

Abstract
Fibrosis is a dynamic process with the potential for reversibility and restoration of near-normal tissue architecture and organ function. Herein, we review mechanisms for resolution of organ fibrosis, in particular that involving the lung, with an emphasis on the critical roles of myofibroblast apoptosis and clearance of deposited matrix.
Introduction
Organ fibrosis is characterized by loss of cellular homeostasis and disruption of normal tissue architecture. In most organs, this tissue response develops when there is persistent or recurrent injury to the epithelium or endothelium. The etiological agent that induces such injury may range from infectious agents to toxic/metabolic exposures to autoimmunity, and, in some cases, the cause of the injury is never identified. When regeneration of the epithelium/endothelium is insufficient or incomplete, the subtending mesenchyme becomes activated, resulting in exuberant deposition of extracellular matrix (ECM). Although transient activation of the mesenchyme in response to tissue injury may represent an evolutionarily conserved, adaptive response to facilitate wound-repair and to contain microbial pathogens, maladaptive repair responses can culminate in organ fibrosis (165). Restoration of tissue homeostasis following injury requires tightly orchestrated temporal and spatial responses by multiple resident and recruited cells that communicate through an array of soluble factors under the influence of a dynamic ECM. In some cases, non-resolving fibrosis may result from persistent or recurrent extrinsic injuries, whereas, in others, such maladaptive fibrotic reactions may develop from an autonomous and self-propagating process. Thus, even when the inciting cause of injury is identified and eliminated, the resultant fibrosis may persist and progress (79, 175).
Recent estimates suggest that fibrotic disease is responsible for almost half of all deaths in Western developed countries (182). The mechanisms that resolve established fibrosis and restore tissue function are incompletely understood. The capacity for fibrosis resolution may differ depending on the organ involved, the nature and chronicity of the injurious stimulus, and host-specific factors, including age and genetic predisposition (13, 49, 165). Nevertheless, accumulating studies from humans and from animal models suggest that the potential to reverse fibrotic changes exists in most organs/tissues studied, including the liver, lung, kidney, skeletal muscle, heart, and bone marrow. In this review, we discuss the mechanisms that are currently known to regulate the resolution of normal wound repair and how these mechanisms are perturbed in fibrotic diseases. We broadly explore potential therapeutic strategies to facilitate the resolution of fibrosis and restore tissue homeostasis. In consideration of the topic, we submit that, although fibrosis is a stereotypical response with common mechanisms across various organs, there are also notable differences that determine both the capacity for, and the mechanisms of, fibrosis resolution. Thus our goal for this review is to address general themes that may apply to all organs, although we focus on lung fibrosis as an exemplar of fibrotic repair with the recognition that the capacity for resolution of fibrosis may be organ-specific (13). Additionally, the time required for the resolution of fibrosis may vary depending on the organ involved, the cause of injury, and host-specific factors such as age, genetic and epigenetic background, and immune competence. Furthermore, each organ or tissue may be subject to a “point of no return” at which any ability to resolve a fibrotic wound becomes markedly diminished, as has been shown in the carbon tetrachloride model of murine liver fibrosis (70). Only two pharmacological agents (pirfenidone and nintedanib) have been shown to alter the natural history of idiopathic pulmonary fibrosis (IPF), the most common and severe form of lung fibrosis that is associated with the worst outcomes. In IPF, although each drug slows the rate of decline in lung function, neither has been shown to reverse existing fibrosis based on physiological, histological, or radiological assessments. Interestingly, a recent analysis revealed that a subpopulation of patients with IPF treated with nintedanib had an improvement in lung physiology, challenging the notion that IPF is irreversible (35). Further studies are warranted to define the extent to which fibrosis is reversible in different human fibrotic disorders.
Wound Repair
The host response to tissue injury is a complex but evolutionarily conserved stereotypic process that involves the precise temporal and spatial orchestration of resident and recruited inflammatory cells (FIGURE 1). In addition to a number of soluble mediators, the changing biochemical composition and mechanical properties of the ECM determine the eventual outcome of the repair process. Our understanding of normal wound repair comes largely from studies of skin wounds, although the core cellular and molecular mechanisms are fundamentally similar in other organs (34, 46, 149, 167).
FIGURE 1.
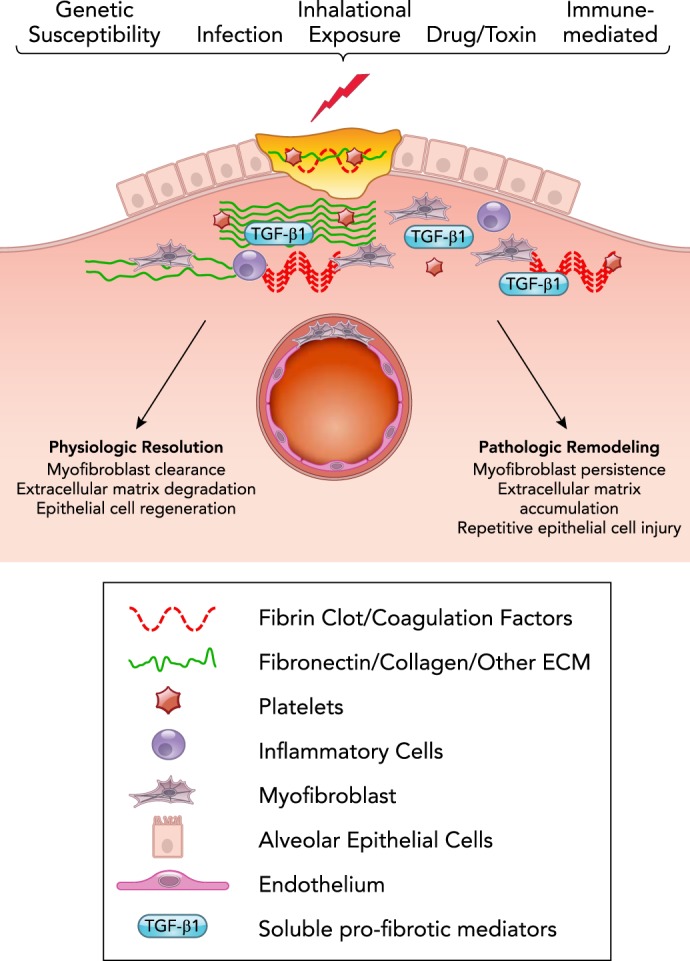
Homeostatic and fibrotic wound repair
The host response to injury is a complex interplay of epithelial cells, mesenchymal cells, vascular endothelium, and inflammatory cells that are regulated by plasma proteins, coagulation factors, other soluble mediators synthesized from cells and/or liberated from protein-bound niches in the extracellular matrix, and mechanical cues transmitted to cells from the environment. Resolution of wound repair requires the highly orchestrated spatial and temporal regulation of these processes, which, when successful, culminate in the reestablishment of an intact epithelium coupled with degradation of extracellular matrix components and clearance of activated myofibroblasts. Intrinsic and extrinsic factors, including genetic/epigenetic background, the nature of the stimulus for injury, and the extent of the initial injury are additional factors that determine the outcome of wound repair.
Briefly, in response to tissue injury, there are several overlapping “phases” of repair. The first phase is characterized by initiation of the clotting cascade and the influx of platelets leading to the formation of a fibrin clot that is also rich in fibronectin. Platelet aggregation assists in hemostasis, and platelet degranulation serves as a rich source of cytokines and growth factors, including TGF-β. This is rapidly followed by recruitment of neutrophils and monocytes/macrophages with amplification of the acute inflammatory response, efferocytosis of dead/dying cells, phagocytosis of damaged tissue, and protection from microbial invasion (34, 46, 149). The second phase of repair is characterized by proliferation and effector cell activation. Epithelial cells proliferate and migrate on the provisional matrix to reestablish barrier function. Endothelial cells proliferate, and angiogenesis is evident. Fibroblasts derived from different progenitor populations are recruited. Next comes an effector phase of ECM deposition and remodeling in which the accumulated fibroblasts predominate. These cells differentiate into “myofibroblasts,” which are smooth muscle-like contractile cells characterized by the neo-expression of organized “stress fibers” composed of alpha-smooth muscle actin and myosin filaments; these myofibroblasts are primarily responsible for the deposition, remodeling, organization, and maturation of scar tissue (167). The deposition and remodeling of ECM leads to formation of a physiological scar that is rich in cross-linked type-1 collagen (46). With the conclusion of the normal wound-repair process, myofibroblasts are cleared via apoptosis, although the in vivo trigger(s) have not been defined (29). Studies also demonstrate the ability of myofibroblasts to de-differentiate, and one study of liver repair indicates that the de-differentiated cells can persist in a quiescent state and remain primed for activation when confronted with a subsequent injury stimulus (39, 52, 88, 188).
Fibrosis: Failed Resolution of the Repair Response
Fibrosis represents the consequence of a dysregulated repair response characterized by persistent epithelial injury, death, and failed regeneration coupled with the accumulation of activated myofibroblasts. In some cases, such as chronic viral hepatitis, connective tissue disease, or environmental/occupational exposures to inhaled particulates or antigens, there is an identifiable stimulus for ongoing epithelial injury. In other instances, such as IPF or fibrostenotic Crohn’s disease that progress despite anti-inflammatory therapy, the repair response is seen in the absence of a clearly persistent exogenous stimulus (44, 175). In these cases, interactions between the myofibroblasts and the juxtaposed epithelium are thought to maintain a vicious cycle of continued epithelial damage and fibroblast activation (65, 187). Interactions between each of these cell types and a biomechanically stiffened ECM can perpetuate a positive feedback loop by directly contributing to fibroblast survival and epithelial cell apoptosis (31, 64, 77, 173, 177, 178).
Myofibroblast Apoptosis
Myofibroblast apoptosis heralds the resolution phase of normal wound repair, although the temporal and mechanistic triggers have not been determined (42, 46). In contrast, fibrosis is characterized by a distinct absence of myofibroblast apoptosis despite persistent epithelial cell injury and apoptosis (29, 56, 57, 72, 162, 170). There is growing evidence that this is related to the acquisition of an apoptosis-resistant phenotype of myofibroblasts (42, 80, 162). It is likely that these factors function synergistically in a manner that involves both cell autonomous and non-autonomous mechanisms. Apoptosis of any cell is influenced by a number of factors, including the nature of the stimulus, which can be “extrinsic” (death receptor/ligand-mediated) or “intrinsic” (cellular response to stress without a specific receptor-ligand interaction) (162). Additionally, apoptosis is regulated by the ability of a cell to sense an external stimulus, the ability to propagate a receptor-mediated signal intracellularly, the balance of pro- and anti-apoptotic BCL-2 family proteins that regulate mitochondrial release of cytochrome-c and the apoptosome, and the levels of intracellular anti-apoptotic proteins that can inhibit the caspase-mediated execution phase of the apoptosis program. Each of these mechanisms can, in turn, be controlled by genetic, epigenetic, transcriptional, and posttranslational regulation. Thus the apoptotic susceptibility of a cell, or a population of cells, represents a tightly regulated balance of pro- and anti-apoptotic factors, which can be skewed to favor survival or programmed death in response to environmental cues (33, 162). Despite the complexity of the interacting and somewhat redundant mechanisms regulating fibroblast apoptosis and survival in the context of wound repair and fibrosis, several consistent themes have emerged.
The Resolution of Fibrosis is Associated with Myofibroblast Apoptosis
Consistent with the critical role of myofibroblast persistence in the maintenance and progression of fibrosis, in vivo studies demonstrate that fibrosis resolution is coupled with myofibroblast apoptosis. This association has been most firmly established in murine models of liver fibrosis (18, 70, 73, 74, 88, 116, 136). In the carbon tetrachloride model of liver fibrosis, withdrawal of the stimulus (carbon tetrachloride) within 4 wk of the onset of exposure is associated with spontaneous myofibroblast apoptosis and ECM degradation with restoration of normal tissue architecture within 4 wk of the withdrawal (70). Similar evidence has emerged to support the association between myofibroblast apoptosis and the resolution of established fibrosis in murine models of lung fibrosis (4, 53, 77, 144, 195). For example, in young mice exposed to a single dose of intratracheal bleomycin, lung fibrosis reached a peak between 3 and 4 wk, and then significantly diminished between 2 and 4 mo post-injury (53). Aged mice had significantly impaired fibrosis resolution in association with increased markers of myofibroblast senescence and decreased evidence of apoptosis in the lungs at 3 wk post-injury (53). Similar findings have been reported in animal models of skin fibrosis, with one study showing enhanced myofibroblast apoptosis and diminished fibrosis within 2 wk of treatment with a pan-BCL2 inhibitor, and another study showing resolution of hypertrophic scars associated with myofibroblast apoptosis within 1–2 wk of TGF-β inhibition (95, 192). In 2-yr-old transgenic mice with skeletal muscle fibrosis due to a dystrophin deficiency, inhibition of myostatin led to myofibroblast apoptosis and a reduction in fibrosis over 6 wk (15).
Soluble Pro-Fibrotic Mediators Induce Myofibroblast Resistance to Apoptosis
A number of soluble mediators, including growth factors, cytokines, and lipid mediators, in addition to matrix alterations have been implicated in the pathobiology of fibrosis through activation of a growing number of signaling mechanisms (FIGURE 2). Most of the soluble mediators implicated in fibrogenesis have been found to promote myofibroblast activation and resistance to apoptosis, and, in many cases, common signaling mechanisms have been identified. TGF-β1 has been strongly and broadly implicated in the pathophysiology of fibrosis (14, 56, 149). Beyond its role in myofibroblast differentiation and activation, TGF-β1 diminishes fibroblast susceptibility to apoptosis by activation of pro-survival protein kinase pathways involving PI3K/AKT and focal adhesion kinase (FAK) (60, 61, 63, 124), upregulation of NADPH oxidase 4 (Nox4) (53, 54), upregulation of the inhibitor of apoptosis-family proteins (IAPs) survivin and XIAP (X-linked IAP) (2, 6, 155), suppression of Fas (CD-95) expression (33, 58, 119), induction of Rho-kinase mediated nuclear localization of myocardin-related transcription factor-A (MRTF-A) (139, 154, 195), and regulation of BCL2 family protein expression (60, 95, 130, 190) (FIGURE 2). Many of these same signaling pathways are activated by other soluble mediators implicated in lung fibrosis, including endothelin-1 (59, 63) and lysophasphatidic acid (135, 147).
FIGURE 2.
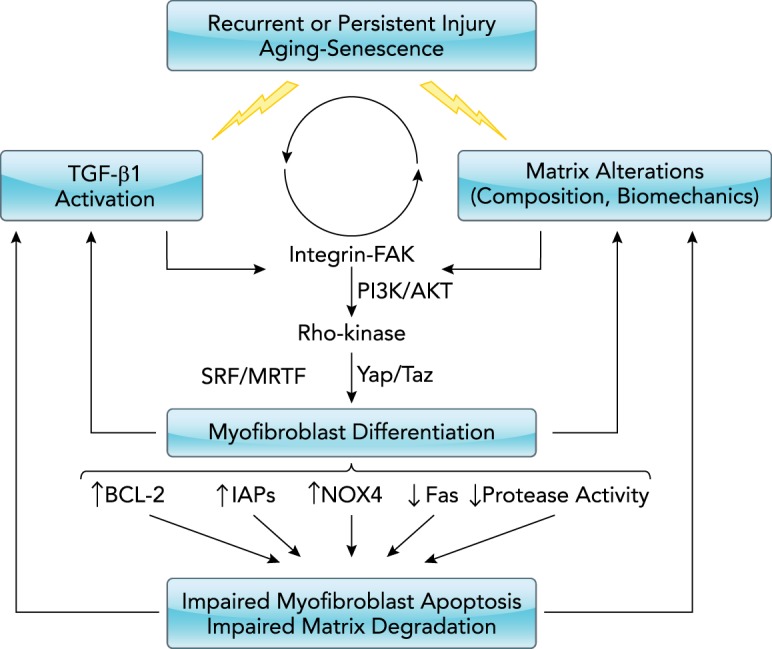
Mechanisms of non-resolving fibrosis
Resolution of fibrosis requires the orchestrated apoptosis of myofibroblasts, and the degradation and clearance of deposited matrix molecules. Persistent TGF-β1 activation and/or altered matrix biochemistry/biomechanics can lead to the activation of signaling pathways that promote both differentiation and inhibit apoptosis of myofibroblasts.
Biochemical and Biomechanical Properties of the Extracellular Matrix Regulate Fibroblast Susceptibility to Apoptosis
Cell-matrix interactions and signals generated in response to cell adhesion are well recognized to regulate survival/apoptosis of many cell types; a form of apoptosis, termed “anoikis,” results from the loss of cell adhesion or disruption of adhesion-mediated signals (36, 37). Focal adhesion kinase (FAK) activity is a critical mediator of adhesion-mediated survival signaling and EMT (epithelial-mesenchymal transition) responses in epithelial cells (31), and is required for TGF-β1-induced myofibroblast differentiation and survival (63, 163). Consistently, interruption of fibroblast-matrix interactions using soluble peptides to competitively inhibit cell interactions with RGD motifs on ECM proteins was sufficient to reduce FAK phosphorylation and induce fibroblast apoptosis (48, 60, 63, 186). In a similar manner, plasmin-mediated fibronectin proteolysis (64) and inhibition of the collagen binding discoidin domain receptor-2 (DDR2) are also associated with fibroblast apoptosis (77).
Over the last decade, it has become increasingly evident that matrix-mediated regulation of fibroblast phenotypes extends beyond the biochemical composition of the matrix and that biomechanical cues are critical to the maintenance and persistence of myofibroblasts (55, 105, 168). A series of studies showed that polymerized, but not monomeric, collagen functions to suppress proliferation in normal human lung fibroblasts and that fibroblasts isolated from IPF lung tissue elude suppressive signals from polymerized collagen and maintain their proliferative phenotype (128, 183–185). Additionally, in vitro model systems employing polyacrylamide hydrogels with varying stiffness and decellularized lung matrices from patients with fibrotic lung disease have shown that fibroblasts exposed to “stiff” substrates have decreased basal rates of apoptosis and decreased susceptibility to extrinsic apoptotic stimuli compared with cells cultured on more compliant substrates (16, 33, 104, 105). This relationship is also consistent with evidence demonstrating that increased substrate stiffness promotes Rho-kinase-mediated nuclear translocation of MRTF-A (also known as MKL1) (195). Importantly, matrix stiffness was found to be a key mechanism for the activation of latent TGF-β, suggesting that increased matrix stiffness can, by itself, perpetuate a feed-forward cycle of fibroblast activation, survival, and ECM production (179). Increased matrix-stiffness was found to be associated with suppression of PGE2, which generally antagonizes TGF-β in the regulation of fibroblast phenotypes (66, 105, 115, 166). Thus matrix stiffness-mediated suppression of PGE2 could functionally amplify the impact of active TGF-β in the cellular microenvironment. However, some studies have also shown that the matrix-mediated effects on fibroblast phenotype are independent of TGF-β activation (16). Although our understanding continues to grow and evolve, the importance of biomechanical signals supporting fibroblast survival in the context of fibrosis is well substantiated. Interestingly, increased tissue stiffness has been shown to precede substantial increases in collagen production in murine models of liver fibrosis (40), suggesting that these biomechanical signals contribute not only to fibrosis progression but to its initiation.
Interruption of Pro-Survival Signaling Pathways Enhances Myofibroblast Susceptibility to Apoptosis and Facilitates the Resolution of Fibrosis
There is now substantial evidence from in vivo models of lung fibrosis indicating that interruption of the signaling mechanisms that mediate fibroblast resistance to apoptosis diminishes the severity of lung fibrosis and may even reverse established fibrosis. In lung fibrosis, for example, administration of a pharmacological inhibitor that suppresses AKT and FAK activation 8 days after administration of intratracheal bleomycin (i.e., during the late-inflammatory, early fibrotic phase of the model) was associated with a significant reduction in lung collagen accumulation by day 15 (171). Similarly, pharmacological and siRNA-mediated inhibition of FAK was shown to diminish lung fibrosis induced by bleomycin administration and by endothelin overexpression (86, 93, 94). Conversely, deficiency of FAK-related non-kinase (FRNK), which functions as an endogenous inhibitor of FAK and is decreased in the lungs of patients with IPF, exacerbates lung fibrosis, whereas overexpression of FRNK diminishes lung fibrosis in mice (30). Inhibition of AKT signaling alone or in combination with inhibition of MAP-kinase signaling through MEK was shown to diminish lung fibrosis induced by TGF-α overexpression (91, 98, 112), bleomycin (113), and TGF-β overexpression (81). Notably, FAK and/or AKT activation also have been implicated in hypertrophic scar formation (138), liver fibrosis (78, 137), and kidney fibrosis (51, 83). In addition to FAK, inhibition of downstream mechanotransduction-regulated signaling intermediates, including Rho-kinase and MRTF-A, have been shown to reduce fibrosis in the lung (9, 90, 154, 195) and skin (47, 68).
The resolution of fibrosis that is enhanced by targeting fibroblast survival signaling extends to pathways beyond mechanotransduction. Recently, targeted inhibition of pro-survival BCL2 family proteins was shown to promote myofibroblast apoptosis and to diminish bleomycin-induced skin fibrosis (95). Increased expression of pro-fibrotic BCL2 family proteins was also identified in apoptosis-resistant fibroblasts isolated from patients with non-resolving acute respiratory distress syndrome (60). Consistently, inhibition of pro-survival IAP family proteins reduces bleomycin-induced lung fibrosis (4), as does upregulation of Fas expression induced by quercetin (58) or administration of TNF-α (145). Outside of the lung, inhibition of myostatin was shown to induce myofibroblast apoptosis and to reverse established skeletal muscle fibrosis in a murine model (15). In the kidney, treatment of mice with an ALK3 agonist led to the resolution of established fibrosis in complementary murine models of nephrotoxin-induced fibrosis and diabetic glomerulosclerosis (158).
Senescence and Aging in the Regulation of Fibroblast Survival/Apoptosis
IPF is a disease of aging (143, 164); this has motivated investigators to identify age-related factors that predispose to lung fibrosis or contribute to diminished capacity for fibrosis resolution (53, 144, 159). Cellular senescence is a hallmark of aging (111). The induction of a senescence program in cells is intricately linked to co-activation of survival signaling and the acquisition of an anti-apoptotic phenotype (87). Indeed, explanted fibroblasts from patients with IPF have been shown to impair proliferative capacity compared with fibroblasts from normal lungs, which was associated with increased AKT activation and decreased Fas expression. In vitro, the senolytic drug quercetin reduced AKT activation, increased Fas expression, and sensitized fibroblasts to undergo apoptosis. Moreover, treatment of aged mice with quercetin led to diminished bleomycin-induced lung fibrosis and decreased lung levels of senescence-related proteins (58). Additionally, survival signaling mediated by BCL2 family proteins has been used to target senescent cells, including fibroblasts, to undergo apoptosis (22, 174).
The accumulation of apoptosis-resistant senescent myofibroblasts contributes to a non-resolving fibrosis in an aging model of lung fibrosis in mice, and inhibition of the senescence-inducing, oxidant-generating enzyme Nox4 reverses established lung fibrosis in aged mice (53). Nox4 is induced in replication-induced senescent fibroblasts, and gene silencing of Nox4 mitigates this senescence phenotype (151). Similar to Nox4 inhibition, strategies to target fibroblast and epithelial cell senescence using “senolytic” cocktails are being developed as potential anti-fibrotic strategies for recalcitrant fibrotic disorders such as IPF (100, 152).
Extracellular Matrix Degradation
In addition to myofibroblast apoptosis, the degradation and clearance of ECM from tissues is essential for resolution of fibrosis. ECM deposition and degradation are dynamic processes, with the balance shifting during the development vs. recovery phases (12, 25, 84, 92, 142, 169, 193). Fibrosis is characterized by the persistence of increased levels of ECM proteins, particularly type 1 collagen; biochemical measurements of collagen are a standard experimental endpoint of fibrosis, whereas collagen mRNA and protein are commonly reported as surrogates for fibroblast activation (76). In vivo studies have estimated that, under homeostatic conditions, new collagen is synthesized at rates between 1 and 2% per week and as high as 10% per day in the lung, and between 3 and 5% per day in the skin, heart, and skeletal muscle (5, 12, 97). Non-collagen ECM proteins were estimated to have an even higher turnover rate ranging from 10 to 30% per day, depending on the organ (97). However, the relationship between synthesis and deposition is not linear, since there is also a high rate of intracellular degradation before secretion (144). Nevertheless, secreted collagen can increase up to 100% in the context of an acute injury, and, to prevent progressive accumulation, any increase in collagen deposition must be matched by degradation of extracellular collagen fibers (5, 10, 11, 120, 121, 125, 129, 193). The importance of this equilibrium is evidenced by the demonstration that transgenic mice expressing a collagenase-resistant mutant collagen develop skin fibrosis (107) and have impaired resolution of liver fibrosis that is associated with decreased stellate cell apoptosis (75). Although the mechanisms that promote collagen synthesis by fibroblasts have been extensively studied, the mechanisms regulating collagen degradation, and how these mechanisms are impaired in the context of fibrosis, have received relatively little attention (5, 13, 161, 194).
Proteases and Their Inhibitors in Matrix Degradation and Fibrosis
Degradation of collagen that is incorporated into ECM fibers is largely accomplished by sequential proteolysis mediated by matrix metalloproteinases (MMPS) and antagonized by tissue-inhibitors of MMPs (TIMPS) (121). Additionally, cathepsins, mepins, and plasmin have been implicated in degradation of collagen, fibronectin, and other matrix components (146). There is an extensive literature on MMPs, TIMPs, and their relative expression in the context of fibrosis and several studies support impaired collagenolysis in fibrotic diseases (121, 131). For example, samples taken from sites of skin involvement in patients with scleroderma had reduced collagenase activity compared with samples from clinically uninvolved skin (17). However, MMPs have diverse effects in addition to ECM proteolysis; MMPs can activate latent growth factors, including TGF-β1, that reside in the matrix, thereby increasing the availability of trophic factors during repair and remodeling (1). In line with this concept, studies variably show that, in some cases, MMPs can contribute to fibrosis, and several MMPs have been reported to promote lung fibrosis in murine models (121, 123, 146). Consistent with a pro-fibrotic role for some MMPs, a broad-spectrum MMP inhibitor prevented lung fibrosis by reducing MMP-2 and MMP-9 activity (1, 27, 123). Similarly, mice lacking TIMP3 develop increased renal fibrosis (82). Moreover, the physiological roles of MMPs and TIMPs in wound repair extend beyond catalytic effects on matrix and matrix-bound growth factors since they may also regulate inflammatory responses to injury (41).
Collectively, these studies indicate that, despite some evidence supporting a paradigm of skewed MMP/TIMP balance favoring an anti-proteolytic environment in fibrotic disease, the conceptualization of fibrosis as the consequence of an imbalance between collagen proteases and their inhibitors is an over-simplification (1, 3, 38, 71, 103, 126, 153, 189). The complexity and diversity cells that secrete and respond to MMPs, the overlapping specificity and redundancy of MMPs and their substrates, and the biological activities of cleaved matrix peptides (matrikines) highlight the temporal and contextual actions of MMPs in lung injury repair. Thus a more nuanced mechanistic understanding is required if MMPs and/or TIMPS are to be targeted for the development of precision therapeutics to treat fibrotic disease (1, 146).
In addition to MMPs/TIMPs, fibrosis has been strongly associated with a dysregulated balance in serine proteases and their inhibitors, although the role of these in matrix proteolysis is less clear. Among the serine proteases, activation of urokinase plasminogen activator (uPA) in the lung has been shown to protect mice from bleomycin-induced lung fibrosis, whereas excessive levels of plasminogen activator-inhibitor 1 (PAI-1) promote fibrosis in the lung (157). Surprisingly, however, the pro-fibrotic effects of PAI-1 have been found to be largely independent of its anti-protease activity (28). Nevertheless, uPA-mediated plasminogen activation itself can diminish fibrosis, and several mechanisms have been identified (67, 123, 156). Plasmin liberates hepatocyte growth factor (HGF) from the ECM and promotes HGF-associated anti-fibrotic actions, including increased PGE2 synthesis (7, 118, 132). Additionally, plasminogen activation by fibroblasts promotes pericellular fibronectin degradation in association with induction of fibroblast apoptosis (23, 64). Signaling from an intact ECM impacts this plasminogen activation system, since matrix stiffness-regulated activation of Hippo-pathway transcription factors and nuclear localization of myocardin-related transcription factor A (MRTF-A) have both been shown to increase production of PAI-1 and to promote lung fibrosis in vivo (104, 154, 195). Although the precise role of the plasminogen-activation system in ECM proteolysis remains unclear, evidence indicates that signaling from a rigid matrix supports a pro-fibrotic perturbation of the plasminogen activation and that ECM proteolysis could enhance the restoration of a homeostatic balance between plasminogen activators and inhibitors.
Extracellular Matrix Cross-Linking
Extracellular matrix cross-linking, primarily mediated by transglutaminase (Tg) enzymes and lysyl oxidase (LOX) or its homologs, lysyl oxidase-like (LOXL) enzymes, is increasingly recognized as a critical regulatory checkpoint in fibrosis development and resolution (80). First, cross-linking stabilizes the matrix and promotes resistance to proteolysis (42, 121, 141, 146). Second, cross-linking promotes fibroblast adhesion and proliferation, possibly by increasing matrix stiffness and engaging integrin-mediated signaling (101). As with MMPs, there is a broad recognition that tissue transglutaminase and LOX/LOXL proteins are critical to lung, liver, cardiac, biliary, and dermal fibrosis (146). In the liver, transglutaminase-mediated cross-linking stabilizes matrix and promotes liver fibrosis (45), and Tg-mediated cross-linking accounts, at least in part, for incomplete resolution of fibrosis in the carbon tetrachloride model (74). In the lung, tissue transglutaminase-2 expression and activity were increased in patients with IPF, and mice lacking Tg2 or treated with Tg inhibitors had decreased fibrosis following bleomycin administration (133, 134).
Lysyl oxidase (LOX) and its homologous LOX-like (LOXL) proteins have been strongly implicated in fibroblast activity and fibrosis (69, 106, 150), and LOX enzymes have been evaluated as a biomarker for fibrotic disease in patients with scleroderma and IPF (26, 148). Pro-fibrotic stimuli increase cardiac and kidney fibroblast LOX expression (43, 172), whereas deficiency in, or inhibition of, these enzymes reduces fibrosis in the lung, heart, and peritoneum (8, 50, 117). Other types of ECM cross-linking reactions, such as those involving advanced glycation end-product protein adducts (180) and dityrosine cross-linking (96, 140), require further study in the context of progressive, non-resolving fibrosis.
Intracellular Processing of Cleaved Collagen
Matrix degradation does not conclude with extracellular proteolysis, and the cleaved products generated, including collagen peptides, can function as biologically active “matrikines” (176). Such matrikines have been associated with a wide range of biological processes, including wound repair and inflammation, although they have not been extensively studied in the pathobiology of fibrosis (21, 176). Collagen peptides are taken up by macrophages, fibrocytes, and fibroblasts, which further degrade those peptides via the lysosomal pathway (5, 42, 89, 99, 121). The specific contribution of intracellular collagen processing by each of these cell types during normal tissue homeostasis, physiological wound repair, and fibrosis is unclear. The importance of intracellular collagen degradation is established by studies demonstrating an anti-fibrotic role for uPARP/endo180, one of the receptors that mediates collagen uptake and delivery to lysosomes, in models of lung, kidney, and liver models (20, 110, 114). Similarly, milk fat globule epidermal growth factor 8 (Mfge8) expression on macrophages was shown to mediate cellular uptake of collagen, and mice lacking Mfge8 developed increased lung fibrosis following bleomycin administration (114). In another study, an unbiased assessment of mechanisms regulating collagen internalization in phagocytic cells identified a critical role for beclin-1, the mammalian homolog of the autophagy protein ATG6 (99). Given the emerging role of autophagy in fibrosis, this finding suggests that insufficient autophagy may promote dysregulated intracellular collagen processing and cellular toxicity (32, 102, 108, 109, 191, 194). A mechanistic association between autophagy, collagen processing, and fibrosis is supported by a study demonstrating increased collagen accumulation in the kidneys of transgenic mice with reduced levels of beclin-1 and increased intracellular collagen in mesangial cells in which beclin-1 levels have been suppressed by siRNA (85). Notably, however, there are also reports indicating that autophagy can promote the development of fibrosis, further highlighting the need to understand the cell-specific, temporal, and spatial regulation of this process during the evolution of fibrosis development and resolution (24, 122, 181) (FIGURE 3).
FIGURE 3.

Therapeutic strategies to promote fibrosis resolution
Strategies that may stimulate or accelerate the resolution of fibrosis may include the restoration of myofibroblast sensitivity to apoptosis, killing/elimination of senescent cells, disruption of collagen-matrix cross-links, augmentation of matrix proteolytic activity, and the clearance of degraded matrix molecules by cellular uptake and autophagy.
Conclusions
Disruption of tissue architecture and reduced organ function due to fibrosis across diverse organ systems are major causes of morbidity and mortality. Historically, fibrosis has been viewed as an irreversible process, such that attempts to identify and eradicate the inciting stimulus was the only hope in halting progression. More recently, evidence has emerged to suggest that fibrosis may be, at least partly, reversible (42, 80, 149). However, the extent to which reversibility can be achieved may be organ-/tissue- and stimulus-dependent. As outlined in this review, strategies to promote fibrosis resolution may include restoration of myofibroblast susceptibility to apoptosis, enhancing mechanisms of ECM degradation/clearance, inhibition of collagen cross-linking, and elimination of senescent cells (FIGURE 3).
To date, clinical trials of drugs targeting specific mechanisms in human fibrotic diseases such as IPF have been disappointing, and none have been shown to “reverse” fibrosis. The two drugs that have received FDA approval for treatment of IPF slow disease progression. Most compounds tested in clinical trials have had no clear impact on primary end points in clinical trials, and some interventions that were based on studies suggesting potential benefits were subsequently found to be detrimental (127). Collectively, these studies highlight not only the complexity and heterogeneity of wound repair and fibrosis in humans but also the substantial challenges in translating basic and pre-clinical studies into therapies in humans. In doing so, these studies accentuate the necessity of continued investment in efforts to understand the relevant mechanisms of persistent fibrosis within individual patients during the evolution of aberrant wound repair (19, 62, 160). Along with this improved understanding, the translational application of precision-based interventions necessitates the identification of mechanistic biomarkers and surrogates of fibrosis resolution that will facilitate the assessment of fibrosis and its resolution.
Acknowledgments
This work was supported by National Institutes of Health Grants R01 HL-141195 (to J.C.H.), P01 HL-114470, and R01 AG-046210, and VA Merit Award I01BX003056 (to V.J.T.).
J.C.H. has received research funding from Boehringer-Ingelheim. V.J.T. has consulted for Boehringer-Ingelheim and Mistral Therapeutics, and has received research funding from Genkyotex.
J.C.H. and V.J.T. analyzed data; J.C.H. and V.J.T. interpreted results of experiments; J.C.H. and V.J.T. prepared figures; J.C.H. and V.J.T. drafted manuscript; J.C.H. and V.J.T. edited and revised manuscript; J.C.H. and V.J.T. approved final version of manuscript.
References
- 1.Afratis NA, Selman M, Pardo A, Sagi I. Emerging insights into the role of matrix metalloproteases as therapeutic targets in fibrosis. Matrix Biol 68-69: 167–179, 2018. doi: 10.1016/j.matbio.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Ajayi IO, Sisson TH, Higgins PD, Booth AJ, Sagana RL, Huang SK, White ES, King JE, Moore BB, Horowitz JC. X-linked inhibitor of apoptosis regulates lung fibroblast resistance to Fas-mediated apoptosis. Am J Respir Cell Mol Biol 49: 86–95, 2013. doi: 10.1165/rcmb.2012-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol 44–46: 247–254, 2015. doi: 10.1016/j.matbio.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Ashley SL, Sisson TH, Wheaton AK, Kim KK, Wilke CA, Ajayi IO, Subbotina N, Wang S, Duckett CS, Moore BB, Horowitz JC. Targeting inhibitor of apoptosis proteins protects from bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol 54: 482–492, 2016. doi: 10.1165/rcmb.2015-0148OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atabai K, Jame S, Azhar N, Kuo A, Lam M, McKleroy W, Dehart G, Rahman S, Xia DD, Melton AC, Wolters P, Emson CL, Turner SM, Werb Z, Sheppard D. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J Clin Invest 119: 3713–3722, 2009. doi: 10.1172/JCI40053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai L, Bernard K, Tang X, Hu M, Horowitz JC, Thannickal VJ, Sanders YY. Glutaminolysis epigenetically regulates anti-apoptotic gene expression in IPF fibroblasts. Am J Respir Cell Mol Biol. In press. doi: 10.1165/rcmb.2018-0180OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauman KA, Wettlaufer SH, Okunishi K, Vannella KM, Stoolman JS, Huang SK, Courey AJ, White ES, Hogaboam CM, Simon RH, Toews GB, Sisson TH, Moore BB, Peters-Golden M. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J Clin Invest 120: 1950–1960, 2010. doi: 10.1172/JCI38369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bellaye PS, Shimbori C, Upagupta C, Sato S, Shi W, Gauldie J, Ask K, Kolb M. Lysyl Oxidase-like 1 protein deficiency protects mice from adenoviral transforming growth factor-β1-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 58: 461–470, 2018. doi: 10.1165/rcmb.2017-0252OC. [DOI] [PubMed] [Google Scholar]
- 9.Bernau K, Ngam C, Torr EE, Acton B, Kach J, Dulin NO, Sandbo N. Megakaryoblastic leukemia-1 is required for the development of bleomycin-induced pulmonary fibrosis. Respir Res 16: 45, 2015. doi: 10.1186/s12931-015-0206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bienkowski RS, Baum BJ, Crystal RG. Fibroblasts degrade newly synthesised collagen within the cell before secretion. Nature 276: 413–416, 1978. doi: 10.1038/276413a0. [DOI] [PubMed] [Google Scholar]
- 11.Bienkowski RS, Cowan MJ, McDonald JA, Crystal RG. Degradation of newly synthesized collagen. J Biol Chem 253: 4356–4363, 1978. [PubMed] [Google Scholar]
- 12.Blaauboer ME, Emson CL, Verschuren L, van Erk M, Turner SM, Everts V, Hanemaaijer R, Stoop R. Novel combination of collagen dynamics analysis and transcriptional profiling reveals fibrosis-relevant genes and pathways. Matrix Biol 32: 424–431, 2013. doi: 10.1016/j.matbio.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Blackwell TS, Tager AM, Borok Z, Moore BB, Schwartz DA, Anstrom KJ, Bar-Joseph Z, Bitterman P, Blackburn MR, Bradford W, Brown KK, Chapman HA, Collard HR, Cosgrove GP, Deterding R, Doyle R, Flaherty KR, Garcia CK, Hagood JS, Henke CA, Herzog E, Hogaboam CM, Horowitz JC, King TE Jr, Loyd JE, Lawson WE, Marsh CB, Noble PW, Noth I, Sheppard D, Olsson J, Ortiz LA, O’Riordan TG, Oury TD, Raghu G, Roman J, Sime PJ, Sisson TH, Tschumperlin D, Violette SM, Weaver TE, Wells RG, White ES, Kaminski N, Martinez FJ, Wynn TA, Thannickal VJ, Eu JP. Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report. Am J Respir Crit Care Med 189: 214–222, 2014. doi: 10.1164/rccm.201306-1141WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med 342: 1350–1358, 2000. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 15.Bo Li Z, Zhang J, Wagner KR. Inhibition of myostatin reverses muscle fibrosis through apoptosis. J Cell Sci 125: 3957–3965, 2012. doi: 10.1242/jcs.090365. [DOI] [PubMed] [Google Scholar]
- 16.Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, Weiss K, Horowitz JC, Fiore VF, Barker TH, Moore BB, Martinez FJ, Niklason LE, White ES. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med 186: 866–876, 2012. doi: 10.1164/rccm.201204-0754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brady AH. Collagenase in scleroderma. J Clin Invest 56: 1175–1180, 1975. doi: 10.1172/JCI108194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brea R, Motiño O, Francés D, García-Monzón C, Vargas J, Fernández-Velasco M, Boscá L, Casado M, Martín-Sanz P, Agra N. PGE2 induces apoptosis of hepatic stellate cells and attenuates liver fibrosis in mice by downregulating miR-23a-5p and miR-28a-5p. Biochim Biophys Acta Mol Basis Dis 1864: 325–337, 2018. doi: 10.1016/j.bbadis.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Brownell R, Kaminski N, Woodruff PG, Bradford WZ, Richeldi L, Martinez FJ, Collard HR. Precision medicine: the new frontier in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 193: 1213–1218, 2016. doi: 10.1164/rccm.201601-0169CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bundesmann MM, Wagner TE, Chow YH, Altemeier WA, Steinbach T, Schnapp LM. Role of urokinase plasminogen activator receptor-associated protein in mouse lung. Am J Respir Cell Mol Biol 46: 233–239, 2012. doi: 10.1165/rcmb.2010-0485OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burgess JK, Weckmann M. Matrikines and the lungs. Pharmacol Ther 134: 317–337, 2012. doi: 10.1016/j.pharmthera.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22: 78–83, 2016. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang W, Wei K, Jacobs SS, Upadhyay D, Weill D, Rosen GD. SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of beta-catenin. J Biol Chem 285: 8196–8206, 2010. doi: 10.1074/jbc.M109.025684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Yu Y, Li S, Liu Y, Zhou S, Cao S, Yin J, Li G. MicroRNA-30a ameliorates hepatic fibrosis by inhibiting Beclin1-mediated autophagy. J Cell Mol Med 21: 3679–3692, 2017. doi: 10.1111/jcmm.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chesnutt AN, Matthay MA, Tibayan FA, Clark JG. Early detection of type III procollagen peptide in acute lung injury. Pathogenetic and prognostic significance. Am J Respir Crit Care Med 156: 840–845, 1997. doi: 10.1164/ajrccm.156.3.9701124. [DOI] [PubMed] [Google Scholar]
- 26.Chien JW, Richards TJ, Gibson KF, Zhang Y, Lindell KO, Shao L, Lyman SK, Adamkewicz JI, Smith V, Kaminski N, O’Riordan T. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur Respir J 43: 1430–1438, 2014. doi: 10.1183/09031936.00141013. [DOI] [PubMed] [Google Scholar]
- 27.Corbel M, Caulet-Maugendre S, Germain N, Molet S, Lagente V, Boichot E. Inhibition of bleomycin-induced pulmonary fibrosis in mice by the matrix metalloproteinase inhibitor batimastat. J Pathol 193: 538–545, 2001. doi: 10.1002/path.826. [DOI] [PubMed] [Google Scholar]
- 28.Courey AJ, Horowitz JC, Kim KK, Koh TJ, Novak ML, Subbotina N, Warnock M, Xue B, Cunningham AK, Lin Y, Goldklang MP, Simon RH, Lawrence DA, Sisson TH. The vitronectin-binding function of PAI-1 exacerbates lung fibrosis in mice. Blood 118: 2313–2321, 2011. doi: 10.1182/blood-2010-12-324574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desmoulière A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 146: 56–66, 1995. [PMC free article] [PubMed] [Google Scholar]
- 30.Ding Q, Cai GQ, Hu M, Yang Y, Zheng A, Tang Q, Gladson CL, Hayasaka H, Wu H, You Z, Southern BD, Grove LM, Rahaman SO, Fang H, Olman MA. FAK-related nonkinase is a multifunctional negative regulator of pulmonary fibrosis. Am J Pathol 182: 1572–1584, 2013. doi: 10.1016/j.ajpath.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding Q, Subramanian I, Luckhardt TR, Che P, Waghray M, Zhao XK, Bone N, Kurundkar AR, Hecker L, Hu M, Zhou Y, Horowitz JC, Vittal R, Thannickal VJ. Focal adhesion kinase signaling determines the fate of lung epithelial cells in response to TGF-β. Am J Physiol Lung Cell Mol Physiol 312: L926–L935, 2017. doi: 10.1152/ajplung.00121.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding Y, Kim S, Lee SY, Koo JK, Wang Z, Choi ME. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol 25: 2835–2846, 2014. doi: 10.1681/ASN.2013101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dodi AE, Ajayi IO, Chang C, Beard M, Ashley SL, Huang SK, Thannickal VJ, Tschumperlin DJ, Sisson TH, Horowitz JC. Regulation of fibroblast Fas expression by soluble and mechanical pro-fibrotic stimuli. Respir Res 19: 91, 2018. doi: 10.1186/s12931-018-0801-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med 6: 265sr6, 2014. doi: 10.1126/scitranslmed.3009337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flaherty KR, Kolb M, Vancheri C, Tang W, Conoscenti CS, Richeldi L. Stability or improvement in forced vital capacity with nintedanib in patients with idiopathic pulmonary fibrosis. Eur Respir J 52: 1702593, 2018. doi: 10.1183/13993003.02593-2017. [DOI] [PubMed] [Google Scholar]
- 36.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 124: 619–626, 1994. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol 134: 793–799, 1996. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.García-Alvarez J, Ramirez R, Checa M, Nuttall RK, Sampieri CL, Edwards DR, Selman M, Pardo A. Tissue inhibitor of metalloproteinase-3 is up-regulated by transforming growth factor-beta1 in vitro and expressed in fibroblastic foci in vivo in idiopathic pulmonary fibrosis. Exp Lung Res 32: 201–214, 2006. doi: 10.1080/01902140600817481. [DOI] [PubMed] [Google Scholar]
- 39.Garrison G, Huang SK, Okunishi K, Scott JP, Kumar Penke LR, Scruggs AM, Peters-Golden M. Reversal of myofibroblast differentiation by prostaglandin E(2). Am J Respir Cell Mol Biol 48: 550–558, 2013. doi: 10.1165/rcmb.2012-0262OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, Mick R, Janmey PA, Furth EE, Wells RG. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol 293: G1147–G1154, 2007. doi: 10.1152/ajpgi.00032.2007. [DOI] [PubMed] [Google Scholar]
- 41.Gill SE, Huizar I, Bench EM, Sussman SW, Wang Y, Khokha R, Parks WC. Tissue inhibitor of metalloproteinases 3 regulates resolution of inflammation following acute lung injury. Am J Pathol 176: 64–73, 2010. doi: 10.2353/ajpath.2010.090158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glasser SW, Hagood JS, Wong S, Taype CA, Madala SK, Hardie WD. Mechanisms of Lung Fibrosis Resolution. Am J Pathol 186: 1066–1077, 2016. doi: 10.1016/j.ajpath.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goto Y, Uchio-Yamada K, Anan S, Yamamoto Y, Ogura A, Manabe N. Transforming growth factor-beta1 mediated up-regulation of lysyl oxidase in the kidneys of hereditary nephrotic mouse with chronic renal fibrosis. Virchows Arch 447: 859–868, 2005. doi: 10.1007/s00428-005-0001-8. [DOI] [PubMed] [Google Scholar]
- 44.Govani SM, Stidham RW, Higgins PD. How early to take arms against a sea of troubles? The case for aggressive early therapy in Crohn’s disease to prevent fibrotic intestinal strictures. J Crohn’s Colitis 7: 923–927, 2013. doi: 10.1016/j.crohns.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 45.Grenard P, Bresson-Hadni S, El Alaoui S, Chevallier M, Vuitton DA, Ricard-Blum S. Transglutaminase-mediated cross-linking is involved in the stabilization of extracellular matrix in human liver fibrosis. J Hepatol 35: 367–375, 2001. doi: 10.1016/S0168-8278(01)00135-0. [DOI] [PubMed] [Google Scholar]
- 46.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature 453: 314–321, 2008. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 47.Haak AJ, Tsou PS, Amin MA, Ruth JH, Campbell P, Fox DA, Khanna D, Larsen SD, Neubig RR. Targeting the myofibroblast genetic switch: inhibitors of myocardin-related transcription factor/serum response factor-regulated gene transcription prevent fibrosis in a murine model of skin injury. J Pharmacol Exp Ther 349: 480–486, 2014. doi: 10.1124/jpet.114.213520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hadden HL, Henke CA. Induction of lung fibroblast apoptosis by soluble fibronectin peptides. Am J Respir Crit Care Med 162: 1553–1560, 2000. doi: 10.1164/ajrccm.162.4.2001015. [DOI] [PubMed] [Google Scholar]
- 49.Hagood JS. Beyond the genome: epigenetic mechanisms in lung remodeling. Physiology (Bethesda) 29: 177–185, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harlow CR, Wu X, van Deemter M, Gardiner F, Poland C, Green R, Sarvi S, Brown P, Kadler KE, Lu Y, Mason JI, Critchley HOD, Hillier SG. Targeting lysyl oxidase reduces peritoneal fibrosis. PLoS One 12: e0183013, 2017. doi: 10.1371/journal.pone.0183013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayashida T, Wu MH, Pierce A, Poncelet AC, Varga J, Schnaper HW. MAP-kinase activity necessary for TGFbeta1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397. J Cell Sci 120: 4230–4240, 2007. doi: 10.1242/jcs.03492. [DOI] [PubMed] [Google Scholar]
- 52.Hecker L, Jagirdar R, Jin T, Thannickal VJ. Reversible differentiation of myofibroblasts by MyoD. Exp Cell Res 317: 1914–1921, 2011. doi: 10.1016/j.yexcr.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, Meldrum E, Sanders YY, Thannickal VJ. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med 6: 231ra47, 2014. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 15: 1077–1081, 2009. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herrera J, Henke CA, Bitterman PB. Extracellular matrix as a driver of progressive fibrosis. J Clin Invest 128: 45–53, 2018. doi: 10.1172/JCI93557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol 170: 1807–1816, 2007. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 180: 1340–1355, 2012. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hohmann MS, Habiel DM, Coelho AL, Verri WA Jr, Hogaboam CM. Quercetin Enhances Ligand-induced Apoptosis in Senescent IPF Fibroblasts and Reduces Lung Fibrosis In Vivo. Am J Respir Cell Mol Biol. In press. doi: 10.1165/rcmb.2017-0289OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horowitz JC, Ajayi IO, Kulasekaran P, Rogers DS, White JB, Townsend SK, White ES, Nho RS, Higgins PD, Huang SK, Sisson TH. Survivin expression induced by endothelin-1 promotes myofibroblast resistance to apoptosis. Int J Biochem Cell Biol 44: 158–169, 2012. doi: 10.1016/j.biocel.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horowitz JC, Cui Z, Moore TA, Meier TR, Reddy RC, Toews GB, Standiford TJ, Thannickal VJ. Constitutive activation of prosurvival signaling in alveolar mesenchymal cells isolated from patients with nonresolving acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 290: L415–L425, 2006. doi: 10.1152/ajplung.00276.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H, Cui Z, Thannickal VJ. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem 279: 1359–1367, 2004. doi: 10.1074/jbc.M306248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Horowitz JC, Osterholzer JJ, Marazioti A, Stathopoulos GT. “Scar-cinoma”: viewing the fibrotic lung mesenchymal cell in the context of cancer biology. Eur Respir J 47: 1842–1854, 2016. doi: 10.1183/13993003.01201-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Horowitz JC, Rogers DS, Sharma V, Vittal R, White ES, Cui Z, Thannickal VJ. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal 19: 761–771, 2007. doi: 10.1016/j.cellsig.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horowitz JC, Rogers DS, Simon RH, Sisson TH, Thannickal VJ. Plasminogen activation induced pericellular fibronectin proteolysis promotes fibroblast apoptosis. Am J Respir Cell Mol Biol 38: 78–87, 2008. doi: 10.1165/rcmb.2007-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horowitz JC, Thannickal VJ. Epithelial-mesenchymal interactions in pulmonary fibrosis. Semin Respir Crit Care Med 27: 600–612, 2006. doi: 10.1055/s-2006-957332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang SK, White ES, Wettlaufer SH, Grifka H, Hogaboam CM, Thannickal VJ, Horowitz JC, Peters-Golden M. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J 23: 4317–4326, 2009. doi: 10.1096/fj.08-128801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang WT, Akhter H, Jiang C, MacEwen M, Ding Q, Antony V, Thannickal VJ, Liu RM. Plasminogen activator inhibitor 1, fibroblast apoptosis resistance, and aging-related susceptibility to lung fibrosis. Exp Gerontol 61: 62–75, 2015. doi: 10.1016/j.exger.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hutchings KM, Lisabeth EM, Rajeswaran W, Wilson MW, Sorenson RJ, Campbell PL, Ruth JH, Amin A, Tsou PS, Leipprandt JR, Olson SR, Wen B, Zhao T, Sun D, Khanna D, Fox DA, Neubig RR, Larsen SD. Pharmacokinetic optimitzation of CCG-203971: Novel inhibitors of the Rho/MRTF/SRF transcriptional pathway as potential antifibrotic therapeutics for systemic scleroderma. Bioorg Med Chem Lett 27: 1744–1749, 2017. doi: 10.1016/j.bmcl.2017.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ikenaga N, Peng ZW, Vaid KA, Liu SB, Yoshida S, Sverdlov DY, Mikels-Vigdal A, Smith V, Schuppan D, Popov YV. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 66: 1697–1708, 2017. doi: 10.1136/gutjnl-2016-312473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest 117: 539–548, 2007. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iredale JP. Tissue inhibitors of metalloproteinases in liver fibrosis. Int J Biochem Cell Biol 29: 43–54, 1997. doi: 10.1016/S1357-2725(96)00118-5. [DOI] [PubMed] [Google Scholar]
- 72.Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, Hovell C, Arthur MJ. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 102: 538–549, 1998. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Issa R, Williams E, Trim N, Kendall T, Arthur MJ, Reichen J, Benyon RC, Iredale JP. Apoptosis of hepatic stellate cells: involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut 48: 548–557, 2001. doi: 10.1136/gut.48.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MD, Sands E, Suliman I, Trim N, Knorr A, Arthur MJ, Benyon RC, Iredale JP. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology 126: 1795–1808, 2004. doi: 10.1053/j.gastro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 75.Issa R, Zhou X, Trim N, Millward-Sadler H, Krane S, Benyon C, Iredale J. Mutation in collagen-1 that confers resistance to the action of collagenase results in failure of recovery from CCl4-induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. FASEB J 17: 47–49, 2003. doi: 10.1096/fj.02-0494fje. [DOI] [PubMed] [Google Scholar]
- 76.Jenkins RG, Moore BB, Chambers RC, Eickelberg O, Königshoff M, Kolb M, Laurent GJ, Nanthakumar CB, Olman MA, Pardo A, Selman M, Sheppard D, Sime PJ, Tager AM, Tatler AL, Thannickal VJ, White ES; ATS Assembly on Respiratory Cell and Molecular Biology . An Official American Thoracic Society Workshop Report: Use of animal models for the preclinical assessment of potential therapies for pulmonary fibrosis. Am J Respir Cell Mol Biol 56: 667–679, 2017. doi: 10.1165/rcmb.2017-0096ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jia S, Agarwal M, Yang J, Horowitz JC, White ES, Kim KK. Discoidin domain receptor 2 signaling regulates fibroblast apoptosis through PDK1/Akt. Am J Respir Cell Mol Biol 59: 295–305, 2018. doi: 10.1165/rcmb.2017-0419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang HQ, Zhang XL, Liu L, Yang CC. Relationship between focal adhesion kinase and hepatic stellate cell proliferation during rat hepatic fibrogenesis. World J Gastroenterol 10: 3001–3005, 2004. doi: 10.3748/wjg.v10.i20.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson LA, Luke A, Sauder K, Moons DS, Horowitz JC, Higgins PD. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: impact of a “Top-Down” approach to intestinal fibrosis in mice. Inflamm Bowel Dis 18: 460–471, 2012. doi: 10.1002/ibd.21812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jun JI, Lau LF. Resolution of organ fibrosis. J Clin Invest 128: 97–107, 2018. doi: 10.1172/JCI93563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kang HR, Lee CG, Homer RJ, Elias JA. Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary fibrosis. J Exp Med 204: 1083–1093, 2007. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kassiri Z, Oudit GY, Kandalam V, Awad A, Wang X, Ziou X, Maeda N, Herzenberg AM, Scholey JW. Loss of TIMP3 enhances interstitial nephritis and fibrosis. J Am Soc Nephrol 20: 1223–1235, 2009. doi: 10.1681/ASN.2008050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, Rossi JJ, Natarajan R. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol 11: 881–889, 2009. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kelley J, Chrin L, Shull S, Rowe DW, Cutroneo KR. Bleomycin selectively elevates mRNA levels for procollagen and fibronectin following acute lung injury. Biochem Biophys Res Commun 131: 836–843, 1985. doi: 10.1016/0006-291X(85)91315-4. [DOI] [PubMed] [Google Scholar]
- 85.Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J Biol Chem 287: 11677–11688, 2012. doi: 10.1074/jbc.M111.308460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kinoshita K, Aono Y, Azuma M, Kishi J, Takezaki A, Kishi M, Makino H, Okazaki H, Uehara H, Izumi K, Sone S, Nishioka Y. Antifibrotic effects of focal adhesion kinase inhibitor in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Cell Mol Biol 49: 536–543, 2013. doi: 10.1165/rcmb.2012-0277OC. [DOI] [PubMed] [Google Scholar]
- 87.Kirkland JL, Tchkonia T. Cellular Senescence: A Translational Perspective. EBioMedicine 21: 21–28, 2017. doi: 10.1016/j.ebiom.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, Iwaisako K, Moore-Morris T, Scott B, Tsukamoto H, Evans SM, Dillmann W, Glass CK, Brenner DA. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA 109: 9448–9453, 2012. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kleaveland KR, Velikoff M, Yang J, Agarwal M, Rippe RA, Moore BB, Kim KK. Fibrocytes are not an essential source of type I collagen during lung fibrosis. J Immunol 193: 5229–5239, 2014. doi: 10.4049/jimmunol.1400753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Knipe RS, Probst CK, Lagares D, Franklin A, Spinney JJ, Brazee PL, Grasberger P, Zhang L, Black KE, Sakai N, Shea BS, Liao JK, Medoff BD, Tager AM. The Rho kinase isoforms ROCK1 and ROCK2 each contribute to the development of experimental pulmonary fibrosis. Am J Respir Cell Mol Biol 58: 471–481, 2018. doi: 10.1165/rcmb.2017-0075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Korfhagen TR, Le Cras TD, Davidson CR, Schmidt SM, Ikegami M, Whitsett JA, Hardie WD. Rapamycin prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 41: 562–572, 2009. doi: 10.1165/rcmb.2008-0377OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kulkarni T, O’Reilly P, Antony VB, Gaggar A, Thannickal VJ. Matrix remodeling in pulmonary fibrosis and emphysema. Am J Respir Cell Mol Biol 54: 751–760, 2016. doi: 10.1165/rcmb.2015-0166PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lagares D, Busnadiego O, García-Fernández RA, Kapoor M, Liu S, Carter DE, Abraham D, Shi-Wen X, Carreira P, Fontaine BA, Shea BS, Tager AM, Leask A, Lamas S, Rodríguez-Pascual F. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum 64: 1653–1664, 2012. doi: 10.1002/art.33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lagares D, Busnadiego O, García-Fernández RA, Lamas S, Rodríguez-Pascual F. Adenoviral gene transfer of endothelin-1 in the lung induces pulmonary fibrosis through the activation of focal adhesion kinase. Am J Respir Cell Mol Biol 47: 834–842, 2012. doi: 10.1165/rcmb.2011-0446OC. [DOI] [PubMed] [Google Scholar]
- 95.Lagares D, Santos A, Grasberger PE, Liu F, Probst CK, Rahimi RA, Sakai N, Kuehl T, Ryan J, Bhola P, Montero J, Kapoor M, Baron M, Varelas X, Tschumperlin DJ, Letai A, Tager AM. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci Transl Med 9: eaal3765, 2017. doi: 10.1126/scitranslmed.aal3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Larios JM, Budhiraja R, Fanburg BL, Thannickal VJ. Oxidative protein cross-linking reactions involving L-tyrosine in transforming growth factor-beta1-stimulated fibroblasts. J Biol Chem 276: 17437–17441, 2001. doi: 10.1074/jbc.M100426200. [DOI] [PubMed] [Google Scholar]
- 97.Laurent GJ. Rates of collagen synthesis in lung, skin and muscle obtained in vivo by a simplified method using [3H]proline. Biochem J 206: 535–544, 1982. doi: 10.1042/bj2060535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Le Cras TD, Korfhagen TR, Davidson C, Schmidt S, Fenchel M, Ikegami M, Whitsett JA, Hardie WD. Inhibition of PI3K by PX-866 prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Pathol 176: 679–686, 2010. doi: 10.2353/ajpath.2010.090123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee TH, McKleroy W, Khalifeh-Soltani A, Sakuma S, Lazarev S, Riento K, Nishimura SL, Nichols BJ, Atabai K. Functional genomic screen identifies novel mediators of collagen uptake. Mol Biol Cell 25: 583–593, 2014. doi: 10.1091/mbc.e13-07-0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, Ota C, Costa R, Schiller HB, Lindner M, Wagner DE, Günther A, Königshoff M. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J 50: 1602367, 2017. doi: 10.1183/13993003.02367-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139: 891–906, 2009. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li H, Peng X, Wang Y, Cao S, Xiong L, Fan J, Wang Y, Zhuang S, Yu X, Mao H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 12: 1472–1486, 2016. doi: 10.1080/15548627.2016.1190071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li YY, McTiernan CF, Feldman AM. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc Res 46: 214–224, 2000. doi: 10.1016/S0008-6363(00)00003-1. [DOI] [PubMed] [Google Scholar]
- 104.Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, Probst CK, Hiemer SE, Sisson TH, Horowitz JC, Rosas IO, Fredenburgh LE, Feghali-Bostwick C, Varelas X, Tager AM, Tschumperlin DJ. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 308: L344–L357, 2015. doi: 10.1152/ajplung.00300.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol 190: 693–706, 2010. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu SB, Ikenaga N, Peng ZW, Sverdlov DY, Greenstein A, Smith V, Schuppan D, Popov Y. Lysyl oxidase activity contributes to collagen stabilization during liver fibrosis progression and limits spontaneous fibrosis reversal in mice. FASEB J 30: 1599–1609, 2016. doi: 10.1096/fj.14-268425. [DOI] [PubMed] [Google Scholar]
- 107.Liu X, Wu H, Byrne M, Jeffrey J, Krane S, Jaenisch R. A targeted mutation at the known collagenase cleavage site in mouse type I collagen impairs tissue remodeling. J Cell Biol 130: 227–237, 1995. doi: 10.1083/jcb.130.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu X, Zhang Y, Shi M, Wang Y, Zhang F, Yan R, Liu L, Xiao Y, Guo B. Notch1 regulates PTEN expression to exacerbate renal tubulointerstitial fibrosis in diabetic nephropathy by inhibiting autophagy via interactions with Hes1. Biochem Biophys Res Commun 497: 1110–1116, 2018. doi: 10.1016/j.bbrc.2018.02.187. [DOI] [PubMed] [Google Scholar]
- 109.Lodder J, Denaës T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM, Lotersztajn S, Teixeira-Clerc F. Macrophage autophagy protects against liver fibrosis in mice. Autophagy 11: 1280–1292, 2015. doi: 10.1080/15548627.2015.1058473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.López-Guisa JM, Cai X, Collins SJ, Yamaguchi I, Okamura DM, Bugge TH, Isacke CM, Emson CL, Turner SM, Shankland SJ, Eddy AA. Mannose receptor 2 attenuates renal fibrosis. J Am Soc Nephrol 23: 236–251, 2012. doi: 10.1681/ASN.2011030310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 153: 1194–1217, 2013. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Madala SK, Edukulla R, Phatak M, Schmidt S, Davidson C, Acciani TH, Korfhagen TR, Medvedovic M, Lecras TD, Wagner K, Hardie WD. Dual targeting of MEK and PI3K pathways attenuates established and progressive pulmonary fibrosis. PLoS One 9: e86536, 2014. doi: 10.1371/journal.pone.0086536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Madala SK, Maxfield MD, Davidson CR, Schmidt SM, Garry D, Ikegami M, Hardie WD, Glasser SW. Rapamycin regulates bleomycin-induced lung damage in SP-C-deficient mice. Pulm Med 2011: 653524, 2011. doi: 10.1155/2011/653524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Madsen DH, Jürgensen HJ, Ingvarsen S, Melander MC, Vainer B, Egerod KL, Hald A, Rønø B, Madsen CA, Bugge TH, Engelholm LH, Behrendt N. Endocytic collagen degradation: a novel mechanism involved in protection against liver fibrosis. J Pathol 227: 94–105, 2012. doi: 10.1002/path.3981. [DOI] [PubMed] [Google Scholar]
- 115.Maher TM, Evans IC, Bottoms SE, Mercer PF, Thorley AJ, Nicholson AG, Laurent GJ, Tetley TD, Chambers RC, McAnulty RJ. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 182: 73–82, 2010. doi: 10.1164/rccm.200905-0674OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marsillach J, Ferré N, Camps J, Rull A, Beltran R, Joven J. Changes in the expression of genes related to apoptosis and fibrosis pathways in CCl4-treated rats. Mol Cell Biochem 308: 101–109, 2008. doi: 10.1007/s11010-007-9617-0. [DOI] [PubMed] [Google Scholar]
- 117.Martínez-Martínez E, Rodríguez C, Galán M, Miana M, Jurado-López R, Bartolomé MV, Luaces M, Islas F, Martínez-González J, López-Andrés N, Cachofeiro V. The lysyl oxidase inhibitor (β-aminopropionitrile) reduces leptin profibrotic effects and ameliorates cardiovascular remodeling in diet-induced obesity in rats. J Mol Cell Cardiol 92: 96–104, 2016. doi: 10.1016/j.yjmcc.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 118.Matsuoka H, Sisson TH, Nishiuma T, Simon RH. Plasminogen-mediated activation and release of hepatocyte growth factor from extracellular matrix. Am J Respir Cell Mol Biol 35: 705–713, 2006. doi: 10.1165/rcmb.2006-0006OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Matsushima S, Ishiyama J. MicroRNA-29c regulates apoptosis sensitivity via modulation of the cell-surface death receptor, Fas, in lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 311: L1050–L1061, 2016. doi: 10.1152/ajplung.00252.2016. [DOI] [PubMed] [Google Scholar]
- 120.McAnulty RJ, Laurent GJ. Collagen synthesis and degradation in vivo. Evidence for rapid rates of collagen turnover with extensive degradation of newly synthesized collagen in tissues of the adult rat. Coll Relat Res 7: 93–104, 1987. doi: 10.1016/S0174-173X(87)80001-8. [DOI] [PubMed] [Google Scholar]
- 121.McKleroy W, Lee TH, Atabai K. Always cleave up your mess: targeting collagen degradation to treat tissue fibrosis. Am J Physiol Lung Cell Mol Physiol 304: L709–L721, 2013. doi: 10.1152/ajplung.00418.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Meng Y, Pan M, Zheng B, Chen Y, Li W, Yang Q, Zheng Z, Sun N, Zhang Y, Li X. Autophagy attenuates angiotensin ii-induced pulmonary fibrosis by inhibiting redox imbalance-mediated NOD-like receptor family pyrin domain containing 3 inflammasome activation. Antioxid Redox Signal. In press. doi: 10.1089/ars.2017.7261. [DOI] [PubMed] [Google Scholar]
- 123.Menou A, Duitman J, Crestani B. The impaired proteases and anti-proteases balance in idiopathic pulmonary fibrosis. Matrix Biol 68-69: 382–403, 2018. doi: 10.1016/j.matbio.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 124.Mercer PF, Woodcock HV, Eley JD, Platé M, Sulikowski MG, Durrenberger PF, Franklin L, Nanthakumar CB, Man Y, Genovese F, McAnulty RJ, Yang S, Maher TM, Nicholson AG, Blanchard AD, Marshall RP, Lukey PT, Chambers RC. Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax 71: 701–711, 2016. doi: 10.1136/thoraxjnl-2015-207429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Molnar JA, Alpert N, Burke JF, Young VR. Synthesis and degradation rates of collagens in vivo in whole skin of rats, studied with 1802 labelling. Biochem J 240: 431–435, 1986. doi: 10.1042/bj2400431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Montaño M, Ramos C, González G, Vadillo F, Pardo A, Selman M. Lung collagenase inhibitors and spontaneous and latent collagenase activity in idiopathic pulmonary fibrosis and hypersensitivity pneumonitis. Chest 96: 1115–1119, 1989. doi: 10.1378/chest.96.5.1115. [DOI] [PubMed] [Google Scholar]
- 127.Mora AL, Rojas M, Pardo A, Selman M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat Rev Drug Discov 16: 810, 2017. doi: 10.1038/nrd.2017.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nho RS, Peterson M, Hergert P, Henke CA. FoxO3a (Forkhead Box O3a) deficiency protects idiopathic pulmonary fibrosis (IPF) fibroblasts from type I polymerized collagen matrix-induced apoptosis via caveolin-1 (cav-1) and Fas. PLoS One 8: e61017, 2013. doi: 10.1371/journal.pone.0061017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nimni ME, De Guia E, Bavetta LA. Synthesis and turnover of collagen precursors in rabbit skin. Biochem J 102: 143–147, 1967. doi: 10.1042/bj1020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Novo E, Marra F, Zamara E, Valfrè di Bonzo L, Monitillo L, Cannito S, Petrai I, Mazzocca A, Bonacchi A, De Franco RS, Colombatto S, Autelli R, Pinzani M, Parola M. Overexpression of Bcl-2 by activated human hepatic stellate cells: resistance to apoptosis as a mechanism of progressive hepatic fibrogenesis in humans. Gut 55: 1174–1182, 2006. doi: 10.1136/gut.2005.082701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Okazaki I, Maruyama K. Collagenase activity in experimental hepatic fibrosis. Nature 252: 49–50, 1974. doi: 10.1038/252049a0. [DOI] [PubMed] [Google Scholar]
- 132.Okunishi K, Sisson TH, Huang SK, Hogaboam CM, Simon RH, Peters-Golden M. Plasmin overcomes resistance to prostaglandin E2 in fibrotic lung fibroblasts by reorganizing protein kinase A signaling. J Biol Chem 286: 32231–32243, 2011. doi: 10.1074/jbc.M111.235606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Olsen KC, Epa AP, Kulkarni AA, Kottmann RM, McCarthy CE, Johnson GV, Thatcher TH, Phipps RP, Sime PJ. Inhibition of transglutaminase 2, a novel target for pulmonary fibrosis, by two small electrophilic molecules. Am J Respir Cell Mol Biol 50: 737–747, 2014. doi: 10.1165/rcmb.2013-0092OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Olsen KC, Sapinoro RE, Kottmann RM, Kulkarni AA, Iismaa SE, Johnson GV, Thatcher TH, Phipps RP, Sime PJ. Transglutaminase 2 and its role in pulmonary fibrosis. Am J Respir Crit Care Med 184: 699–707, 2011. doi: 10.1164/rccm.201101-0013OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Orosa B, González A, Mera A, Gómez-Reino JJ, Conde C. Lysophosphatidic acid receptor 1 suppression sensitizes rheumatoid fibroblast-like synoviocytes to tumor necrosis factor-induced apoptosis. Arthritis Rheum 64: 2460–2470, 2012. doi: 10.1002/art.34443. [DOI] [PubMed] [Google Scholar]
- 136.Paik YH, Kim JK, Lee JI, Kang SH, Kim DY, An SH, Lee SJ, Lee DK, Han KH, Chon CY, Lee SI, Lee KS, Brenner DA. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut 58: 1517–1527, 2009. doi: 10.1136/gut.2008.157420. [DOI] [PubMed] [Google Scholar]
- 137.Parsons CJ, Takashima M, Rippe RA. Molecular mechanisms of hepatic fibrogenesis. J Gastroenterol Hepatol 22, Suppl 1: S79–S84, 2007. doi: 10.1111/j.1440-1746.2006.04659.x. [DOI] [PubMed] [Google Scholar]
- 138.Paterno J, Vial IN, Wong VW, Rustad KC, Sorkin M, Shi Y, Bhatt KA, Thangarajah H, Glotzbach JP, Gurtner GC. Akt-mediated mechanotransduction in murine fibroblasts during hypertrophic scar formation. Wound Repair Regen 19: 49–58, 2011. doi: 10.1111/j.1524-475X.2010.00643.x. [DOI] [PubMed] [Google Scholar]
- 139.Penke LR, Huang SK, White ES, Peters-Golden M. Prostaglandin E2 inhibits α-smooth muscle actin transcription during myofibroblast differentiation via distinct mechanisms of modulation of serum response factor and myocardin-related transcription factor-A. J Biol Chem 289: 17151–17162, 2014. doi: 10.1074/jbc.M114.558130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pennathur S, Vivekanandan-Giri A, Locy ML, Kulkarni T, Zhi D, Zeng L, Byun J, de Andrade JA, Thannickal VJ. Oxidative modifications of protein tyrosyl residues are increased in plasma of human subjects with interstitial lung disease. Am J Respir Crit Care Med 193: 861–868, 2016. doi: 10.1164/rccm.201505-0992OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Philp CJ, Siebeke I, Clements D, Miller S, Habgood A, John AE, Navaratnam V, Hubbard RB, Jenkins G, Johnson SR. Extracellular matrix cross-linking enhances fibroblast growth and protects against matrix proteolysis in lung fibrosis. Am J Respir Cell Mol Biol 58: 594–603, 2018. doi: 10.1165/rcmb.2016-0379OC. [DOI] [PubMed] [Google Scholar]
- 142.Raghow R, Lurie S, Seyer JM, Kang AH. Profiles of steady state levels of messenger RNAs coding for type I procollagen, elastin, and fibronectin in hamster lungs undergoing bleomycin-induced interstitial pulmonary fibrosis. J Clin Invest 76: 1733–1739, 1985. doi: 10.1172/JCI112163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 174: 810–816, 2006. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 144.Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, Locy ML, Ravi S, Deshane J, Mannon RB, Abraham E, Darley-Usmar V, Thannickal VJ, Zmijewski JW. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med 24: 1121–1127, 2018. doi: 10.1038/s41591-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Redente EF, Keith RC, Janssen W, Henson PM, Ortiz LA, Downey GP, Bratton DL, Riches DW. Tumor necrosis factor-α accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am J Respir Cell Mol Biol 50: 825–837, 2014. doi: 10.1165/rcmb.2013-0386OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ricard-Blum S, Baffet G, Théret N. Molecular and tissue alterations of collagens in fibrosis. Matrix Biol 68-69: 122–149, 2018. doi: 10.1016/j.matbio.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 147.Riches DW, Backos DS, Redente EF. ROCK and Rho: Promising therapeutic targets to ameliorate pulmonary fibrosis. Am J Pathol 185: 909–912, 2015. doi: 10.1016/j.ajpath.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rimar D, Rosner I, Nov Y, Slobodin G, Rozenbaum M, Halasz K, Haj T, Jiries N, Kaly L, Boulman N, Daood R, Vadasz Z. Brief report: lysyl oxidase is a potential biomarker of fibrosis in systemic sclerosis. Arthritis Rheumatol 66: 726–730, 2014. doi: 10.1002/art.38277. [DOI] [PubMed] [Google Scholar]
- 149.Rockey DC, Bell PD, Hill JA. Fibrosis–a common pathway to organ injury and failure. N Engl J Med 372: 1138–1149, 2015. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 150.Rosini S, Pugh N, Bonna AM, Hulmes DJS, Farndale RW, Adams JC. Thrombospondin-1 promotes matrix homeostasis by interacting with collagen and lysyl oxidase precursors and collagen cross-linking sites. Sci Signal 11: eaar2566, 2018. doi: 10.1126/scisignal.aar2566. [DOI] [PubMed] [Google Scholar]
- 151.Sanders YY, Liu H, Liu G, Thannickal VJ. Epigenetic mechanisms regulate NADPH oxidase-4 expression in cellular senescence. Free Radic Biol Med 79: 197–205, 2015. doi: 10.1016/j.freeradbiomed.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 152.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8: 14532, 2017. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Selman M, Ruiz V, Cabrera S, Segura L, Ramírez R, Barrios R, Pardo A. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am J Physiol Lung Cell Mol Physiol 279: L562–L574, 2000. doi: 10.1152/ajplung.2000.279.3.L562. [DOI] [PubMed] [Google Scholar]
- 154.Sisson TH, Ajayi IO, Subbotina N, Dodi AE, Rodansky ES, Chibucos LN, Kim KK, Keshamouni VG, White ES, Zhou Y, Higgins PD, Larsen SD, Neubig RR, Horowitz JC. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol 185: 969–986, 2015. doi: 10.1016/j.ajpath.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sisson TH, Maher TM, Ajayi IO, King JE, Higgins PD, Booth AJ, Sagana RL, Huang SK, White ES, Moore BB, Horowitz JC. Increased survivin expression contributes to apoptosis-resistance in IPF fibroblasts. Adv Biosci Biotechnol 3: 657–664, 2012. doi: 10.4236/abb.2012.326085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sisson TH, Nguyen MH, Yu B, Novak ML, Simon RH, Koh TJ. Urokinase-type plasminogen activator increases hepatocyte growth factor activity required for skeletal muscle regeneration. Blood 114: 5052–5061, 2009. doi: 10.1182/blood-2008-12-196212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sisson TH, Simon RH. The plasminogen activation system in lung disease. Curr Drug Targets 8: 1016–1029, 2007. doi: 10.2174/138945007781662319. [DOI] [PubMed] [Google Scholar]
- 158.Sugimoto H, LeBleu VS, Bosukonda D, Keck P, Taduri G, Bechtel W, Okada H, Carlson W Jr, Bey P, Rusckowski M, Tampe B, Tampe D, Kanasaki K, Zeisberg M, Kalluri R. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat Med 18: 396–404, 2012. doi: 10.1038/nm.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Thannickal VJ. Mechanistic links between aging and lung fibrosis. Biogerontology 14: 609–615, 2013. doi: 10.1007/s10522-013-9451-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Thannickal VJ, Antony VB. Is personalized medicine a realistic goal in idiopathic pulmonary fibrosis? Expert Rev Respir Med 12: 441–443, 2018. doi: 10.1080/17476348.2018.1464913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Thannickal VJ, Henke CA, Horowitz JC, Noble PW, Roman J, Sime PJ, Zhou Y, Wells RG, White ES, Tschumperlin DJ. Matrix biology of idiopathic pulmonary fibrosis: a workshop report of the national heart, lung, and blood institute. Am J Pathol 184: 1643–1651, 2014. doi: 10.1016/j.ajpath.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc 3: 350–356, 2006. doi: 10.1513/pats.200601-001TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 278: 12384–12389, 2003. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 164.Thannickal VJ, Murthy M, Balch WE, Chandel NS, Meiners S, Eickelberg O, Selman M, Pardo A, White ES, Levy BD, Busse PJ, Tuder RM, Antony VB, Sznajder JI, Budinger GR. Blue journal conference. Aging and susceptibility to lung disease. Am J Respir Crit Care Med 191: 261–269, 2015. doi: 10.1164/rccm.201410-1876PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Thannickal VJ, Zhou Y, Gaggar A, Duncan SR. Fibrosis: ultimate and proximate causes. J Clin Invest 124: 4673–4677, 2014. doi: 10.1172/JCI74368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thomas PE, Peters-Golden M, White ES, Thannickal VJ, Moore BB. PGE(2) inhibition of TGF-beta1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am J Physiol Lung Cell Mol Physiol 293: L417–L428, 2007. doi: 10.1152/ajplung.00489.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3: 349–363, 2002. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 168.Tschumperlin DJ, Ligresti G, Hilscher MB, Shah VH. Mechanosensing and fibrosis. J Clin Invest 128: 74–84, 2018. doi: 10.1172/JCI93561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tsukui T, Ueha S, Abe J, Hashimoto S, Shichino S, Shimaoka T, Shand FH, Arakawa Y, Oshima K, Hattori M, Inagaki Y, Tomura M, Matsushima K. Qualitative rather than quantitative changes are hallmarks of fibroblasts in bleomycin-induced pulmonary fibrosis. Am J Pathol 183: 758–773, 2013. doi: 10.1016/j.ajpath.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 170.van der Poorten D, George J. Disease-specific mechanisms of fibrosis: hepatitis C virus and nonalcoholic steatohepatitis. Clin Liver Dis 12: 805–824, 2008. doi: 10.1016/j.cld.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 171.Vittal R, Horowitz JC, Moore BB, Zhang H, Martinez FJ, Toews GB, Standiford TJ, Thannickal VJ. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am J Pathol 166: 367–375, 2005. doi: 10.1016/S0002-9440(10)62260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Voloshenyuk TG, Hart AD, Khoutorova E, Gardner JD. TNF-α increases cardiac fibroblast lysyl oxidase expression through TGF-β and PI3Kinase signaling pathways. Biochem Biophys Res Commun 413: 370–375, 2011. doi: 10.1016/j.bbrc.2011.08.109. [DOI] [PubMed] [Google Scholar]
- 173.Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, Thannickal VJ. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J 19: 854–856, 2005. doi: 10.1096/fj.04-2882fje. [DOI] [PubMed] [Google Scholar]
- 174.Wang E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 55: 2284–2292, 1995. [PubMed] [Google Scholar]
- 175.Wells AU, Brown KK, Flaherty KR, Kolb M, Thannickal VJ; IPF Consensus Working Group . What’s in a name? That which we call IPF, by any other name would act the same. Eur Respir J 51: 1800692, 2018. doi: 10.1183/13993003.00692-2018. [DOI] [PubMed] [Google Scholar]
- 176.Wells JM, Gaggar A, Blalock JE. MMP generated matrikines. Matrix Biol 44-46: 122–129, 2015. doi: 10.1016/j.matbio.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wheaton AK, Agarwal M, Jia S, Kim KK. Lung epithelial cell focal adhesion kinase signaling inhibits lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol 312: L722–L730, 2017. doi: 10.1152/ajplung.00478.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wheaton AK, Velikoff M, Agarwal M, Loo TT, Horowitz JC, Sisson TH, Kim KK. The vitronectin RGD motif regulates TGF-β-induced alveolar epithelial cell apoptosis. Am J Physiol Lung Cell Mol Physiol 310: L1206–L1217, 2016. doi: 10.1152/ajplung.00424.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol 179: 1311–1323, 2007. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wood LK, Kayupov E, Gumucio JP, Mendias CL, Claflin DR, Brooks SV. Intrinsic stiffness of extracellular matrix increases with age in skeletal muscles of mice. J Appl Physiol (1985) 117: 363–369, 2014. doi: 10.1152/japplphysiol.00256.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wu J, Xing C, Zhang L, Mao H, Chen X, Liang M, Wang F, Ren H, Cui H, Jiang A, Wang Z, Zou M, Ji Y. Autophagy promotes fibrosis and apoptosis in the peritoneum during long-term peritoneal dialysis. J Cell Mol Med 22: 1190–1201, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 4: 583–594, 2004. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Xia H, Diebold D, Nho R, Perlman D, Kleidon J, Kahm J, Avdulov S, Peterson M, Nerva J, Bitterman P, Henke C. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med 205: 1659–1672, 2008. doi: 10.1084/jem.20080001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Xia H, Khalil W, Kahm J, Jessurun J, Kleidon J, Henke CA. Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. Am J Pathol 176: 2626–2637, 2010. doi: 10.2353/ajpath.2010.091117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Xia H, Nho R, Kleidon J, Kahm J, Henke CA. Polymerized collagen inhibits fibroblast proliferation via a mechanism involving the formation of a beta1 integrin-protein phosphatase 2A-tuberous sclerosis complex 2 complex that suppresses S6K1 activity. J Biol Chem 283: 20350–20360, 2008. doi: 10.1074/jbc.M707489200. [DOI] [PubMed] [Google Scholar]
- 186.Xia H, Nho RS, Kahm J, Kleidon J, Henke CA. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem 279: 33024–33034, 2004. doi: 10.1074/jbc.M313265200. [DOI] [PubMed] [Google Scholar]
- 187.Yang J, Velikoff M, Canalis E, Horowitz JC, Kim KK. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am J Physiol Lung Cell Mol Physiol 306: L786–L796, 2014. doi: 10.1152/ajplung.00243.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yang X, Chen B, Liu T, Chen X. Reversal of myofibroblast differentiation: a review. Eur J Pharmacol 734: 83–90, 2014. doi: 10.1016/j.ejphar.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 189.Zaoui P, Cantin JF, Alimardani-Bessette M, Monier F, Halimi S, Morel F, Cordonnier D. Role of metalloproteases and inhibitors in the occurrence and progression of diabetic renal lesions. Diabetes Metab 26, Suppl 4: 25–29, 2000. [PubMed] [Google Scholar]
- 190.Zhang HY, Phan SH. Inhibition of myofibroblast apoptosis by transforming growth factor beta(1). Am J Respir Cell Mol Biol 21: 658–665, 1999. doi: 10.1165/ajrcmb.21.6.3720. [DOI] [PubMed] [Google Scholar]
- 191.Zhang Y, Zhao S, Wu D, Liu X, Shi M, Wang Y, Zhang F, Ding J, Xiao Y, Guo B. MicroRNA-22 promotes renal tubulointerstitial fibrosis by targeting PTEN and suppressing autophagy in diabetic nephropathy. J Diabetes Res 2018: 4728645, 2018. doi: 10.1155/2018/4728645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhao R, Yan Q, Huang H, Lv J, Ma W. Transdermal siRNA-TGFβ1-337 patch for hypertrophic scar treatment. Matrix Biol 32: 265–276, 2013. doi: 10.1016/j.matbio.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 193.Zhou S, Salisbury J, Preedy VR, Emery PW. Increased collagen synthesis rate during wound healing in muscle. PLoS One 8: e58324, 2013. doi: 10.1371/journal.pone.0058324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhou Y, Horowitz JC, Naba A, Ambalavanan N, Atabai K, Balestrini J, Bitterman PB, Corley RA, Ding BS, Engler AJ, Hansen KC, Hagood JS, Kheradmand F, Lin QS, Neptune E, Niklason L, Ortiz LA, Parks WC, Tschumperlin DJ, White ES, Chapman HA, Thannickal VJ. Extracellular matrix in lung development, homeostasis and disease. Matrix Biol 73: 77–104, 2018. doi: 10.1016/j.matbio.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, Jin TH, Desai L, Bernard K, Thannickal VJ. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest 123: 1096–1108, 2013. doi: 10.1172/JCI66700. [DOI] [PMC free article] [PubMed] [Google Scholar]