

Abstract
The MUC1 tumor-associated antigen is overexpressed in the majority of human carcinomas and several hematologic malignancies. Much attention has been paid to the hypoglycosylated variable number of tandem repeats (VNTR) region of the N-terminus of MUC1 as a vaccine target, and recombinant viral vector vaccines are also being evaluated that express the entire MUC1 transgene. While previous studies have described MUC1 as a tumor-associated tissue differentiation antigen, studies have now determined that the C-terminus of MUC1 (MUC1-C) is an oncoprotein, and its expression is an indication of poor prognosis in numerous tumor types. We report here the identification of nine potential CD8+ cytotoxic T lymphocyte epitopes of MUC1, seven in the C-terminus and two in the VNTR region, and have identified enhancer agonist peptides for each of these epitopes. These epitopes span HLA-A2, HLA-A3, and HLA-A24 major histocompatibility complex (MHC) class I alleles, which encompass the majority of the population. The agonist peptides, compared to the native peptides, more efficiently (a) generate T-cell lines from the peripheral blood mononuclear cells of cancer patients, (b) enhance the production of IFN-γ by peptide-activated human T cells, and (c) lyse human tumor cell targets in an MHC-restricted manner. The agonist epitopes described here can be incorporated into various vaccine platforms and for the ex vivo generation of human T cells. These studies provide the rationale for the T-cell-mediated targeting of the oncogenic MUC1-C, which has been shown to be an important factor in both drug resistance and poor prognosis for numerous tumor types.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-013-1494-7) contains supplementary material, which is available to authorized users.
Keywords: Vaccines, Oncogene, T cells, Agonist epitopes, MUC1-C
Introduction
MUC1 (CD227) is a large transmembrane glycoprotein, normally expressed at the apical surface of glandular epithelial cells [1]. In adenocarcinoma (e.g., breast, prostate, colorectal, ovarian, lung, bladder, and pancreatic cancer), it is overexpressed and aberrantly glycosylated [2, 3]. Loss of epithelial cell polarization also results in its expression throughout on the cell surface. Therefore, the MUC1 tumor-associated antigen (TAA) has been recognized as a potential target for immunotherapy [4]. MUC1 is also expressed in hematologic malignancies such as B-cell lymphoma, chronic myelogenous leukemia, and multiple myeloma [5–7].
The MUC1 molecule has two distinct regions, formed by autocleavage with a SEA domain (Supplemental Fig. 1). The N-terminal (MUC1-N) is the large extracellular domain, which consists of the variable number of tandem repeats region (VNTR) and the non-VNTR region. MUC1-N is shed from the cells and can be found in the circulation of patients with advanced cancer. It is used as a tumor marker (CA15.3) in breast cancer patients [8]. The C-terminal region of MUC1 (MUC1-C) has three distinctive parts: a small extracellular domain that is covalently bound to MUC1-N, a single transmembrane domain, and a cytoplasmic tail [9]. The cytoplasmic tail contains sites for interaction with signaling proteins, such as β-catenin, epidermal growth factor receptor (EGFR), and Src [10]. Since these proteins are situated at the basolateral part of healthy cells, protein–MUC1 interactions are not believed to be significant. However, loss of polarity in human tumor cells allows the cytoplasmic tail to be exposed to the signaling proteins, and interaction can occur [11]. The MUC1-C region has been shown to act as an oncogene, leading to transformation of human cells when MUC1-C binds to β-catenin [12–14]. Moreover, MUC1-C transfection has been demonstrated to be sufficient to induce transformation and confer oncogenic activities previously attributed to the full-length MUC1 protein, such as increased growth rate, anchorage-independent cell growth, and resistance to chemotherapy agents [15]. In addition, MUC1-C signaling activated by c-Src is involved in the disruption of both E-cadherin adherens junctions and integrin focal adhesions that stimulate cancer cell motility, invasion, and metastasis, suggesting a possible role for MUC1-C in epithelial to mesenchymal transition (EMT) [16]. Overexpression of genes related to MUC1 has also been found to be highly associated with poor prognosis in patients with lung and breast cancer [17] and with drug resistance [15].
Numerous clinical trials have evaluated MUC1 as a potential target for vaccine therapy of a range of human tumors. The majority of these have employed polypeptides of the VNTR region [18–21]. Vector-based trials, some of which are ongoing, have employed recombinant poxviral vectors expressing the MUC1 transgene. These include MVA-MUC1-IL-2 [22] and a diversified prime-boost approach employing vaccinia and fowlpox CEA-MUC1-TRICOM recombinants, designated PANVAC [23–25]. PANVAC consists of recombinant vaccinia (V) and fowlpox (F) vectors containing transgenes for CEA, MUC1, and three costimulatory molecules (B7.1, intercellular adhesion molecule 1, and lymphocyte function-associated antigen 3).
There are numerous reports of putative T-cell epitopes in the N-terminus of MUC1 (both in the VNTR and non-VNTR regions). To our knowledge, however, there is only one report [26] of a T-cell epitope in the C-terminus MUC1 region; this was reported to strongly bind to the major histocompatibility complex (MHC), but did not induce peptide-specific responses such as IFN-γ production or tumor cell lysis.
One method that has been shown to enhance the ability of a vaccine to be more efficacious is to make alterations in the amino acid sequence of putative T-cell epitopes, which in turn can hopefully enhance T-cell activation and specific T-cell killing of tumor cells [27, 28]. Not all substitutions of an amino acid of a potential cytotoxic T lymphocyte (CTL) epitope, however, will lead to an enhancer agonist epitope, and some substitutions will lead to antagonist epitopes. Moreover, the generation of a putative agonist epitope of a TAA may well lead to enhanced T-cell activation by IFN-γ production, but will be useless unless the activated T cell will recognize the endogenous (native) epitope expressed in the context of the MHC on the surface of human tumor cells and consequently lyse those tumor cells (see Tables 1 and 2).
Table 1.
MUC1 HLA-A2-, HLA-A3-, and HLA-A24-binding peptides and potential agonists, with predicted binding and T2-cell binding assay
| Peptide | Location | Position | Sequence | Class I allele | Predicted binding* | Actual binding# |
|---|---|---|---|---|---|---|
| C1 | C domain | 1172–1181 | ALAIVYLIAL | A2 | 49 | 249 |
| C1A | YLAIVYLIAL | 226 | 245 | |||
| C2 | C domain | 1177–1186 | YLIALAVCQC | A2 | 52 | 211 |
| C2A | YLIALAVCQV | 736 | 299 | |||
| C3 | C domain | 1240–1248 | SLSYTNPAV | A2 | 70 | 326 |
| C3A | YLSYTNPAV | 320 | 342 | |||
| V1 | VNTR region | 150–158 | STAPPAHGV | A2 | 1 | 166 |
| V1A | YLAPPAHGV | 320 | 486 | |||
| V2 | VNTR region | 141–149 | APDTRPAPG | A2 | 0 | 210 |
| V2A | YLDTRPAPV | 128 | 647 | |||
| C4 | C domain | 432–441 | ALAIVYLIAL | A3 | 5 | NA |
| C4A | ALFIVYLIAK | 900 | NA | |||
| C5 | C domain | 483–491 | STDRSPYEK | A3 | 3 | NA |
| C5A | SLFRSPYEK | 300 | NA | |||
| C6 | C domain | 462–471 | TYHPMSEYPT | A24 | 6 | NA |
| C6A | KYHPMSEYAL | 480 | NA | |||
| C7 | C domain | 502–510 | SYTNPAVAA | A24 | 5 | NA |
| C7A | KYTNPAVAL | 400 | NA |
Amino acids that were changed to generate an agonist epitope are in bold
* Predicted binding on the basis of reported motif [32]; score estimate of half time of disassociation of a molecule containing this sequence.
#Peptides were used at a concentration of 12.5 μg/ml in a binding assay with T2-A2 cells. Results are expressed as mean fluorescence intensity (MFI). NA: A functional binding assay was not available for HLA-A3 and HLA-A24 peptides
Table 2.
Inability of the predicted HLA-A2 binding to predict the biologic activity of MUC1 peptides
| Peptide | Position | Sequence | Predicted binding to HLA-A2 | T2-A2 binding | Level of killing by peptide-specific CTL | Level of IFN-γ produced by peptide-specific CTL |
|---|---|---|---|---|---|---|
| C1A | 1172 | YLAIVYLIAL | 5 | 5 | 2 | 3 |
| C2A | 1177 | YLIALAVCQV | 3 | 3 | 1 | 1 |
| C3A | 1240 | YLSYTNPAV | 4 | 1 | 3 | 2 |
| C8A | 1135 | YLSDVSVSDV | 1 | 2 | Negative | 4 |
| C9A | 1162 | YLLVLVCVLV | 2 | 4 | Negative | Negative |
Comparison and ranking of predicted and actual binding, as well as peptide-specific killing and IFN-γ production for potential agonist epitope peptides for MUC1-C. We found that the predicted binding of an epitope did not always correspond to the actual binding to T2-A2 cells and also that the epitope with the best binding affinity did not always generate T cells with the most efficient tumor cell killing or IFN-γ production. 1 = highest level, 5 = lowest level. The agonist epitopes C8A and C9A were not among those evaluated in the other figures and tables
CTL cytotoxic T lymphocytes
To date, three agonist epitopes of MUC1 have been reported, all three being in the N-terminus non-VNTR region. One agonist epitope (P93L) was shown, compared to the native epitope, to enhance the generation of T cells that can also more efficiently lyse human tumor cells [29]. Two other potential agonist epitopes in this region were shown to enhance T-cell cytokine production, but no tumor cell killing was reported [30]. We report here the identification of nine novel enhancer agonist peptides of MUC1: seven in the C-terminus and two in the VNTR region. These epitopes span human leukocyte antigen (HLA)-A2, HLA-A3, and HLA-A24 MHC class I alleles, which encompass the majority of the population. The agonist peptides, compared to their respective native peptide counterparts, are shown to more efficiently (a) generate T-cell lines from the peripheral blood mononuclear cells (PBMCs) of cancer patients, (b) enhance the production of IFN-γ by peptide-activated human T cells, and (c) lyse human tumor cell targets endogenously expressing the native epitope in an MHC-restricted manner. These studies thus form the rationale for the use of these agonist peptides in various vaccination modalities or for the ex vivo generation of T cells. The information encoding these agonist epitopes can also be employed as transgenes to potentially enhance the efficacy of recombinant vector-based vaccines.
Materials and methods
Patients
For the experiments using HLA-A2- and HLA-A3-specific peptides, we utilized PBMCs from cancer patients enrolled in a previously described clinical trial of a CEA- and MUC1-based viral vaccine (PANVAC-V/F) [23]. PANVAC consists of recombinant vaccinia (V) and fowlpox (F) vectors containing transgenes for CEA, MUC1, and three costimulatory molecules (B7.1, intercellular adhesion molecule 1, and lymphocyte function-associated antigen 3, designated TRICOM). Recombinant PANVAC-V was given as a prime, with PANVAC-F as boost injections every other week for the first month, and monthly thereafter. GM-CSF was given on four consecutive days with each vaccination. Patients #1 and #3 had colon carcinoma, and patient #2 had breast carcinoma. For the experiments using HLA-A24-specific peptides, we used PBMCs from two patients (#4 and #5) with prostate cancer enrolled in a previously described clinical trial of PSA-TRICOM vaccine in combination with ipilimumab [31]. The time to progression (TTP) and overall survival (OS) for patient #1 were 69 and 227 days, respectively; TTP and OS for patient #2 were 76 and 242 days, respectively; TTP and OS for patient #3 were >30 and >30 months, respectively; TTP and OS for patient #4 were 178 and 753 days, respectively; and TTP and OS for patient #5 were 166 and 1,116 days, respectively. An institutional review board of the National Institutes of Health (NIH) Clinical Center had approved the procedures, and informed consent was obtained in accordance with the Declaration of Helsinki. All injections were given at the NIH Clinical Center (Bethesda, MD, USA).
Peptides
The MUC1 amino acid sequence was scanned for matches to consensus motifs for HLA-A2-, HLA-A3-, and HLA-A24-binding peptides. We used the computer algorithm developed by Parker et al. [32] to rank potential MHC-binding peptides according to the predicted one-half-time dissociation of peptide/MHC complexes. American Peptide Company (Sunnyvale, CA) synthesized 9-mer and 10-mer peptide analogs from the MUC1-C and VNTR regions of MUC1 with single amino acid substitutions in order to increase the binding affinity (Tables 1,2). The purity of the peptides was >90 %.
Affinity and avidity assays
The affinity of the native and agonist epitope peptides was investigated in an assay determining the mean fluorescence intensity (MFI) of the peptide–HLA-A2 molecule complexes on T2 cells after overnight incubation with peptide (50, 25, 12.5, 6.25, and 3,12 μg/ml) [33]. MFI was measured by flow cytometry. In an additional experiment, the binding of C2A peptide was compared to that of Flu-A2 peptide (GILGFVFTL) at the same concentrations. The avidity of the native and agonist epitope peptides was investigated in an assay determining the stability of the peptide–HLA-A2 molecule complexes on T2 cells. The frequency of remaining complexes at different time points was measured as MFI by flow cytometry at 0, 2, 4, 6, 8, and 10 h and compared to the MFI at 0 h. Despite numerous attempts to establish binding assays for HLA-A3 and HLA-A24 peptides using T2-A3 and T2-A24 cells, we were not able to establish reliable assays for these alleles. Therefore, these peptides were evaluated based solely on the ability to lyse cells pulsed with the corresponding peptide and tumor cells expressing the native peptide.
Generation of dendritic cells from PBMCs
Peripheral blood was collected from patients, and PBMCs were isolated by centrifugation on a density gradient (Lymphocyte Separation Medium, ICN Biochemicals, Aurora, VA). Dendritic cells (DCs) were generated using a modification of the previously described procedure [34]. DCs were grown in AIM-V medium containing 100 ng/ml GM-CSF and 20 ng/ml IL-4 (PeproTech, Rocky Hill, NJ). After 5 days in culture, the DCs were matured by the addition of 1 μg/ml CD40L and 1 μg/ml enhancer (Enzo Life Sciences, Farmingdale, NY) for 24 h. They were then either used immediately for the first in vitro stimulation of PBMCs (IVS1) or frozen in aliquots for future use.
Establishment of T-cell lines
A modified version of the protocol described by Tsang et al. [34] was used to generate MUC1-specific CTLs. Irradiated autologous DCs were pulsed with 20 μg/ml of peptide for 2 h, and then, PBMCs were added at a 10:1 ratio. After 3 days, human IL-2 (20 Cetus units/ml) was added. Cells were restimulated every 7 days. After the third IVS, cells were restimulated using autologous Epstein–Barr virus-transformed B cells as antigen-presenting cells (APCs) at a ratio of 2:1 and maintained in medium containing IL-7 (10 ng/ml) and IL-15 (10 ng/ml).
Detection of cytokines
Autologous B cells pulsed with peptides at different concentrations (25, 12.5, 6.25, 3.13, and 1.56 μg/ml) were incubated with MUC1-specific T-cell lines at a 2:1 ratio for 24 h. The supernatants were analyzed for IFN-γ by ELISA (Invitrogen, Frederick, MD).
Tetramer staining
Phycoerythrin (PE)-labeled HLA-A2 and HLA-A3 tetramers were prepared for all agonist epitopes by the NIH/NIAID MHC Tetramer Core Facility (Atlanta, GA), and PE-labeled MHC class I human negative tetramer (Class I iTAg MHC Tetramer) was obtained from Beckman Coulter Inc. (Sykesville, MD). The negative tetramer has no known specificity and does not bind to human CD8+ T cells of any HLA allele. The tetramers were used at a 1:100 dilution, and cells were stained for 45 min at 4 °C. For all flow cytometry, 1 × 105 cells were acquired on an LSRII (BD, Becton–Dickinson, San Jose, CA), and data were analyzed using FlowJo 9.0.1 software (Tree Star Inc, Ashland, OR).
Tumor cell cultures
The human breast carcinoma cell line MCF-7 (HLA-A2+, MUC1+), pancreatic carcinoma cell line CFPAC-1 (HLA-A2+, HLA-A3+, MUC1+), melanoma cell line SK-Mel (HLA-A2+, MUC1neg), pancreatic carcinoma cell line ASPC-1 (HLA-A3neg, HLA-A24neg, MUC1+), ovarian cancer cell line SKOV3 (HLA-A3+, MUC1+), colon cancer cell line SW620 (HLA-A24+, MUC1+), and prostate cancer cell line PC3 (HLA-A24+, MUC1+) were purchased from American Type Culture Collection (Manassas, VA). All cell cultures were free of mycoplasma and maintained in complete medium (RPMI 1640 supplemented with 10 % fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM l-glutamine) (Mediatech, Herndon, VA). K562-A2.1 cells were obtained from Dr. C Britten (Johannes Gutenberg University, Mainz, Germany) and maintained in complete medium supplemented with 0.5 mg/ml of G418 (Mediatech). The 174CEM-T2 cell line (T2) [35] transport deletion mutant was provided by Dr. Peter Cresswell (Yale University School of Medicine, New Haven, CT) and cultured in Iscove’s modified Dulbecco’s complete medium (Mediatech).
Cytotoxicity assay, cold target inhibition, and antibody blocking of tumor cell lysis
To determine T-cell-mediated killing, a 16-h 111Indium release assay was used [34]. 2 × 106 target cells were labeled with 60 μCi 111In oxide (GE Health care, Vienna, VA) at 37 °C for 20 min and used at 3,000 cells/well in 96-well round-bottom culture plates. T cells were added at different ratios. All assays were performed in RPMI medium substituted with 10 % human AB serum (Omega Scientific, Tarzana, CA), glutamine, and antibiotics (Mediatech, Manassas, VA). Spontaneous release was determined by incubating target cells with medium alone, and complete lysis was determined by incubation with 2.5 % Triton X-100. Lysis was calculated using the formula:
A cold target inhibition assay was performed by adding K562-A2.1 or K562-A3 cells, with or without prior pulsing with the corresponding peptide, at a ratio of 1:10 to the wells [34]. Antibody blocking was performed by pre-incubating tumor cells with 10 μg/ml of anti-HLA-A2, anti-HLA-A3 or anti-HLA-A24 antibody, or isotype control antibody (UPC10).
Results
Identification of HLA-A2-binding T-cell epitopes of MUC1-C and the VNTR region of MUC1
Fourteen potential CTL epitopes (9 and 10 mer) of the MUC1 C-terminus and three potential epitopes of the VNTR region of MUC1 were selected on the basis of computer algorithms for MHC class I binding [32] and were synthesized. Potential enhancer agonist epitopes of each peptide were also synthesized. Agonists were developed by substitution of a single amino acid residue at the HLA class I-binding site. Sequences of the native and potential agonist epitopes are given in Table 1. The MUC1-C amino acid sequence (from residue 1098 to 1254) and the VNTR region (from residue 141, repeats of 20 amino acids) were scanned for matches to consensus motifs for HLA-A2-binding peptides to identify possible CD8+ T-cell epitopes. Two of the five HLA-A2 peptide agonists showed decreased binding to T2-A2 cells and were not further investigated (Table 2). Three HLA-A2-binding peptides for the MUC1-C domain, designated C1 (position 1172), C2 (position 1177), and C3 (position 1240), were identified, as well as their corresponding potential agonists with higher predicted binding affinity (designated C1A, C2A, and C3A, respectively). Two HLA-A2-binding peptides were also identified for the VNTR region, designated V1 (position 150) and V2 (position 141); also identified were the corresponding potential agonists with higher predicted binding affinity (designated V1A and V2A, respectively). Table 1 shows the amino acid sequences and positions of these peptides, their predicted binding using the computer algorithm developed by Parker et al. [32], and their actual binding to T2 cells. Supplemental Fig. 1 shows the location of these epitopes in the MUC1 molecule.
The native and potential agonist peptides were investigated for their ability to bind and form stable complexes with HLA-A2 molecules in a T2 cell binding and stability assay. The prostate peptide NGEP [36], which has a high affinity for HLA-A2, was used as a positive control, and the CEA-specific peptide CAP7 [34] (specific for HLA-A3) was used as a negative control with a binding value of < 1. The agonist epitopes C2A, V1A, and V2A displayed higher binding affinity than their corresponding native peptides (Table 1). Despite higher predicted binding, peptides C1A and C3A did not bind to HLA-A2 with greater affinity than the corresponding native peptides. Studies were also conducted to compare the binding affinity of the MUC1 agonist C2A with that of the known strong HLA-A2-binding peptide of influenza (GILGFVFTL, denoted here as flu peptide). Using the T2-A2 binding assay described in “Materials and methods,” 10–16-fold more C2A peptide versus flu peptide was required to obtain a given MFI binding intensity.
The stability of the MHC–peptide complexes was then investigated by determining the frequency of remaining complexes at different time points after the addition of brefeldin A. The MUC1-C agonist peptides showed binding stability similar to their native peptides, whereas the VNTR epitope V2A displayed slightly higher avidity than its native epitope (Supplemental Fig. 2).
Peptide-specific T-cell lines produced IFN-γ when stimulated with their respective peptides
The immunogenicity of the MUC1 native and agonist peptides was then investigated by evaluating their ability to generate specific human T-cell lines in vitro. T-cell lines were established from patients enrolled on a PANVAC (rV, rF-CEA-MUC1-TRICOM) vaccine trial. PBMCs from a patient (#1) with colon carcinoma were used to generate the C1, C1A, C2, C2A, V1A, and V2A T cell lines; due to limitations in the number of PBMCs available, we used PBMCs from another patient on that trial with breast carcinoma (#2) to generate T-cell lines C3 and C3A. To evaluate the specificity of the T-cell lines, IFN-γ production was measured by ELISA after 24 h of stimulation with different amounts of each native peptide and its corresponding potential agonist peptide. Autologous B cells (see “Materials and methods”) were used as APCs. Each of the T-cell lines generated with the MUC1-C agonist peptides (T-C1A, T-C2A, and T-C3A) produced higher levels of IFN-γ at several different peptide concentrations than the corresponding native peptide T-cell lines (Fig. 1a–c). T-cell lines could not be generated using the native VNTR region epitopes V1 and V2, but were successfully generated concurrently from the same patient’s PBMC with agonist peptides V1A and V2A. These T-cell lines both produced appreciable IFN-γ upon stimulation at various peptide concentrations (Fig. 1d, e).
Fig. 1.
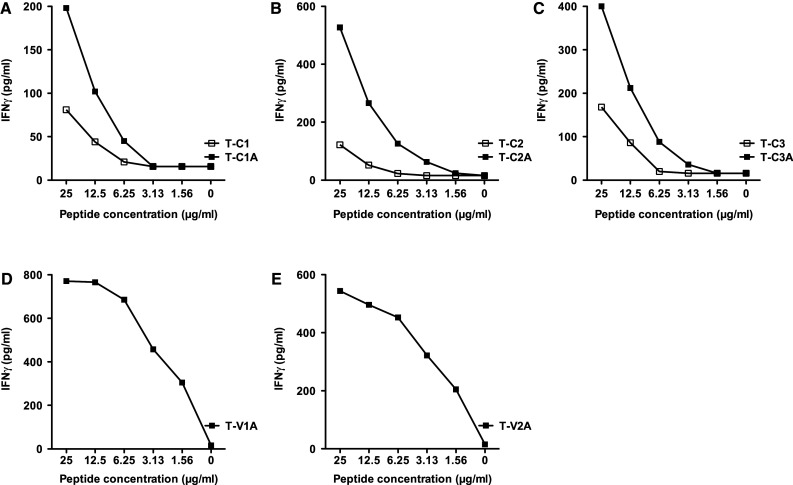
IFN-γ production from peptide-specific T-cell lines. Specific T-cell lines were established for the native and agonist MUC1 peptides as described in “Materials and methods,” from patients enrolled on a PANVAC vaccine trial, and used at IVS4. PBMCs from a patient (#1) with colon carcinoma were used to establish the C1, C1A, C2, C2A, V1A, and V2A T-cell lines. For the C3 and C3A T-cell lines, PBMCs from a patient with breast carcinoma (#2) were used. The IFN-γ production of the specific T-cell lines was measured by ELISA after 24 h of stimulation with different amounts of peptide. Autologous B cells were used as antigen-presenting cells (see “Materials and methods”). a Peptide C1 was used to stimulate the T-C1 T-cell line, and peptide C1A was used to stimulate the T-C1A T-cell line. b Peptide C2 was used to stimulate the T-C2 T-cell line, and peptide C2A was used to stimulate the T-C2A T-cell line. c Peptide C3 was used to stimulate the T-C3 T-cell line, and peptide C3A was used to stimulate the T-C3A T-cell line. d Peptide V1A was used to stimulate the T-V1A T-cell line. e Peptide V2A was used to stimulate the T-V2A T-cell line. T-cell lines could not be established from the native VNTR region epitopes V1 and V2. All MUC1-C agonist epitope T-cell lines produced higher levels of IFN-γ than the corresponding native peptide T-cell lines
Tumor cell lysis
The MUC1-specific T-cell lines were then evaluated in cytotoxicity assays using as a target the human breast carcinoma cell line MCF-7 (86 % HLA-A2+, 48 % MUC1+) and melanoma cell line SK-Mel (100 % HLA-A2+, MUC1neg) as a negative control target. The expression of HLA-A2 and MUC1 was evaluated by flow cytometry. Using two effector:target cell (E:T) ratios, the C1A and C3A agonist T-cell lines more efficiently lysed MCF7 tumor cells than did their native T-cell line counterparts (Table 3). There was only a minor difference in tumor lysis between the C2 and C2A T-cell lines at the lower E:T ratio. There was no lysis of SK-Mel using any of the C-terminus-directed T-cell lines, indicating the specificity for MUC1. As mentioned above, it was not possible to generate T-cell lines using the native peptides V1 and V2. Both of the agonist VNTR region T-cell lines, however, efficiently lysed MCF-7, but not SK-Mel (Table 3).
Table 3.
MUC1 native and agonist epitope-specific T-cell lines lyse tumor cell lines expressing native MUC1 and HLA-A2, HLA-A3, or HLA-A24
| T-cell line HLA-A2 | E:T ratio | MCF-7 MUC1+HLA-A2+ | CF-PAC1 MUC1+HLA-A2+ | SK-Mel MUC1negHLA-A2+ |
|---|---|---|---|---|
| T-C1 | 50:1 | 26.4 | NA | 8.0 |
| 25:1 | 3.9 | NA | NA | |
| T-C1A | 50:1 | 40.7 | NA | 0 |
| 25:1 | 25.5 | 18.1 | NA | |
| T-C2 | 50:1 | 53.6 | NA | 4.6 |
| 25:1 | 38.5 | NA | NA | |
| T-C2A | 50:1 | 54.4 | NA | 0 |
| 25:1 | 46.2 | 21.8 | NA | |
| T-C3 | 50:1 | 9.2 | NA | NA |
| 25:1 | 8.4 | NA | 0.8 | |
| T-C3A | 50:1 | 25.9 | NA | NA |
| 25:1 | 20.8 | 16.6 | 1.3 | |
| T-V1 | 25:1 | NA | NA | NA |
| 12.5:1 | NA | NA | NA | |
| T-V1A | 25:1 | 42.2 | 38.3 | 5.0 |
| 12.5:1 | 24.6 | NA | 0 | |
| T-V2 | 25:1 | NA | NA | NA |
| 12.5:1 | NA | NA | NA | |
| T-V2A | 25:1 | 53.4 | 25.8 | 0 |
| 12.5:1 | 45.1 | NA | 1.5 |
| T-cell line HLA-A3 | E:T ratio | CF-PAC1 MUC1+HLA-A3+ | SKOV3 MUC1+HLA-A3+ | ASPC-1 MUC1+HLA-A3neg |
|---|---|---|---|---|
| T-C4 | 50:1 | 18.8 | NA | 0 |
| 25:1 | NA | NA | NA | |
| T-C4A | 50:1 | 29.5 | 37.6 | 0 |
| 25:1 | 24 | 36.3 | NA | |
| T-C5 | 50:1 | 15.2 | NA | 0 |
| 25:1 | NA | NA | NA | |
| T-C5A | 50:1 | 39 | 37.9 | 0 |
| 25:1 | 32.2 | 35.4 | NA |
| T-cell line HLA-A24 | E:T ratio | SW620 MUC1+HLA-A24+ | PC3 MUC1+HLA-A24+ | ASPC-1 MUC1+HLA-A24neg |
|---|---|---|---|---|
| T-C6 | 25:1 | NA | NA | NA |
| 12.5:1 | NA | NA | NA | |
| T-C6A | 25:1 | 41.2 | 35.5 | 2.4 |
| 12.5:1 | 26.0 | 22.8 | 1.9 | |
| T-C7 | 25:1 | 22.2 | NA | 0 |
| 12.5:1 | 13.7 | NA | NA | |
| T-C7A | 25:1 | 41.9 | 22.6 | 3.4 |
| 12.5:1 | 32.6 | NA | 2.1 |
Specific T-cell lines were established as described in “Materials and methods” from patients enrolled on the PANVAC vaccine trial and the PSA-TRICOM and ipilimumab trial for the native and agonist MUC1 peptides and used at IVS4. T-cell lines for the native epitopes V1, V2, and C6 could not be established. The MUC1-specific T-cell lines were evaluated in cytotoxicity assays using the human tumor cell lines MCF-7 (breast carcinoma, HLA-A2+, MUC1+), SK-Mel (melanoma, HLA-A2+, MUC1neg), CF-PAC1 (pancreatic cancer, HLA-A2+, HLA-A3+, MUC1+), ASPC-1 (pancreatic cancer, HLA-A3neg, HLA-A24neg, MUC1+), SKOV3 (ovarian cancer, HLA-A3+, MUC1+), SW620 (colon cancer, HLA-A24+, MUC1+), and PC3 (prostate cancer, HLA-A24+, MUC1+) as targets and control targets, as described in “Materials and methods”. Results are expressed as % specific lysis. The assays were performed at two effector (E)-to-target (T) cell ratios. NA Not available. Agonists are in bold
To further investigate tumor cell lysis with a second human tumor cell line and to evaluate HLA-A2 restriction, the MUC1 T-cell lines were evaluated in a cold target inhibition assay with 111Indium-treated human pancreatic carcinoma cell line CF-PAC-1 (100 % HLA-A2+, 76 % MUC1+) as a target (see “Materials and methods”). The tumor cell lysis was inhibited by the addition of APCs pulsed with the corresponding peptide. When APCs were added without peptide, there was no inhibition (Fig. 2a–e). The addition of anti-HLA-A2 antibody inhibited tumor lysis, but the addition of UPC-10, a control antibody, did not (Fig. 2f–h). The above results thus provide evidence that the MUC1-C and VNTR region-specific T cells generated using agonist epitopes could lyse human tumor cells that endogenously express native MUC1 in an antigen-specific and HLA-A2 restricted manner.
Fig. 2.
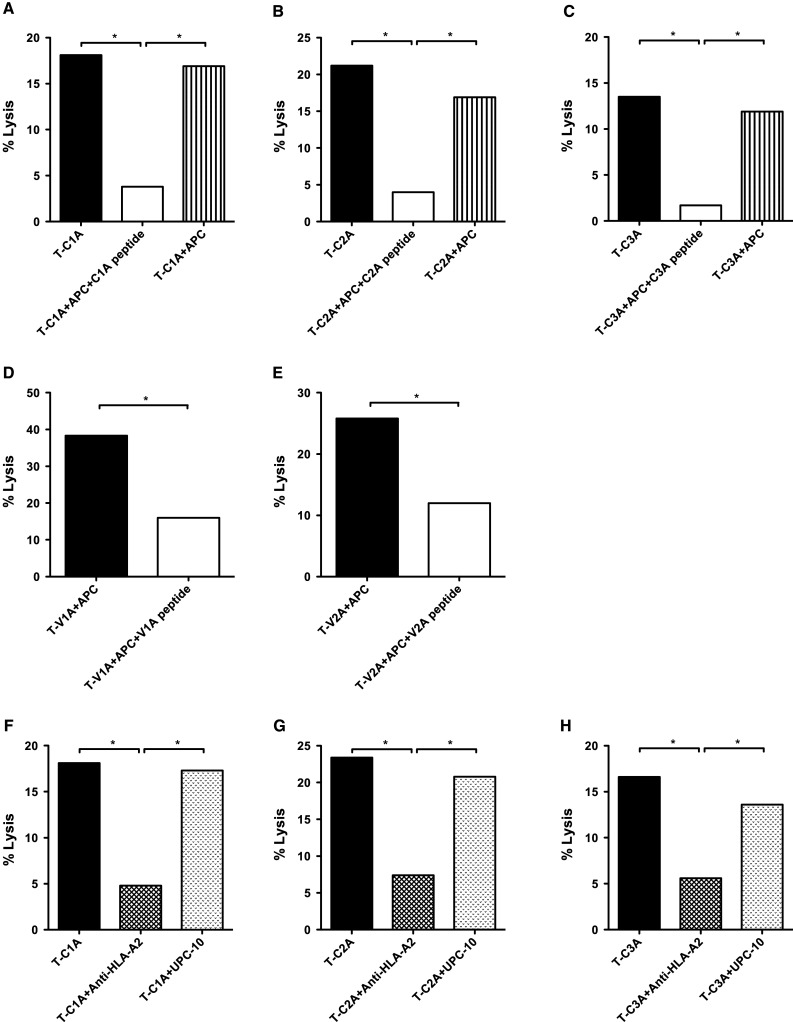
Tumor cell lysis by the MUC1 specific T-cell lines was HLA-A2 restricted. Specific T-cell lines were established for the MUC1 agonist epitopes as described in “Materials and methods,” from patients enrolled on a PANVAC vaccine trial. PBMCs from patients (#1 and #3) with colon carcinoma were used to establish the C1A, C2A, V1A, and V2A T-cell lines. For the C3A T-cell line, PBMCs from a patient (#2) with breast carcinoma were used. The MUC1 agonist-specific T-cell lines were evaluated in a cytotoxicity assay with 111Indium-treated human pancreatic carcinoma cell line CFPAC-1 (100 % HLA-A2+, 76 % MUC1+) as a target. The tumor lysis was inhibited by the addition of peptide-pulsed APCs (K562-A2 cells) at a 1:10 ratio. When APCs were added without peptide, there was no inhibition. a T-cell line specific for C1A; b T-cell line specific for C2A; c T-cell line specific for C3A; d T-cell line specific for V1A; e T-cell line specific for V2A. Moreover, the addition of anti-HLA-A2 antibody inhibited cytotoxicity, but the addition of UPC-10, a control antibody, did not. f T-cell line specific for C1A; g T-cell line specific for C2A; h T-cell line specific for C3A. The effector: target cell ratio was 25:1. *indicates a significant difference
Tetramer binding
MUC1 native and agonist epitopes were also used to stimulate PBMCs from patients #1 and 2 enrolled in the PANVAC vaccine trial. The frequency of MUC1 peptide-specific CD8+ T cells was measured after two IVS (see “Materials and methods”) for binding directed against tetramers synthesized with each of the corresponding agonist epitopes. A class I iTAg MHC tetramer (Beckman Coulter) was used as a negative control. For all three MUC1-C domain peptides, the agonist epitope-stimulated PBMCs displayed ≥6.0 % tetramer binding, which in each case was higher than the PBMCs stimulated with native epitopes (Supplemental Table 1). As mentioned above, T-cell lines could not be established for the VNTR native peptides, but both agonist peptides efficiently stimulated PBMCs that bound to the V1A and V2A tetramers (5.4 and 2.4 %, respectively, Supplemental Table 1). The lower panels of Supplemental Table 1 show representative FACS plots. To demonstrate specificity of the tetramer binding, the C2A agonist T-cell line was shown to bind to the C2A agonist tetramer; the binding was only minimally inhibited by the addition of one, two, or four tetramers specific for antigens other than MUC1.
Analysis of HLA-A3 MUC1-C native and agonist epitopes
Six native epitopes were selected based on HLA-A3-binding algorithms, and the 9 and 10 mer peptides and their corresponding potential enhancer agonist epitope peptides were synthesized. Numerous attempts to develop a HLA class I A3 binding assay failed using T2-A3 cells and C1RA3 cells. Thus, T-cell lines were generated from PBMCs of HLA-A3 patients previously vaccinated with PANVAC vaccine using the six potential agonist peptides. Each cell line produced IFN-γ when stimulated with peptide versus control peptide. However, lysis of C1RA3 cells pulsed with the corresponding peptide was seen with only two of the six lines, subsequently designated C4A and C5A (Table 1).
T-cell lines from PBMCs of a patient with colon cancer (patient #3) vaccinated with PANVAC vaccine were generated using native peptides C4 and C5 and their potential agonist epitopes C4A and C5A, respectively. Following multiple attempts, the T-cell lines generated with both native peptides grew poorly and thus limited experiments could be carried out using these lines. In contrast, T-cell lines from the same patient could be derived using the agonist peptides C4A and C5A. Enough cells from the native peptide-derived T-cell lines were available to compare lysis of the human pancreatic tumor cell line CF-PAC1 (MUC1+, HLA-A3+) with the human pancreatic tumor cell line ASPC-1 (MUC1+, HLA-A3neg). As seen in Table 3, both agonist derived T-cell lines gave higher lysis of CF-PAC1 than the native derived lines (albeit at only one E:T ratio due to the limited amounts of T cells derived using the two native peptides).
Lysis of the ovarian carcinoma cell line SKOV3 (MUC1+, HLA-A3+) was also evaluated using T-cell lines derived from both a colorectal cancer patient and an ovarian cancer patient vaccinated with PANVAC vaccine. Cell lines derived using agonist peptides C4A and C5A from both patients efficiently lysed SKOV3 cells endogenously expressing the native epitopes at numerous E:T ratios and showed no lysis of the MUC1+, HLA-A3neg ASPC-1 tumor cell line (Table 3). The HLA-A3 restricted lysis seen by both T-cell lines derived with the agonist epitopes is shown in Table 4, with anti-HLA-A3 inhibition of lysis of the pancreatic tumor line. Additional cold target inhibition studies confirmed the lysis of tumor cells endogenously expressing the native HLA-A3 epitope and the HLA-A3 restriction of the lysis (Table 4). T-cell lines generated with the C4A and C5A agonist epitopes showed 20.2 and 16.2 % binding to their corresponding tetramers and <0.04 % binding to the control tetramer (Supplemental Fig. 3).
Table 4.
MUC1 HLA-A3 and HLA-A24 agonist epitope-specific T-cell lines lyse tumor cell lines expressing native MUC1 in a HLA-restricted manner
| T-cell line (A3) | Blocking | % Lysis of CF-PAC1 MUC1+HLA-A3+ |
|---|---|---|
| T-C4A | – | 59.6 |
| Anti-HLA-A3 | 17.2 | |
| Isotype control | 45.7 | |
| K562-A3 + C4A peptide | 13.8 | |
| K562-A3 alone | 52.1 | |
| T-C5A | – | 66.3 |
| Anti-HLA-A3 | 10.3 | |
| Isotype control | 58.5 | |
| K562-A3 + C5A peptide | 7.1 | |
| K562-A3 alone | 49.3 |
| T-cell line (A24) | Blocking | % Lysis of SW620 MUC1+HLA-A24+ | % Lysis of PC3 MUC1+HLA-A24+ |
|---|---|---|---|
| T-C6A | – | 41.2 | 22.8 |
| Anti-HLA-A24 | 14.6 | 10.2 | |
| Isotype control | 37.0 | 20.1 | |
| T-C7A | – | 22.7 | 22.6 |
| Anti-HLA-A24 | 8.6 | 3.1 | |
| Isotype control | 17.9 | 19.7 |
The MUC1-specific T-cell lines were evaluated in cytotoxicity assays using the human tumor cell lines CF-PAC1 (pancreatic cancer, HLA-A2+, HLA-A3+, MUC1+), SW620 (colon cancer, HLA-A24+, MUC1+) and PC3 (prostate cancer, HLA-A24+, MUC1+) as targets, and blocking by anti-HLA-A3 or anti-HLA-A24 antibody, as described in “Materials and methods.” In addition, a cold target inhibition assay was performed for the HLA-A3-specific peptides C4A and C5A, using K562-A3 cells pulsed with the corresponding peptide as cold targets. Results are expressed as % specific lysis. The assays were performed at an E:T ratio of 25:1, except the T-C6A lysis of PC3 cells, which was performed at a ratio of 12.5:1
T-cell lines were generated from the same patient (#3) with the C4 native peptide or the agonist C4A peptide. Both were then stimulated with the agonist peptide and analyzed for cytokine production. As seen in Table 5, the agonist peptide-derived T-cell line produced higher levels of the Type I cytokines IFN-γ, GM-CSF, TNF-α, and IL-2. Similar results were seen with T-cell lines produced using the native C5 peptide and its corresponding agonist peptide C5A. Upon stimulation of each of these T-cell lines with autologous B cells pulsed with the C5A peptide, more type I cytokines were produced by the C5A-derived T-cell line.
Table 5.
MUC1 HLA-A3 and HLA-A24 agonist epitope-specific T-cell lines produce Type I cytokines upon stimulation
| T-cell line | Peptide | IFN-γ | GM-CSF | IL-2 | TNFα | IL-8 | IL-6 | IL-10 |
|---|---|---|---|---|---|---|---|---|
| A3 | ||||||||
| T-C4 | C4A | 2,777 | 1,135 | 4 | 119 | 20 | 71 | 27 |
| T-C4A | C4A | >10,000 | >10,000 | 170 | 3,804 | 279 | 192 | 79 |
| T-C5 | C5A | >10,000 | 6,355 | 347 | 2,576 | 165 | 138 | 16 |
| T-C5A | C5A | >10,000 | >10,000 | 1,266 | 7,546 | 2,738 | 414 | 74 |
| A24 | ||||||||
| T-C7 | C7 | 750 | 237 | <2.4 | 21 | 6.8 | 6.4 | 25 |
| C7A | 1,279 | 300 | <2.4 | 30 | 7.3 | 7.7 | 45 | |
| T-C7A | C7 | 680 | 215 | <2.4 | 30 | 112 | <2.4 | 92 |
| C7A | 2,000 | 910 | <2.4 | 70 | 360 | 40 | 375 | |
The MUC1 specific T-cell lines were evaluated for cytokine production upon stimulation with autologous B cells pulsed with the corresponding native and agonist peptides. For the HLA-A3 peptide epitopes, T-cell lines were established from PBMCs from a patient with colon cancer treated with PANVAC vaccine. The T-cell lines were used at IVS5 (T-C4 and T-C4A) and IVS4 (T-C5 and T-C5A), stimulated with B cells pulsed with the corresponding peptides for 24 h, and cytokine levels were analyzed in the supernatant. 1 × 106 T cells/ml, values expressed as pg/ml. For the HLA-A24 peptide epitopes, T-cell lines were established from PBMCs from a patient with prostate cancer treated with PSA-TRICOM and ipilimumab. Despite several attempts, it was not possible to establish a T-cell line using the native C6 peptide, although a T-cell line could be established from the same patient with the corresponding agonist peptide. The native T-C7 and agonist T-C7A T-cell lines were stimulated for 24 h with B cells pulsed with both the native and the agonist peptides, and cytokine levels in the supernatant were analyzed. Results are expressed as pg/ml/2.5 × 105 T cells. The levels of IL-12p70 and IL-1β were < 100 pg/ml for all native and agonist epitopes
Analysis of HLA-A24 MUC1-C agonist epitopes
The algorithm for HLA-A24 class I-binding peptides in the MUC1-C region revealed no potential A24 binders. Changes in anchor residues revealed the potential for three HLA-A24 agonists. Studies with two of these agonists (C6A and C7A, Table 1) are described. (The third potential agonist is not shown because a T-cell line generated did not lyse tumor cells.)
Attempts to generate T-cell lines with the native peptide designated C6 were unsuccessful using PBMCs from two different vaccinated cancer patients (patients #4 and #5 were from a trial employing Prostvac vaccine and ipilimumab). T-cell lines, however, could be generated from these same patients using APCs pulsed with the corresponding agonist peptide C6A. The T-cell line derived from APCs pulsed with the C6A peptide was evaluated for lysis versus two different MUC1+, HLA-A24+ tumor cell lines (SW620, colon cancer, and PC3, prostate cancer) and the ASPC-1 pancreatic cancer cell line (MUC1+, HLA-A24neg). Lysis of both of the HLA-A24+ cell lines was seen (Table 3), in contrast to the HLA-A24neg line. The T-cell line derived with the native C7 peptide grew poorly, but enough cells were available to evaluate this T-cell line in a cytotoxicity assay using the colon cancer cell line SW620. As can be seen in Table 4, the T-cell line derived with the agonist C7A peptide lysed SW620 cells more efficiently than the T-cell line derived with the native C7 peptide. Neither T-cell line lysed the ASPC-1 tumor cell line. The addition of an anti-HLA-A24 antibody greatly reduced the lysis of tumor cells, demonstrating the MHC restriction of the lysis for both the C6A- and C7A-specific T-cell lines (Table 4).
The T-cell line generated with the native C6 peptide grew poorly. Stimulation of the T-cell line generated with the C6A agonist peptide produced high levels (pg/ml/105 cells) of IFN-γ (2,651), GM-CSF (> 10,000), IL-8 (> 10,000), and TNF-α (372) and low levels (< 50) of IL-2, IL-6, IL-10, and IL-12.
T-cell lines could be generated from the same patient using autologous APCs pulsed with the native C7 or agonist C7A peptides. Each cell line was then stimulated with either the native C7 or agonist C7A peptide. As seen in Table 5, the T-cell line generated with the native peptide produced more Type I cytokine IFN-γ when simulated with the agonist C7A versus the native C7 peptide. Additionally, when the T-cell line generated with the agonist C7A peptide was stimulated with both native and agonist peptides, more IFN-γ, GM-CSF, IL-8, IL-10, and TNF-α were produced by stimulation with APCs pulsed with agonist C7A peptide versus the native C7 peptide (Table 5).
Discussion
The studies reported here demonstrate that while the use of an algorithm to determine potential CD8+ T-cell epitopes and thus potential agonist epitopes of a human tumor antigen can be useful, the algorithm does not necessarily predict actual MHC binding (Table 2). As shown in Table 2, the HLA-A2 binding also does not necessarily predict the ability to activate human T cells to produce interferon. Most importantly, none of the above ranked the agonist epitopes evaluated in terms of killing tumor cells endogenously expressing the native antigen (Table 2).
The C-terminus of MUC1 has been shown by several groups to be extremely important in the initiation and progression of a range of human neoplasms. It has been shown to play a part in the epithelial stress response, the coordinated sequence of signals that induces normal epithelial cells to temporarily undergo loss of polarity, which triggers growth promotion, survival, and repair of damage to the epithelial layer [4, 5, 11, 37–39]. Overexpression of MUC1-C makes it possible for malignant cells of epithelial or hematopoietic origin to exploit this physiologic stress response and thus stimulate their expansion and survival. It was previously shown that expression of mutations in the cytoplasmic tail of MUC1-C could block anchorage-independent growth and tumorigenicity of human carcinoma cells [4].
The MUC1-C oncoprotein has also been shown to induce tamoxifen and herceptin resistance in human breast tumor cells [40, 41]. Inhibition of MUC1-C has been shown to be synergistic with cytotoxic agents in the treatment of breast cancer cells [42]. MUC1-C-induced transcriptional programs have also been shown to be associated with tumorigenesis and predict outcome in breast and lung cancer patients [43–45]. The androgen receptor has been shown to regulate the MUC1-C oncoprotein expression in human prostate cancer cells [46], while the MUC1-C oncoprotein has been shown to confer androgen-independent growth in human prostate cancer cells [47]. MUC1-C has also been shown to regulate survival of pancreatic cancer cells [48] and enhance invasiveness of pancreatic cancer cells by inducing EMT [49].
The MUC1-C oncoprotein has also recently been shown to play an important role in several hematopoietic malignancies. The survival of human multiple myeloma cells was shown to be dependent on MUC1-C-terminal transmembrane subunit oncogenic function, and inhibition of the MUC1-C oncoprotein induces multiple myeloma cell death; conversely, expression of the MUC1-C oncoprotein was shown to promote growth and survival of human multiple myeloma cells [6]. The MUC1-C oncoprotein suppresses terminal differentiation of acute myelogenous leukemia cells [50] and chronic myelogenous leukemia cells [7] and also regulates Bcr-Ab1 stability in chronic myelogenous leukemia [5]. These studies render the C-terminus of MUC1 a biologically relevant target for immunotherapy of a range of human cancers.
The C-terminus of MUC1 has also been shown to play a role in tumor rejection in murine models [51–53]. A portion of both the tandem repeat and the C-terminus contributed to the CD4+ T-cell rejection of MUC1-expressing murine B16 melanoma cells, but not in the rejection of MUC1-expressing Panc02 tumor cells. The C-terminus MUC1 epitope was shown to be necessary for CD8+-mediated rejection of Panc02 tumors. Results also showed that MUC1 C-terminus epitope vaccination could prolong survival in MUC1-transgenic mice challenged with MUC1-expressing tumors, without evidence of autoimmune responses [51–53].
The studies reported here identify seven novel potential CTL epitopes in the MUC1-C region of MUC1, but more importantly, identify enhancer agonists for each of these epitopes. This was demonstrated by the ability of the agonist, compared to its corresponding native epitope, to generate MUC1-C-specific T-cell lines, enhance IFN-γ production by T cells, and lyse human tumor cell targets endogenously expressing the native epitope in an MHC-restricted manner. These epitopes span class I MHC HLA-A2, HLA-A3, and HLA-A24, which encompass the majority of the population. Two additional agonist epitopes were also identified in the VNTR region of MUC1. In these studies, T-cell lines were able to be generated from PBMCs of nine different cancer patients, employing the MUC1 agonist peptides described. The results obtained from five of these patients are shown in the “Results” section.
Numerous preclinical studies and recent clinical studies have demonstrated the importance of the induction of CD8+ T-cell responses in vaccine-mediated anti-tumor immunity. Both the number and avidity of T cells can contribute to tumor cell lysis. Indeed, it has been shown that high avidity T cells can lyse targets with up to 1,000-fold lower peptide–MHC complexes than low avidity T cells [54–56]. Since the majority of tumor antigens are “self-antigens,” they will by nature induce lower avidity T cells. Even some gene products of somatic mutations, such as point mutated ras, will generate T cells of much lower avidity when compared to T cells induced by microbial antigens such as influenza. For this reason, strategies have been undertaken to enhance both the number and avidity of T cells to tumor-associated antigens. One such strategy, the design of enhancer agonist epitopes, was employed in the studies reported here. Complementary strategies to enhance T-cell numbers and avidity include the use of viral, bacterial, or yeast-based recombinant vaccines, or peptide-pulsed DCs, to enhance the vaccine-mediated immune milieu; other strategies include the use of vaccines in combination with other immune stimulants such as cytokines, with immune checkpoint inhibitors, and/or with the use of specific chemotherapeutic agents, radiation, or specific small molecule targeted therapeutics.
The studies reported here provide the rationale for immunotherapy clinical studies employing the agonist epitopes of both the C-terminus and VNTR of MUC1. These include the use of peptides alone, on DCs, with classical or novel adjuvant formulation, or with a range of biologic adjuvants, or cytokines such as IL-12, GM-CSF, or IL-15. These agonist peptides can also be used to activate T cells in vitro in adoptive T-cell therapy approaches. The T-cell receptors directed against these agonist epitopes can also be used in genetically engineered T-cell adoptive transfer studies. Longer peptide or the MUC1 protein itself containing the agonist epitopes can also be employed as described above. Finally, recombinant vector-based vaccines can be employed, which encode for the MUC1 transgene and include the sequences for these agonist epitopes.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
Grant support was provided by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. The authors thank Diane J. Poole for technical assistance and Debra Weingarten for editorial assistance in the preparation of this manuscript.
Conflict of interest
The authors declare that they have no conflicts of interest.
Abbreviations
- APC
Antigen-presenting cell
- CTL
Cytotoxic T lymphocyte
- DC
Dendritic cell
- EGFR
Epidermal growth factor receptor
- EMT
Epithelial to mesenchymal transition
- HLA
Human leukocyte antigen
- IVS
In vitro stimulation
- MHC
Major histocompatibility complex
- MUC1-C
C-terminal region of MUC1
- MUC1-N
N-terminal region of MUC1
- OS
Overall survival
- PBMC
Peripheral blood mononuclear cell
- TAA
Tumor-associated antigen
- TTP
Time to progression
- VNTR
Variable number of tandem repeats
Footnotes
J. Schlom and K.-Y. Tsang contributed equally to this paper.
References
- 1.Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, Pemberton L, Lalani EN, Wilson D. Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J Biol Chem. 1990;265:15286–15293. [PubMed] [Google Scholar]
- 2.Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004;4:45–60. doi: 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- 3.Kufe D, Inghirami G, Abe M, Hayes D, Justi-Wheeler H, Schlom J. Differential reactivity of a novel monoclonal antibody (DF3) with human malignant versus benign breast tumors. Hybridoma. 1984;3:223–232. doi: 10.1089/hyb.1984.3.223. [DOI] [PubMed] [Google Scholar]
- 4.Kufe DW. Functional targeting of the MUC1 oncogene in human cancers. Cancer Biol Ther. 2009;8:1197–1203. doi: 10.4161/cbt.8.13.8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawano T, Ito M, Raina D, Wu Z, Rosenblatt J, Avigan D, Stone R, Kufe D. MUC1 oncoprotein regulates Bcr-Abl stability and pathogenesis in chronic myelogenous leukemia cells. Cancer Res. 2007;67:11576–11584. doi: 10.1158/0008-5472.CAN-07-2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin L, Ahmad R, Kosugi M, Kufe T, Vasir B, Avigan D, Kharbanda S, Kufe D. Survival of human multiple myeloma cells is dependent on MUC1 C-terminal transmembrane subunit oncoprotein function. Mol Pharmacol. 2010;78:166–174. doi: 10.1124/mol.110.065011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin L, Kufe D. MUC1-C oncoprotein blocks terminal differentiation of chronic myelogenous leukemia cells by a ROS-mediated mechanism. Genes Cancer. 2011;2:56–64. doi: 10.1177/1947601911405044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayes DF, Zurawski VR, Jr, Kufe DW. Comparison of circulating CA15-3 and carcinoembryonic antigen levels in patients with breast cancer. J Clin Oncol. 1986;4:1542–1550. doi: 10.1200/JCO.1986.4.10.1542. [DOI] [PubMed] [Google Scholar]
- 9.Lan MS, Batra SK, Qi WN, Metzgar RS, Hollingsworth MA. Cloning and sequencing of a human pancreatic tumor mucin cDNA. J Biol Chem. 1990;265:15294–15299. [PubMed] [Google Scholar]
- 10.Li Y, Ren J, Yu W, Li Q, Kuwahara H, Yin L, Carraway KL, 3rd, Kufe D. The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and beta-catenin. J Biol Chem. 2001;276:35239–35242. doi: 10.1074/jbc.C100359200. [DOI] [PubMed] [Google Scholar]
- 11.Vermeer PD, Einwalter LA, Moninger TO, Rokhlina T, Kern JA, Zabner J, Welsh MJ. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature. 2003;422:322–326. doi: 10.1038/nature01440. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Liu D, Chen D, Kharbanda S, Kufe D. Human DF3/MUC1 carcinoma-associated protein functions as an oncogene. Oncogene. 2003;22:6107–6110. doi: 10.1038/sj.onc.1206732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raina D, Ahmad R, Joshi MD, Yin L, Wu Z, Kawano T, Vasir B, Avigan D, Kharbanda S, Kufe D. Direct targeting of the mucin 1 oncoprotein blocks survival and tumorigenicity of human breast carcinoma cells. Cancer Res. 2009;69:5133–5141. doi: 10.1158/0008-5472.CAN-09-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei X, Xu H, Kufe D. Human mucin 1 oncoprotein represses transcription of the p53 tumor suppressor gene. Cancer Res. 2007;67:1853–1858. doi: 10.1158/0008-5472.CAN-06-3063. [DOI] [PubMed] [Google Scholar]
- 15.Ren J, Agata N, Chen D, Li Y, Yu WH, Huang L, Raina D, Chen W, Kharbanda S, Kufe D. Human MUC1 carcinoma-associated protein confers resistance to genotoxic anticancer agents. Cancer Cell. 2004;5:163–175. doi: 10.1016/S1535-6108(04)00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu XF, Yang E, Li J, Xing PX. MUC1 cytoplasmic tail: a potential therapeutic target for ovarian carcinoma. Expert Rev Anticancer Ther. 2006;6:1261–1271. doi: 10.1586/14737140.6.8.1261. [DOI] [PubMed] [Google Scholar]
- 17.Khodarev NN, Pitroda SP, Beckett MA, MacDermed DM, Huang L, Kufe DW, Weichselbaum RR. MUC1-induced transcriptional programs associated with tumorigenesis predict outcome in breast and lung cancer. Cancer Res. 2009;69:2833–2837. doi: 10.1158/0008-5472.CAN-08-4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beatty PL, Narayanan S, Gariepy J, Ranganathan S, Finn OJ. Vaccine against MUC1 antigen expressed in inflammatory bowel disease and cancer lessens colonic inflammation and prevents progression to colitis-associated colon cancer. Cancer Prev Res (Phila) 2010;3:438–446. doi: 10.1158/1940-6207.CAPR-09-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butts C, Maksymiuk A, Goss G, Soulieres D, Marshall E, Cormier Y, Ellis PM, Price A, Sawhney R, Beier F, Falk M, Murray N. Updated survival analysis in patients with stage IIIB or IV non-small-cell lung cancer receiving BLP25 liposome vaccine (L-BLP25): phase IIB randomized, multicenter, open-label trial. J Cancer Res Clin Oncol. 2011;137:1337–1342. doi: 10.1007/s00432-011-1003-3. [DOI] [PubMed] [Google Scholar]
- 20.Lepisto AJ, Moser AJ, Zeh H, Lee K, Bartlett D, McKolanis JR, Geller BA, Schmotzer A, Potter DP, Whiteside T, Finn OJ, Ramanathan RK. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008;6:955–964. [PMC free article] [PubMed] [Google Scholar]
- 21.Ramanathan RK, Lee KM, McKolanis J, Hitbold E, Schraut W, Moser AJ, Warnick E, Whiteside T, Osborne J, Kim H, Day R, Troetschel M, Finn OJ. Phase I study of a MUC1 vaccine composed of different doses of MUC1 peptide with SB-AS2 adjuvant in resected and locally advanced pancreatic cancer. Cancer Immunol Immunother. 2005;54:254–264. doi: 10.1007/s00262-004-0581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramlau R, Quoix E, Rolski J, Pless M, Lena H, Levy E, Krzakowski M, Hess D, Tartour E, Chenard MP, Limacher JM, Bizouarne N, Acres B, Halluard C, Velu T. A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV non-small cell lung cancer. J Thorac Oncol. 2008;3:735–744. doi: 10.1097/JTO.0b013e31817c6b4f. [DOI] [PubMed] [Google Scholar]
- 23.Gulley JL, Arlen PM, Tsang KY, Yokokawa J, Palena C, Poole DJ, Remondo C, Cereda V, Jones JL, Pazdur MP, Higgins JP, Hodge JW, Steinberg SM, Kotz H, Dahut WL, Schlom J. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin Cancer Res. 2008;14:3060–3069. doi: 10.1158/1078-0432.CCR-08-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohebtash M, Tsang KY, Madan RA, Huen NY, Poole DJ, Jochems C, Jones J, Ferrara T, Heery CR, Arlen PM, Steinberg SM, Pazdur M, Rauckhorst M, Jones EC, Dahut WL, Schlom J, Gulley JL. A pilot study of MUC-1/CEA/TRICOM poxviral-based vaccine in patients with metastatic breast and ovarian cancer. Clin Cancer Res. 2011;17:7164–7173. doi: 10.1158/1078-0432.CCR-11-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morse MA, Niedzwiecki D, Marshall JL, Garrett C, Chang DZ, Aklilu M, Crocenzi TS, Cole DJ, Dessureault S, Hobeika AC, Osada T, Onaitis M, Clary BM, Hsu D, Devi GR, Bulusu A, Annechiarico RP, Chadaram V, Clay TM, Lyerly HK (2013) A randomized phase II study of immunization with dendritic cells modified with poxvectors encoding CEA and MUC1 compared with the same poxvectors plus GM-CSF for resected metastatic colorectal cancer. Ann Surg doi:10.1097/SLA.0b013e318292919e [DOI] [PMC free article] [PubMed]
- 26.Heukamp LC, van der Burg SH, Drijfhout JW, Melief CJ, Taylor-Papadimitriou J, Offringa R. Identification of three non-VNTR MUC1-derived HLA-A*0201-restricted T-cell epitopes that induce protective anti-tumor immunity in HLA-A2/K(b)-transgenic mice. Int J Cancer. 2001;91:385–392. doi: 10.1002/1097-0215(200002)9999:9999<::AID-IJC1051>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 27.Grey HM, Ruppert J, Vitiello A, Sidney J, Kast WM, Kubo RT, Sette A. Class I MHC-peptide interactions: structural requirements and functional implications. Cancer Surv. 1995;22:37–49. [PubMed] [Google Scholar]
- 28.Terasawa H, Tsang KY, Gulley J, Arlen P, Schlom J. Identification and characterization of a human agonist cytotoxic T-lymphocyte epitope of human prostate-specific antigen. Clin Cancer Res. 2002;8:41–53. [PubMed] [Google Scholar]
- 29.Tsang KY, Palena C, Gulley J, Arlen P, Schlom J. A human cytotoxic T-lymphocyte epitope and its agonist epitope from the nonvariable number of tandem repeat sequence of MUC-1. Clin Cancer Res. 2004;10:2139–2149. doi: 10.1158/1078-0432.CCR-1011-03. [DOI] [PubMed] [Google Scholar]
- 30.Mitchell MS, Lund TA, Sewell AK, Marincola FM, Paul E, Schroder K, Wilson DB, Kan-Mitchell J. The cytotoxic T cell response to peptide analogs of the HLA-A*0201-restricted MUC1 signal sequence epitope, M1.2. Cancer Immunol Immunother. 2007;56:287–301. doi: 10.1007/s00262-006-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madan RA, Mohebtash M, Arlen PM, Vergati M, Rauckhorst M, Steinberg SM, Tsang KY, Poole DJ, Parnes HL, Wright JJ, Dahut WL, Schlom J, Gulley JL. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:501–508. doi: 10.1016/S1470-2045(12)70006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152:163–175. [PubMed] [Google Scholar]
- 33.Nijman HW, Houbiers JG, Vierboom MP, van der Burg SH, Drijfhout JW, D’Amaro J, Kenemans P, Melief CJ, Kast WM. Identification of peptide sequences that potentially trigger HLA-A2.1-restricted cytotoxic T lymphocytes. Eur J Immunol. 1993;23:1215–1219. doi: 10.1002/eji.1830230603. [DOI] [PubMed] [Google Scholar]
- 34.Tsang KY, Zaremba S, Nieroda CA, Zhu MZ, Hamilton JM, Schlom J. Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. J Natl Cancer Inst. 1995;87:982–990. doi: 10.1093/jnci/87.13.982. [DOI] [PubMed] [Google Scholar]
- 35.Hogan KT, Shimojo N, Walk SF, Engelhard VH, Maloy WL, Coligan JE, Biddison WE. Mutations in the alpha 2 helix of HLA-A2 affect presentation but do not inhibit binding of influenza virus matrix peptide. J Exp Med. 1988;168:725–736. doi: 10.1084/jem.168.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cereda V, Poole DJ, Palena C, Das S, Bera TK, Remondo C, Gulley JL, Arlen PM, Yokokawa J, Pastan I, Schlom J, Tsang KY. New gene expressed in prostate: a potential target for T cell-mediated prostate cancer immunotherapy. Cancer Immunol Immunother. 2010;59:63–71. doi: 10.1007/s00262-009-0723-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kufe D. Oncogenic function of the MUC1 receptor subunit in gene regulation. Oncogene. 2010;29:5663–5666. doi: 10.1038/onc.2010.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9:874–885. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin L, Kharbanda S, Kufe D. MUC1 oncoprotein promotes autophagy in a survival response to glucose deprivation. Int J Oncol. 2009;34:1691–1699. doi: 10.3892/ijo_00000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fessler SP, Wotkowicz MT, Mahanta SK, Bamdad C. MUC1* is a determinant of trastuzumab (Herceptin) resistance in breast cancer cells. Breast Cancer Res Treat. 2009;118:113–124. doi: 10.1007/s10549-009-0412-3. [DOI] [PubMed] [Google Scholar]
- 41.Kharbanda A, Rajabi H, Jin C, Raina D, Kufe D. MUC1-C oncoprotein induces tamoxifen resistance in human breast cancer cells. Mol Cancer Res. 2013 doi: 10.1158/1541-7786.MCR-12-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uchida Y, Raina D, Kharbanda S, Kufe D. Inhibition of the MUC1-C oncoprotein is synergistic with cytotoxic agents in the treatment of breast cancer cells. Cancer Biol Ther. 2013;14:127–134. doi: 10.4161/cbt.22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lacunza E, Baudis M, Colussi AG, Segal-Eiras A, Croce MV, Abba MC. MUC1 oncogene amplification correlates with protein overexpression in invasive breast carcinoma cells. Cancer Genet Cytogenet. 2010;201:102–110. doi: 10.1016/j.cancergencyto.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 44.MacDermed DM, Khodarev NN, Pitroda SP, Edwards DC, Pelizzari CA, Huang L, Kufe DW, Weichselbaum RR. MUC1-associated proliferation signature predicts outcomes in lung adenocarcinoma patients. BMC Med Genomics. 2010;3:16. doi: 10.1186/1755-8794-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pitroda SP, Khodarev NN, Beckett MA, Kufe DW, Weichselbaum RR. MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proc Natl Acad Sci U S A. 2009;106:5837–5841. doi: 10.1073/pnas.0812029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajabi H, Joshi MD, Jin C, Ahmad R, Kufe D. Androgen receptor regulates expression of the MUC1-C oncoprotein in human prostate cancer cells. Prostate. 2011;71:1299–1308. doi: 10.1002/pros.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajabi H, Ahmad R, Jin C, Joshi MD, Guha M, Alam M, Kharbanda S, Kufe D. MUC1-C oncoprotein confers androgen-independent growth of human prostate cancer cells. Prostate. 2012;72:1659–1668. doi: 10.1002/pros.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banerjee S, Mujumdar N, Dudeja V, Mackenzie T, Krosch TK, Sangwan V, Vickers SM, Saluja AK. MUC1c regulates cell survival in pancreatic cancer by preventing lysosomal permeabilization. PLoS ONE. 2012;7:e43020. doi: 10.1371/journal.pone.0043020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roy LD, Sahraei M, Subramani DB, Besmer D, Nath S, Tinder TL, Bajaj E, Shanmugam K, Lee YY, Hwang SI, Gendler SJ, Mukherjee P. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene. 2011;30:1449–1459. doi: 10.1038/onc.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin L, Wu Z, Avigan D, Rosenblatt J, Stone R, Kharbanda S, Kufe D. MUC1-C oncoprotein suppresses reactive oxygen species-induced terminal differentiation of acute myelogenous leukemia cells. Blood. 2011;117:4863–4870. doi: 10.1182/blood-2010-10-296632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kohlgraf KG, Gawron AJ, Higashi M, VanLith ML, Shen X, Caffrey TC, Anderson JM, Hollingsworth MA. Tumor-specific immunity in MUC1.Tg mice induced by immunization with peptide vaccines from the cytoplasmic tail of CD227 (MUC1) Cancer Immunol Immunother. 2004;53:1068–1084. doi: 10.1007/s00262-004-0557-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sivinski CL, Kohlgraf KG, VanLith ML, Morikane K, Tempero RM, Hollingsworth MA. Molecular requirements for CD8-mediated rejection of a MUC1-expressing pancreatic carcinoma: implications for tumor vaccines. Cancer Immunol Immunother. 2002;51:327–340. doi: 10.1007/s00262-002-0277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.VanLith ML, Kohlgraf KG, Sivinski CL, Tempero RM, Hollingsworth MA. MUC1-specific anti-tumor responses: molecular requirements for CD4-mediated responses. Int Immunol. 2002;14:873–882. doi: 10.1093/intimm/dxf053. [DOI] [PubMed] [Google Scholar]
- 54.Derby M, Alexander-Miller M, Tse R, Berzofsky J. High-avidity CTL exploit two complementary mechanisms to provide better protection against viral infection than low-avidity CTL. J Immunol. 2001;166:1690–1697. doi: 10.4049/jimmunol.166.3.1690. [DOI] [PubMed] [Google Scholar]
- 55.Hodge JW, Chakraborty M, Kudo-Saito C, Garnett CT, Schlom J. Multiple costimulatory modalities enhance CTL avidity. J Immunol. 2005;174:5994–6004. doi: 10.4049/jimmunol.174.10.5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oh S, Hodge JW, Ahlers JD, Burke DS, Schlom J, Berzofsky JA. Selective induction of high avidity CTL by altering the balance of signals from APC. J Immunol. 2003;170:2523–2530. doi: 10.4049/jimmunol.170.5.2523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.