endogenous ERK protein expression were determined using immunoblotting. B: ERK2-L4A-MEK1 fusion protein accumulates in the nucleus and cytoplasm of PASMC. The indicated fusion proteins were localized in CS54 cells using an anti-Myc antibody and immunofluorescence. Shown are wide-field images, with the focal plane centered in the mid-nuclear region, as determined by diamidino-2-phenylindole (DAPI)-mediated DNA staining. C: nuclear ERK decreases sGCα1 mRNA expression. CS54 cells were transfected with plasmids encoding the indicated fusion proteins, serum restricted 48 h later, and then treated with 0 or 2 µM SCH772984 for 1 h. The cells were lysed after 6 h, and the relative sGCα1 mRNA expression levels were determined and normalized to that of the cells not expressing the fusion proteins; n = 6/group; *P < 0.05.
Pulmonary injury increases ERK activation and decreases sGC protein expression in the developing lung.
Previous studies suggest that during normal pulmonary development, ERK primarily regulates the formation of the conducting airway structures. Although ERK is expressed in the lung throughout gestation in the rat, ERK phosphorylation greatly decreases after the canalicular phase of fetal lung development (45), nearly 1 wk after the branching of conducting airway structures is completed. Before the levels decrease, p-ERK is localized in endothelial, smooth muscle, and epithelial cells, predominantly in mitotic cells (45). Moreover, studies show that inhibition of MEK/ERK signaling in the early developing lung inhibits conducting airway formation. In vitro treatment of fetal rat lung explants with MEK inhibitors was observed to decrease branching morphogenesis of airway structures and to cause mesenchymal cell apoptosis (46). Also, in vivo mesenchymal and epithelial deletion of MEK function in the mouse was found to inhibit the branching of conductive airways and cause defective tracheal cartilage development (12). Although ERK does not appear to regulate normal acinar lung development, some studies suggest that its activation during lung injury disrupts pulmonary alveolar development. For example, inhalation of high levels of oxygen (95%) has been shown to increase ERK phosphorylation in newborn rats during the saccular phase of lung development and to inhibit alveolarization (38, 81).
Previously, we determined that exposure to a lower level of O2 (85%) activates pulmonary TGFβ and thereby decreases sGC expression in the newborn lung (6, 67). Because our data detailed above suggested a role of ERK in decreasing sGCα1 expression in PASMC, we tested whether O2-induced pulmonary injury increases ERK activation in the newborn lung. As shown in Fig. 7, breathing 85% O2 increases ERK phosphorylation and decreases sGC expression in the mouse pup lung. Whereas almost no p-ERK was detected in the mouse pup lung during this stage of lung development, it was greatly increased in a variety of cells by the lung injury. p-ERK was detected in cells residing in the blood vessel wall, the lung parenchyma, and the epithelium. In particular, p-ERK was detected in PASMC in the injured newborn lung. As shown in Fig. 7B, reacting sequential sections of oxygen-injured pup with antibodies that detect p-ERK and smoothelin, a marker of SMC, mapped p-ERK expression to PASMC. In agreement with past work (6), this form of pulmonary injury was determined to decrease sGCα1 expression in PASMC and parenchymal cells in the lung (Fig. 7, C and D). The specificity of the anti-sGCα1 antibody used in this work was confirmed by its detection of a single immunoreactive protein band of the expected molecular weight during immunoblotting studies using soluble proteins obtained from wild-type mouse pup lung lysates but not using proteins obtained from the lungs of sGCα1-knockout pups (Fig. 7E).
Fig. 7.

Lung injury increases ERK activation and decreases stimulated soluble guanylate cyclase (sGC) expression in the mouse pup. A: lung injury increases ERK phosphorylation in mouse pup parenchymal cells. Newborn mouse pups breathed either air or 85% O2 for 10 days, and then phosphorylated (p)-ERK protein was detected in lung tissue sections using immunohistochemistry and a colorimetric substrate (brown). The sections were counterstained with Gill’s hematoxylin. Cells in the blood vessel wall (arrows) and interstitial (*) and epithelial cells (**) are identified in the images, which are representative of lungs of 5 pups. B: p-ERK is detected in PASMC in the injured pup lung. p-ERK or smoothelin, a SMC marker protein, was detected in consecutive, 5-µm-thick sections of 85% O2-treated mouse pup lungs using antibodies and immunohistochemistry (brown). PASMC expressing p-ERK are identified (arrow) based on their localization within the blood vessel wall and reactivity with the anti-smoothelin antibody; an adluminal endothelial cell exhibiting ERK activation is also shown (arrowhead). The images are typical of 2 pups. C and D: pulmonary injury increases p-ERK and decreases sGCα1 protein expression in the newborn lung. sGCα1 protein expression was detected in air- or 85% O2-treated mouse pup lungs using an anti-sGCα1 antibody and immunohistochemistry (brown). sGCα1 and p-ERK protein expression were quantified in pup lung lysates using immunoblotting and densitometry; n = 4 each group; *P < 0.05. E: the anti-sGCα1 antibody specificity is shown by its inability to react with soluble proteins obtained from sGCα1-knockout (KO) in comparison with wild-type (WT) mouse pup lungs by immunoblotting. The secondary antibody specificity is supported by its lack of reactivity to WT pup lungs treated with rabbit IgG rather than the rabbit anti-sGCα1 antibody.
ERK decreases sGCα1 protein expression in TGFβ-treated primary mouse PASMC.
To determine the in vivo relevance of the results that we obtained with the CS54 cells, we treated hyperoxic mouse pups with an ERK inhibitor to examine a role of p-ERK in decreasing sGCα1 protein expression during lung injury. However, the pups did not tolerate systemic ERK inhibition. Therefore, we tested whether ERK regulates sGCα1 expression in TGFβ-treated early passage primary PASMC derived from the mouse pups. Primary PASMC were isolated from mouse pup lungs and identified by their characteristic morphology and reactivity with an anti-smoothelin antibody. We found that TGFβ-1 treatment decreased sGCα1 immunoreactivity in the primary PASMC (Fig. 8A). Moreover, pretreating the cells with an ERK inhibitor prevented the decrease in sGCα1 expression in the TGFβ-treated cells. Because vascular SMC rapidly lose sGC expression with passaging (57), we determined the changes in sGCα1 protein expression using quantitative IF. As shown in Fig. 8B, TGFβ-1 treatment decreased sGC protein expression in the mouse pup PASMC in an ERK-dependent manner.
Fig. 8.
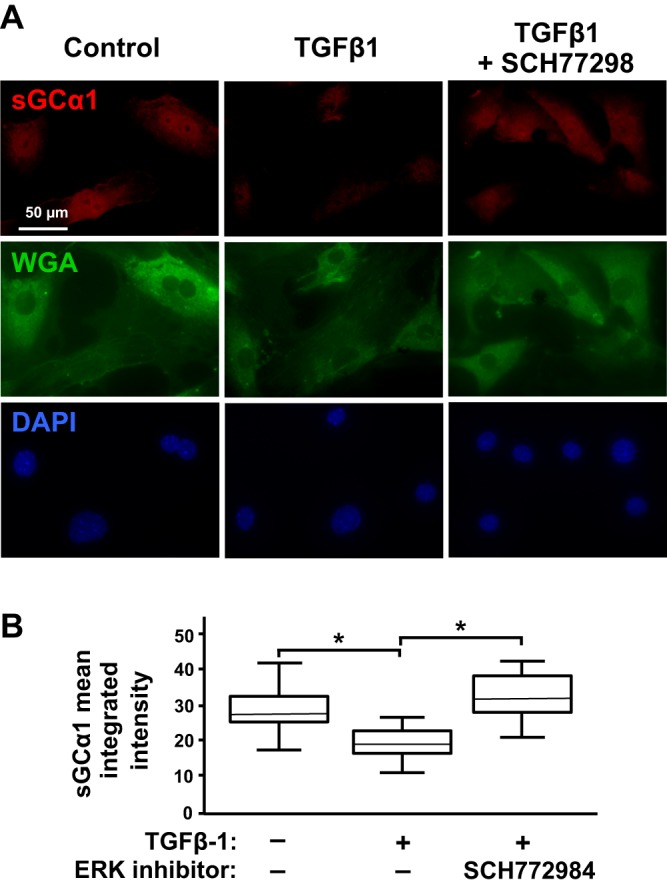
Transforming growth factor-β (TGFβ) decreases soluble guanylate cyclase (Sgc) protein expression in primary mouse pup pulmonary artery smooth muscle cells (mPASMC) via ERK activity. mPASMC were treated with 0 or 2 µM SCH772984 for 1 h and then 0 or 10 ng/ml TGFβ-1 for 6 h before sGCα1 protein expression was detected using immunofluorescence, and cell area was determined using wheat germ agglutinin (WGA)-conjugated with a fluoroprobe. Nuclear DNA was identified using DAPI. Cellular images were captured using wide-field fluorescence microscopy. A: representative images of mPASMC, treated with TGFβ-1 and the ERK inhibitor with sGCα1 antibody and WGA reactivity are shown. B: mean cellular sGCα1 integrated intensity within the cellular area of ∼50 cells was determined using the fluorescence images by investigators masked with respect to the cell treatment groups. *P < 0.015.
DISCUSSION
Previous studies indicate that TGFβ decreases sGC expression in the injured newborn lung and in PASMC (6, 67). In that work, treatment with TGFβ-neutralizing antibodies inhibited intracellular TGFβ signaling and protected sGC expression in hyperoxic mouse pup lungs. Moreover, the regulatory effect of TGFβ on sGC expression appeared to be direct; TGFβ was found to inhibit sGC subunit mRNA expression in cultured primary PASMC. However, the mechanisms by which TGFβ inhibits sGC mRNA expression were unknown. The objective of the current investigation was to identify the intracellular pathways utilized by TGFβ to decrease sGC expression in PASMC. Because sGC plays a pivotal role in regulating newborn lung development and pulmonary vascular tone, the results of this work might identify pathways that could be targeted to protect cGMP signaling during newborn lung injury.
The mechanisms employed by TGFβ to regulate gene expression and cellular phenotype depend on the TGFβ dose, cell type, and cellular context. TGFβ activation of TGFβR1 controls canonical, Smad2/3-dependent, and a variety of noncanonical signaling systems. The Smad2/3-independent mechanisms include the stimulation of TAK1 and subsequent activation of p38 MAPK, JNK, and IKKβ pathways and activation of TAK1-independent PI3K/Akt and MEK and ERK signaling pathways. Recent studies show that TGFβ can also stimulate Smad1/5 in some cultured cells (54). This later mechanism appears to be relevant in the newborn lung. This is because mixed Smad1/2 and Smad1/5 complexes, which are indicative of this TGFβ-stimulated Smad1/5 activation (28), have been detected in the lungs of newborn mice (103). Moreover, TGFβ has been determined to stimulate Smad1/5 phosphorylation in primary PASMC obtained from these newborn animals (103). In the work presented here, we report that TGFβ levels detected in the newborn lung (47) regulate sGC expression in PASMC. Moreover, studies employing two well-characterized kinase inhibitors indicated that TGFβR1 mediates the diminished sGC expression by TGFβ. Although in many instances TGFβR1-stimulated canonical Smads play an important role in regulating intracellular TGFβ signaling, we determined in PASMC that effective knockdown of Smad1/2 did not mediate TGFβ’s regulation of sGC expression. Moreover, a small-molecule kinase inhibitor screen, using established doses of the inhibitors, implicated MEK in the regulation of sGC by TGFβ.
MEK and ERK occupy a central role in regulating mitogen-activated signaling systems. MEK becomes phosphorylated and activated in response to stimulation by several growth-factor signaling systems. In turn, activated MEK phosphorylates ERK, and activated ERK stimulates a variety of cytosolic and nuclear targets that regulate gene expression (72, 96). To determine the interplay between TGFβ, MEK, and ERK signaling and sGC expression in our model PASMC system, we next tested whether TGFβ-1 increases MEK and ERK phosphorylation in the CS54 cells. Previous work by others has shown that TGFβ causes sustained ERK phosphorylation in systemic vascular SMC (82). In our studies in PASMC, we determined that physiological levels of TGFβ increased the activation of these proteins. Importantly, MEK and ERK inhibition protected sGCα1 mRNA expression in the CS54 cells. We also tested whether this regulation of sGC mRNA expression by TGFβ-stimulated ERK was associated with changes in enzyme function. For these studies, we employed a newly developed in vivo cGMP sensor and examined the role of TGFβ and ERK in sGC function using a HEK-293 cell model. In this case, the TGFβ-mediated decreased sGC enzyme activity that we detected in the cells was protected by ERK inhibition. The potential in vivo relevance of the TGFβ-MEK/ERK-sGC signaling system detected during the cell studies is supported by the observation that oxygen-induced lung injury, which has been shown to activate TGFβ (67), stimulates ERK phosphorylation in PASMC and interstitial cells and decreases sGC protein expression in the mouse pup lungs. Demonstrating that ERK inhibition protects sGC protein expression in TGFβ-treated primary mouse pup PASMC suggested further potential relevance of this signaling system in vivo.
Activated ERK mediates its effects by phosphorylating several intracellular protein targets (79). Furthermore, ERK compartmentation plays an important role in regulating its access to phosphorylation targets and downstream activities. Previous studies in thrombin-stimulated vascular SMC show that activated ERK accumulates in the nucleus, where it phosphorylates transcription factors residing there, such as Elk-1 and c-myc (84). Moreover, serotonin was observed to stimulate nuclear p-ERK localization in bovine PASMC (55). However, activated ERK can also phosphorylate proteins residing in the cytosol and stimulate their movement into the nucleus. For example, p-ERK was determined to phosphorylate RSK in the cytosol, stimulating p-RSK migration into the nucleus and gene expression regulation (16). To gain more insight into the mechanisms by which ERK regulates sGC expression in PASMC, we expressed a constitutively active ERK2-MEK1 fusion protein, with and without a mutation in the MEK nuclear export motif, in the CS54 cells and assessed sGC expression. Whereas expression of a ERK2-MEK1 fusion protein that accumulates in the nucleus and cytosol was sufficient to decrease sGC mRNA levels in the PASMC, we determined that expression of the fusion protein that resided primarily in the cytosol did not. In addition, we found that treatment with an ERK inhibitor decreased the effect of the nuclear ERK2-MEK1 fusion protein on sGCα1 mRNA levels, confirming a direct effect of nuclear ERK in downregulating sGC expression. This work indicates that nuclear but not cytosolic targets of activated ERK regulate sGC expression in PASMC.
The results of the studies detailed here provide additional information about the mechanisms that regulate sGC expression. The sGC gene promoter has been cloned and characterized in the mouse (85, 91), rat (39), and human (58). Several transcription factor-binding sites have been characterized within this TATA-less promoter. In agreement with our determination that Smad1/2 does not mediate the downregulation of sGC expression by TGFβ, a Smad-dependent TGFβ-response element has not been identified in the sGCα1 promoter region in these studies. Moreover, our determination that ERK is sufficient to decrease sGC expression in PASMC might also have relevance in investigations about how other systems inhibit the expression of this enzyme. For example, reactive oxygen species (ROS) have been reported to decrease sGC expression in fetal ovine PASMC and vascular SMC (95). Although the mechanism was not defined during these studies, previous work in fibroblasts shows that ROS can stimulate ERK activity (33) in part by Fyn and JAK2-mediated Ras activation (1). Our data support investigations about the role of ERK signaling in ROS-mediated sGC downregulation.
The nature of our newborn mouse pup model introduces some limitations in our work. Although our studies indicate that ERK mediates TGFβ’s downregulation of sGCα1 protein expression in primary PASMC obtained from mouse pups, we were unable to demonstrate that this mechanism regulates sGC expression in vivo. We could not sustain ERK inhibition in this model; we found that systemic delivery of ERK inhibitors was lethal in the mouse pup within a few days of treatment. This likely reflects the importance of ERK signaling during mouse pup growth and development because ERK inhibitors are tolerated by adult animals (e.g., see Ref. 53). Because we were unable to directly determine the extent by which ERK decreases sGC expression in the injured newborn lung, it is possible that other mechanisms might also regulate sGC expression during pulmonary injury. Moreover, we did not determine how TGFβ activates ERK in PASMC. However, others show that TGFβ can induce Ras activation, which appears to be commensurate with a low level of ERK activation in some cells (65). Also, others have demonstrated that TGFβ activates ERK by TGFβR1-mediated recruitment and direct phosphorylation of ShcA, thereby inducing its association with Grb2/Sos in epithelial cell lines (50).
In summary, this work indicates that MEK and ERK mediate how TGFβ downregulates sGCα1 expression in PASMC. The work suggests that selective targeting of active TGFβ, TGFβR1, or pulmonary MEK and ERK signaling might provide a way to protect cGMP signing in the injured newborn lung.
GRANTS
This work was supported by a grant from the National Heart, Lung, and Blood Institute (HL-125715, to J. D. Roberts, Jr.) and the Massachusetts General Hospital Department of Anesthesia, Critical Care, and Pain Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.D. and J.D.R. conceived and designed research; L.D. and J.D.R. performed experiments; L.D. and J.D.R. analyzed data; L.D. and J.D.R. interpreted results of experiments; L.D. and J.D.R. prepared figures; L.D. and J.D.R. drafted manuscript; L.D. and J.D.R. edited and revised manuscript; L.D. and J.D.R. approved final version of manuscript.
REFERENCES
- 1.Abe J, Berk BC. Fyn and JAK2 mediate Ras activation by reactive oxygen species. J Biol Chem 274: 21003–21010, 1999. doi: 10.1074/jbc.274.30.21003. [DOI] [PubMed] [Google Scholar]
- 2.Abman SH, Chatfield BA, Hall SL, McMurtry IF. Role of endothelium-derived relaxing factor during transition of pulmonary circulation at birth. Am J Physiol 259: H1921–H1927, 1990. doi: 10.1152/ajpheart.1990.259.6.H1921. [DOI] [PubMed] [Google Scholar]
- 3.Alejandre-Alcázar MA, Kwapiszewska G, Reiss I, Amarie OV, Marsh LM, Sevilla-Pérez J, Wygrecka M, Eul B, Köbrich S, Hesse M, Schermuly RT, Seeger W, Eickelberg O, Morty RE. Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 292: L537–L549, 2007. doi: 10.1152/ajplung.00050.2006. [DOI] [PubMed] [Google Scholar]
- 4.Ambalavanan N, Nicola T, Hagood J, Bulger A, Serra R, Murphy-Ullrich J, Oparil S, Chen YF. Transforming growth factor-beta signaling mediates hypoxia-induced pulmonary arterial remodeling and inhibition of alveolar development in newborn mouse lung. Am J Physiol Lung Cell Mol Physiol 295: L86–L95, 2008. doi: 10.1152/ajplung.00534.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bachiller PR, Cornog KH, Kato R, Buys ES, Roberts JD Jr. Soluble guanylate cyclase modulates alveolarization in the newborn lung. Am J Physiol Lung Cell Mol Physiol 305: L569–L581, 2013. doi: 10.1152/ajplung.00401.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bachiller PR, Nakanishi H, Roberts JD Jr. Transforming growth factor-beta modulates the expression of nitric oxide signaling enzymes in the injured developing lung and in vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 298: L324–L334, 2010. doi: 10.1152/ajplung.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat 29: 1165–1188, 2001. [Google Scholar]
- 8.Black SM, Johengen MJ, Soifer SJ. Coordinated regulation of genes of the nitric oxide and endothelin pathways during the development of pulmonary hypertension in fetal lambs. Pediatr Res 44: 821–830, 1998. doi: 10.1203/00006450-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Black SM, Sanchez LS, Mata-Greenwood E, Bekker JM, Steinhorn RH, Fineman JR. sGC and PDE5 are elevated in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 281: L1051–L1057, 2001. doi: 10.1152/ajplung.2001.281.5.L1051. [DOI] [PubMed] [Google Scholar]
- 10.Bland RD, Ling CY, Albertine KH, Carlton DP, MacRitchie AJ, Day RW, Dahl MJ. Pulmonary vascular dysfunction in preterm lambs with chronic lung disease. Am J Physiol Lung Cell Mol Physiol 285: L76–L85, 2003. doi: 10.1152/ajplung.00395.2002. [DOI] [PubMed] [Google Scholar]
- 11.Bloch KD, Filippov G, Sanchez LS, Nakane M, de la Monte SM. Pulmonary soluble guanylate cyclase, a nitric oxide receptor, is increased during the perinatal period. Am J Physiol 272: L400–L406, 1997. doi: 10.1152/ajplung.1997.272.3.L400. [DOI] [PubMed] [Google Scholar]
- 12.Boucherat O, Nadeau V, Bérubé-Simard FA, Charron J, Jeannotte L. Crucial requirement of ERK/MAPK signaling in respiratory tract development. Development 141: 3197–3211, 2014. doi: 10.1242/dev.110254. [DOI] [PubMed] [Google Scholar]
- 13.Casteel DE, Zhang T, Zhuang S, Pilz RB. cGMP-dependent protein kinase anchoring by IRAG regulates its nuclear translocation and transcriptional activity. Cell Signal 20: 1392–1399, 2008. doi: 10.1016/j.cellsig.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaussade C, Rewcastle GW, Kendall JD, Denny WA, Cho K, Grønning LM, Chong ML, Anagnostou SH, Jackson SP, Daniele N, Shepherd PR. Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. Biochem J 404: 449–458, 2007. doi: 10.1042/BJ20070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Roberts JD Jr. cGMP-dependent protein kinase I gamma encodes a nuclear localization signal that regulates nuclear compartmentation and function. Cell Signal 26: 2633–2644, 2014. doi: 10.1016/j.cellsig.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen RH, Sarnecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol 12: 915–927, 1992. doi: 10.1128/MCB.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiche JD, Schlutsmeyer SM, Bloch DB, de la Monte SM, Roberts JD Jr, Filippov G, Janssens SP, Rosenzweig A, Bloch KD. Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J Biol Chem 273: 34263–34271, 1998. doi: 10.1074/jbc.273.51.34263. [DOI] [PubMed] [Google Scholar]
- 18.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett 364: 229–233, 1995. doi: 10.1016/0014-5793(95)00357-F. [DOI] [PubMed] [Google Scholar]
- 19.D’Angelis CA, Nickerson PA, Steinhorn RH, Morin FC III. Heterogeneous distribution of soluble guanylate cyclase in the pulmonary vasculature of the fetal lamb. Anat Rec 250: 62–69, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 20.DaCosta Byfield S, Major C, Laping NJ, Roberts AB. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol Pharmacol 65: 744–752, 2004. doi: 10.1124/mol.65.3.744. [DOI] [PubMed] [Google Scholar]
- 21.Dasgupta C, Sakurai R, Wang Y, Guo P, Ambalavanan N, Torday JS, Rehan VK. Hyperoxia-induced neonatal rat lung injury involves activation of TGF-beta and Wnt signaling and is protected by rosiglitazone. Am J Physiol Lung Cell Mol Physiol 296: L1031–L1041, 2009. doi: 10.1152/ajplung.90392.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denninger JW, Marletta MA. Guanylate cyclase and the. NO/cGMP signaling pathway. Biochim Biophys Acta 1411: 334–350, 1999. doi: 10.1016/S0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 23.Deruelle P, Balasubramaniam V, Kunig AM, Seedorf GJ, Markham NE, Abman SH. BAY 41-2272, a direct activator of soluble guanylate cyclase, reduces right ventricular hypertrophy and prevents pulmonary vascular remodeling during chronic hypoxia in neonatal rats. Biol Neonate 90: 135–144, 2006. doi: 10.1159/000092518. [DOI] [PubMed] [Google Scholar]
- 24.Dzamko N, Inesta-Vaquera F, Zhang J, Xie C, Cai H, Arthur S, Tan L, Choi H, Gray N, Cohen P, Pedrioli P, Clark K, Alessi DR. The IkappaB kinase family phosphorylates the Parkinson’s disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS One 7: e39132, 2012. doi: 10.1371/journal.pone.0039132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ebelt ND, Kaoud TS, Edupuganti R, Van Ravenstein S, Dalby KN, Van Den Berg CL. A c-Jun N-terminal kinase inhibitor, JNK-IN-8, sensitizes triple negative breast cancer cells to lapatinib. Oncotarget 8: 104894–104912, 2017. doi: 10.18632/oncotarget.20581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fineman JR, Wong J, Morin FC III, Wild LM, Soifer SJ. Chronic nitric oxide inhibition in utero produces persistent pulmonary hypertension in newborn lambs. J Clin Invest 93: 2675–2683, 1994. doi: 10.1172/JCI117281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finlay GA, Thannickal VJ, Fanburg BL, Paulson KE. Transforming growth factor-beta 1-induced activation of the ERK pathway/activator protein-1 in human lung fibroblasts requires the autocrine induction of basic fibroblast growth factor. J Biol Chem 275: 27650–27656, 2000. doi: 10.1074/jbc.M000893200. [DOI] [PubMed] [Google Scholar]
- 28.Flanders KC, Heger CD, Conway C, Tang B, Sato M, Dengler SL, Goldsmith PK, Hewitt SM, Wakefield LM. Brightfield proximity ligation assay reveals both canonical and mixed transforming growth factor-β/bone morphogenetic protein Smad signaling complexes in tissue sections. J Histochem Cytochem 62: 846–863, 2014. doi: 10.1369/0022155414550163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forster B, Van De Ville D, Berent J, Sage D, Unser M. Complex wavelets for extended depth-of-field: a new method for the fusion of multichannel microscopy images. Microsc Res Tech 65: 33–42, 2004. doi: 10.1002/jemt.20092. [DOI] [PubMed] [Google Scholar]
- 30.Friebe A, Koesling D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ Res 93: 96–105, 2003. doi: 10.1161/01.RES.0000082524.34487.31. [DOI] [PubMed] [Google Scholar]
- 31.Gudi T, Huvar I, Meinecke M, Lohmann SM, Boss GR, Pilz RB. Regulation of gene expression by cGMP-dependent protein kinase. Transactivation of the c-fos promoter. J Biol Chem 271: 4597–4600, 1996. doi: 10.1074/jbc.271.9.4597. [DOI] [PubMed] [Google Scholar]
- 32.Gudi T, Lohmann SM, Pilz RB. Regulation of gene expression by cyclic GMP-dependent protein kinase requires nuclear translocation of the kinase: identification of a nuclear localization signal. Mol Cell Biol 17: 5244–5254, 1997. doi: 10.1128/MCB.17.9.5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem 271: 4138–4142, 1996. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 34.Hasan A, Danker KY, Wolter S, Bähre H, Kaever V, Seifert R. Soluble adenylyl cyclase accounts for high basal cCMP and cUMP concentrations in HEK293 and B103 cells. Biochem Biophys Res Commun 448: 236–240, 2014. doi: 10.1016/j.bbrc.2014.04.099. [DOI] [PubMed] [Google Scholar]
- 35.Hata A, Chen YG. TGF-β Signaling from Receptors to Smads. Cold Spring Harb Perspect Biol 8: a022061, 2016. doi: 10.1101/cshperspect.a022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He L, Sabet A, Djedjos S, Miller R, Sun X, Hussain MA, Radovick S, Wondisford FE. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 137: 635–646, 2009. doi: 10.1016/j.cell.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Idriss SD, Gudi T, Casteel DE, Kharitonov VG, Pilz RB, Boss GR. Nitric oxide regulation of gene transcription via soluble guanylate cyclase and type I cGMP-dependent protein kinase. J Biol Chem 274: 9489–9493, 1999. doi: 10.1074/jbc.274.14.9489. [DOI] [PubMed] [Google Scholar]
- 38.Jiang JS, Lang YD, Chou HC, Shih CM, Wu MY, Chen CM, Wang LF. Activation of the renin-angiotensin system in hyperoxia-induced lung fibrosis in neonatal rats. Neonatology 101: 47–54, 2012. doi: 10.1159/000329451. [DOI] [PubMed] [Google Scholar]
- 39.Jiang Y, Stojilkovic SS. Molecular cloning and characterization of alpha1-soluble guanylyl cyclase gene promoter in rat pituitary cells. J Mol Endocrinol 37: 503–515, 2006. doi: 10.1677/jme.1.02180. [DOI] [PubMed] [Google Scholar]
- 40.Kahan C, Seuwen K, Meloche S, Pouysségur J. Coordinate, biphasic activation of p44 mitogen-activated protein kinase and S6 kinase by growth factors in hamster fibroblasts. Evidence for thrombin-induced signals different from phosphoinositide turnover and adenylylcyclase inhibition. J Biol Chem 267: 13369–13375, 1992. [PubMed] [Google Scholar]
- 41.Kato S, Chen J, Cornog KH, Zhang H, Roberts JD Jr. The Golgi apparatus regulates cGMP-dependent protein kinase I compartmentation and proteolysis. Am J Physiol Cell Physiol 308: C944–C958, 2015. doi: 10.1152/ajpcell.00199.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kato S, Zhang R, Roberts JD Jr. Proprotein convertases play an important role in regulating PKGI endoproteolytic cleavage and nuclear transport. Am J Physiol Lung Cell Mol Physiol 305: L130–L140, 2013. doi: 10.1152/ajplung.00391.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim K, Ryu K, Ko Y, Park C. Effects of nuclear factor-kappaB inhibitors and its implication on natural killer T-cell lymphoma cells. Br J Haematol 131: 59–66, 2005. doi: 10.1111/j.1365-2141.2005.05720.x. [DOI] [PubMed] [Google Scholar]
- 44.Kim SI, Kwak JH, Na HJ, Kim JK, Ding Y, Choi ME. Transforming growth factor-beta (TGF-beta1) activates TAK1 via TAB1-mediated autophosphorylation, independent of TGF-beta receptor kinase activity in mesangial cells. J Biol Chem 284: 22285–22296, 2009. doi: 10.1074/jbc.M109.007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kling DE, Brandon KL, Sollinger CA, Cavicchio AJ, Ge Q, Kinane TB, Donahoe PK, Schnitzer JJ. Distribution of ERK1/2 and ERK3 during normal rat fetal lung development. Anat Embryol (Berl) 211: 139–153, 2006. doi: 10.1007/s00429-005-0063-z. [DOI] [PubMed] [Google Scholar]
- 46.Kling DE, Lorenzo HK, Trbovich AM, Kinane TB, Donahoe PK, Schnitzer JJ. MEK-1/2 inhibition reduces branching morphogenesis and causes mesenchymal cell apoptosis in fetal rat lungs. Am J Physiol Lung Cell Mol Physiol 282: L370–L378, 2002. doi: 10.1152/ajplung.00200.2001. [DOI] [PubMed] [Google Scholar]
- 47.Kotecha S, Wangoo A, Silverman M, Shaw RJ. Increase in the concentration of transforming growth factor beta-1 in bronchoalveolar lavage fluid before development of chronic lung disease of prematurity. J Pediatr 128: 464–469, 1996. doi: 10.1016/S0022-3476(96)70355-4. [DOI] [PubMed] [Google Scholar]
- 48.Kumarasamy A, Schmitt I, Nave AH, Reiss I, van der Horst I, Dony E, Roberts JD Jr, de Krijger RR, Tibboel D, Seeger W, Schermuly RT, Eickelberg O, Morty RE. Lysyl oxidase activity is dysregulated during impaired alveolarization of mouse and human lungs. Am J Respir Crit Care Med 180: 1239–1252, 2009. doi: 10.1164/rccm.200902-0215OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J 26: 3957–3967, 2007. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lehners M, Dobrowinski H, Feil S, Feil R. cGMP signaling and vascular smooth muscle cell plasticity. J Cardiovasc Dev Dis 5: E20, 2018. doi: 10.3390/jcdd5020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li D, Zhou N, Johns RA. Soluble guanylate cyclase gene expression and localization in rat lung after exposure to hypoxia. Am J Physiol 277: L841–L847, 1999. doi: 10.1152/ajplung.1999.277.4.L841. [DOI] [PubMed] [Google Scholar]
- 53.Li LF, Liao SK, Ko YS, Lee CH, Quinn DA. Hyperoxia increases ventilator-induced lung injury via mitogen-activated protein kinases: a prospective, controlled animal experiment. Crit Care 11: R25, 2007. doi: 10.1186/cc5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu X, Yue J, Frey RS, Zhu Q, Mulder KM. Transforming growth factor beta signaling through Smad1 in human breast cancer cells. Cancer Res 58: 4752–4757, 1998. [PubMed] [Google Scholar]
- 55.Liu Y, Suzuki YJ, Day RM, Fanburg BL. Rho kinase-induced nuclear translocation of ERK1/ERK2 in smooth muscle cell mitogenesis caused by serotonin. Circ Res 95: 579–586, 2004. doi: 10.1161/01.RES.0000141428.53262.a4. [DOI] [PubMed] [Google Scholar]
- 56.Luo C, Lim JH, Lee Y, Granter SR, Thomas A, Vazquez F, Widlund HR, Puigserver P. A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature 537: 422–426, 2016. doi: 10.1038/nature19347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marczin N, Antonov A, Papapetropoulos A, Munn DH, Virmani R, Kolodgie FD, Gerrity R, Catravas JD. Monocyte-induced downregulation of nitric oxide synthase in cultured aortic endothelial cells. Arterioscler Thromb Vasc Biol 16: 1095–1103, 1996. doi: 10.1161/01.ATV.16.9.1095. [DOI] [PubMed] [Google Scholar]
- 58.Marro ML, Peiró C, Panayiotou CM, Baliga RS, Meurer S, Schmidt HH, Hobbs AJ. Characterization of the human alpha1 beta1 soluble guanylyl cyclase promoter: key role for NF-kappaB(p50) and CCAAT-binding factors in regulating expression of the nitric oxide receptor. J Biol Chem 283: 20027–20036, 2008. doi: 10.1074/jbc.M801223200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsuda S, Harada K, Ito M, Takizawa M, Wongso D, Tsuboi T, Kitaguchi T. Generation of a cGMP indicator with an expanded dynamic range by optimization of amino acid linkers between a fluorescent protein and PDE5α. ACS Sens 2: 46–51, 2017. doi: 10.1021/acssensors.6b00582. [DOI] [PubMed] [Google Scholar]
- 60.McCurnin DC, Pierce RA, Chang LY, Gibson LL, Osborne-Lawrence S, Yoder BA, Kerecman JD, Albertine KH, Winter VT, Coalson JJ, Crapo JD, Grubb PH, Shaul PW. Inhaled NO improves early pulmonary function and modifies lung growth and elastin deposition in a baboon model of neonatal chronic lung disease. Am J Physiol Lung Cell Mol Physiol 288: L450–L459, 2005. doi: 10.1152/ajplung.00347.2004. [DOI] [PubMed] [Google Scholar]
- 61.Meloche S, Seuwen K, Pagès G, Pouysségur J. Biphasic and synergistic activation of p44mapk (ERK1) by growth factors: correlation between late phase activation and mitogenicity. Mol Endocrinol 6: 845–854, 1992. doi: 10.1210/mend.6.5.1603090. [DOI] [PubMed] [Google Scholar]
- 62.Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M, Bland RD. Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol 298: L23–L35, 2010. doi: 10.1152/ajplung.00251.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moreno L, Gonzalez-Luis G, Cogolludo A, Lodi F, Lopez-Farre A, Tamargo J, Villamor E, Perez-Vizcaino F. Soluble guanylyl cyclase during postnatal porcine pulmonary maturation. Am J Physiol Lung Cell Mol Physiol 288: L125–L130, 2005. doi: 10.1152/ajplung.00244.2004. [DOI] [PubMed] [Google Scholar]
- 64.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, Long B, Liu J, Dinunzio E, Windsor W, Zhang R, Zhao S, Angagaw MH, Pinheiro EM, Desai J, Xiao L, Shipps G, Hruza A, Wang J, Kelly J, Paliwal S, Gao X, Babu BS, Zhu L, Daublain P, Zhang L, Lutterbach BA, Pelletier MR, Philippar U, Siliphaivanh P, Witter D, Kirschmeier P, Bishop WR, Hicklin D, Gilliland DG, Jayaraman L, Zawel L, Fawell S, Samatar AA. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov 3: 742–750, 2013. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 65.Mulder KM. Role of Ras and Mapks in TGFbeta signaling. Cytokine Growth Factor Rev 11: 23–35, 2000. doi: 10.1016/S1359-6101(99)00026-X. [DOI] [PubMed] [Google Scholar]
- 66.Nakane M, Arai K, Saheki S, Kuno T, Buechler W, Murad F. Molecular cloning and expression of cDNAs coding for soluble guanylate cyclase from rat lung. J Biol Chem 265: 16841–16845, 1990. [PubMed] [Google Scholar]
- 67.Nakanishi H, Sugiura T, Streisand JB, Lonning SM, Roberts JD Jr. TGF-beta-neutralizing antibodies improve pulmonary alveologenesis and vasculogenesis in the injured newborn lung. Am J Physiol Lung Cell Mol Physiol 293: L151–L161, 2007. doi: 10.1152/ajplung.00389.2006. [DOI] [PubMed] [Google Scholar]
- 68.Nasim MT, Ogo T, Chowdhury HM, Zhao L, Chen CN, Rhodes C, Trembath RC. BMPR-II deficiency elicits pro-proliferative and anti-apoptotic responses through the activation of TGFβ-TAK1-MAPK pathways in PAH. Hum Mol Genet 21: 2548–2558, 2012. doi: 10.1093/hmg/dds073. [DOI] [PubMed] [Google Scholar]
- 69.Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M, Mihara M, Tsuchiya M, Matsumoto K. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem 278: 18485–18490, 2003. doi: 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- 70.Pera T, Sami R, Zaagsma J, Meurs H. TAK1 plays a major role in growth factor-induced phenotypic modulation of airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 301: L822–L828, 2011. doi: 10.1152/ajplung.00017.2011. [DOI] [PubMed] [Google Scholar]
- 71.Phyu SM, Tseng CC, Fleming IN, Smith TA. Probing the PI3K/Akt/mTor pathway using 31P-NMR spectroscopy: routes to glycogen synthase kinase 3. Sci Rep 6: 36544, 2016. doi: 10.1038/srep36544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pouysségur J, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Eur J Biochem 270: 3291–3299, 2003. doi: 10.1046/j.1432-1033.2003.03707.x. [DOI] [PubMed] [Google Scholar]
- 73.R Core Team A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2012. [Google Scholar]
- 74.Rasband WS. ImageJ (Online) Bethesda, MD: National Institutes of Health, 1997–2007. https://imagej.nih.gov/ij/. [Google Scholar]
- 75.Roberts JD Jr, Chen TY, Kawai N, Wain J, Dupuy P, Shimouchi A, Bloch K, Polaner D, Zapol WM. Inhaled nitric oxide reverses pulmonary vasoconstriction in the hypoxic and acidotic newborn lamb. Circ Res 72: 246–254, 1993. doi: 10.1161/01.RES.72.2.246. [DOI] [PubMed] [Google Scholar]
- 76.Roberts JD Jr, Chiche JD, Weimann J, Steudel W, Zapol WM, Bloch KD. Nitric oxide inhalation decreases pulmonary artery remodeling in the injured lungs of rat pups. Circ Res 87: 140–145, 2000. doi: 10.1161/01.RES.87.2.140. [DOI] [PubMed] [Google Scholar]
- 77.Roberts JD Jr, Roberts CT, Jones RC, Zapol WM, Bloch KD. Continuous nitric oxide inhalation reduces pulmonary arterial structural changes, right ventricular hypertrophy, and growth retardation in the hypoxic newborn rat. Circ Res 76: 215–222, 1995. doi: 10.1161/01.RES.76.2.215. [DOI] [PubMed] [Google Scholar]
- 78.Robinson MJ, Stippec SA, Goldsmith E, White MA, Cobb MH. A constitutively active and nuclear form of the MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr Biol 8: 1141–1152, 1998. doi: 10.1016/S0960-9822(07)00485-X. [DOI] [PubMed] [Google Scholar]
- 79.Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 66: 105–143, 2012. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 80.Rothman A, Kulik TJ, Taubman MB, Berk BC, Smith CW, Nadal-Ginard B. Development and characterization of a cloned rat pulmonary arterial smooth muscle cell line that maintains differentiated properties through multiple subcultures. Circulation 86: 1977–1986, 1992. doi: 10.1161/01.CIR.86.6.1977. [DOI] [PubMed] [Google Scholar]
- 81.Sakurai R, Villarreal P, Husain S, Liu J, Sakurai T, Tou E, Torday JS, Rehan VK. Curcumin protects the developing lung against long-term hyperoxic injury. Am J Physiol Lung Cell Mol Physiol 305: L301–L311, 2013. doi: 10.1152/ajplung.00082.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Samarakoon R, Higgins SP, Higgins CE, Higgins PJ. TGF-beta1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscle cells requires pp60(c-src)/EGFR(Y845) and Rho/ROCK signaling. J Mol Cell Cardiol 44: 527–538, 2008. doi: 10.1016/j.yjmcc.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sastre AP, Grossmann S, Reusch HP, Schaefer M. Requirement of an intermediate gene expression for biphasic ERK1/2 activation in thrombin-stimulated vascular smooth muscle cells. J Biol Chem 283: 25871–25878, 2008. doi: 10.1074/jbc.M800949200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schauwienold D, Plum C, Helbing T, Voigt P, Bobbert T, Hoffmann D, Paul M, Reusch HP. ERK1/2-dependent contractile protein expression in vascular smooth muscle cells. Hypertension 41: 546–552, 2003. doi: 10.1161/01.HYP.0000054213.37471.84. [DOI] [PubMed] [Google Scholar]
- 85.Sharina IG, Krumenacker JS, Martin E, Murad F. Genomic organization of alpha1 and beta1 subunits of the mammalian soluble guanylyl cyclase genes. Proc Natl Acad Sci USA 97: 10878–10883, 2000. doi: 10.1073/pnas.190331697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sugiura T, Nakanishi H, Roberts JD Jr. Proteolytic processing of cGMP-dependent protein kinase I mediates nuclear cGMP signaling in vascular smooth muscle cells. Circ Res 103: 53–60, 2008. doi: 10.1161/CIRCRESAHA.108.176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tamemoto H, Kadowaki T, Tobe K, Ueki K, Izumi T, Chatani Y, Kohno M, Kasuga M, Yazaki Y, Akanuma Y. Biphasic activation of two mitogen-activated protein kinases during the cell cycle in mammalian cells. J Biol Chem 267: 20293–20297, 1992. [PubMed] [Google Scholar]
- 88.Tan L, Nomanbhoy T, Gurbani D, Patricelli M, Hunter J, Geng J, Herhaus L, Zhang J, Pauls E, Ham Y, Choi HG, Xie T, Deng X, Buhrlage SJ, Sim T, Cohen P, Sapkota G, Westover KD, Gray NS. Discovery of type II inhibitors of TGFβ-activated kinase 1 (TAK1) and mitogen-activated protein kinase kinase kinase kinase 2 (MAP4K2). J Med Chem 58: 183–196, 2015. doi: 10.1021/jm500480k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tzao C, Nickerson PA, Russell JA, Gugino SF, Steinhorn RH. Pulmonary hypertension alters soluble guanylate cyclase activity and expression in pulmonary arteries isolated from fetal lambs. Pediatr Pulmonol 31: 97–105, 2001. doi:. [DOI] [PubMed] [Google Scholar]
- 90.Uhl M, Aulwurm S, Wischhusen J, Weiler M, Ma JY, Almirez R, Mangadu R, Liu YW, Platten M, Herrlinger U, Murphy A, Wong DH, Wick W, Higgins LS, Weller M. SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res 64: 7954–7961, 2004. doi: 10.1158/0008-5472.CAN-04-1013. [DOI] [PubMed] [Google Scholar]
- 91.Vazquez-Padron RI, Pham SM, Pang M, Li S, Aïtouche A. Molecular dissection of mouse soluble guanylyl cyclase alpha1 promoter. Biochem Biophys Res Commun 314: 208–214, 2004. doi: 10.1016/j.bbrc.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 92.Ventura E, Weller M, Macnair W, Eschbach K, Beisel C, Cordazzo C, Claassen M, Zardi L, Burghardt I. TGF-β induces oncofetal fibronectin that, in turn, modulates TGF-β superfamily signaling in endothelial cells. J Cell Sci 131: jcs209619, 2018. doi: 10.1242/jcs.209619. [DOI] [PubMed] [Google Scholar]
- 93.Vermeersch P, Buys E, Pokreisz P, Marsboom G, Ichinose F, Sips P, Pellens M, Gillijns H, Swinnen M, Graveline A, Collen D, Dewerchin M, Brouckaert P, Bloch KD, Janssens S. Soluble guanylate cyclase-alpha1 deficiency selectively inhibits the pulmonary vasodilator response to nitric oxide and increases the pulmonary vascular remodeling response to chronic hypoxia. Circulation 116: 936–943, 2007. doi: 10.1161/CIRCULATIONAHA.106.677245. [DOI] [PubMed] [Google Scholar]
- 94.Warner BB, Stuart LA, Papes RA, Wispé JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol 275: L110–L117, 1998. doi: 10.1152/ajplung.1998.275.1.L110. [DOI] [PubMed] [Google Scholar]
- 95.Wedgwood S, Steinhorn RH, Bunderson M, Wilham J, Lakshminrusimha S, Brennan LA, Black SM. Increased hydrogen peroxide downregulates soluble guanylate cyclase in the lungs of lambs with persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol 289: L660–L666, 2005. doi: 10.1152/ajplung.00369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wortzel I, Seger R. The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer 2: 195–209, 2011. doi: 10.1177/1947601911407328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wrana JL, Attisano L, Cárcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massagué J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell 71: 1003–1014, 1992. doi: 10.1016/0092-8674(92)90395-S. [DOI] [PubMed] [Google Scholar]
- 98.Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol 39: 23–31, 2011. doi: 10.3892/ijo.2011.1015. [DOI] [PubMed] [Google Scholar]
- 99.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, Thomas G, Garrett MD, Sullivan DM, de Bono JS, Tolcher AW. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol 29: 4688–4695, 2011. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 100.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, Marlow A, Hurley B, Lyssikatos J, Lee PA, Winkler JD, Koch K, Wallace E. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res 13: 1576–1583, 2007. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 101.Yoshida T, Yamashita M, Horimai C, Hayashi M. Smooth muscle-selective inhibition of nuclear factor-κB attenuates smooth muscle phenotypic switching and neointima formation following vascular injury. J Am Heart Assoc 2: e000230, 2013. doi: 10.1161/JAHA.113.000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol 4: 33–41, 2008. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang H, Du L, Zhong Y, Flanders KC, Roberts JD Jr. Transforming growth factor-β stimulates Smad1/5 signaling in pulmonary artery smooth muscle cells and fibroblasts of the newborn mouse through ALK1. Am J Physiol Lung Cell Mol Physiol 313: L615–L627, 2017. doi: 10.1152/ajplung.00079.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, Laughlin JD, Park H, LoGrasso PV, Patricelli M, Nomanbhoy TK, Sorger PK, Alessi DR, Gray NS. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol 19: 140–154, 2012. doi: 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]