

Abstract
Giant cell arteritis (GCA) is the most common idiopathic systemic vasculitis in persons aged 50 years or greater. Treatment options for GCA, to-date, have been limited and have consisted primarily of glucocorticoids. Significant advances in the understanding of the genetic and cellular mechanisms in GCA are leading to identification of potential pathogenic targets. The recent success of interleukin-6 blockade in the treatment of GCA has opened the landscape to targeted biologic therapy. T cells, particularly T helper 1 and T helper 17 cell lineages have been identified as key inflammatory cells in both active and chronic vascular inflammatory lesions. Therapeutic agents, including abatacept and ustekinumab, which can impede both vasculitogenic cell lines are of particular interest. Inhibition of signalling pathways, including the janus kinase-signal tranducers and activation of transcription (JAK-STAT) and Notch pathways are evolving options. Tocilizumab has shown clear benefit in both newly diagnosed and relapsing patients with GCA and approval of this medication for treatment of GCA has led to rapid incorporation into treatment regimens. More information is required to understand the long-term outcomes of tocilizumab and other investigational targeted therapeutics in the treatment of GCA.
Electronic supplementary material
The online version of this article (10.1186/s41927-017-0004-5) contains supplementary material, which is available to authorized users.
Keywords: Vasculitis, Giant cell arteritis, Pathogenesis, Therapeutics, Biologics
Background
Giant cell arteritis (GCA) is the most common idiopathic systemic vasculitis in persons aged 50 years or older [1] and demonstrates a predilection for involvement of the aorta and its primary branches. The clinical presentation of GCA is variable and is based on the distribution of vascular involvement with most patients demonstrating classical cranial manifestations (headache, scalp tenderness, jaw claudication, vision loss) while others present primarily with constitutional and/or musculoskeletal symptoms. Less commonly, arterial occlusive disease of the extremities leads to symptoms of vascular claudication. Histopathologic examination of a temporal artery biopsy (TAB) specimen remains the gold-standard method of diagnosis, although vascular imaging studies are increasingly used in conjunction with, or instead of, an invasive biopsy. The aetiology and pathogenesis underscoring the development of GCA are still incompletely understood. Nevertheless, several advances in genetic and immunology research have led to a greater insight into the pathomechanisms that initiate and sustain vascular inflammation in GCA. Despite the availability of effective biologic agents for other autoimmune conditions, the treatment options for GCA, to-date, have been limited. Although glucocorticoids (GC) have been the standard-of-care for over six decades, treatment is not curative and is associated with significant morbidity. Therefore, the ongoing identification of key immune mediators in disease pathogenesis is encouraging the pursuit of clinical trials with targeted therapeutic agents. The aim of this article is to summarize the fundamental pathogenic mechanisms and new potential therapeutic options for the treatment of GCA. In order to accomplish this, we searched PubMed for the following search terms: “giant cell arteritis”, “large vessel vasculitis”, “temporal arteritis”, “arteritis” and “vasculitis”. Publications from the past 10 years were analyzed for pathogenic and therapeutic studies. Case reports were not included, unless providing unique insights into pathogenic pathways. The date of the last search was 1 July 2017.
Main text
Genetic associations
Several independent studies have implicated associations between GCA susceptibility and certain human leukocyte antigen (HLA) class I and class II alleles. In particular, carriage of HLA-DRB1*0401 and DRB1*0404 haplotypes have been consistently identified in multiple GCA cohorts [2–7]. A large-scale, international, genome-wide association study has further reinforced the importance of the HLA class II region [8]. Comparison of 2134 patients with GCA to 9125 controls demonstrated strong independent signals associated with conferring predisposition to GCA located in the HLA regions between HLA-DRA and HLA-DRB1 as well as between HLA-DQA1 and HLA-DQA2 [8]. Over-representation of these HLA class II genes strongly suggests that GCA is mediated by an antigen-driven immune response.
While HLA class II genetic factors have demonstrated the strongest association with risk of GCA development, several single nucleotide polymorphism risk signals in loci among non-HLA regions have also been recognized (Table 1). However, it should be noted that most studies evaluating genetic associations in GCA have included small groups of patients and will require validation in larger patient cohorts. Nevertheless, the wide array of potentially involved cellular pathways associated with GCA susceptibility underscores the polygenic nature and complex immunopathogenesis employing both the innate and adaptive immune response in this condition.
Table 1.
Non-HLA genetic loci associated with giant cell arteritis
| Non-HLA Locus | Function |
|---|---|
| Sample size ≥ 1000a | |
| Plasminogen (PLG) [8] | Lymphocyte recruitment, wound healing, fibrinolysis, angiogenesis |
| Prolyl 4-hydroxylase subunit alpha 2 (P4HA2) [8] | Collagen biosynthesis, folding of procollagen chains, hypoxia response |
| Tyrosine phosphatase non-receptor type 22 (PTPN22) [102] | Regulation of T and B cell receptor signaling |
| Interleukin-12B [8] | Th1 differentiation |
| Interleukin-17A ([103]; [104]) | Th17 lymphocyte differentiation and maintenance |
| Interleukin-33 [105] | Th2 lymphocyte and mast cell activation, endothelial cell activation |
| Sample size ≥ 500 | |
| NLR family pyrin domain containing 1 (NLRP1) [106] | Inflammasome assembly, activation of proinflammatory cytokines IL-1β, IL-18, IL-33 |
| Sample size ≥ 250 | |
| CC chemokine receptor 5 (CCR5) [107] | Proinflammatory chemokine activating cellular chemotaxis of macrophages, T lymphocytes, dendritic cells |
| Vascular endothelial growth factor (VEGF) ([108]; [109]; [110]) | Neoangiogenesis, vascular remodeling |
| Interleukin-6 ([110]; [111]; [112]) | Pleotropic pro-inflammatory cytokine |
| Sample size < 250 | |
| Endothelial nitric oxide synthase (eNOS) ([113]; [114]) | Synthesis of nitric oxide; regulation of endothelial cell vascular tone, cellular proliferation, platelet aggregation, and leukocyte adhesion |
| Tumor necrosis factor-a2 (TNFa2) [115] | Pleotropic pro-inflammatory cytokine |
| Interleukin-10 [116] | Regulation of Th1 and Th2 immunity, skews immune response to Th2 phenotype by inhibition of IL-12 production |
| Interleukin-18 [117] | Th1 differentiation |
aIf more than one study listed, then the sample size refers to the combined total of patients evaluated
Infection
Isolation of identical T cell clones from different vasculitic sites suggests a response to a specific antigenic stimulus [9] and studies have proposed that arterial wall dendritic cells may be activated by environmental infectious agents or autoantigens [10]. Several microorganisms have been suggested as a possible infectious trigger including Chlamydia pneumoniae [11, 12], Mycoplasma pneumoniae [13], Burkholderia pseudomallei [14], parvovirus B19 [15, 16], herpes simplex virus [17] and Ebstein-Barr virus [18]. Although infection-induced autoimmunity leading to loss of self-tolerance through mechanisms of molecular mimicry, bystander T-cell activation and epitope spreading is plausible, direct evidence of such remains elusive. Indeed, attempts to identify pathologic organisms in temporal artery biopsy specimens have produced inconsistent results for any specific causal infectious agent [15, 19–21].
Varicella zoster virus (VZV) has received recent focus as a potential associated infectious aetiology. The presence of VZV antigen by immunohistochemistry was identified in 68 of 93 (73%) patients with histologically confirmed GCA and 45 of 70 (64%) patients with biopsy-negative GCA, compared to only 11 of 49 (22%) normal controls [22]. The same investigators identified VZV DNA by PCR amplification in a blinded analysis in 3 of 3 TAB-positive GCA patients and 4 of 6 TAB-negative GCA patients [23]. These investigators have proposed that the VZV is transported along the afferent nerves to the temporal artery inciting an inflammatory process resulting in arteritis. Consequently, Gilden et al. have advocated for use of the antiviral medication acyclovir in the treatment of patients with active or refractory GCA [24]. The presence of VZV as a causative agent for GCA, however, has not been substantiated by other groups. Muratore and colleagues evaluated 79 formalin-fixed and fresh-frozen temporal artery biopsies (34 TAB-positive GCA, 15 TAB-negative GCA, and 30 controls) by immunohistochemistry and PCR analysis [25]. Only 1 of 34 patients with TAB-positive GCA had evidence of VZV antigen whereas VZV antigen was not detected among any of the TAB-negative GCA patients or controls. Furthermore, VZV DNA was not found in any of the formalin-fixed or fresh-frozen TAB samples. In a recent prospective study, Procop and colleagues similarly did not identify VZV DNA from surgically sterile temporal artery and thoracic aortic samples from patients with large-vessel vasculitis [26].
In addition to histopathology evaluations, population level studies have failed to show a causal role of VZV in GCA. In comparing 204 cases of incident GCA diagnosed between 1950 and 2004 to 408 matched controls from the same geographic location, Schäfer and colleagues found no associated risk of incident VZV among patients with GCA compared to the general population [27]. Rhee et al. performed a population-based case-control study evaluating a larger sample of patients with GCA (n = 4559) and controls (n = 22,795) and similarly concluded there was minimal-to-no association of clinically overt VZV with GCA [28]. At current, conclusive evidence does not support direct infection with VZV as a causal process for the development of GCA and the use of acyclovir as an adjunct to, or in lieu of, immunosuppression is unsubstantiated and not recommended.
Innate immune system
Vascular dendritic cells
Although the specific immunostimulatory trigger(s) is unknown, the immunopathology of GCA appears to originate from a dysregulated interaction between the vessel wall and both the innate and adaptive immune systems [29, 30]. Unlike small vessels which rely primarily on oxygen through luminal diffusion, large vessels require a microvascular network (vasa vasorum) to distribute oxygen to the media-adventia vascular cell layers. Arteries with vasa vasorum contain vascular dendritic cells (vasDCs) at the media-advential border where they are thought to participate in immune surveillance. In normal arteries, vasDCs are immature and lack the capacity to stimulate T cells [31] allowing arteries to maintain immune privilege and self-tolerance. In vasculitic lesions immune privilege is lost and vasDCs become activated via Toll-like receptors (TLRs), redistributing throughout the vessel wall [32]. Activated vasDCs are able to attract and activate T lymphocytes and macrophages through production of specific chemokine and cytokine signatures, providing a microenvironment necessary for initiating and sustaining arterial inflammation and granuloma formation [29]. This central role of vasDCs in arteritis has been demonstrated using a model in which human temporal arteries were engrafted into immunodeficient mice, following which targeted depletion of CD83+ vasDCs resulted in marked reduction of observed vasculitis. Due to their role early in the disease process, prevention of initial vasDCs activation is an unlikely therapeutic target. However therapies addressing persistently activated vasDCs remain a viable option.
Macrophages
GCA is a granulomatous vasculitis and multinucleated giant cells, a key feature of macrophage involvement, are a considered a pathognomonic hallmark of arterial lesions. Giant cell formation is present in approximately 50% of positive temporal artery biopsies. Macrophages recruited by activated vasDCs and T lymphocytes infiltrate the arterial wall through the vasa vasorum and further differentiate into M1 and M2 phenotypes according to the arterial microenvironment. In the adventitia, activated M1 macrophages primarily secrete proinflammatory cytokines (IL-1 and IL-6) [33] while M1 macrophages in the medial layer degrade the arterial matrix through secretion of matrix metalloproteinases and damage vascular smooth muscle cells (VSMCs) and endothelial cells (ECs) through local oxidative stress mechanisms [34]. On the contrary, M2 macrophages co-localize to the intima-media border and produce proangiogenic growth factors (vascular endothelial growth factor, fibroblast growth factor and platelet-derived growth factor) which results in myofibroblast proliferation, relocation, and the marked thickening of the arterial intima.
The emerging paradigm of personalized medicine is encouraging development of nanoparticle-based theranostic agents, which integrate therapeutic and imaging functionalities. These molecules are being explored in cancer and inflammatory diseases [35]. Selective targeting of specific macrophage phenotypes may overcome the toxicity associated with macrophage inhibition through non-selective cytotoxic drugs. Preclinical models for imaging and treatment of pathogenic macrophages have shown initial promise [36]; however, further advances in this evolving field will be needed prior to consideration of selective macrophage ablation, inhibition, or phenotype modulation in GCA.
Adaptive immune system
T lymphocytes
Normal arteries are devoid of CD4+ T cell infiltration. In GCA, T cells are recruited via chemokines secreted by activated vasDCs, enter the arterial layer initially through the vasa vasorum, polarize into effector cell types and infiltrate the arterial layers. Two distinct CD4+ T effector cell subtypes have been identified as key regulators in vasculitic lesions of GCA; type 17 helper T cells (Th17) and type 1 helper T cells (Th1) [37]. These T cell lineages are likely to be stimulated by independent signals from distinct antigen presenting cells (APC) with Th17 cells dependent on IL-1β, IL-6, IL-21 and IL-23, while the Th1 pathway requires APCs secreting IL-12 and IL-18 [30, 37].
Studies conducted in patients with active, untreated GCA show that both Th1 and Th17 lineages are present in the inflammatory arterial infiltrates and are expanded in the peripheral circulation [38, 39]. The Th17 pathway appears to be very responsive to treatment and glucocorticoids (GCs) rapidly reduce the Th17 effector cytokine production of IL-1, IL-6, IL-17 and IL-23 [30, 38] with simultaneous depletion of both circulating and tissue infiltrative Th17 cells.
The rapid decline in these cytokines upon GC initiation contributes to the prompt decrease of systemic inflammatory features. Despite the effective reduction of the Th17 pathway, a Th1 cell response persists, both in blood samples and arterial specimens from patients treated with high-dose GC [38]. The Th1 cytokine signature identified in chronic vasculitis in GCA is associated with production of IL-2 and interferon-gamma (IFN- γ) and is poorly susceptible to GCs. It is likely that persistence of vascular Th1 cellular infiltrates despite prolonged courses of GCs is responsible for the chronicity and relapsing nature observed in GCA [30].
In addition to Th1/Th17 effector T cells, regulatory T cells (Tregs) appears to have an important role in GCA pathogenesis. Tregs, which are tasked with maintaining tolerance and prevention of autoimmunity, are decreased in the blood and arterial lesions of patients with GCA [39]. Compared to healthy controls, Tregs in patients with GCA appear to be defective and potentially pathogenic, exhibiting decreased proliferation and increased production of IL-17 [40]. While GC appear to be ineffective in modifying Treg dysfunction in GCA [39], IL-6 blockade was recently shown to revert the Treg abnormalities detected in active GCA [40].
Further confirmation that T cells are implicated in the development of large-vessel vasculitis is provided by the demonstration of T cell immune check point dysregulation in GCA. Checkpoint molecules, particularly cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death-1 (PD-1), play a key role in downregulating T cell activation, maintaining self-tolerance and limiting autoimmunity. T cell activation requires antigen recognition through the T-cell receptor as well as a second signal delivered by co-stimulatory molecules. CTLA-4 and its homologous protein, cluster of differentiation 28 (CD28), are expressed on activated T cells and participate in the co-stimulatory check point. Binding of CD28 to the CD80/86 molecules on APCs results in T cell activation and clonal expansion. On the contrary, down regulation of the immune response occurs through binding of CTLA4 to CD80/86, which results in T cell anergy. Checkpoint blockade immunotherapy is now being utilized in cancer treatments [41, 42]. Competitive inhibition of CTLA-4 by the immunostimulatory medication, ipilimumab, increases cytokine production, leads to persistent T cell activation and enhances T cell mediated immune responses to tumors [43]. However, use of ipilimumab has been associated with development of autoimmune conditions and organ-specific inflammation, including drug-induced giant cell arteritis [44].
The receptor molecule PD-1 provides inhibitory signals by binding to programmed cell death ligand 1 and 2 (PD-L1 and PD-L2), resulting in T cell anergy, apoptosis, or polarization to Tregs [45]. Recent transcriptome analysis of temporal arteries positive for GCA has demonstrated an inefficiency of the PD-1/PD-L1 checkpoint. Specifically, inflamed arteries of patients with GCA lack the inhibitory ligand PD-L1 and are enriched for PD-1–expressing T cells [46]. PD-1 blockade in a mouse model with engrafted temporal arteries worsened vascular inflammation, promoted PD-1+ effector T cells and increased effector T cell cytokines IL-17, IL-21, and IFN-γ [46]. These pre-clinical findings have been corroborated with recent reports of GCA developing in patients treated with pembrolizumab, a monoclonal antibody directed against PD-1 [47, 48].
B cells
Currently, the role of B cells in GCA is not fully understood. While plasma cells have been observed in positive temporal artery specimens in up to 83% of cases, the functional role of these cells remains indeterminate [49]. Associations with humoral immunity have been suggested by the presence of auto-antibodies directed against cardiolipin [50], ferritin [51, 52], endothelial cells [53] and recently 14–3-3 proteins [54]. However detection of auto-antibodies overall lacks specificity, has not been universally replicated, and is of unknown significance in the pathogenesis of GCA.
Although a major immunologic role of B cells is the production of antibodies, B cells can also regulate T cell responses in auto-immunity through the secretion of both pro-inflammatory (TNF-α and IL-6) and anti-inflammatory (IL-10) cytokines [55]. In a prospective study, patients with active GCA were observed to have low levels of circulating B cells, particularly IL-6 secreting effector B cells, with a return to near normal levels during GC-induced remission [55]. It is hypothesized that the B cells are distributed into secondary lymphoid organs and/or inflamed tissues and then on normalization of the inflammatory process return to the peripheral blood. However, the reservoir for B cells during active disease is incompletely known and remains an area of ongoing research.
One potential location for B cell redistribution during disease is within artery tertiary lymphoid organs (ATLO). Ciccia and colleagues evaluated temporal artery biopsies from fifty patients with active, untreated GCA and found distinct artery tertiary lymphoid organ structures in the medial layer among 60% of patients with biopsies positive for GCA [56]. These structures were organized into specific T cell areas, B cell follicles and associated with a network of follicular dendritic cells. Compared to patients without ATLOs and healthy controls, arteries with ATLOs had a high level of B cell activating factor (BAFF) and a proliferation inducing ligand (APRIL), proteins which are strongly correlated with the differentiation and proliferation of B cells. Production of these proteins was primarily driven by endothelial cells and vascular smooth muscle cells highlighting an interaction between myointimal cells and B cells, an area that requires further exploration and confirmation [56].
Although a potential role of B cells exists in the pathogenesis of GCA, the efficacy of B cell depletion is largely unknown. Rituximab, an anti-CD20 monoclonal antibody, has only been reported in two patients with GCA with observed benefit [57, 58]. Further investigation of this agent in prospective trials is needed before considering as a viable treatment option in GCA.
Endothelial cells
Endothelial cells, the interface between the circulating blood and vessel wall, are active participants in the regulation of inflammation and angiogenesis. The intercellular adhesion molecules expressed on endothelial cells mediate leukocyte trafficking, migration, and development of inflammatory infiltrates. Increased expression of endothelin-1 and endothelin-B receptor immunoreactivity has been seen in higher concentration in the medial layer of temporal arteries from patients with GCA compared to controls and this immunoreactivity has been observed to correlate with the degree of systemic inflammation [59]. In addition, the endothelin system is increased at the protein level in temporal artery lesions in patients with GCA, creating a microvascular environment that is predisposed to develop ischemic complications, which is not immediately abrogated by glucocorticoid administration [60]. Indeed, upregulation of endothelin-1 in GCA lesions promotes migration of vascular smooth muscle cells to migrate towards the intima, leading to development of intimal hyperplasia and resulting in vascular occlusion [61]. Blockade of endothelin receptors using macitentan, a dual endothelin-A/endothelin-B receptor antagonist, has been shown to reduce vascular smooth muscle proliferation in biopsy-proven GCA and may prove to be a promising future adjunct in the treatment of GCA [62].
Novel treatments
For over six decades, treatment with high-dose GCs has been the mainstay for both induction to remission and maintenance therapy in GCA. Unfortunately, GCs contribute substantial toxicity and GC-associated adverse events are nearly universal [63, 64]. In addition, despite adequate treatment with GCs, relapses are frequent [65] and chronic arterial inflammation can persist despite clinical and biochemical remission [38, 49]. Therefore, a great unmet need has existed for safe and effective GC-sparing agents for both induction and maintenance therapy in GCA.
Methotrexate was evaluated in 3 small clinical trials for treatment of GCA, and results were variable. According to expert consensus, methotrexate has been generally recommended as a second-line agent after GC in patients with relapsing or severely active disease or in patients with high-risk for GC-associated adverse events [66]. However, an individual patient data meta-analysis of the three clinical trials evaluating methotrexate found only a modest benefit in decreasing relapse risk and reducing GC exposure. Moreover, the effect of methotrexate on reducing relapse risk only became significant after about 48 weeks of treatment, and GC-related adverse effects were not diminished [67]. Limited to no benefit has been observed among other oral conventional immunosuppressive agents including azathioprine [68], leflunomide [69, 70] and mycophenolate mofetil [71].
Due to the limited success of conventional immunosuppressive agents, targeted therapeutics have gained greater interest and investigation for control of GCA. Early reports of increased expression of tumor necrosis factor (TNF)-alpha in temporal artery specimens among patients with GCA [72] and dramatic benefit of TNF-inhibitors in other autoimmune diseases (e.g. rheumatoid arthritis) led to evaluation of TNF-alpha blockade in this condition. Unfortunately placebo-controlled trials utilizing TNF-inhibition with infliximab [73], etanercept [74], and adalimumab [75] have failed to show significant therapeutic efficacy in patients with GCA and did not result in significant reduction of GC use or adverse events. Despite the inadequate results of TNF-alpha inhibition, several other targeted therapeutics are being investigated (Fig. 1).
Fig. 1.
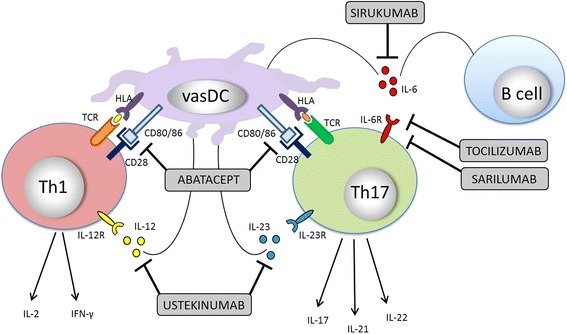
Vascular dendritic cells (vasDC) activate T helper 1 (Th1) and T helper 17 (Th17) cells by presenting antigen in the context of human leukocyte antigen (HLA) molecules. Abatacept can bind to CD80/86 and therefore block the required co-stimulatory signal required for T cell activation. Both B cells and vasDCs secrete interleukin (IL)-6 which stimulates Th17 cells via binding to its respective the T cell receptor (TCR). Sirukumab blocks soluble IL-6 whereas tocilizumab and sarilumab target the soluble and membrane-bound IL-6 receptor (IL-6R). IL-12 and IL-23 are secreted by vasDCs and antigen presenting cells and stimulate Th1 and Th17 cells, respectively. Ustekinumab binds the p40 subunits of both IL-12 and IL-23. Major effector cytokines produced by Th1 cells include IL-2 and interferon gamma (IFN-γ) and cytokines produced by Th17 cells include IL-17, IL-21 and IL-22
Interleukin-6 inhibitors
IL-6 is produced locally in the arterial vasculitic lesion [9], and is expressed in circulating monocytes from patients with GCA [33]. IL-6 is known to be a critical factor in the induction of acute-phase proteins, which are typically markedly elevated in patients with GCA and are used in clinical practice to monitor disease activity. Moreover, although IL-6 is highly elevated in untreated GCA, levels drop promptly with GC therapy. Indeed, IL-6 levels correlate with disease activity and can be a useful biomarker in evaluating disease activity [76, 77]. Tocilizumab (TCZ), a humanized anti IL-6 receptor antibody has shown efficacy in both newly diagnosed and relapsing patients in several small observational series [78–85] and two double-blind, placebo-controlled trials [86, 87].
In the first reported double-blind, placebo-controlled phase 2 trial evaluating TCZ in GCA, Villiger and colleagues randomized 30 patients with GCA (23 newly diagnosed, 7 relapsing) to receive prednisolone taper with intravenous tocilizumab (8 mg/kg/4 weeks) or prednisolone taper with placebo infusions [87]. Among the 20 patients receiving TCZ, complete remission at 12 weeks (85% vs. 40%) and relapse-free survival at 1 year (85% vs 20%) were significantly higher in the treatment group compared to placebo. On the contrary, serious adverse events were less frequent in the TCZ group (35% vs. 50%). Only one relapse was observed in the TCZ group and no relapses occurred after discontinuation of prednisolone in patients receiving TCZ.
The benefit and safety profile of TCZ has further been confirmed in the tocilizumab in GCA (GiACTA) trial [86]. GiACTA constitutes the largest international, multi-center, placebo-controlled trial in GCA to-date and the first to employ a blinded prednisone taper and a dual-assessor strategy to avoid bias created by knowledge of inflammatory markers [86]. In this phase 3 trial, Stone and colleagues recruited 251 patients (119 newly diagnosed, 132 relapsing) to one of four arms: weekly TCZ (162 mg, subcutaneous) plus a 26-week prednisone taper, every other week TCZ plus a 26-week prednisone taper, weekly placebo plus a 26-week prednisone taper, or weekly placebo plus a 52-week prednisone taper. Patients were diagnosed based on modifications to the 1990 American College of Rheumatology criteria for classification of GCA [88], which permitted inclusion of patients based on evidence of large-vessel vasculitis (LVV) on cross-sectional imaging. Whereas 62% of patients had a positive temporal biopsy, 37% of patients were ultimately enrolled on the basis of radiographic findings of LVV, regardless of temporal artery biopsy results.
The primary outcome, sustained remission at 52-weeks, was significantly higher among patients receiving weekly TCZ (56%) and every other week TCZ (53%) compared to the placebo groups (placebo plus 26-week prednisone taper, 14%; placebo plus 52-week prednisone taper, 18%; respectively). Disease flares occurred less frequently in patients receiving weekly TCZ (23%) and every other week TCZ (26%) compared to the 26-week (68%) and 52-week (49%) placebo arms. Over the 12-month trial, GC exposure in the treatment arms was approximately half compared to the placebo arms. Overall adverse events were frequent in all groups (92–98%); however, serious adverse events were lower among patients receiving weekly TCZ (15%) and every other week TCZ (14%) compared to placebo plus 26-week prednisone taper (22%) or 52-week prednisone taper (25%). Despite the accelerated GC taper, permanent vision loss did not occur in any arm and only one patient receiving every other week TCZ had transient visual loss, which recovered with glucocorticoids.
The landmark findings of TCZ safety and efficacy reported in GiACTA have led to the United States Food and Drug Administration to approve TCZ 162 mg administered subcutaneously weekly for the treatment of GCA; the first-ever medication to receive such distinction. The successful results of TCZ have prompted further investigation into additional novel anti-IL-6 molecules in this disease. A phase 3 trial utilizing sirukumab (human anti-IL-6 monoclonal antibody) is currently in progress (ClinicalTrials.gov Identifier NCT02531633) and an upcoming trial of sarilumab (anti-IL-6 receptor monoclonal antibody), is planned. The results of these studies will strongly influence whether additional candidate IL-6 blockers will be evaluated in GCA; including clazakizumab, olokizumab, and vobarilizumab. While the benefit of IL-6 blockade in treatment of GCA is apparent, 44–47% of patients receiving TCZ in GiACTA were still unable to reach sustained remission at 52 weeks [86]. In an earlier report, a patient with GCA who achieved apparent clinical remission on TCZ was noted to have evidence of persistent and active vascular inflammation at autopsy, despite normalization of inflammatory markers [85]. Active disease despite IL-6 blockade highlights that other immunologic pathomechanisms are present in ongoing inflammatory states in GCA and further investigation into other novel therapeutics is still needed.
Abatacept
Whereas the checkpoint inhibitor ipilimumab has led to the generation of arterial inflammation, inhibition of co-stimulatory T cell signalling via CTLA-4 is a possible target for controlling GCA. Abatacept is a fusion protein composed of the Fc region of the immunoglobulin IgG1 fused to the extracellular domain of CTLA-4 and blocks the co-stimulatory signal required for T cell activation. Langford and colleagues investigated the safety and efficacy of intravenous abatacept (10 mg/kg) in patients with newly diagnosed or relapsing GCA [89]. Following a remission induction phase, 41 patients in remission at week 12 were randomized to receive either placebo infusions or monthly abatacept. Relapse-free survival at 12 months, the primary outcome, was achieved by 48% on abatacept and 31% receiving placebo (p = 0.049). In addition, the duration of remission was on average 6 months longer in patients receiving abatacept compared to placebo. No difference in adverse events was observed. Although considered statistically and clinically significant findings, the effectiveness of abatacept in GCA needs to be confirmed in larger cohorts prior to incorporation of this agent in routine clinical care.
Ustekinumab
Tocilizumab, through blockade of IL-6R, mediates its effect largely on the Th17/Treg imbalance, without significant impact on the Th1 cellular pathway. On the contrary, ustekinumab, a monoclonal antibody directed against IL-12/23p40 has the potential to disrupt both Th1 (IL-12) and Th17 (IL-23) pathways. The effect of modulating the Th1/Th17/Treg imbalance has been demonstrated by Samson and colleagues in a refractory GCA patient treated with ustekinumab [90]. Compared to baseline, peripheral blood mononuclear cells evaluated after three injections of ustekinumab (45 mg weeks 0, 4, 16) demonstrated that Th1 and Th17 cells each fell by 50%, cytotoxic T lymphocytes reduced by 64%, and Tregs increased 5-fold. This dramatic improvement in T cell homeostasis is promising and the benefit of ustekinumab has been observed in a small open-label study. Conway and colleagues studied 12 patients with relapsing GCA and administered ustekinumab 90 mg at weeks 0, 4, and then every 12 weeks for a median of 8 months [91]. During the course of treatment GC requirements were noticeably reduced (median 23 mg prior, 5 mg after). Three patients were able to discontinue GC and eight patients were able to stop additional baseline immunosuppressives. No relapses were observed during treatment with ustekinumab but two of three patients experienced a flare following cessation of therapy. These findings demonstrate the potential efficacy of ustekinumab as a treatment option for GCA and a phase 2 trial to confirm these results is ongoing (ClinicalTrials.gov Identifier NCT02955147).
Janus kinase / signal transducers and activators of transcription inhibitors
The Janus kinase–signal transducers and activators of transcription (JAK-STAT) signalling pathway is involved in cellular regulation and has been implicated in the pathogenesis of several inflammatory and autoimmune conditions, including rheumatoid arthritis, inflammatory bowel disease, and psoriasis [92]. Ligand binding of immune relevant mediators (e.g. IL-2, IL-4, IL-6, IL-7, IL-9, IL-12, IL-15, IL-21, IL-23, IL-27, type 1 interferon, interferon-gamma) to their cell surface receptors leads to activation of associated JAKs [93]. The activated JAKs increase their kinase activity, recruit, bind and activate STATs. The STAT molecules then form hetero- or homo-dimers which translocate to the nucleus and induce transcription and expression of target genes, which in the context of pro-inflammatory cytokines can lead to increased inflammation and autoimmunity [94]. As a result, targeted intervention of the JAK-STAT signalling cascade is attractive and being investigated for several autoimmune and cancer disorders.
JAK-STAT signalling has been identified as having a potential role in sustaining vascular inflammation. In particular, STAT-1 signalling appears to regulate the activity of vasDCs as well as controlling T cell trafficking and retention of inflammatory T cells in the vascular lesions [95]. Hartmann and colleagues evaluated the presence of STAT-1 transcripts in an experimentally induced vasculitis of human temporal arteries grafted in immunodeficient mice and found STAT-1 abundantly expressed in arteritic tissue lesions. Interferon-gamma, the major inducer of STAT-1, was, 10-fold higher in patients affected by GCA compared to controls. In the experimental model, while high-dose GCs effectively suppressed the activity of vasDCs and reduced IL-6 in the lesional tissue, the Th1 cellular infiltrate was not affected. Conversely, administration of the JAK/STAT inhibitor, tofacitinib, effectively prevented Th1 cell accumulation in the vessel wall and reduced interferon-gamma production [95]. Compared to TCZ, which mediates its effect primarily through the IL-6/gp130/STAT3 signalling axis [96]; JAK inhibitors can exert control over multiple pathogenic cellular lineages by impeding the proinflammatory signal of more than one cytokine signalling pathway (Fig. 2). The benefit of JAK inhibition in patients with GCA is currently unknown but a phase 2 trial evaluating baricitinib (oral selective JAK1, JAK2 inhibitor) in patients with relapsing GCA is currently ongoing (ClinicalTrials.gov Identifier NCT03026504).
Fig. 2.
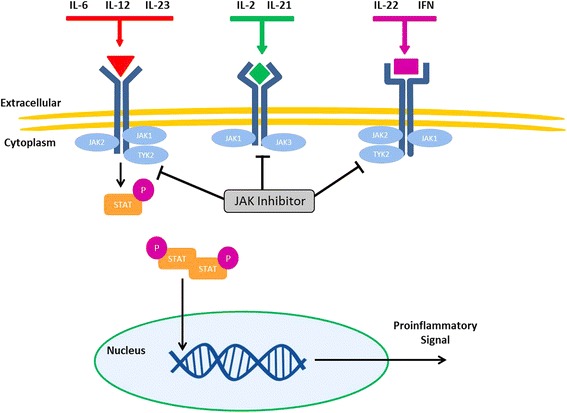
Effector cytokines can stimulate transcription of proinflammatory signals through the Janus kinase–signal transducers and activators of transcription (JAK-STAT) signalling pathway. Intracellular JAK and tyrosine kinase (TYK) proteins are activated when a ligand binds to its receptor which induces phosphorylation and activation, which in turn activate STAT proteins. The STAT proteins dimerize and translocate to the nucleus where they bind STAT-specific response elements in target gene promotors and regulate gene transcription; including proinflammatory signals. JAK inhibitors (e.g. baricitinib and tofacitinib) are able to abrogate signalling cascades from more than one effector cytokine pathway
Future treatment options
The Notch signalling pathway is critical for regulating cellular proliferation, differentiation, apoptosis, and homeostasis. Dysregulation of this highly preserved pathway has been associated with several malignancies and autoimmune conditions [97, 98]. Interaction between lymphocytes and vascular smooth muscles cells and endothelial cells involves the activity of the Notch pathway. Immunohistochemical and gene expression analysis of inflamed temporal arteries have identified overexpression of Notch receptors and their associated ligands (Jagged1, Delta1). Blockade of Notch signalling in a humanized vasculitis mouse model effectively depleted both Th1 and Th17 cells from vascular infiltrates [99]. In addition, it has been shown that systemic vascular endothelial growth factor (VEGF) can trigger aberrant Notch signalling through upregulation of the Notch ligand Jagged1 and induces pathogenic effector cell function via adventitial microvascular endothelial cells [100]. The arterial microvascular network appears to be essential in the recruitment of activated T cells and may therefore provide additional therapeutic targets. Multiple phase 1 and phase 2 trials evaluating different classes of Notch inhibition are underway in treatment of various forms of cancer, but Notch inhibition as of yet has not been attempted in patients with GCA.
The role of the pro-inflammatory cytokine IL-1-beta in GCA is uncertain; however, expression of IL-1-beta has been found in 60–80% of circulating monocytes in patients with untreated GCA and 20% of macrophages in temporal artery lesions produce this cytokine [33]. Limited information is available to determine the clinical efficacy of IL-1 blockade in patients with GCA but a small case series utilizing the IL-1 inhibitor, anakinra, showed benefit in three refractory cases [101]. A randomized, double-blind, placebo-controlled study evaluating gevokizumab (monoclonal antibody targeting IL-1 beta) for the treatment of patients with relapsing GCA is ongoing and has recruited 13 patients to date (EudraCT number 2013–002778-38).
Conclusion
Recent advances have provided a greater understanding of the pathomechanisms involved in GCA; however, further studies are required to fully elucidate the aetiology and pathogenesis of this inflammatory vasculopathy. The complex interaction of genetics, vascular factors and immunologic pathways in this disease is responsible for the variability in both the clinical presentation and response to immunosuppressive therapy. After decades of treating GCA almost exclusively with glucocorticoids, clinicians are finally able to reach for novel and targeted biologic agents. IL-6 is a key mediator in GCA and tocilizumab is the first agent to show a profound effect on disease control and GC reduction. This agent is quickly being incorporated into the treatment algorithm of both newly diagnosed and relapsing patients with GCA. More information is needed to understand tocilizumab’s effect on long-term outcomes and the optimal length of therapy required for maintenance in this disease. Abatacept and ustekinumab have shown preliminary evidence of potential efficacy in the treatment of GCA but confirmation in larger trials is needed before either is utilized in clinical care. The proposed benefit of JAK-STAT inhibition seen in preclinical studies of vasculitis is encouraging and the evaluation of baricitinib in patient trials is underway. The era of glucocorticoid monotherapy for GCA may be coming to a close, a most welcome prospect for patients with this condition.
Additional file
Reviewer reports and AU response to reviewers. (DOCX 16 kb)
Acknowledgements
We wish to thank those who reviewed the manuscript for their constructive comments (Additional file 1).
Funding
None.
Availability of data and materials
Not applicable.
Author’s contributions
Manuscript design/conception: MJK, KJW. Drafting of manuscript: MJK, KJW. Critical revision: MJK, KJW.
Approval of manuscript: MJK, KJW. Both authors read and approved the final manuscript.
Abbreviations
- APC
antigen presenting cell
- CTLA-4
cytotoxic T-lymphocyte associated protein-4
- GC
glucocorticoids
- GCA
giant cell arteritis
- HLA
human leukocyte antigen
- IL
interleukin
- JAK-STAT
Janus kinase–signal transducers and activators of transcription
- LVV
large vessel vasculitis
- PD-1
programmed cell death-1
- TAB
temporal artery biopsy
- TCZ
tocilizumab
- Th1
type 1 helper T cell
- Th17
type 17 helper T cell
- vasDC
vascular dendritic cell
- VZV
varicella zoster virus
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Matthew J. Koster, Email: koster.matthew@mayo.edu
Kenneth J. Warrington, Email: warrington.kenneth@mayo.edu
References
- 1.Weyand CM, Goronzy JJ. Clinical practice. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med. 2014;371(1):50–57. doi: 10.1056/NEJMcp1214825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Combe B, Sany J, Le Quellec A, Clot J, Eliaou JF: Distribution of HLA-DRB1 alleles of patients with polymyalgia rheumatica and giant cell arteritis in a Mediterranean population. J Rheumatol. 1998;25(1):94-98. [PubMed]
- 3.Dababneh A, Gonzalez-Gay MA, Garcia-Porrua C, Hajeer A, Thomson W, Ollier W. Giant cell arteritis and polymyalgia rheumatica can be differentiated by distinct patterns of HLA class II association. J Rheumatol. 1998;25(11):2140–2145. [PubMed] [Google Scholar]
- 4.Jacobsen S, Baslund B, Madsen HO, Tvede N, Svejgaard A, Garred P. Mannose-binding lectin variant alleles and HLA-DR4 alleles are associated with giant cell arteritis. J Rheumatol. 2002;29(10):2148–2153. [PubMed] [Google Scholar]
- 5.Martinez-Taboda VM, Bartolome MJ, Lopez-Hoyos M, Blanco R, Mata C, Calvo J, Corrales A, Rodriguez-Valverde V. HLA-DRB1 allele distribution in polymyalgia rheumatica and giant cell arteritis: influence on clinical subgroups and prognosis. Semin Arthritis Rheum. 2004;34(1):454–464. doi: 10.1016/j.semarthrit.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Salvarani C, Boiardi L, Mantovani V, Ranzi A, Cantini F, Olivieri I, Viggiani M, Bragliani M, Macchioni P. HLA-DRB1, DQA1, and DQB1 alleles associated with giant cell arteritis in northern Italy. J Rheumatol. 1999;26(11):2395–2399. [PubMed] [Google Scholar]
- 7.Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. The HLA-DRB1 locus as a genetic component in giant cell arteritis. Mapping of a disease-linked sequence motif to the antigen binding site of the HLA-DR molecule. J Clin Invest. 1992;90(6):2355–2361. doi: 10.1172/JCI116125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carmona FD, Vaglio A, Mackie SL, Hernandez-Rodriguez J, Monach PA, Castaneda S, Solans R, Morado IC, Narvaez J, Ramentol-Sintas M, et al. A genome-wide association study identifies risk alleles in Plasminogen and P4HA2 associated with Giant cell Arteritis. Am J Hum Genet. 2017;100(1):64–74. doi: 10.1016/j.ajhg.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weyand CM, Schonberger J, Oppitz U, Hunder NN, Hicok KC, Goronzy JJ. Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes. J Exp Med. 1994;179(3):951–960. doi: 10.1084/jem.179.3.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duhaut P, Bosshard S, Ducroix JP. Is giant cell arteritis an infectious disease? Biological and epidemiological evidence. Presse Med. 2004;33(19 Pt 2):1403–1408. doi: 10.1016/S0755-4982(04)98939-7. [DOI] [PubMed] [Google Scholar]
- 11.Ljungstrom L, Franzen C, Schlaug M, Elowson S, Viidas U. Reinfection with Chlamydia pneumoniae may induce isolated and systemic vasculitis in small and large vessels. Scand J Infect Dis Suppl. 1997;104:37–40. [PubMed] [Google Scholar]
- 12.Wagner AD, Gerard HC, Fresemann T, Schmidt WA, Gromnica-Ihle E, Hudson AP, Zeidler H. Detection of Chlamydia pneumoniae in giant cell vasculitis and correlation with the topographic arrangement of tissue-infiltrating dendritic cells. Arthritis Rheum. 2000;43(7):1543–1551. doi: 10.1002/1529-0131(200007)43:7<1543::AID-ANR19>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 13.Elling P, Olsson AT, Elling H. Synchronous variations of the incidence of temporal arteritis and polymyalgia rheumatica in different regions of Denmark; association with epidemics of Mycoplasma pneumoniae infection. J Rheumatol. 1996;23(1):112–119. [PubMed] [Google Scholar]
- 14.Koening CL, Katz BJ, Hernandez-Rodriguez J. Identification of a Burkholderia-like strain from temporal arteries of subjects with giant cell arteritis. Arthritis Rheum. 2012;64:S373. [Google Scholar]
- 15.Alvarez-Lafuente R, Fernandez-Gutierrez B, Jover JA, Judez E, Loza E, Clemente D, Garcia-Asenjo JA, Lamas JR. Human parvovirus B19, varicella zoster virus, and human herpes virus 6 in temporal artery biopsy specimens of patients with giant cell arteritis: analysis with quantitative real time polymerase chain reaction. Ann Rheum Dis. 2005;64(5):780–782. doi: 10.1136/ard.2004.025320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabriel SE, Espy M, Erdman DD, Bjornsson J, Smith TF, Hunder GG. The role of parvovirus B19 in the pathogenesis of giant cell arteritis: a preliminary evaluation. Arthritis Rheum. 1999;42(6):1255–1258. doi: 10.1002/1529-0131(199906)42:6<1255::AID-ANR23>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 17.Powers JF, Bedri S, Hussein S, Salomon RN, Tischler AS. High prevalence of herpes simplex virus DNA in temporal arteritis biopsy specimens. Am J Clin Pathol. 2005;123(2):261–264. doi: 10.1309/2996TT2CTLTKN0KT. [DOI] [PubMed] [Google Scholar]
- 18.Giardina A, Rizzo A, Ferrante A, Capra G, Triolo G, Ciccia F. Giant cell arteritis associated with chronic active Epstein-Barr virus infection. Reumatismo. 2013;65(1):36–39. doi: 10.4081/reumatismo.2013.36. [DOI] [PubMed] [Google Scholar]
- 19.Cooper RJ, D'Arcy S, Kirby M, Al-Buhtori M, Rahman MJ, Proctor L, Bonshek RE. Infection and temporal arteritis: a PCR-based study to detect pathogens in temporal artery biopsy specimens. J Med Virol. 2008;80(3):501–505. doi: 10.1002/jmv.21092. [DOI] [PubMed] [Google Scholar]
- 20.Helweg-Larsen J, Tarp B, Obel N, Baslund B. No evidence of parvovirus B19, Chlamydia pneumoniae or human herpes virus infection in temporal artery biopsies in patients with giant cell arteritis. Rheumatology (Oxford) 2002;41(4):445–449. doi: 10.1093/rheumatology/41.4.445. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez-Pla A, Bosch-Gil JA, Echevarria-Mayo JE, Rossello-Urgell J, Solans-Laque R, Huguet-Redecilla P, Stone JH, Vilardell-Tarres M. No detection of parvovirus B19 or herpesvirus DNA in giant cell arteritis. J Clin Virol. 2004;31(1):11–15. doi: 10.1016/j.jcv.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Nagel MA, White T, Khmeleva N, Rempel A, Boyer PJ, Bennett JL, Haller A, Lear-Kaul K, Kandasmy B, Amato M, et al. Analysis of Varicella-zoster virus in temporal arteries biopsy positive and negative for Giant cell Arteritis. JAMA Neurol. 2015;72(11):1281–1287. doi: 10.1001/jamaneurol.2015.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilden D, White T, Khmeleva N, Katz BJ, Nagel MA. Blinded search for varicella zoster virus in giant cell arteritis (GCA)-positive and GCA-negative temporal arteries. J Neurol Sci. 2016;364:141–143. doi: 10.1016/j.jns.2016.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilden D, Nagel MA. Varicella zoster virus and giant cell arteritis. Curr Opin Infect Dis. 2016;29(3):275–279. doi: 10.1097/QCO.0000000000000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muratore F, Croci S, Tamagnini I, Zerbini A, Bellafiore S, Belloni L, Boiardi L, Bisagni A, Pipitone N, Parmeggiani M, et al. No detection of varicella-zoster virus in temporal arteries of patients with giant cell arteritis. Semin Arthritis Rheum. 2017. [DOI] [PubMed]
- 26.Procop GW, Eng C, Clifford A, Villa-Forte A, Calabrese LH, Roselli E, Svensson L, Johnston D, Pettersson G, Soltesz E, et al. Varicella zoster virus and large vessel Vasculitis, the absence of an association. Pathog Immun. 2017;2(2):228–238. doi: 10.20411/pai.v2i2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schafer VS, Kermani TA, Crowson CS, Hunder GG, Gabriel SE, Ytterberg SR, Matteson EL, Warrington KJ. Incidence of herpes zoster in patients with giant cell arteritis: a population-based cohort study. Rheumatology (Oxford) 2010;49(11):2104–2108. doi: 10.1093/rheumatology/keq200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhee RL, Grayson PC, Merkel PA, Tomasson G. Infections and the risk of incident giant cell arteritis: a population-based, case-control study. Ann Rheum Dis. 2017;76(6):1031–1035. doi: 10.1136/annrheumdis-2016-210152. [DOI] [PubMed] [Google Scholar]
- 29.Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med. 2003;349(2):160–169. doi: 10.1056/NEJMra022694. [DOI] [PubMed] [Google Scholar]
- 30.Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. 2013;9(12):731–740. doi: 10.1038/nrrheum.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med. 2004;199(2):173–183. doi: 10.1084/jem.20030850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krupa WM, Dewan M, Jeon MS, Kurtin PJ, Younge BR, Goronzy JJ, Weyand CM. Trapping of misdirected dendritic cells in the granulomatous lesions of giant cell arteritis. Am J Pathol. 2002;161(5):1815–1823. doi: 10.1016/S0002-9440(10)64458-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner AD, Goronzy JJ, Weyand CM. Functional profile of tissue-infiltrating and circulating CD68+ cells in giant cell arteritis. Evidence for two components of the disease. J Clin Invest. 1994;94(3):1134–1140. doi: 10.1172/JCI117428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rittner HL, Kaiser M, Brack A, Szweda LI, Goronzy JJ, Weyand CM. Tissue-destructive macrophages in giant cell arteritis. Circ Res. 1999;84(9):1050–1058. doi: 10.1161/01.RES.84.9.1050. [DOI] [PubMed] [Google Scholar]
- 35.Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62(11):1064–1079. doi: 10.1016/j.addr.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel SK, Janjic JM. Macrophage targeted theranostics as personalized nanomedicine strategies for inflammatory diseases. Theranostics. 2015;5(2):150–172. doi: 10.7150/thno.9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weyand CM, Liao YJ, Goronzy JJ. The immunopathology of giant cell arteritis: diagnostic and therapeutic implications. J Neuroophthalmol. 2012;32(3):259–265. doi: 10.1097/WNO.0b013e318268aa9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. 2010;121(7):906–915. doi: 10.1161/CIRCULATIONAHA.109.872903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, Ciudad M, Leguy V, Berthier S, Vinit J, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum. 2012;64(11):3788–3798. doi: 10.1002/art.34647. [DOI] [PubMed] [Google Scholar]
- 40.Miyabe C, Miyabe Y, Strle K, Kim ND, Stone JH, Luster AD, Unizony S. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann Rheum Dis. 2017;76(5):898–905. doi: 10.1136/annrheumdis-2016-210070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 43.Tarhini A, Lo E, Minor DR. Releasing the brake on the immune system: ipilimumab in melanoma and other tumors. Cancer Biother Radiopharm. 2010;25(6):601–613. doi: 10.1089/cbr.2010.0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldstein BL, Gedmintas L, Todd DJ. Drug-associated polymyalgia rheumatica/giant cell arteritis occurring in two patients after treatment with ipilimumab, an antagonist of ctla-4. Arthritis Rheumatol. 2014;66(3):768–769. doi: 10.1002/art.38282. [DOI] [PubMed] [Google Scholar]
- 45.Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, Joyce RM, Wellenstein K, Keefe W, Schickler M, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother. 2011;34(5):409–418. doi: 10.1097/CJI.0b013e31821ca6ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, Goronzy JJ, Weyand CM. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci U S A. 2017;114(6):E970–E979. doi: 10.1073/pnas.1616848114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse events associated with immune checkpoint blockade in patients with cancer: a systematic review of case reports. PLoS One. 2016;11(7):e0160221. doi: 10.1371/journal.pone.0160221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Micaily I, Chernoff M. An unknown reaction to Pembrolizumab: Giant cell Arteritis. Ann Oncol. 2017. [DOI] [PubMed]
- 49.Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, Weyand CM. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod Pathol. 2017;30(6):788–796. doi: 10.1038/modpathol.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Espinoza LR, Jara LJ, Silveira LH, Martinez-Osuna P, Zwolinska JB, Kneer C, Aguilar JL. Anticardiolipin antibodies in polymyalgia rheumatica-giant cell arteritis: association with severe vascular complications. Am J Med. 1991;90(4):474–478. doi: 10.1016/0002-9343(91)80088-4. [DOI] [PubMed] [Google Scholar]
- 51.Baerlecken NT, Linnemann A, Gross WL, Moosig F, Vazquez-Rodriguez TR, Gonzalez-Gay MA, Martin J, Kotter I, Henes JC, Melchers I, et al. Association of ferritin autoantibodies with giant cell arteritis/polymyalgia rheumatica. Ann Rheum Dis. 2012;71(6):943–947. doi: 10.1136/annrheumdis-2011-200413. [DOI] [PubMed] [Google Scholar]
- 52.Grosse K, Schmidt RE, Witte T, Baerlecken NT. Epitope mapping of antibodies against ferritin heavy chain in giant cell arteritis and polymyalgia rheumatica. Scand J Rheumatol. 2013;42(3):215–219. doi: 10.3109/03009742.2012.733959. [DOI] [PubMed] [Google Scholar]
- 53.Navarro M, Cervera R, Font J, Reverter JC, Monteagudo J, Escolar G, Lopez-Soto A, Ordinas A, Ingelmo M. Anti-endothelial cell antibodies in systemic autoimmune diseases: prevalence and clinical significance. Lupus. 1997;6(6):521–526. doi: 10.1177/096120339700600608. [DOI] [PubMed] [Google Scholar]
- 54.Kistner A, Bigler MB, Glatz K, Egli SB, Baldin FS, Marquardsen FA, Mehling M, Rentsch KM, Staub D, Aschwanden M, et al. Characteristics of autoantibodies targeting 14-3-3 proteins and their association with clinical features in newly diagnosed giant cell arteritis. Rheumatology (Oxford) 2017;56(5):829–834. doi: 10.1093/rheumatology/kew469. [DOI] [PubMed] [Google Scholar]
- 55.van der Geest KS, Abdulahad WH, Chalan P, Rutgers A, Horst G, Huitema MG, Roffel MP, Roozendaal C, Kluin PM, Bos NA, et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol. 2014;66(7):1927–1938. doi: 10.1002/art.38625. [DOI] [PubMed] [Google Scholar]
- 56.Ciccia F, Rizzo A, Maugeri R, Alessandro R, Croci S, Guggino G, Cavazza A, Raimondo S, Cannizzaro A, Iacopino DG, et al. Ectopic expression of CXCL13, BAFF, APRIL and LT-beta is associated with artery tertiary lymphoid organs in giant cell arteritis. Ann Rheum Dis. 2017;76(1):235–243. doi: 10.1136/annrheumdis-2016-209217. [DOI] [PubMed] [Google Scholar]
- 57.Bhatia A, Ell PJ, Edwards JC. Anti-CD20 monoclonal antibody (rituximab) as an adjunct in the treatment of giant cell arteritis. Ann Rheum Dis. 2005;64(7):1099–1100. doi: 10.1136/ard.2005.036533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mayrbaeurl B, Hinterreiter M, Burgstaller S, Windpessl M, Thaler J. The first case of a patient with neutropenia and giant-cell arteritis treated with rituximab. Clin Rheumatol. 2007;26(9):1597–1598. doi: 10.1007/s10067-007-0684-0. [DOI] [PubMed] [Google Scholar]
- 59.Dimitrijevic I, Andersson C, Rissler P, Edvinsson L. Increased tissue endothelin-1 and endothelin-B receptor expression in temporal arteries from patients with giant cell arteritis. Ophthalmology. 2010;117(3):628–636. doi: 10.1016/j.ophtha.2009.07.043. [DOI] [PubMed] [Google Scholar]
- 60.Lozano E, Segarra M, Corbera-Bellalta M, Garcia-Martinez A, Espigol-Frigole G, Pla-Campo A, Hernandez-Rodriguez J, Cid MC. Increased expression of the endothelin system in arterial lesions from patients with giant-cell arteritis: association between elevated plasma endothelin levels and the development of ischaemic events. Ann Rheum Dis. 2010;69(2):434–442. doi: 10.1136/ard.2008.105692. [DOI] [PubMed] [Google Scholar]
- 61.Planas-Rigol E, Terrades-Garcia N, Corbera-Bellalta M, Lozano E, Alba MA, Segarra M, Espigol-Frigole G, Prieto-Gonzalez S, Hernandez-Rodriguez J, Preciado S, et al. Endothelin-1 promotes vascular smooth muscle cell migration across the artery wall: a mechanism contributing to vascular remodelling and intimal hyperplasia in giant-cell arteritis. Ann Rheum Dis. 2017;76(9):1624–1634. doi: 10.1136/annrheumdis-2016-210792. [DOI] [PubMed] [Google Scholar]
- 62.Regent A, Ly KH, Groh M, Khifer C, Lofek S, Clary G, Chafey P, Baud V, Broussard C, Federici C, et al. Molecular analysis of vascular smooth muscle cells from patients with giant cell arteritis: targeting endothelin-1 receptor to control proliferation. Autoimmun Rev. 2017;16(4):398–406. doi: 10.1016/j.autrev.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 63.Dunstan E, Lester SL, Rischmueller M, Dodd T, Black R, Ahern M, Cleland LG, Roberts-Thomson P, Hill CL. Epidemiology of biopsy-proven giant cell arteritis in South Australia. Intern Med J. 2014;44(1):32–39. doi: 10.1111/imj.12293. [DOI] [PubMed] [Google Scholar]
- 64.Proven A, Gabriel SE, Orces C, O'Fallon WM, Hunder GG. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum. 2003;49(5):703–708. doi: 10.1002/art.11388. [DOI] [PubMed] [Google Scholar]
- 65.Labarca C, Koster MJ, Crowson CS, Makol A, Ytterberg SR, Matteson EL, Warrington KJ. Predictors of relapse and treatment outcomes in biopsy-proven giant cell arteritis: a retrospective cohort study. Rheumatology (Oxford) 2016;55(2):347–356. doi: 10.1093/rheumatology/kev348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dasgupta B, Borg FA, Hassan N, Alexander L, Barraclough K, Bourke B, Fulcher J, Hollywood J, Hutchings A, James P, et al. BSR and BHPR guidelines for the management of giant cell arteritis. Rheumatology (Oxford) 2010;49(8):1594–1597. doi: 10.1093/rheumatology/keq039a. [DOI] [PubMed] [Google Scholar]
- 67.Mahr AD, Jover JA, Spiera RF, Hernandez-Garcia C, Fernandez-Gutierrez B, Lavalley MP, Merkel PA. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56(8):2789–2797. doi: 10.1002/art.22754. [DOI] [PubMed] [Google Scholar]
- 68.De Silva M, Hazleman BL. Azathioprine in giant cell arteritis/polymyalgia rheumatica: a double-blind study. Ann Rheum Dis. 1986;45(2):136–138. doi: 10.1136/ard.45.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adizie T, Christidis D, Dharmapaliah C, Borg F, Dasgupta B. Efficacy and tolerability of leflunomide in difficult-to-treat polymyalgia rheumatica and giant cell arteritis: a case series. Int J Clin Pract. 2012;66(9):906–909. doi: 10.1111/j.1742-1241.2012.02981.x. [DOI] [PubMed] [Google Scholar]
- 70.Diamantopoulos AP, Hetland H, Myklebust G. Leflunomide as a corticosteroid-sparing agent in giant cell arteritis and polymyalgia rheumatica: a case series. Biomed Res Int. 2013;2013:120638. doi: 10.1155/2013/120638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sciascia S, Piras D, Baldovino S, Russo A, Naretto C, Rossi D, Alpa M, Roccatello D. Mycophenolate mofetil as steroid-sparing treatment for elderly patients with giant cell arteritis: report of three cases. Aging Clin Exp Res. 2012;24(3):273–277. doi: 10.1007/BF03325257. [DOI] [PubMed] [Google Scholar]
- 72.Hernandez-Rodriguez J, Segarra M, Vilardell C, Sanchez M, Garcia-Martinez A, Esteban MJ, Queralt C, Grau JM, Urbano-Marquez A, Palacin A, et al. Tissue production of pro-inflammatory cytokines (IL-1beta, TNFalpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatology (Oxford) 2004;43(3):294–301. doi: 10.1093/rheumatology/keh058. [DOI] [PubMed] [Google Scholar]
- 73.Hoffman GS, Cid MC, Rendt-Zagar KE, Merkel PA, Weyand CM, Stone JH, Salvarani C, Xu W, Visvanathan S, Rahman MU, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146(9):621–630. doi: 10.7326/0003-4819-146-9-200705010-00004. [DOI] [PubMed] [Google Scholar]
- 74.Martinez-Taboada VM, Rodriguez-Valverde V, Carreno L, Lopez-Longo J, Figueroa M, Belzunegui J, Mola EM, Bonilla G. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis. 2008;67(5):625–630. doi: 10.1136/ard.2007.082115. [DOI] [PubMed] [Google Scholar]
- 75.Seror R, Baron G, Hachulla E, Debandt M, Larroche C, Puechal X, Maurier F, de Wazieres B, Quemeneur T, Ravaud P, et al. Adalimumab for steroid sparing in patients with giant-cell arteritis: results of a multicentre randomised controlled trial. Ann Rheum Dis. 2014;73(12):2074–2081. doi: 10.1136/annrheumdis-2013-203586. [DOI] [PubMed] [Google Scholar]
- 76.Dasgupta B, Panayi GS. Interleukin-6 in serum of patients with polymyalgia rheumatica and giant cell arteritis. Br J Rheumatol. 1990;29(6):456–458. doi: 10.1093/rheumatology/29.6.456. [DOI] [PubMed] [Google Scholar]
- 77.Weyand CM, Fulbright JW, Hunder GG, Evans JM, Goronzy JJ. Treatment of giant cell arteritis: interleukin-6 as a biologic marker of disease activity. Arthritis Rheum. 2000;43(5):1041–1048. doi: 10.1002/1529-0131(200005)43:5<1041::AID-ANR12>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 78.Beyer C, Axmann R, Sahinbegovic E, Distler JH, Manger B, Schett G, Zwerina J. Anti-interleukin 6 receptor therapy as rescue treatment for giant cell arteritis. Ann Rheum Dis. 2011;70(10):1874–1875. doi: 10.1136/ard.2010.149351. [DOI] [PubMed] [Google Scholar]
- 79.Loricera J, Blanco R, Castaneda S, Humbria A, Ortego-Centeno N, Narvaez J, Mata C, Melchor S, Aurrecoechea E, Calvo-Alen J, et al. Tocilizumab in refractory aortitis: study on 16 patients and literature review. Clin Exp Rheumatol. 2014;32(3 Suppl 82):S79–S89. [PubMed] [Google Scholar]
- 80.Lurati A, Bertani L, Re KA, Marrazza M, Bompane D, Scarpellini M. Successful treatment of a patient with giant cell vasculitis (horton arteritis) with tocilizumab a humanized anti-interleukin-6 receptor antibody. Case Rep Rheumatol. 2012;2012:639612. doi: 10.1155/2012/639612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pazzola G, Padovano I, Boiardi L, Versari A, Pipitone N, Catanoso M, Pulsatelli L, Meliconi R, Salvarani C. Tocilizumab in glucocorticoid-naive large-vessel vasculitis. Clin Exp Rheumatol. 2013;31(1 Suppl 75):S59–S61. [PubMed] [Google Scholar]
- 82.Salvarani C, Magnani L, Catanoso M, Pipitone N, Versari A, Dardani L, Pulsatelli L, Meliconi R, Boiardi L. Tocilizumab: a novel therapy for patients with large-vessel vasculitis. Rheumatology (Oxford) 2012;51(1):151–156. doi: 10.1093/rheumatology/ker296. [DOI] [PubMed] [Google Scholar]
- 83.Sciascia S, Rossi D, Roccatello D. Interleukin 6 blockade as steroid-sparing treatment for 2 patients with giant cell arteritis. J Rheumatol. 2011;38(9):2080–2081. doi: 10.3899/jrheum.110496. [DOI] [PubMed] [Google Scholar]
- 84.Seitz M, Reichenbach S, Bonel HM, Adler S, Wermelinger F, Villiger PM. Rapid induction of remission in large vessel vasculitis by IL-6 blockade. A case series. Swiss Med Wkly. 2011;w131(56):141. doi: 10.4414/smw.2011.13156. [DOI] [PubMed] [Google Scholar]
- 85.Unizony S, Arias-Urdaneta L, Miloslavsky E, Arvikar S, Khosroshahi A, Keroack B, Stone JR, Stone JH. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res (Hoboken) 2012;64(11):1720–1729. doi: 10.1002/acr.21750. [DOI] [PubMed] [Google Scholar]
- 86.Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, Brouwer E, Cid MC, Dasgupta B, Rech J, et al. Trial of Tocilizumab in Giant-cell Arteritis. N Engl J Med. 2017;377(4):317–328. doi: 10.1056/NEJMoa1613849. [DOI] [PubMed] [Google Scholar]
- 87.Villiger PM, Adler S, Kuchen S, Wermelinger F, Dan D, Fiege V, Butikofer L, Seitz M, Reichenbach S. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10031):1921–1927. doi: 10.1016/S0140-6736(16)00560-2. [DOI] [PubMed] [Google Scholar]
- 88.Hunder GG, Bloch DA, Michel BA, Stevens MB, Arend WP, Calabrese LH, Edworthy SM, Fauci AS, Leavitt RY, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33(8):1122–1128. doi: 10.1002/art.1780330810. [DOI] [PubMed] [Google Scholar]
- 89.Langford CA, Cuthbertson D, Ytterberg SR, Khalidi N, Monach PA, Carette S, Seo P, Moreland LW, Weisman M, Koening CL, et al. A randomized, double-blind trial of Abatacept (CTLA-4Ig) for the treatment of Giant cell Arteritis. Arthritis Rheumatol. 2017;69(4):837–845. doi: 10.1002/art.40044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Samson M, Ghesquiere T, Berthier S, Bonnotte B. Ustekinumab inhibits Th1 and Th17 polarisation in a patient with giant cell arteritis. Ann Rheum Dis. 2017. [DOI] [PubMed]
- 91.Conway R, O'Neill L, O'Flynn E, Gallagher P, McCarthy GM, Murphy CC, Veale DJ, Fearon U, Molloy ES. Ustekinumab for the treatment of refractory giant cell arteritis. Ann Rheum Dis. 2016;75(8):1578–1579. doi: 10.1136/annrheumdis-2016-209351. [DOI] [PubMed] [Google Scholar]
- 92.Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77(5):521–546. doi: 10.1007/s40265-017-0701-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57(12):5023–5038. doi: 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]
- 94.Gadina M. Janus kinases: an ideal target for the treatment of autoimmune diseases. J Investig Dermatol Symp Proc. 2013;16(1):S70–S72. doi: 10.1038/jidsymp.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hartmann B, Mohan SV, Goronzy JJ, Weyand CM: JAK/STAT-signaling in giant cell arteritis. Circulation Conference: American Heart Association. 2013, 128(22 SUPPL. 1).
- 96.Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol. 2015;34:75–82. doi: 10.1016/j.coi.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 97.Shang Y, Smith S, Hu X. Role of Notch signaling in regulating innate immunity and inflammation in health and disease. Protein Cell. 2016;7(3):159–174. doi: 10.1007/s13238-016-0250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S, GS W, Wu K. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369(1):20–27. doi: 10.1016/j.canlet.2015.07.048. [DOI] [PubMed] [Google Scholar]
- 99.Piggott K, Deng J, Warrington K, Younge B, Kubo JT, Desai M, Goronzy JJ, Weyand CM. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation. 2011;123(3):309–318. doi: 10.1161/CIRCULATIONAHA.110.936203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, Liao YJ, Goronzy JJ, Weyand CM. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med. 2017;9(399) [DOI] [PMC free article] [PubMed]
- 101.Ly KH, Stirnemann J, Liozon E, Michel M, Fain O, Fauchais AL. Interleukin-1 blockade in refractory giant cell arteritis. Joint Bone Spine. 2014;81(1):76–78. doi: 10.1016/j.jbspin.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 102.Carmona FD, Mackie SL, Martin JE, Taylor JC, Vaglio A, Eyre S, Bossini-Castillo L, Castaneda S, Cid MC, Hernandez-Rodriguez J, et al. A large-scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility. Am J Hum Genet. 2015;96(4):565–580. doi: 10.1016/j.ajhg.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Espigol-Frigole G, Corbera-Bellalta M, Planas-Rigol E, Lozano E, Segarra M, Garcia-Martinez A, Prieto-Gonzalez S, Hernandez-Rodriguez J, Grau JM, Rahman MU, et al. Increased IL-17A expression in temporal artery lesions is a predictor of sustained response to glucocorticoid treatment in patients with giant-cell arteritis. Ann Rheum Dis. 2013;72(9):1481–1487. doi: 10.1136/annrheumdis-2012-201836. [DOI] [PubMed] [Google Scholar]
- 104.Marquez A, Hernandez-Rodriguez J, Cid MC, Solans R, Castaneda S, Fernandez-Contreras ME, Ramentol M, Morado IC, Narvaez J, Gomez-Vaquero C, et al. Influence of the IL17A locus in giant cell arteritis susceptibility. Ann Rheum Dis. 2014;73(9):1742–1745. doi: 10.1136/annrheumdis-2014-205261. [DOI] [PubMed] [Google Scholar]
- 105.Marquez A, Solans R, Hernandez-Rodriguez J, Cid MC, Castaneda S, Ramentol M, Rodriguez-Rodriguez L, Narvaez J, Blanco R, Ortego-Centeno N, et al. A candidate gene approach identifies an IL33 genetic variant as a novel genetic risk factor for GCA. PLoS One. 2014;9(11):e113476. doi: 10.1371/journal.pone.0113476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Serrano A, Carmona FD, Castaneda S, Solans R, Hernandez-Rodriguez J, Cid MC, Prieto-Gonzalez S, Miranda-Filloy JA, Rodriguez-Rodriguez L, Morado IC, et al. Evidence of association of the NLRP1 gene with giant cell arteritis. Ann Rheum Dis. 2013;72(4):628–630. doi: 10.1136/annrheumdis-2012-202609. [DOI] [PubMed] [Google Scholar]
- 107.Carmona FD, Rodriguez-Rodriguez L, Castaneda S, Miranda-Filloy JA, Morado IC, Narvaez J, Mari-Alfonso B, Unzurrunzaga A, Rios-Fernandez R, Blanco R, et al. Role of the CCR5/Delta32CCR5 polymorphism in biopsy-proven giant cell arteritis. Hum Immunol. 2011;72(5):458–461. doi: 10.1016/j.humimm.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 108.Boiardi L, Casali B, Nicoli D, Farnetti E, Chen Q, Macchioni P, Catanoso MG, Pulsatelli L, Meliconi R, Salvarani C. Vascular endothelial growth factor gene polymorphisms in giant cell arteritis. J Rheumatol. 2003;30(10):2160–2164. [PubMed] [Google Scholar]
- 109.Rueda B, Lopez-Nevot MA, Lopez-Diaz MJ, Garcia-Porrua C, Martin J, Gonzalez-Gay MA. A functional variant of vascular endothelial growth factor is associated with severe ischemic complications in giant cell arteritis. J Rheumatol. 2005;32(9):1737–1741. [PubMed] [Google Scholar]
- 110.Enjuanes A, Benavente Y, Hernandez-Rodriguez J, Queralt C, Yague J, Jares P, de Sanjose S, Campo E, Cid MC. Association of NOS2 and potential effect of VEGF, IL6, CCL2 and IL1RN polymorphisms and haplotypes on susceptibility to GCA--a simultaneous study of 130 potentially functional SNPs in 14 candidate genes. Rheumatology (Oxford) 2012;51(5):841–851. doi: 10.1093/rheumatology/ker429. [DOI] [PubMed] [Google Scholar]
- 111.Salvarani C, Casali B, Farnetti E, Pipitone N, Nicoli D, Macchioni P, Cimino L, Bajocchi G, Catanoso MG, Boiardi L. Interleukin-6 promoter polymorphism at position −174 in giant cell arteritis. J Rheumatol. 2005;32(11):2173–2177. [PubMed] [Google Scholar]
- 112.Gonzalez-Gay MA, Hajeer AH, Dababneh A, Garcia-Porrua C, Mattey DL, Amoli MM, Thomson W, Ollier WE. IL-6 promoter polymorphism at position −174 modulates the phenotypic expression of polymyalgia rheumatica in biopsy-proven giant cell arteritis. Clin Exp Rheumatol. 2002;20(2):179–184. [PubMed] [Google Scholar]
- 113.Amoli MM, Garcia-Porrua C, Llorca J, Ollier WE, Gonzalez-Gay MA. Endothelial nitric oxide synthase haplotype associations in biopsy-proven giant cell arteritis. J Rheumatol. 2003;30(9):2019–2022. [PubMed] [Google Scholar]
- 114.Salvarani C, Casali B, Nicoli D, Farnetti E, Macchioni P, Catanoso MG, Chen Q, Bajocchi G, Boiardi L. Endothelial nitric oxide synthase gene polymorphisms in giant cell arteritis. Arthritis Rheum. 2003;48(11):3219–3223. doi: 10.1002/art.11307. [DOI] [PubMed] [Google Scholar]
- 115.Mattey DL, Hajeer AH, Dababneh A, Thomson W, Gonzalez-Gay MA, Garcia-Porrua C, Ollier WE. Association of giant cell arteritis and polymyalgia rheumatica with different tumor necrosis factor microsatellite polymorphisms. Arthritis Rheum. 2000;43(8):1749–1755. doi: 10.1002/1529-0131(200008)43:8<1749::AID-ANR11>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 116.Boiardi L, Casali B, Farnetti E, Pipitone N, Nicoli D, Macchioni P, Cimino L, Bajocchi G, Catanoso MG, Pattacini L, et al. Interleukin-10 promoter polymorphisms in giant cell arteritis. Arthritis Rheum. 2006;54(12):4011–4017. doi: 10.1002/art.22218. [DOI] [PubMed] [Google Scholar]
- 117.Palomino-Morales RJ, Vazquez-Rodriguez TR, Torres O, Morado IC, Castaneda S, Miranda-Filloy JA, Callejas-Rubio JL, Fernandez-Gutierrez B, Gonzalez-Gay MA, Martin J. Association between IL-18 gene polymorphisms and biopsy-proven giant cell arteritis. Arthritis Res Ther. 2010;12(2):R51. doi: 10.1186/ar2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reviewer reports and AU response to reviewers. (DOCX 16 kb)
Data Availability Statement
Not applicable.