

Abstract
Nicotine is a highly addictive principal component of both tobacco and electronic cigarette that is readily absorbed in blood. Nicotine-containing electronic cigarettes are promoted as a safe alternative to cigarette smoking. However, the isolated effects of inhaled nicotine are largely unknown. Here we report a novel rat model of aerosolized nicotine with a particle size (~1 μm) in the respirable diameter range. Acute nicotine inhalation caused increased pulmonary edema and lung injury as measured by enhanced bronchoalveolar lavage fluid protein, IgM, lung wet-to-dry weight ratio, and high-mobility group box 1 (HMGB1) protein and decreased lung E-cadherin protein. Immunohistochemical analysis revealed congested blood vessels and increased neutrophil infiltration. Lung myeloperoxidase mRNA and protein increased in the nicotine-exposed rats. Complete blood counts also showed an increase in neutrophils, white blood cells, eosinophils, and basophils. Arterial blood gas measurements showed an increase in lactate. Lungs of nicotine-inhaling animals revealed increased mRNA levels of IL-1A and CXCL1. There was also an increase in IL-1α protein. In in vitro air-liquid interface cultures of airway epithelial cells, there was a dose dependent increase in HMGB1 release with nicotine treatment. Air-liquid cultures exposed to nicotine also resulted in a dose-dependent loss of barrier as measured by transepithelial electrical resistance and a decrease in E-cadherin expression. Nicotine also caused a dose-dependent increase in epithelial cell death and an increase in caspase-3/7 activities. These results show that the nicotine content of electronic cigarettes may have adverse pulmonary and systemic effects.
Keywords: aerosol, lung injury, nicotine, pulmonary edema, rats
INTRODUCTION
Tobacco cigarette smoking is on the decline, as its adverse health effects are increasingly being recognized (49). It is, however, being replaced by electronic cigarettes (e-cigs) that are assumed to be safe, as they do not involve smoke and tar, the known toxic components of tobacco. Although e-cigs may have a significant contribution in cigarette smoking reduction, the health effects of its ingredients are largely unknown (23). Nicotine is the principal component in both tobacco and e-cigs. Inhaled nicotine is highly addictive as it is readily absorbed in blood and modifies reward/pleasure regions in brain. The urge to continue smoking causes further increases in blood nicotine levels. Nicotine content of e-cigs is on the rise as is its popularity (22, 52, 77). Nicotine content of up to 5% (50 mg/ml) is commonly reported in the e-cig refills, but the labels are frequently misleading (22). E-cig fluids with nicotine content as high as 10% are also readily available on the internet (20). Elevated blood nicotine from tobacco smoking can contribute to development of lung diseases such as chronic obstructive pulmonary disease (COPD), lung cancer, and heart diseases (38, 60). Nicotine when administered at higher concentrations may also promote carcinogenesis (33, 41). With the increase in use of e-cigs, the incidence of accidental nicotine poisoning has also increased tremendously (15, 25, 71, 76). While uncommon, reports of nicotine-related fatalities are not rare (15, 16, 18, 67). However, studies on health effects of nicotine present in e-cigs that is inhaled as aerosol are scant. Inhaled nicotine has distinct pharmacokinetics and undergoes less metabolic processing. Since the levels of nicotine in e-cigs are highly variable and unregulated, e-cigs may deliver much higher doses of aerosolized nicotine, further raising significant concerns of safety and long-term effects.
The cardiopulmonary effects of inhaled nicotine are not clear. The effects could be influenced by the concentration of nicotine, particle size distribution, duration of exposure, and systemic absorption, among factors. In a long-term study of inhaled nicotine in rats, there was no adverse effect reported (73). Although pulmonary effects were not investigated in this study, these nicotine-exposed rats had consistently low body weight. In another very short-term study (5 min inhalation) carried out in humans, there was a dose-dependent increase in cough and airway resistance along with an increase in heart rate and blood pressure measured over a 30-min period (31). The pulmonary effects of inhaled nicotine are beginning to emerge. Most of the toxicity studies using e-cig vapor are often compounded by the fact that it includes other ingredients along with nicotine. E-cig exposure studies in rodents show increases in inflammatory cells, inflammatory cytokines, markers of oxidative stress and DNA damage, and increased susceptibility to infections (60). While implicated, the contribution of nicotine in unclear. Chronic exposure of mice to e-cigs, with or without nicotine, caused development of COPD-like features (24). Therefore, the effects of nicotine are less clear. Tobacco companies however continue to promote and report that inhaled nicotine has no adverse effect (54). It is imperative to mention that these studies were carried out at nicotine levels far less than what is used in e-cig refills.
A number of environmental factors including cigarette smoke can cause lung injury. Acute lung injury (ALI) is characterized by increased tissue inflammation, disruption of the alveolar-capillary barrier, and neutrophil recruitment to the lungs and hypoxemia, among other symptoms. (47). Cigarette smoke exposures are known to disrupt alveolar-epithelial barrier integrity leading to increase proteins in bronchoalveolar lavage fluid and lung edema and an increase inflammation and hypoxemia (12, 44, 75) In humans, cigarette smoke has also been associated with increased susceptibility to acute respiratory distress syndrome and its severity, which is closely correlated with the level of smoking (36, 44). Additionally, in a prospective study, the level of cotinine, a nicotine metabolite, measured on patients upon arrival, was closely correlated with the severity of trauma-induced ALI (13). These studies underscore the potential role of nicotine in inducing ALI.
The goal of this study is to characterize the acute effects of inhaled nicotine on the lung. To address this, a nose-only inhalation system was developed that very closely mimics nicotine delivery by e-cigs, in terms of concentration of aerosolized vapor, its particle size distribution and the flow rate at which they are delivered. However, in contrast to e-cigs that contain nicotine along with other ingredients, which is first vaporized by heating, nicotine aerosol in saline was used to study effects of nicotine alone. These studies will also be relevant to the next generation e-cig products, which are being developed as an aerosol using the capillary aerosol generator (CAG) technology, where heating is not involved (37).
MATERIALS AND METHODS
Animals.
All animal procedures were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee. Male Sprague-Dawley rats weighing 275–300 g were procured from Envigo and used in this study. The animals were fed ad libitum and maintained at 25°C in a 12-h light-dark cycle room.
Nicotine inhalation system.
Nicotine exposures were carried out using the Jaeger nose-only inhalation system (CH Technologies). The system is equipped with the Bioaerosol Nebulizer Generator (BANG) modules for generation of aerosol. Additionally, a seven-stage Mercer impactor provided the necessary measurements to characterize the nicotine test atmosphere at the animal-breathing zone. Anesthetized rats [cocktail of ketamine (75 mg/kg), xylazine (7.5 mg/kg), and acepromazine (1.5 mg/kg)] were connected to the plenum of the inhalation system. Animals were anesthetized to ensure a uniform breathing pattern and a uniform delivery of aerosol. The syringe pump with syringe containing saline or nicotine solution (nicotine diluted in sterile saline) was turned on, and animals were exposed for 15 min while respiration was monitored. For exposure, dilution air was delivered at 6 l/min and aerosolized air was added at 6 l/min. This delivers aerosol at a rate of 1 l/min per port, a value close to the puffing flow rate (7). A schematic of the system is shown in Fig. 1B.
Fig. 1.

Nicotine aerosol generation, characterization, and exposure of rats. Sprague-Dawley rats were anesthetized and exposed to saline or nicotine aerosol for 15 min and transferred to room air. A: schematic representation of our exposure paradigm. Rats were monitored by pulse oximetry and clinical scoring after nose-only exposure nicotine aerosol. At the end of exposure rats were euthanized and necropsy was performed to evaluate parameters as shown in the experimental design (top). B: schematic of the CH-technology liquid particle generator and exposure system that utilizes Bio-Aerosol Nebulizing Generator (BANG) device to produce appropriate nicotine particles for alveolar delivery. C: plot showing particle size distribution of a 5% solution of nicotine in ethanol. The solution was injected into the BANG and an air flow of 6 l/min and a pressure of 60 psi was used to generate the aerosol. MMAD, mass median aerodynamic diameter; GSD, geometric standard deviation. D: cotinine, a nicotine metabolite was estimated in the plasma of rats exposed to aerosolized nicotine (15 min using 5 or 10% nicotine in normal saline). Plasma was collected and analyzed for cotinine 6 h after exposure. Data shown are means ± SE (n = 6). *P < 0.05 from the unexposed controls.
Characterization of particle size distribution of aerosolized nicotine.
Since particle size has a bearing on the site of deposition of nicotine in the lung (9, 39), these studies characterized size and concentration of particles using the BANG. Our goal was to assess the effects of aerosolized nicotine that would more closely mimic the particle sizes generated by e-cigs and mainstream smoking and that could affect both the upper and lower airways (6, 26). With the use of this device, particle sizes appropriate for alveolar delivery were generated. For characterization of particle size, nicotine solution was made in ethanol to avoid interference from the salt in saline. A 5% solution of nicotine in ethanol was used to determine particle size distribution and total particulate matter. The total particulate matter was determined using the gravimetric analyzer attached to the exposure system. Particle size distribution was determined using a mercer impactor by weighing the solute collected on the different stages. Since particle size can be best characterized in liquids that evaporate, and to avoid other interfering solute particle, ethanol was used as diluent for measurements (10). However, all animal experiments with nicotine were carried out using saline as diluent.
Pulse oximetry and arterial blood gas analysis.
For noninvasive oxygen saturation measurements, a MouseOx small animal oximeter (Starr Life Sciences, Oakmont, PA) in unanesthetized rats was used with a large CollarClip sensor. Blood was collected from the descending aorta under ketamine/xylazine anesthesia, immediately before euthanasia by exsanguination plus bilateral thoracotomy. Blood was placed into a precalibrated test card and analyzed using the EPOC-Vet Blood Analysis System (Epocal) (81).
Lung wet-to-dry weight ratios.
Lungs were excised intact after exsanguination, and wet weights were measured immediately using a precision balance. Dry weights were determined after lungs (right upper lobe) were dried for 48 h (or until the weights are constant) in an oven at a temperature of 98°C.
Histology.
Lungs were inflation fixed at 20 cmH2O with 4% paraformaldehyde in PBS for 30 min and embedded in paraffin. Five-micrometer sections were made from paraffin-embedded lung tissues and stained with hematoxylin and eosin. Morphometric analysis was carried out using the National Institutes of Health ImageJ software.
Western blots.
Western blots were performed as described in detail earlier with tissue or cell lysates (3). Anti-rat HMGB1 antibody (cat. no. AB 18258) and anti-rat-E-cadherin (cat. no. Ab 76055) were purchased from Abcam. Anti-rat myeloperoxidase (MPO) antibody (cat. no. NBP1−4291) was procured from Novus Biological. IL-1α protein was measured by the University of Alabama at Birmingham Consolidated Flow Cytometry Core laboratory using Luminex assay.
Complete blood counts.
Blood was collected from tail vein at different time points, and CBCs were determined using a Hemavet 950 FS hematology analyzer.
Cell culture, transepithelial electrical resistance, and cell death assays.
Normal human bronchial epithelial cells, 16HBE cell line, and alveolar epithelial cell line (A549) were cultured as described by us before (4, 58). Primary human airway epithelial cells isolated from cadaver or transplant discard tracheobronchial tissues (under approved Institutional Review Board protocol) were seeded on collagen coated Snapwells (Corning) and cultured in bronchoepithelial growth media. After the cells reached confluence they were maintained at air-liquid interface as described before (4). A thin layer (50 μl) of media or nicotine-containing media was added apically and apical media were collected to estimate HMGB1. Transepithelial electrical resistance (TEER) was measured at various time durations after exposure. TEER measurements, cell death assays, and caspase-3/7 release were determined as described by us previously (5).
Statistical analysis.
Prism 6 software (GraphPad, La Jolla, CA) was used, with one-way ANOVA followed by Tukey’s post hoc analysis, unless otherwise indicated. Data reported are means ± SE. P < 0.05 was considered significant.
RESULTS
Characterization of the nicotine delivery.
The in vivo experimental design is outlined in Fig. 1A. To develop a noninvasive method of delivering nicotine to rodents through aerosol inhalation, we utilized a nose-only inhalation system (Fig. 1B), which is equipped with a BANG nebulizer. This nebulizer is utilized for the generation of aqueous aerosols at a low air-flow rate. While the BANG is based on the aerosolization principle of the collision nebulizer, the new design includes a modification to minimize foaming of solutions, thus maintaining stability of substances while maximizing aerosol output. With the use of BANG, particle sizes generated were ~1 μm, which suggests potential delivery to the upper and lower respiratory tracts including alveolar regions (Fig. 1C). This is consistent with particle size determinations of e-cig vapors that were characterized by other methods and found to be in this range (57, 64). Nicotine solution in saline has also been shown to generate droplets in the respirable diameter range (64, 65).
Inhaled nicotine causes increase of its metabolite in plasma.
A 5 or 10% solution of nicotine (in sterile saline) was used for our studies, as these concentrations are normally found in commercial e-cigs. To assess efficiency of nicotine aerosolization, we determined plasma levels of cotinine, a nicotine metabolite (Fig. 1D). We chose cotinine as it is more stable than nicotine and its levels are easily detectable up to 24 h. As expected, inhaled nicotine caused a dose-dependent increase in plasma cotinine, which is comparable to the cotinine levels of light smokers and nicotine-containing e-cig users (43, 45). These values are also comparable to the serum cotinine levels reported in rats exposed to e-cig in whole body exposure chamber (28).
Inhaled nicotine causes pulmonary injury.
Sprague-Dawley rats exposed to nicotine aerosol as described above were monitored for heart rate, breath rate, and oxygen saturations using a pulse oximeter at various time intervals postexposure. The heart rate was decreased acutely at earlier time points in rats exposed to nicotine (5 and 10%) (Fig. 2A). The breath rate and the tissue oxygen saturations were not significantly different (not shown). Pulmonary injury and impairment in the alveolar-capillary barrier were also measured by protein and IgM content in the bronchoalveolar fluid (BALF) and by measuring the lung wet-to-dry weight ratios. The protein contents of BALF of animals exposed to aerosols of 10% nicotine solutions were significantly increased (Fig. 2B), and IgM content was increased in both 5 and 10% nicotine-exposed animals indicating a disruption of the alveolar-capillary barrier (Fig. 2C). Alveolar dysfunction and impaired fluid clearance after nicotine inhalation were also evident by the significantly increased lung wet-to-dry weight ratios in the nicotine-exposed animals (Fig. 2D). These data indicate pulmonary damage after acute exposures to inhaled nicotine at concentrations found in e-cigs and at plasma cotinine levels similar to smokers. Arterial blood gas analysis revealed trends of decreased partial pressure of oxygen, although it was not statistically significant (not shown). Arterial blood gas measurements also showed an increase in lactate, an independent risk factor for morbidity and mortality in ALI (Fig. 2E). However, there was no significant difference in blood glucose levels though (Fig. 2F). Immunohistochemical analysis of 10% nicotine-exposed lungs at 24 h revealed extensive congestion in the vessels (Fig. 2, G and H). There was also evidence of neutrophils in the air spaces (Fig. 2G, bottom).
Fig. 2.

Acute nicotine inhalation causes depression in heart rate and pulmonary injury. Sprague-Dawley rats were exposed to aerosolized saline or nicotine for 15 min at 2 different concentrations (5 or 10%) of nicotine in normal saline. A: the heart rate (HR) was monitored at various time intervals by pulse oximetry (-■-: saline, -●-: 5% nicotine; -▲-: 10% nicotine group). P < 0.05 from HR before exposure of the same animal in A and from unexposed controls. B and C: animals were euthanized at 6 or 24 h postexposure and BALF was collected and protein (B) and IgM (C) content determined as described in the methods. D: lung wet-to-dry (W/D) weight ratios were also evaluated. The right upper lobe (RUL) was used to determine the weights. In B–D, the open bars represent saline, black bars represent 5% nicotine and gray bars represent 10% saline. Data shown are means ± SE (n = 6). * P < 0.05 from saline controls. E and F: arterial blood gas measurements showing blood lactate (E) and glucose (F) are shown. G: representative hematoxylin and eosin-stained paraffin embedded lung sections from saline or nicotine (10%, 24 h)-exposed rats at ×10 (top) or ×20 (bottom) resolution are shown. Arrowhead demonstrates RBC congested areas that are quantified at bottom (H). The arrows demonstrate neutrophils. *P < 0.05 from saline controls.
Nicotine-induces systemic and pulmonary inflammation.
Cigarette smoke and e-cigs trigger inflammation through neutrophil recruitment and activation (34). Cigarette smoke may also cause endothelial activation and vasodilator dysfunction, associated with the expression of adhesion molecules and chemokines that can facilitate vascular inflammation. However, the role of nicotine in causing these systemic or pulmonary neutrophil infiltrations is unknown. Figure 3 shows the CBC values of 5 or 10% nicotine-exposed rats at various time intervals. At early time points of 3 and 6 h, there were increases in neutrophil counts in the circulation of rats exposed to aerosolized nicotine (Fig. 3A). A similar increase in white blood cell counts was also observed in rats exposed to nicotine (Fig. 3B) when observed for up to 24 h. Similarly, eosinophils and basophils were increased acutely at the 3- and 6-h time points after nicotine aerosol exposure. The BALF neutrophil content was increased in nicotine-exposed animals but was not significant (not shown). However, gene expression analysis revealed significantly increased MPO in the lungs (Fig. 4A). Western blot of lung tissue lysates showed increased MPO as well (Fig. 4, B and C). An increase in mRNA levels of IL-1A and CXCL1 was also observed in the animals exposed to 10% nicotine at 24 h postexposure (Fig. 4, D and E). While there was no increase in protein levels of CXCL1, there was an increase in IL-1α protein (Fig. 4F).
Fig. 3.

Complete blood counts following nicotine inhalation. Rats were exposed to aerosols of saline or 5 or 10% solution of nicotine. Blood was collected from the tail vein at different time intervals and complete blood counts (CBCs) were determined using a Hemavet 950S differential cell counter. A–D: neutrophils (A), white blood cells (WBC) (B), eosinophils (C), and basophils (D) were plotted. In A–D, the open bars represent saline, black bars represent 5% nicotine and gray bars represent 10% nicotine. Data shown are means ± SE (n = 6). *P < 0.05 from saline control.
Fig. 4.

Inhaled nicotine caused increase in expression of genes in the inflammatory pathway. Rats were exposed to aerosols of saline or 10% solution of nicotine. Lungs were harvested after 24 h and total RNA was extracted. A–C: mRNA encoding Myeloperoxidase (MPO; A) was estimated by using real-time RT-PCR analysis (B), MPO protein was measured by Western blot analysis and quantified (C). mRNA levels of IL-1a (D) and CXCL1 (E) were also estimated. F: lysates were prepared of the right middle lobe and analyzed by Luminex (IL-1α shown). Data shown are means ± SE (n = 5). *P < 0.05 from saline control.
HMGB1, the most well-characterized damage-associated molecular pattern, recruits neutrophils, monocytes, and macrophages to sites of inflammation as demonstrated in models of ALI (1, 21). Nicotine has previously been shown to prevent the release of HMGB1 (50, 74). We therefore sought to evaluate HMGB1 in the BALF of nicotine aerosol-exposed rats. There was an increase in HMGB1 levels in the BALF supernatant of rats at 24 h after nicotine inhalation, providing evidence of proinflammatory mediator generation early following nicotine inhalation (Fig. 5A). Whether nicotine inhalation causes pulmonary barrier disruption was analyzed by assessing E-cadherin expression in lung tissue lysates. Nicotine inhalation caused a very consistent and significant decrease in E-cadherin expression as compared with the saline-inhaling controls (Fig. 5B).
Fig. 5.

Nicotine causes release of high-mobility group box 1 (HMGB1) and disruption of pulmonary epithelial barrier integrity. A: rats were exposed to saline or 10% nicotine aerosol following which BALF was collected at 24 h and immunoblot for HMGB1 was performed. B: 24 h following nicotine inhalation lungs were harvested and lysates were prepared and analyzed for E-cadherin by Western blots. (+) represents a positive HMGB1 control. The band intensity was measured for quantification, and the representative data are shown as means ± SE (n = 5). *P < 0.05 from saline control.
Effect of nicotine exposure on airway and alveolar epithelial cells.
Nicotine damages the endothelial cell barrier as recently shown (63). However, the effect of nicotine on airway epithelial integrity is unknown. The TEER of air-liquid interface cultures of normal human bronchial epithelial cells was therefore measured to evaluate effects of nicotine on epithelial permeability. We used nicotine concentrations that were >1,000-fold lower than those used in in vivo experiments. Nicotine (0.6–6 mM) treatment at the apical surface caused a loss of TEER after 24 h (Fig. 6A). The loss of TEER was accompanied by a significant decrease in the junctional protein E-cadherin expression after nicotine treatments (Fig. 6B). HMGB1 was also found to be consistently increased in the supernatant media of these cells (Fig. 6C). Whether this decrease could also be related to cell death was investigated in the cytotoxicity studies (Fig. 7). The depth of inhalation can vary in smokers and vapers, which can have a bearing on the cell types affected. We therefore investigated 16HBE cells from the tracheobronchial region of the upper airways and alveolar origin A549 cells. We also used a range of nicotine as nicotine may have a biphasic effect depending on concentrations used. Nicotine concentrations from 0.006 mM onwards caused cell death in both cell lines, with 16-HBE cells being more susceptible (Fig. 7, A and B). The cell death was apoptotic as demonstrated by caspase-3/7 release in A549 cells (Fig. 7C). Nicotine treatment also caused increased IL-6 and VEGF gene expression (Fig. 7, E and F).
Fig. 6.

Nicotine causes loss of cell junction protein, disruption of epithelial barrier integrity and release of high-mobility group box 1 (HMGB1). A: air-liquid interface cultures of primary human bronchial epithelial cells were treated with varying concentrations of nicotine applied as thin layer (100 μl media) on the apical surface. B: transepithelial electrical resistance was measured using an epithelial voltohmmeter electrode after 24 h. C: supernatant media were collected 24 h later and cell lysates were prepared and analyzed for E-cadherin by Western blot. Released HMGB1 was measured using Western blot in the supernatant media. Data shown are means ± SE (n = 4). Figure represents experiments performed in airway epithelial cells isolated from at least 3 different donor tissues. (+) represents a positive HMGB1 control. *P < 0.05 from the values in the absence of nicotine.
Fig. 7.

Nicotine causes airway and alveolar epithelial cell death and expression of proinflammatory genes. A and B: to demonstrate effect of nicotine on airway and alveolar epithelial cells 16HBE (A) and A549 cells (B) were plated on 12 well plates (at 50 k/well) and exposed 24 h later to various concentrations of nicotine. Cell death was assessed 24 h later using Trypan blue dye. C: caspase-3/7 assay was carried out in supernatant media of A549 cells treated with nicotine (3 mM) for 6 h. Data shown are means ± SE (n = 8). *P < 0.05 from the values in the absence of nicotine. Nicotine-treated A549 cells were harvested after 24 h and total RNA was extracted. D and E: steady-state mRNA levels of IL-6 (D) and VEGFA (E) were estimated by real-time RT-PCR analysis using specific Taqman primers and probes. The expression level of each gene was normalized to 18S RNA and reported as fold change. Data shown are means ± SE (n = 6). *P < 0.05 from saline control.
DISCUSSION
Inhaled nicotine is the addictive component of cigarettes that is readily absorbed in blood and modifies reward/pleasure regions in the cortex (46). Cigarette smoke and vapor from e-cigs are aerosols that contain particles of sizes within the respirable range. These particles including nicotine can deposit in the alveoli. Nicotine crosses the alveolar membrane and enters the circulation as suggested by the rapid increases in arterial blood nicotine concentrations (40). Acute e-cig inhalation also leads to enhanced plasma cotinine levels comparable to those of regular tobacco smokers. This also correlates with the rapid physiological effects confirming significant nicotine delivery to the blood stream (72). Therefore, nicotine aerosol by itself could be more potent. E-cigs that were originally developed to treat tobacco addiction are increasingly becoming popular and their nicotine content has exceeded traditional cigarette. The health effects and risks of nicotine inhalation are unknown as the appropriate aerosol-generating systems are not handy.
The droplet size generated in an aerosol can predict the delivery locations in the respiratory tract and whether they are in the respirable diameter range (69). The particle size of nicotine aerosol generated from e-cigs as ascertained by sensitive optical laser spectrometer was found to be in the range of 0.13–0.3 μm (57). More recent estimates have shown that the particle size distribution of most commercial brands range from 0.9 to 1.3 μm (51). Generation of the aerosol with appropriate particle size for airway and alveolar delivery is challenging and is responsible for variations in experimental outcomes (65). The nicotine aerosol generated by a jet collision nebulizer is in the range of 1.95–4 μm (65). Using more advanced BANG technology of aerosol generation, we obtained a relatively uniform distribution of nicotine aerosol particles, which was similar to that reported in most commercial brands of e-cigs. Exposure to these aerosol particles of nicotine resulted in increased plasma cotinine levels in rats. The plasma cotinine values are close to the range found in the plasma of most smokers and e-cig users (40, 43). These values are also comparable to the reported serum cotinine levels in rats exposed to e-cig aerosol in whole body exposure chamber (28) as well as in e-cig-exposed mice (2, 55). This approach enables correlation of effects of inhaled nicotine aerosol in rodent models to human exposures. These data indicate that modern e-cigs are capable of delivering significant amounts of nicotine to the bloodstream.
Studies on effects of inhaled nicotine are emerging but are limited in that pulmonary toxicity has not been thoroughly assessed and in other instances the concentrations used are not relevant to e-cig exposures (47, 65). In one study, 50% of rats inhaling very high concentrations of nicotine (30%) aerosol became acutely apneic and died (65). In another very low-dose study no adverse effects were obtained (54). The nicotine concentrations we used are normally found in e-cigs, and they generate plasma levels that are comparable to e-cig or cigarette smoking users. It has been shown that short-term (e.g., 5 min) e-cig as well as nicotine inhalations exert rapid physiological effects on the cardiovascular system, including elevated heart rate (31, 72). These increases in heart rate were measured just after exposures and over a 1-h period. Our results however indicate a dramatic decrease in heart rate over a 4- to 6-h period, although at 24 h the heart rate returned to normal. We do not rule out an early transient increase in heart rate in our studies. Increased cardiac arrhythmias were observed in pregnant rats inhaling aerosol of 1% nicotine in saline for just 2 min (64). Although compromised alveolar barrier and protein leakage with acute tobacco smoke inhalation has been described before, such damage by nicotine aerosol alone as demonstrated in this report suggests that cigarette smoke-induced barrier disruption could be contributed at least in part by nicotine. Nicotine has been shown in in vitro studies to cause barrier disruption in endothelial cells (63). Our findings are also consistent with other studies where nicotine was found to exacerbate brain edema and cause damage to blood-brain barrier integrity (53, 61). Histopathological alterations of the lungs further supported these findings as significant vascular congestion was noted. It is interesting that rats given nicotine intraperitoneally showed similar congestion of pulmonary vessels (29). The cause of this congestion is unknown but may result from an impaired alveolar-capillary membrane. Blood lactate levels are often increased in a number of diseases including ALI and are strong predictors of outcome. Our findings of increased blood lactate levels in nicotine-exposed animals alongwith disruption of the alveolar-capillary membrane may indicate impaired gas exchange including hypoxemia, which characterize ALI.
Tobacco use is also associated with increased white blood cell and neutrophil counts (8, 32, 62, 66). This association may be of clinical significance as an increased WBC count is an independent risk factor for coronary heart disease and stroke (30). An increase in WBC and neutrophil counts in our studies suggests that nicotine in cigarettes and e-cigs by itself could contribute toward this inflammatory phenotype. Increased inflammation is also reflected by increased neutrophil counts in the blood indicating systemic effects as well. Since nicotine is known to delay spontaneous death of neutrophils (79), it is conceivable that the effects we observe are, in part, due to this phenomenon. A decrease in blood neutrophil counts at a higher concentration and later time-point is plausible because of increased migration and sequestration in the lung (68) or increased death and phagocytosis of neutrophils by macrophages (27).
Interestingly, nicotine has also been shown to be anti-inflammatory, inhibiting the activation of NF-κB pathway and also preventing the release of HMGB1 (74). HMGB1 and its receptor, receptors for advanced glycation end products (RAGE), are both upregulated in alveolar epithelial cells following cigarette smoke exposure (59). Expression of RAGE is also associated with increased lung inflammation (11). It is interesting that the RAGE ligand HMGB1 may inactivate nicotinic acetylcholine receptors in certain neuronal cells (14). Moreover, nicotine treatment at very low doses decreased LPS-induced HMGB1 induction by increasing the nicotinic acetylcholine receptor and hemoxygenase in macrophages and protecting against experimental sepsis (70). It appears that very low doses of nicotine may have beneficial effects. These may however not be relevant in the context of e-cig exposures. Although previous studies have shown that e-cig-induced proinflammatory cytokine production, the role of HMGB1 in such exposures is not known (34, 78). Our findings of increased HMGB1 release along with a decrease in E-cadherin suggest potential increase in inflammation and epithelial cell permeability, similar to situations with other injuries (17, 48). Thus nicotine aerosol exposure via release of HMGB1 likely contributes toward increased epithelial and vascular permeability leading to pulmonary edema. These findings are significant because HMGB1 is also found to be increased in the BALF of COPD patients (56). Increased MPO in the lung along with increases in IL-1α and CXCL1 further indicates acute lung injury and is consistent with other reports (42, 80).
Delivery of e-cig vapors containing nicotine caused apoptosis of airway and alveolar cells in the lungs of mice (24). Our results on nicotine-induced death of airway epithelial cells as well as alveolar epithelial cells further support its toxic effects and are also consistent with a report on colonic epithelial cells (19). Long-term exposure to millimolar concentrations of nicotine results in a steady increase of intracellular calcium, which may also lead to cell damage (35). Taken together, these studies suggest that inhaled nicotine at doses commonly found in e-cigs can cause systemic inflammation, barrier disruption, cell death, and pulmonary edema (Fig. 8). These findings are however different from other reports where no adverse effects were noted in the lung (73, 74). In the study by Waldum et al. (73) whole body nicotine vapor exposures over a 2-yr period caused development of tumors in a number of organs but not in the lungs. These studies were limited in scope and no attempts were made to measure markers of lung injury. In another study by Wang et al. (74) nicotine administered intraperitoneally prevented acute LPS-induced mortality in mice. These studies also showed that nicotine decreased plasma levels of HMGB1 and that it inhibited HMGB1 release from macrophages. It is possible that animal species, exposure model, and cell type differences could account for this divergence from our studies. This could also possibly explain the variable nature of HMGB1 levels in the BALF of nicotine-exposed animals in our study.
Fig. 8.
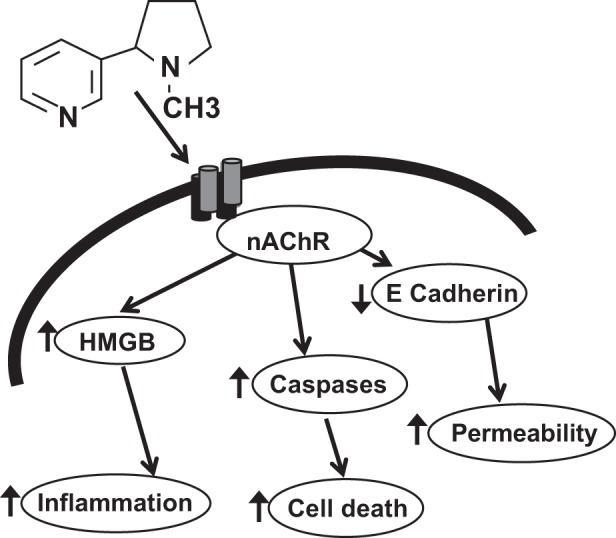
Schematic representation of effects of inhaled nicotine aerosol on the lung. Nicotine acts via nicotinic acetylcholine receptor (nAChR) to cause release of damage associated molecular patterns (DAMPS) such as high-mobility group box 1 (HMGB1) causing increased inflamation, apoptotic cell death and increase cell permeability by decreasing cellular junction protens such as E-cadherin.
In conclusion, inhaled nicotine at concentrations found in e-cigs causes significant pulmonary toxicity. Further studies are warranted to investigate the long-term pulmonary effects of chronic nicotine use at concentrations found in e-cigs.
GRANTS
S. Ahmad is supported by National Institutes of Health Office of the Director (NIH OD), the National Institute of Environmental Health Sciences (NIEHS) Grant U01-ES-025069. S. Ahmad is also supported by intramural funds from Department of Anesthesiology and Perioperative Medicine [University of Alabama at Birmingham (UAB) and a Bridge Fund from the Deans Office, Department of Medicine (UAB). A. Ahmad is supported by the CounterACT Program, NIH OD, NIEHS Grant U01-ES-025069 and National Heart, Lung, and Blood Institute Grant R01-HL-114933.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.A. and S.A. conceived and designed research.S.A., I.Z., N.M., M.H., C.-C.W., and N.V. performed experiments; S.A., I.Z., N.M., M.H., C.-C.W., N.V., I.A.E., and A.A. analyzed data; S.A., I.Z., N.M., C.-C.W., N.V., I.A.E., and A.A. interpreted results of experiments; S.A., I.Z., N.M., M.H., C.-C.W., and A.A. prepared figures; S.A. and A.A. drafted manuscript; S.A., N.M., and A.A. edited and revised manuscript; S.A., I.Z., N.M., M.H., N.V., I.A.E., and A.A. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Yuanqing Edberg for technical assistance and Dritan Xhillari of CH Technologies for providing the flow diagram of the inhalation system.
REFERENCES
- 1.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol 165: 2950–2954, 2000. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 2.Adriani W, Macrì S, Pacifici R, Laviola G. Peculiar vulnerability to nicotine oral self-administration in mice during early adolescence. Neuropsychopharmacology 27: 212–224, 2002. doi: 10.1016/S0893-133X(02)00295-6. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad A, Ahmad S, Glover L, Miller SM, Shannon JM, Guo X, Franklin WA, Bridges JP, Schaack JB, Colgan SP, White CW. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc Natl Acad Sci USA 106: 10684–10689, 2009. doi: 10.1073/pnas.0901326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmad S, Ahmad A, Dremina ES, Sharov VS, Guo X, Jones TN, Loader JE, Tatreau JR, Perraud AL, Schöneich C, Randell SH, White CW. Bcl-2 suppresses sarcoplasmic/endoplasmic reticulum Ca2+-ATPase expression in cystic fibrosis airways: role in oxidant-mediated cell death. Am J Respir Crit Care Med 179: 816–826, 2009. doi: 10.1164/rccm.200807-1104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmad S, Nichols DP, Strand M, Rancourt RC, Randell SH, White CW, Ahmad A. SERCA2 regulates non-CF and CF airway epithelial cell response to ozone. PLoS One 6: e27451, 2011. doi: 10.1371/journal.pone.0027451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrus PG, Rhem R, Rosenfeld J, Dolovich MB. Nicotine microaerosol inhaler. Can Respir J 6: 509–512, 1999. doi: 10.1155/1999/530496. [DOI] [PubMed] [Google Scholar]
- 7.Behar RZ, Hua M, Talbot P. Puffing topography and nicotine intake of electronic cigarette users. PLoS One 10: e0117222, 2015. doi: 10.1371/journal.pone.0117222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Behera D, Dash S, Sen S. Neutrophil count and myeloperoxidase activity in Indian bidi smokers. Respiration 61: 269–273, 1994. doi: 10.1159/000196350. [DOI] [PubMed] [Google Scholar]
- 9.Bennett WD, Brown JS, Zeman KL, Hu SC, Scheuch G, Sommerer K. Targeting delivery of aerosols to different lung regions. J Aerosol Med 15: 179–188, 2002. doi: 10.1089/089426802320282301. [DOI] [PubMed] [Google Scholar]
- 10.Biskos GV, Yurteri CU, Schmidt-Ott A. Generation and sizing of particle for aerosol-based nanotechnology. Kona Powder Particle J 26: 13–35, 2008. doi: 10.14356/kona.26.2008006. [DOI] [Google Scholar]
- 11.Bodine BG, Bennion BG, Leatham E, Jimenez FR, Wright AJ, Jergensen ZR, Erickson CJ, Jones CM, Johnson JP, Knapp SM, Reynolds PR. Conditionally induced RAGE expression by proximal airway epithelial cells in transgenic mice causes lung inflammation. Respir Res 15: 133, 2014. doi: 10.1186/s12931-014-0133-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burns AR, Hosford SP, Dunn LA, Walker DC, Hogg JC. Respiratory epithelial permeability after cigarette smoke exposure in guinea pigs. J Appl Physiol (1985) 66: 2109–2116, 1989. doi: 10.1152/jappl.1989.66.5.2109. [DOI] [PubMed] [Google Scholar]
- 13.Calfee CS, Matthay MA, Eisner MD, Benowitz N, Call M, Pittet JF, Cohen MJ. Active and passive cigarette smoking and acute lung injury after severe blunt trauma. Am J Respir Crit Care Med 183: 1660–1665, 2011. doi: 10.1164/rccm.201011-1802OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandna AR, Nair M, Chang C, Pennington PR, Yamamoto Y, Mousseau DD, Campanucci VA. RAGE mediates the inactivation of nAChRs in sympathetic neurons under high glucose conditions. Eur J Neurosci 41: 341–351, 2015. doi: 10.1111/ejn.12795. [DOI] [PubMed] [Google Scholar]
- 15.Chatham-Stephens K, Law R, Taylor E, Melstrom P, Bunnell R, Wang B, Apelberg B, Schier JG; Centers for Disease Control and Prevention (CDC) . Notes from the field: calls to poison centers for exposures to electronic cigarettes–United States, September 2010-February 2014. MMWR Morb Mortal Wkly Rep 63: 292–293, 2014. [PMC free article] [PubMed] [Google Scholar]
- 16.Chen BC, Bright SB, Trivedi AR, Valento M. Death following intentional ingestion of e-liquid. Clin Toxicol (Phila) 53: 914–916, 2015. doi: 10.3109/15563650.2015.1090579. [DOI] [PubMed] [Google Scholar]
- 17.Chen YC, Statt S, Wu R, Chang HT, Liao JW, Wang CN, Shyu WC, Lee CC. High mobility group box 1-induced epithelial mesenchymal transition in human airway epithelial cells. Sci Rep 6: 18815, 2016. doi: 10.1038/srep18815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corkery JM, Button J, Vento AE, Schifano F. Two UK suicides using nicotine extracted from tobacco employing instructions available on the Internet. Forensic Sci Int 199: e9–e13, 2010. doi: 10.1016/j.forsciint.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Crowley-Weber CL, Dvorakova K, Crowley C, Bernstein H, Bernstein C, Garewal H, Payne CM. Nicotine increases oxidative stress, activates NF-kappaB and GRP78, induces apoptosis and sensitizes cells to genotoxic/xenobiotic stresses by a multiple stress inducer, deoxycholate: relevance to colon carcinogenesis. Chem Biol Interact 145: 53–66, 2003. doi: 10.1016/S0009-2797(02)00162-X. [DOI] [PubMed] [Google Scholar]
- 20.England LJ, Bunnell RE, Pechacek TF, Tong VT, McAfee TA. Nicotine and the developing human: a neglected element in the electronic cigarette debate. Am J Prev Med 49: 286–293, 2015. doi: 10.1016/j.amepre.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erlandsson Harris H, Andersson U. Mini-review: the nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol 34: 1503–1512, 2004. doi: 10.1002/eji.200424916. [DOI] [PubMed] [Google Scholar]
- 22.Etter JF, Bugey A. E-cigarette liquids: constancy of content across batches and accuracy of labeling. Addict Behav 73: 137–143, 2017. doi: 10.1016/j.addbeh.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Farsalinos K. Electronic cigarettes: an aid in smoking cessation, or a new health hazard? Ther Adv Respir Dis 12: 1753465817744960, 2018. doi: 10.1177/1753465817744960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Arcos I, Geraghty P, Baumlin N, Campos M, Dabo AJ, Jundi B, Cummins N, Eden E, Grosche A, Salathe M, Foronjy R. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax 71: 1119–1129, 2016. doi: 10.1136/thoraxjnl-2015-208039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Govindarajan P, Spiller HA, Casavant MJ, Chounthirath T, Smith GA. E-cigarette and liquid nicotine exposures among young children. Pediatrics 141: e20173361, 2018. doi: 10.1542/peds.2017-3361. [DOI] [PubMed] [Google Scholar]
- 26.Gowadia N, Oldham MJ, Dunn-Rankin D. Particle size distribution of nicotine in mainstream smoke from 2R4F, Marlboro Medium, and Quest1 cigarettes under different puffing regimens. Inhal Toxicol 21: 435–446, 2009. doi: 10.1080/08958370802512535. [DOI] [PubMed] [Google Scholar]
- 27.Guzik K, Skret J, Smagur J, Bzowska M, Gajkowska B, Scott DA, Potempa JS. Cigarette smoke-exposed neutrophils die unconventionally but are rapidly phagocytosed by macrophages. Cell Death Dis 2: e131, 2011. doi: 10.1038/cddis.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hage AN, Krause W, Mathues A, Krasner L, Kasten S, Eliason JL, Ghosh A. Comparing the effects of electronic cigarette vapor and cigarette smoke in a novel in vivo exposure system. J Vis Exp 123: e55672, 2017. doi: 10.3791/55672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamza RZ, El-Shenawy NS. Anti-inflammatory and antioxidant role of resveratrol on nicotine-induced lung changes in male rats. Toxicol Rep 4: 399–407, 2017. doi: 10.1016/j.toxrep.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen LK, Grimm RH Jr, Neaton JD. The relationship of white blood cell count to other cardiovascular risk factors. Int J Epidemiol 19: 881–888, 1990. doi: 10.1093/ije/19.4.881. [DOI] [PubMed] [Google Scholar]
- 31.Hansson L, Choudry NB, Karlsson JA, Fuller RW. Inhaled nicotine in humans: effect on the respiratory and cardiovascular systems. J Appl Physiol (1985) 76: 2420–2427, 1994. doi: 10.1152/jappl.1994.76.6.2420. [DOI] [PubMed] [Google Scholar]
- 32.Haswell LE, Papadopoulou E, Newland N, Shepperd CJ, Lowe FJ. A cross-sectional analysis of candidate biomarkers of biological effect in smokers, never-smokers and ex-smokers. Biomarkers 19: 356–367, 2014. doi: 10.3109/1354750X.2014.912354. [DOI] [PubMed] [Google Scholar]
- 33.Haussmann HJ, Fariss MW. Comprehensive review of epidemiological and animal studies on the potential carcinogenic effects of nicotine per se. Crit Rev Toxicol 46: 701–734, 2016. doi: 10.1080/10408444.2016.1182116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higham A, Rattray NJ, Dewhurst JA, Trivedi DK, Fowler SJ, Goodacre R, Singh D. Electronic cigarette exposure triggers neutrophil inflammatory responses. Respir Res 17: 56, 2016. doi: 10.1186/s12931-016-0368-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong W, Peng G, Hao B, Liao B, Zhao Z, Zhou Y, Peng F, Ye X, Huang L, Zheng M, Pu J, Liang C, Yi E, Peng H, Li B, Ran P. Nicotine-induced airway smooth muscle cell proliferation involves TRPC6-dependent calcium influx via α7 nAChR. Cell Physiol Biochem 43: 986–1002, 2017. doi: 10.1159/000481651. [DOI] [PubMed] [Google Scholar]
- 36.Iribarren C, Jacobs DR Jr, Sidney S, Gross MD, Eisner MD. Cigarette smoking, alcohol consumption, and risk of ARDS: a 15-year cohort study in a managed care setting. Chest 117: 163–168, 2000. doi: 10.1378/chest.117.1.163. [DOI] [PubMed] [Google Scholar]
- 37.Iskandar AR, Gonzalez-Suarez I, Majeed S, Marescotti D, Sewer A, Xiang Y, Leroy P, Guedj E, Mathis C, Schaller JP, Vanscheeuwijck P, Frentzel S, Martin F, Ivanov NV, Peitsch MC, Hoeng J. A framework for in vitro systems toxicology assessment of e-liquids. Toxicol Mech Methods 26: 389–413, 2016. doi: 10.3109/15376516.2016.1170251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jerry JM, Collins GB, Streem D. E-cigarettes: safe to recommend to patients? Cleve Clin J Med 82: 521–526, 2015. doi: 10.3949/ccjm.82a.14054. [DOI] [PubMed] [Google Scholar]
- 39.Kleinstreuer C, Feng Y. Lung deposition analyses of inhaled toxic aerosols in conventional and less harmful cigarette smoke: a review. Int J Environ Res Public Health 10: 4454–4485, 2013. doi: 10.3390/ijerph10094454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawson GM, Hurt RD, Dale LC, Offord KP, Croghan IT, Schroeder DR, Jiang NS. Application of serum nicotine and plasma cotinine concentrations to assessment of nicotine replacement in light, moderate, and heavy smokers undergoing transdermal therapy. J Clin Pharmacol 38: 502–509, 1998. doi: 10.1002/j.1552-4604.1998.tb05787.x. [DOI] [PubMed] [Google Scholar]
- 41.Lee HW, Park SH, Weng MW, Wang HT, Huang WC, Lepor H, Wu XR, Chen LC, Tang MS. E-cigarette smoke damages DNA and reduces repair activity in mouse lung, heart, and bladder as well as in human lung and bladder cells. Proc Natl Acad Sci USA 115: E1560–E1569, 2018. doi: 10.1073/pnas.1718185115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lerner CA, Sundar IK, Yao H, Gerloff J, Ossip DJ, McIntosh S, Robinson R, Rahman I. Vapors produced by electronic cigarettes and e-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One 10: e0116732, 2015. doi: 10.1371/journal.pone.0116732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindsay RP, Tsoh JY, Sung HY, Max W. Secondhand smoke exposure and serum cotinine levels among current smokers in the USA. Tob Control 25: 224–231, 2016. doi: 10.1136/tobaccocontrol-2014-051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu Q, Gottlieb E, Rounds S. Effects of cigarette smoke on pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol 314: L743–L756, 2018. doi: 10.1152/ajplung.00373.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marsot A, Simon N. Nicotine and cotinine levels with electronic cigarette: a review. Int J Toxicol 35: 179–185, 2016. doi: 10.1177/1091581815618935. [DOI] [PubMed] [Google Scholar]
- 46.Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, Perkins KA, Picciotto MR, Quik M, Rose JE, Rothenfluh A, Schafer WR, Stolerman IP, Tyndale RF, Wehner JM, Zirger JM. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 190: 269–319, 2007. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- 47.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM; Acute Lung Injury in Animals Study Group . An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2011. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohammad G, Siddiquei MM, Othman A, Al-Shabrawey M, Abu El-Asrar AM. High-mobility group box-1 protein activates inflammatory signaling pathway components and disrupts retinal vascular-barrier in the diabetic retina. Exp Eye Res 107: 101–109, 2013. doi: 10.1016/j.exer.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 49.Ng M, Freeman MK, Fleming TD, Robinson M, Dwyer-Lindgren L, Thomson B, Wollum A, Sanman E, Wulf S, Lopez AD, Murray CJ, Gakidou E. Smoking prevalence and cigarette consumption in 187 countries, 1980-2012. JAMA 311: 183–192, 2014. doi: 10.1001/jama.2013.284692. [DOI] [PubMed] [Google Scholar]
- 50.Ni YF, Tian F, Lu ZF, Yang GD, Fu HY, Wang J, Yan XL, Zhao YC, Wang YJ, Jiang T. Protective effect of nicotine on lipopolysaccharide-induced acute lung injury in mice. Respiration 81: 39–46, 2011. doi: 10.1159/000319151. [DOI] [PubMed] [Google Scholar]
- 51.Oldham MJ, Zhang J, Rusyniak MJ, Kane DB, Gardner WP. Particle size distribution of selected electronic nicotine delivery system products. Food Chem Toxicol 113: 236–240, 2018. doi: 10.1016/j.fct.2018.01.045. [DOI] [PubMed] [Google Scholar]
- 52.Padon AA, Lochbuehler K, Maloney EK, Cappella JN. A Randomized Trial of the Effect of Youth Appealing E-Cigarette Advertising on Susceptibility to Use E-Cigarettes Among Youth. Nicotine Tob Res 20: 954–961, 2018. doi: 10.1093/ntr/ntx155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paulson JR, Yang T, Selvaraj PK, Mdzinarishvili A, Van der Schyf CJ, Klein J, Bickel U, Abbruscato TJ. Nicotine exacerbates brain edema during in vitro and in vivo focal ischemic conditions. J Pharmacol Exp Ther 332: 371–379, 2010. doi: 10.1124/jpet.109.157776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phillips B, Esposito M, Verbeeck J, Boué S, Iskandar A, Vuillaume G, Leroy P, Krishnan S, Kogel U, Utan A, Schlage WK, Bera M, Veljkovic E, Hoeng J, Peitsch MC, Vanscheeuwijck P. Toxicity of aerosols of nicotine and pyruvic acid (separate and combined) in Sprague-Dawley rats in a 28-day OECD 412 inhalation study and assessment of systems toxicology. Inhal Toxicol 27: 405–431, 2015. doi: 10.3109/08958378.2015.1046000. [DOI] [PubMed] [Google Scholar]
- 55.Ponzoni L, Moretti M, Sala M, Fasoli F, Mucchietto V, Lucini V, Cannazza G, Gallesi G, Castellana CN, Clementi F, Zoli M, Gotti C, Braida D. Different physiological and behavioural effects of e-cigarette vapour and cigarette smoke in mice. Eur Neuropsychopharmacol 25: 1775–1786, 2015. doi: 10.1016/j.euroneuro.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 56.Pouwels SD, Heijink IH, ten Hacken NH, Vandenabeele P, Krysko DV, Nawijn MC, van Oosterhout AJ. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol 7: 215–226, 2014. doi: 10.1038/mi.2013.77. [DOI] [PubMed] [Google Scholar]
- 57.Pratte P, Cosandey S, Goujon-Ginglinger C. A scattering methodology for droplet sizing of e-cigarette aerosols. Inhal Toxicol 28: 537–545, 2016. doi: 10.1080/08958378.2016.1224956. [DOI] [PubMed] [Google Scholar]
- 58.Riddle SR, Ahmad A, Ahmad S, Deeb SS, Malkki M, Schneider BK, Allen CB, White CW. Hypoxia induces hexokinase II gene expression in human lung cell line A549. Am J Physiol Lung Cell Mol Physiol 278: L407–L416, 2000. doi: 10.1152/ajplung.2000.278.2.L407. [DOI] [PubMed] [Google Scholar]
- 59.Robinson AB, Stogsdill JA, Lewis JB, Wood TT, Reynolds PR. RAGE and tobacco smoke: insights into modeling chronic obstructive pulmonary disease. Front Physiol 3: 301, 2012. doi: 10.3389/fphys.2012.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rowell TR, Tarran R. Will chronic e-cigarette use cause lung disease? Am J Physiol Lung Cell Mol Physiol 309: L1398–L1409, 2015. doi: 10.1152/ajplung.00272.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sajja RK, Rahman S, Cucullo L. Drugs of abuse and blood-brain barrier endothelial dysfunction: A focus on the role of oxidative stress. J Cereb Blood Flow Metab 36: 539–554, 2015. doi: 10.1177/0271678X15616978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saxena K, Liang Q, Muhammad-Kah R, Sarkar M. Evaluating the relationship between biomarkers of potential harm and biomarkers of tobacco exposure among current, past, and nonsmokers: data from the National Health and Nutrition Examination Survey 2007–2012. Biomarkers 22: 1–10, 2016. doi: 10.1080/1354750X.2016.1201536. [DOI] [PubMed] [Google Scholar]
- 63.Schweitzer KS, Chen SX, Law S, Van Demark M, Poirier C, Justice MJ, Hubbard WC, Kim ES, Lai X, Wang M, Kranz WD, Carroll CJ, Ray BD, Bittman R, Goodpaster J, Petrache I. Endothelial disruptive proinflammatory effects of nicotine and e-cigarette vapor exposures. Am J Physiol Lung Cell Mol Physiol 309: L175–L187, 2015. doi: 10.1152/ajplung.00411.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shao XM, López-Valdés HE, Liang J, Feldman JL. Inhaled nicotine equivalent to cigarette smoking disrupts systemic and uterine hemodynamics and induces cardiac arrhythmia in pregnant rats. Sci Rep 7: 16974, 2017. doi: 10.1038/s41598-017-17301-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shao XM, Xu B, Liang J, Xie XS, Zhu Y, Feldman JL. Nicotine delivery to rats via lung alveolar region-targeted aerosol technology produces blood pharmacokinetics resembling human smoking. Nicotine Tob Res 15: 1248–1258, 2013. doi: 10.1093/ntr/nts261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith KR, Uyeminami DL, Kodavanti UP, Crapo JD, Chang LY, Pinkerton KE. Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidant. Free Radic Biol Med 33: 1106–1114, 2002. doi: 10.1016/S0891-5849(02)01003-1. [DOI] [PubMed] [Google Scholar]
- 67.Sommerfeld K, Łukasik-Głębocka M, Kulza M, Drużdż A, Panieński P, Florek E, Zielińska-Psuja B. Intravenous and oral suicidal e-liquid poisonings with confirmed nicotine and cotinine concentrations. Forensic Sci Int 262: e15–e20, 2016. doi: 10.1016/j.forsciint.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 68.Terashima T, Klut ME, English D, Hards J, Hogg JC, van Eeden SF. Cigarette smoking causes sequestration of polymorphonuclear leukocytes released from the bone marrow in lung microvessels. Am J Respir Cell Mol Biol 20: 171–177, 1999. doi: 10.1165/ajrcmb.20.1.3276. [DOI] [PubMed] [Google Scholar]
- 69.Thomas RJ, Webber D, Sellors W, Collinge A, Frost A, Stagg AJ, Bailey SC, Jayasekera PN, Taylor RR, Eley S, Titball RW. Characterization and deposition of respirable large- and small-particle bioaerosols. Appl Environ Microbiol 74: 6437–6443, 2008. doi: 10.1128/AEM.01194-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsoyi K, Jang HJ, Kim JW, Chang HK, Lee YS, Pae HO, Kim HJ, Seo HG, Lee JH, Chung HT, Chang KC. Stimulation of alpha7 nicotinic acetylcholine receptor by nicotine attenuates inflammatory response in macrophages and improves survival in experimental model of sepsis through heme oxygenase-1 induction. Antioxid Redox Signal 14: 2057–2070, 2011. doi: 10.1089/ars.2010.3555. [DOI] [PubMed] [Google Scholar]
- 71.Vakkalanka JP, Hardison LS Jr, Holstege CP. Epidemiological trends in electronic cigarette exposures reported to U.S. Poison Centers. Clin Toxicol (Phila) 52: 542–548, 2014. doi: 10.3109/15563650.2014.913176. [DOI] [PubMed] [Google Scholar]
- 72.Vansickel AR, Eissenberg T. Electronic cigarettes: effective nicotine delivery after acute administration. Nicotine Tob Res 15: 267–270, 2013. doi: 10.1093/ntr/ntr316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Waldum HL, Nilsen OG, Nilsen T, Rørvik H, Syversen V, Sanvik AK, Haugen OA, Torp SH, Brenna E. Long-term effects of inhaled nicotine. Life Sci 58: 1339–1346, 1996. doi: 10.1016/0024-3205(96)00100-2. [DOI] [PubMed] [Google Scholar]
- 74.Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 10: 1216–1221, 2004. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 75.Ware LB, Lee JW, Wickersham N, Nguyen J, Matthay MA, Calfee CS, California Transplant Donor N; California Transplant Donor Network . Donor smoking is associated with pulmonary edema, inflammation and epithelial dysfunction in ex vivo human donor lungs. Am J Transplant 14: 2295–2302, 2014. doi: 10.1111/ajt.12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weiss D, Tomasallo CD, Meiman JG, Creswell PD, Melstrom PC, Gummin DD, Patel DJ, Michaud NT, Sebero HA, Anderson HA. Electronic cigarette exposure: calls to wisconsin poison control centers, 2010–2015. WMJ 115: 306–310, 2016. [PubMed] [Google Scholar]
- 77.Williams RS, Derrick J, Liebman AK, LaFleur K. Content analysis of e-cigarette products, promotions, prices and claims on Internet tobacco vendor websites, 2013–2014. Tob Control 27: e34–e40, 2018. doi: 10.1136/tobaccocontrol-2017-053762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu Q, Jiang D, Minor M, Chu HW. Electronic cigarette liquid increases inflammation and virus infection in primary human airway epithelial cells. PLoS One 9: e108342, 2014. doi: 10.1371/journal.pone.0108342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu Y, Li H, Bajrami B, Kwak H, Cao S, Liu P, Zhou J, Zhou Y, Zhu H, Ye K, Luo HR. Cigarette smoke (CS) and nicotine delay neutrophil spontaneous death via suppressing production of diphosphoinositol pentakisphosphate. Proc Natl Acad Sci USA 110: 7726–7731, 2013. doi: 10.1073/pnas.1302906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yokohira M, Nakano Y, Hashimoto N, Yamakawa K, Ninomiya F, Kishi S, Saoo K, Imaida K. Toxicity of nicotine by repeated intratracheal instillation to f344 rats. J Toxicol Pathol 25: 257–263, 2012. doi: 10.1293/tox.25.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zaky A, Bradley WE, Lazrak A, Zafar I, Doran S, Ahmad A, White CW, Dell’Italia LJ, Matalon S, Ahmad S. Chlorine inhalation-induced myocardial depression and failure. Physiol Rep 3: 3, 2015. doi: 10.14814/phy2.12439. [DOI] [PMC free article] [PubMed] [Google Scholar]