

Rotavirus is one of the most important pathogens causing severe gastroenteritis in young children worldwide. Here we show that infection with several rotavirus strains induces an alternative splicing of the RNA encoding the stressed-induced transcription factor XBP1. The genetic determinant of XBP1 splicing is the viral RNA translation enhancer NSP3. Since XBP1 is involved in cellular stress and immune responses and since the XBP1 protein made from the alternatively spliced RNA is an active transcription factor, our observations raise the question of whether alternative splicing is a cellular response to rotavirus infection.
KEYWORDS: NSP3, PABP, XBP1, immune response, nuclear transport, rotavirus, splicing, stress response
ABSTRACT
XBP1 is a stress-regulated transcription factor also involved in mammalian host defenses and innate immune response. Our investigation of XBP1 RNA splicing during rotavirus infection revealed that an additional XBP1 RNA (XBP1es) that corresponded to exon skipping in the XBP1 pre-RNA is induced depending on the rotavirus strain used. We show that the translation product of XBP1es (XBP1es) has trans-activation properties similar to those of XBP1 on ER stress response element (ERSE) containing promoters. Using monoreassortant between ES+ (“skipping”) and ES– (“nonskipping”) strains of rotavirus, we show that gene 7 encoding the viral translation enhancer NSP3 is involved in this phenomenon and that exon skipping parallels the nuclear relocalization of cytoplasmic PABP. We further show, using recombinant rotaviruses carrying chimeric gene 7, that the ES+ phenotype is linked to the eIF4G-binding domain of NSP3. Because the XBP1 transcription factor is involved in stress and immunological responses, our results suggest an alternative way to activate XBP1 upon viral infection or nuclear localization of PABP.
IMPORTANCE Rotavirus is one of the most important pathogens causing severe gastroenteritis in young children worldwide. Here we show that infection with several rotavirus strains induces an alternative splicing of the RNA encoding the stressed-induced transcription factor XBP1. The genetic determinant of XBP1 splicing is the viral RNA translation enhancer NSP3. Since XBP1 is involved in cellular stress and immune responses and since the XBP1 protein made from the alternatively spliced RNA is an active transcription factor, our observations raise the question of whether alternative splicing is a cellular response to rotavirus infection.
INTRODUCTION
A variety of cellular insults such as viral infection triggers a coordinated cell adaptive response known as the unfolded protein response (UPR) (1, 2) that increases the endoplasmic reticulum (ER) protein folding capacity and decreases its unfolded protein load. XBP1 is a key mediator of the prosurvival arm of UPR and is activated via an unusual mechanism (1). In unstressed cells, the cytoplasmic transcript XBP1u encodes an inactive unstable transcription factor (XBP1u) with DNA-binding and dimerization domains but without the activation domain required to activate target genes (Fig. 1A) (3, 4). Once activated by stress, the endoribonuclease and protein kinase IRE1α cleaves 26 nucleotides off XBP1u. Subsequent joining of the RNA halves by the RNA ligase RtcB (5–7) produces the XBP1s mRNA and causes a frameshift that enables translation of XBP1s. XBP1s is a stable protein containing the same DNA-binding and dimerization domains as XBP1u but in association with functional transcription-activating and nuclear-localization domains (Fig. 1A). Hence, XBP1s is able to translocate to the nucleus and activate target genes. XBP1s activates a set of genes that contain ER stress response element (ERSE) or unfolded protein response element (UPRE) signals in their promoters (8–10). XBP1s has a major role in the induction of genes encoding chaperones or molecules involved in lipid biosynthesis and ER-associated degradation (9, 11–13).
FIG 1.

Rotavirus infection induces cytoplasmic splicing of XBP1. (A) Schematic structures of the XBP1u and XBP1s mRNAs. The general organization of the XBP1 gene is indicated with exons as gray boxes, noncoding regions are indicated as white boxes, and introns are indicated as lines (not to scale). The mRNA encoding XBP1u is produced by canonical nuclear splicing and then exported to the cytoplasm (black arrow) to be translated (white arrow). The XBP1u protein translated from XBP1u mRNA is 261 amino acids long and contains a DNA-binding domain (hatched box). The mRNA encoding XBP1s is produced by an unconventional cytoplasmic splicing (gray arrow) of XBP1u mRNA at exon 4 (light gray) catalyzed by the IRE1α endoribonuclease. The XBP1s protein is 376 amino acids long and contains DNA-binding and transactivating (black box) domains. (B) Detection of XBP1u splicing by IRE1α. The positions of the primers XBP1dir and XBP1rev on the XBP1 gene and mRNAs are indicated. The RT-PCR products (424 and 398 bp) obtained using these primers on RNA purified from unstressed MA104 cells (lane C) or MA104 cells treated with thapsigargin (400 nM) for 3 and 9 h (T3, T9) are illustrated. (C) The DNA products obtained by RT-PCR from RNA extracted from mock-infected cells or from cells infected (MOI of 10) with rotavirus RF or RRV for the indicated time (in hours) were analyzed by agarose gel electrophoresis. The top panel shows the XBP1 RT-PCR products, and the bottom panel shows the GAPDH RT-PCR products used as a loading control. The 0′ lane corresponds to untreated cells, and the 0 lane corresponds to mock-infected cells. The sizes of the molecular weight markers (MW) are indicated in base pairs on the left side.
IRE1α activation and subsequent XBP1 unconventional splicing are also triggered by the innate immune response (14). Some Toll-like receptors that sense pathogen-associated molecular patterns specifically activate the IRE1α branch and its downstream target XBP1 (15, 16). XBP1 has also been implicated in stimulation of the transcription of the beta interferon gene (17, 18). These observations uncover a critical role of XBP1 in mammalian host defenses and the innate immune response.
Rotavirus is the main cause of gastroenteritis in humans and leads to an estimated 215,000 death each year (19). Rotavirus infection triggers the UPR, but the response is modulated at the translational level (20, 21). Rotavirus infection induces the shutoff cellular protein synthesis in a complex way that includes impairment of nuclear RNA export (22), phosphorylation of eukaryotic translation initiation factor eIF2α (23), and saturation of the translation machinery by viral mRNAs (24). Small interfering RNA (siRNA) experiments have shown that both the translation shutoff and nuclear export arrest are linked to the viral protein NSP3 (22, 25). Rotavirus NSP3 is a viral translation enhancer and surrogate of the cellular cytoplasmic PABP (PABPC) (26) encoded by rotavirus gene 7. Upon homodimerization NSP3 recognizes the 3′ end of viral mRNAs (27–29) and interacts simultaneously with the translation initiation factor eIF4G (30–32), thus strongly stimulating viral mRNA translation (24, 33, 34). Interaction of NSP3 with eIF4G leads to the eviction of PABPC from eIF4G (32, 35) with its subsequent release from poly(A) RNA and relocalization into the nucleus (23, 35, 36).
Investigation of XBP1 activation as an host immune response to rotavirus infection lead us to analyze first the unconventional splicing of XBP1 during a bovine rotavirus infection. This investigation revealed that with several rotavirus strains, XBP1 RNA undergoes an additional alternative, IRE1α-independent splicing. We show that this splicing corresponds to exon 4 skipping during conventional nuclear splicing and that this phenomenon is genetically linked the viral protein NSP3 and, more precisely, to its eIF4G-binding domain. The capability to skip XBP1 exon 4 is linked to the faculty of the viral strain to induce early nuclear relocalization of PABPC. Moreover, we show that the XBP1 mRNA that results from exon 4 skipping produces an active form of the XBP1 transcription factor.
RESULTS
Rotavirus infection induces exon skipping in XBP1 RNA.
The unconventional, cytoplasmic splicing of XBP1 mRNA induced by IRE1α activation occurs in exon 4 and can be monitored by reverse transcription-PCR (RT-PCR) using primers positioned in exons 3 and 5 of the pre-mRNA (Fig. 1A). Our primers (Table 1) yielded two main RT-PCR products. A DNA fragment of 424 bp was mainly observed with RNA from unstressed control cells (Fig. 1B, lane C) and corresponds to the XBP1u (unspliced) mRNA. A 398-bp DNA fragment was observed with RNA from stressed cells (treated with thapsigargin for 3 h [Fig. 1B, lane T3] or 9 h [Fig. 1B, lane T9]) and corresponds to the XBP1s (cytoplasmic spliced) mRNA. A third minor product running above the two others (indicated as “u+s” in all figures) was frequently obtained; this product corresponds to an XBP1u-XBP1s double-stranded hybrid DNA fragment (37).
TABLE 1.
Oligonucleotides used for PCR in this study
| Oligonucleotide | Sequence (5′–3′) | Gene | Usage |
|---|---|---|---|
| XBP1dir | CTGGAACAGCAAGTGGTAGA | XBP-1 | Detection of XBP1u, XBP1s, and XBP1es |
| XBP1rev | CTGGGTCCTTCTGGGTAG | XBP-1 | |
| xbpmacEX2-5dir | TAGCAGCTCAGACTGCCAGA | XBP-1 | Alternative splicing exons 2 to 5 of XBP1 |
| xbpmacEX2-5rev | ACTGGGTCCAAGTTGTCCAG | XBP-1 | |
| xbpmacEX1-3dir | GGGGCCCCTAAAGTACTGCT | XBP-1 | Alternative splicing exons 1 to 3 of XBP1 |
| xbpmacEX1-3rev | TCTTTAGCAACCAGGGCATC | XBP-1 | |
| snoU6up | CCAATGATGAGTTGCCATGC | snoU6 | Control of cell fractionation |
| snoU6lo | GCCCCTCAGATCTTCATGTG | snoU6 | |
| H_SS_475F22_qPCR | CGGAAGCCAAGTCTGATATCCT | XBP-1 | Quantitative detection of XBP1es (junction exons 3 to 5) |
| H_SS_625R21_qPCR | CTGATGACGTCCCCACTGACA | XBP-1 | Quantitative detection of XBP1es (exon 5) |
| GAPDH dir | GGAGCCAAAAGGGTCATCATCTC | GAPDH | Control for PCR loading and cell fractionation |
| GAPDH rev | TCCACAGTCTTCTGGGTGGCAG | GAPDH | |
| NPR2esmacup | CATGTTTGGTGTTTCCAGCTTCC | NPR2FL | Alternative splicing in NPR2FL |
| NPR2esmaclo | CGGGTTAGCTCAATGCGCTT | NPR2FL | |
| GOLGAesmacup | TCAAGAGAACCTACTTAAGCGTTGTAAGG | GOLGA4 | Alternative splicing in GOLGA4 |
| GOLGAesmaclo | TGAGCAATTTCTTCTTCTTTCATTTCC | GOLGA4 | |
| BAZesmacup | TGCTCTGATGGTTTTGGAGTTCC | BAZ1A | Alternative splicing in BAZ1A |
| BAZesmaclo | CGTTTTTGATATCTATACTTTGC | BAZ1A | |
| PAMesmacup | TGTCCCAGTGCCCGGG | PAM | Alternative splicing in PAM |
| PAMesmaclo | GGTGAAATCCACAGCTGACTTGG | PAM | |
| NCOAesmacup | AGGCAACACGACGAAACAGCCATACC | NCOA1 | Alternative splicing in NCOA1 |
| NCOAesmaclo | TCTGGCATAAGATGGCTCTCTGCCC | NCOA1 |
To establish whether XBP1 is activated during rotavirus infection, we performed RT-PCR with RNA purified from cells infected with the RF or RRV strains of rotavirus for 3, 6, or 9 h at a multiplicity of infection (MOI) of 10 (Fig. 1C). For mock-infected cell controls, the RT-PCR product of XBP-u (424 bp) was predominant, showing that IRE1α-dependent XBP1 splicing was not induced and, consequently, that the cells were not stressed by mock infection. Infection with the bovine RF strain of rotavirus induced XBP1 cytoplasmic splicing as early as 3 h postinfection, whereas XBP1 cytoplasmic splicing was observed only after 6 h of infection with the simian RRV strain, suggesting that the two strains could differ in their capacities to induce stress. Surprisingly, a fourth, shorter band of ∼280 bp was observed only with the RF (bovine) strain starting 6 h after infection (noted as “es” in Fig. 1C). This 280-bp band did not originate from cellular DNA since the primers used for PCR were positioned in exons 3 and 5, which are separated by two introns of 884 and 314 bp (Fig. 1A) and therefore would amplify a much larger fragment from cellular DNA. An XBP1 pseudogene exists in the human genome (ENSG00000249947), but its nucleotide sequence is sufficiently divergent that it would not be amplified by the primers used here. The es DNA fragment resulted from the amplification of a reverse transcription (RT) product, since it was not observed in the absence of the RT step (Fig. 2A, lanes 3 and 4). Moreover, the es DNA fragment resulted from the amplification of a cellular polyadenylated RNA because the same PCR product was obtained when an oligo(dT) primer was used (instead of random primers) for the RT reaction (Fig. 2A, lane 2) and because rotavirus genes and mRNAs are not polyadenylated.
FIG 2.

XBP1es is a poly(A) RNA that results from exon skipping, not cytoplasmic splicing. (A) RNA purified from mock (RF–)- or rotavirus RF (RF+)-infected cells was used as the template for a reverse transcription reaction with (RT+) or without (RT–) reverse transcriptase and using oligo(dT) (dT) or random hexanucleotides (Rd) as primers. DNA products obtained by the PCRs using either XBPdir and XBPrev primers or GAPDH primers were analyzed by electrophoresis on the same agarose gel. The sizes of the molecular weight markers (MW) are indicated (in base pairs) on the left side. (B) A chromatogram from Sanger sequencing of the XBP1es RT-PCR DNA product is shown below a schematic representation of the organization of the XBP1 gene and that of XBP1es RNA resulting from exon 4 skipping. The vertical line marks the junction of the exon 3 and 5 sequences. A schematic representation of the putative translation product of XBP1es RNA is shown with the DNA-binding (hatched box) and transactivating domains (black box). The numbers indicate amino acid positions. (C) RNA purified from MA104 cells infected (for 9 h) or not by the RF strain of rotavirus and treated with the IRE1α inhibitor STF-083010 in DMSO (60 μM), with the IRE1α activator thapsigargin (400 nM) or with DMSO (used as vehicle for STF-083010), was subjected to RT-PCR with XBP1 primers and GAPDH primers. The PCR DNA products were analyzed by electrophoresis on two agarose gels. The sizes of the molecular weight markers (MW) are indicated (in base pairs) on the left.
Cloning and sequencing of the 280-bp RT-PCR fragment showed that it was the product of the amplification of an XBP1 RNA lacking exon 4 (Fig. 2B). The 280-bp fragment and the corresponding RNA are indicated by “es” for “exon skipping” here.
To ascertain that XBP1es resulted from a canonical nuclear alternative splicing of the XBP1 pre-mRNA and not from a novel unconventional cytoplasmic splicing by IRE1α, a specific inhibitor of the endonuclease activity of IRE1α (STD 083010 [38]) was used in combination with the UPR inducer thapsigargin and rotavirus infection (Fig. 2C). The basal and thapsigargin-induced IRE1α-dependent splicing of XBP1u were both efficiently inhibited by STD 083010, as indicated by the disappearance of the XBPu (398-bp) RT-PCR product (Fig. 2C, lanes 1 and 3 and lanes 2 and 4). Treatment with dimethyl sulfoxide (DMSO), which was used as the vehicle for STD 083010 and thapsigargin, did not modify the splicing of XBP1u in mock-infected (compare lanes 1 and 5) or RF-infected (lanes 6 and 8) cells. STD 083010 also inhibited the cytoplasmic splicing of XBP1u (to obtain XBP1s) induced by rotavirus infection (lanes 6 and 7) but had no effect on the XBP1es 280-bp RT-PCR DNA fragment (lane 7).
In addition to the nucleotide sequence of this fragment, this last result indicated that the XBP1es 280 bp RT-PCR product corresponded to a conventional nuclear splicing that skipped exon 4 of the XBP1 pre-mRNA.
Exon skipping is specific to XBP1 exon 4.
To assess whether exon skipping occurred in other locations of the XBP1 RNAs, RT-PCR using primers positioned in exons 1 and 3 and exons 2 and 5 (Table 1) was applied to RNA from mock- and RF- infected cells. Only the expected 401-bp RT-PCR product was observed with the pair of primers 1 and 3, showing that skipping of exon 2 was not induced by rotavirus infection. RT-PCR amplification with primers positioned in exons 2 and 5 produced the expected three fragments corresponding to XBP1u, XBP1s, and XBP1es, thus showing that no alternative splicing other than exon 4 skipping occurred in the XBP1 RNAs (data available upon request).
Induction of XBP1 exon skipping depends on the viral strain.
XBP1 exon skipping was induced by bovine RF but not by simian RRV (Fig. 1C). Quantification of the XBP1es RNA by RT-qPCR (Fig. 3A) showed that its expression was 3-fold higher in cells infected with the RF strain than in the mock-infected control or cells infected with the RRV strain. To determine whether XBP1es induction could be modified by the viral load, the same RT-qPCR was applied to RNA extracted from cells infected with MOIs of 1, 5, 10, or 50 for 9 h (Fig. 3B). An MOI of 5 seemed to be optimal for inducing XBP1es with the RF strain. However, increasing the MOI did not increase the level of XBP1es with the RRV simian strain, and the value remained close to that of the mock-infected control. These results firmly established that the induction of XBP1es did not depend on the initial viral load but exclusively on the viral strain.
FIG 3.

XBP1 exon skipping depends on the rotavirus strain but not on the multiplicity of infection. The amount of XPB1es RNA was quantified by RT-qPCR using primers specific for XBP1es (Table 1) and RNA extracted from mock-infected cells or from cells infected with the rotavirus RF or RRV for the indicated time (in hours postpostinfection) (A) or at the indicated MOI (B). The relative amount of XBP1es RNA is presented. The means ± the standard errors of the mean (SEM) for three independent experiments performed in triplicate are shown. Asterisks indicate significant differences from the mock-infected control (P < 0.05) as determined by a two-tailed Student t test.
Induction of XBP1 exon skipping depends on gene 7.
The identification of two strains with different capabilities to induce XBP1 exon skipping (here named “ES phenotypes,” with RF ES+ and RRV ES–) and the segmented nature of the rotavirus genome provided an opportunity to identify the genetic origin of this difference by using viral reassortants. The eleven possible monoreassortant viruses bearing one gene from the RRV strain and 10 genes from the RF strain were isolated from the progenies of an RF and RRV mixed infection and plaque purified twice. The ES+ or ES– phenotype of each virus was established by RT-PCR as described above after 9 h of infection (MOI of 10) of MA104 cells (Fig. 4A). Only the monoreassortant-bearing gene 7 from RRV had an ES– phenotype, indicating that this gene controlled XBP1 exon skipping. To further substantiate this result, the reverse monoreassortant RRV/RF07 was also identified and cloned. As illustrated in Fig. 4B, this monoreassortant had an ES+ phenotype similar to the wild-type (wt) RF virus. This result showed that gene 7 is the main determinant of the ES phenotype.
FIG 4.

Rotavirus gene 7 determines the ES phenotype. (A) RNA purified from MA104 cells infected (9 h, MOI of 10) with the different monoreassortants and parental strains was subjected to RT-PCR with XBP1 and GAPDH primers. The PCR DNA products were analyzed by electrophoresis and ethidium bromide staining on two agarose gels (middle and lower panels). The sizes of the molecular weight markers (MW) in base pairs are indicated on the left. (B) RNA purified from mock-infected MA104 cells (lane 5) or cells infected with either an RRV monoreassortant carrying gene 7 from RF (RRV/RF07 lane 2), an RF monoreassortant carrying gene 7 from RRV (RF/RRV07 lane 3), or the RRV (lane 1) or RF (lane 4) parental strains was subjected to RT-PCR with XBP1 and GAPDH primers. The PCR DNA products were analyzed by electrophoresis and ethidium bromide staining on two agarose gels. The sizes of the molecular weight markers (MW) in base pairs are indicated on the left.
Induction of XBP1 exon skipping depends on the gene 7 genotype.
Rotavirus strains are classified according to the nucleotide sequence of each gene, and at least 16 genotypes have been identified for gene 7 (39–41). To establish whether the ES phenotype is correlated with the gene 7 genotype or with the animal/human origin of a strain, the ES phenotypes of eight cell culture-adapted rotavirus strains belonging to seven T genotypes were established. Two human (Au-1 [T3] and DS1 [T2]), two simian (SA11-4F [T3] and RRV [T5]), and one avian (PO-13 [T4]) strain were identified as ES–, whereas the human WA (T1), porcine OSU (T1), and bovine UK (T7) and RF (T6) strains were identified as ES+ (Fig. 1A and data available upon request). It is interesting that genotypes T1, T6, and T7 (ES+) and T5, T3, T2, and T4 (ES–) belong to the two main phylogenetic groups (40). These results showed that genetic proximity is the determinant of the ES phenotype rather than the animal or human origin of the strain.
XBP1 exon skipping parallels the nuclear localization of PABPC induced by rotavirus infection.
The interaction of rotavirus NSP3 with eIF4G occurs at the expense of PABPC binding to eIF4G, and PABPC is relocalized into the cell nucleus upon rotavirus infection (23, 32, 35). Since PABPC nuclear localization perturbs the transcription (42) and the export and maturation (43, 44) of nuclear RNAs, we investigated whether a difference in PABPC localization could be observed in RF- versus RRV-infected cells and in gene 7 reassortants. At 9 h postinfection (hpi), viruses bearing gene 7 from RF (RF and RRV/RF07 in Fig. 5A) were clearly more efficient at sending PABPC to the nucleus than viruses bearing gene 7 from RRV (RRV and RF/RRV07, Fig. 5A). Quantification of PABPC in the nucleus at different times postinfection (Fig. 5B) showed that the RRV and RF/RRV07 viruses sent PABPC to the nucleus more slowly than the RF and RRV/RF07 viruses. For example, PABPC was not detected in the nucleus before 9 hpi with RRV or RF/RRV07, whereas PABPC was present in the nucleus at 6 hpi with the RF virus. However, with the RRV/RF07 virus, XBP1 exon skipping was delayed in comparison with the parental RF virus, and exon skipping was not observed before 9 hpi (Fig. 5C). Comparing PABPC nuclear localization and the detection of XBP1es in cells infected with the same viruses (Fig. 5B and C) indicated that XBP1 exon skipping was detectable when the ratio of nuclear PABPC reached a threshold (±0.2 in Fig. 5B); this threshold was reached before 6 hpi with the RF virus and between 6 and 9 hpi with the RRV/RF07 reassortant, whereas RRV and RF/RRV07 never exceeded this threshold. This results showed that PABPC nuclear localization kinetics correlates well with the ES phenotype but that the genetic background of gene 7 can influence its onset.
FIG 5.

Nuclear translocation of PABPC and XBP1 exon skipping in parental RF and RRV and gene 7 monoreassortants. (A) Localization of PABPC in rotavirus-infected cells. MA104 cells infected (or mock infected) with bovine RF, rhesus RRV, or gene 7 monoreassortants for 9 h were fixed and incubated with NSP2- and PABPC-specific antibodies. Secondary antibodies coupled to the Alexa fluorophore stains NSP2 (red) and PABPC (green). Nuclei were stained blue with DAPI. (B) Quantification of nuclear PABC1 in rotavirus-infected cells. Images such as those shown in panel A were taken at 3, 6, and 9 h after infection (hpi) with the indicated virus and analyzed. The ratio of nuclear to total green (PABPC) fluorescence (corrected for background) is reported. The results are mean values ± the SEM of three fields with >50 cells. (C) Kinetics of XBP1 splicing. The XBP1 DNA products obtained by RT-PCR of RNA extracted from mock-infected cells or from cells infected (MOI of 10) for 3, 6, and 9 h with the indicated parental or monoreassortant virus were analyzed by agarose gel electrophoresis, together with the GAPDH PCR control. The sizes of the molecular weight markers (MW) are indicated in base pairs on the left.
Induction of XBP1 exon skipping by recombinant chimeric gene 7.
The eIF4G-binding domain of NSP3 is required for the nuclear relocalization of PABPC (35). To establish whether the eIF4G-binding domain of NSP3 determines the ES phenotype, the eIF4G-binding domains (from amino acid 247) of the RRV or RF gene 7 were swapped to construct the chimeras RF/247RRV and RRV/247RF. The chimeras were then introduced into a SA11 genetic background by reverse genetics (45). Recombinant rotavirus carrying the chimeric RRV/247RF gene 7 gave an XBP1es PCR signal at least as strong as the XBP1es signal obtained from RF (Fig. 6, lanes 5 and 2), and the recombinant rotavirus carrying the chimeric RF/247RRV gene 7 was ES–, thus demonstrating that the eIF4G-binding domain of NSP3 RF is a determinant of the ES phenotype.
FIG 6.

ES phenotypes of recombinant viruses bearing NSP3 chimeras The DNA products obtained by RT-PCR (with XBP1 or GAPDH primers) of RNA extracted from mock-infected cells or from cells infected (MOI of 10 [9 hpi]) with RF or RRV or a recombinant virus (SA11 background, bearing RRV-247-RF or RF-247-RRV gene 7 chimeras) were analyzed by agarose gel electrophoresis and ethidium bromide staining, together with the GAPDH PCR control.
Alternative splicing during rotavirus infection.
Exon skipping is the most common mode of alternative splicing of mammalian pre-mRNAs. Is the exon skipping on XBP1 mRNA specific to the XBP1 gene or are other genes affected during rotavirus infection? To address this question, five genes (PAM, NPR2, GOLGA4, NCOA1, and BAZ1A), which are a priori not linked to rotavirus replication but susceptible to alternative splicing (46) and that are transcribed in MA104 cells, were investigated (Fig. 7). NPR2 and GOLGA4 did not show alternative splicing in infected or uninfected MA104 cells, and PAM and BAZ1 alternative splicing was not affected by infection. In the case of NCOA1, alternative splicing was clearly enhanced by rotavirus infection but was independent of the ES phenotype of the virus. Although this study was limited to a small number of genes, the results show that splicing can be perturbed by rotavirus infection.
FIG 7.

Alternative splicing of selected genes in ES– or ES+ rotavirus-infected cells. RNA purified from mock-infected MA104 cells (lanes “M.”) or from MA104 cells infected with the RF (ES+) or RRV (ES–) strain of rotavirus was used for RT-PCR with primers specific for the cellular gene indicated on the top of the lanes. On the left, the sizes of the molecular weight markers (MW) are indicated in base pairs. The expected sizes (in base pairs) of the RT-PCR products are indicated on the right.
Cellular localization of XBP1es RNA.
Rotavirus infection blocks the nucleocytoplasmic trafficking of RNA (22) and perturbs the localization of several cellular proteins (23, 47, 48). To establish whether XBP1es RNA was present in the cytoplasm of infected cells or remained in the nucleus, XBP1es was detected by RT-PCR in nuclear and cytoplasmic fractions of rotavirus-infected cells. To assess the quality of the subcellular fractionation, primers for the (mainly) nuclear U6 RNA and (mainly) cytoplasmic GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA were used as controls (Fig. 8). As expected for efficient subcellular fractionation, all of the XBP1s RNA and most of the GAPDH mRNA was present in the cytoplasm (compare lanes 2 and 3 and lanes 7 and 8), and most of the U6 RNA was present in the nucleus (lanes 4 and 5). A small fraction of XBP1es was detected in the cytoplasmic fraction, showing that the nuclear export of XBP1es RNA was largely impaired.
FIG 8.
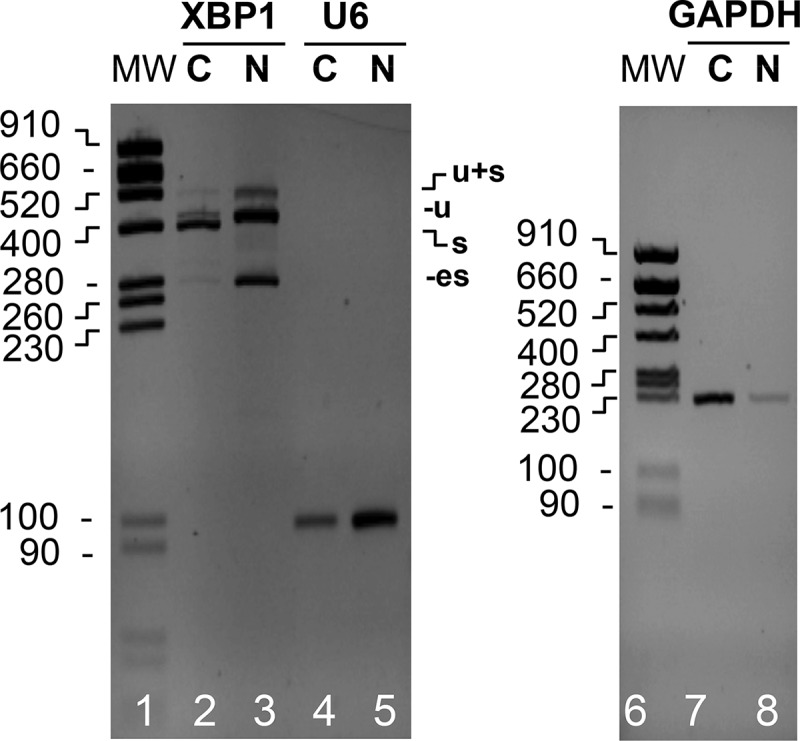
Localization of XBP1es RNA. RNA purified from the cytoplasmic (C) or nuclear (N) fractions of MA104 cells infected with the RF strain of rotavirus (MOI of 10, 9 h postinfection) was subjected to RT-PCR using XBP1, U6, or GAPDH primers. The PCR DNA products were analyzed by electrophoresis and ethidium bromide staining on two agarose gels. The positions of the XBP1u, XBP1s, and XBP1es PCR DNA products and of the molecular weight markers (MW; sizes are denoted in base pairs) are indicated.
Transactivation properties of the XBP1es translation product.
Skipping exon 4 in XBP1u mRNA induces the same frameshift as cytoplasmic processing by IRE1α, and translation of an XBP1 mRNA lacking exon 4 would lead to the production of a XBP1es protein of 336 aa with an NLS and a transactivating domain identical to those of XBP1s (Fig. 9A).
FIG 9.

XBP1es and XBP1s proteins have similar transactivating properties. (A) Expression of XBP1u, XBP1s, and XBP1es in HEK293 cells. On the left, schematic representations of the XBP1u, XBP1s and XBP1es cDNA constructs used are shown together with schematic representations of the proteins they encode (numbers indicate amino acid positions). The functional DNA-binding (hatched box) and transcription-activating domains (black box) are indicated. On the right, lysates of HEK293 cells transfected with XBP1 cDNA constructs were resolved by SDS-PAGE and probed with an antiserum directed against XBP1. The numbers indicate the positions and molecular weights (in kDa) of the markers. The dotted line indicates the expected position of XBP1u. (B) Transactivating properties of XBP1u, XBP1s, and XBP1es. MA104 cells were cotransfected with a combination of three expression plasmids: (i) either one of the XBP1 cDNA constructs described in panel A or a control plasmid encoding eGFP; (ii) a reporter plasmid carrying the firefly luciferase gene under the control of the CHOP, XBP1, GRP94, or GRP78 promoter; and (iii) a normalization plasmid encoding the Renilla luciferase gene under the control of the CMV promoter. Luciferase activities were measured 48 h after transfection and reported as the firefly/Renilla ratio with the GFP control set to 100. The mean values ± the SEM for three independent experiments performed in triplicate are shown. Bars: XBP1u (UNSPL), light gray; XBP1s (SPL), dark gray; XBP1es (ES), black. Asterisks indicate significant differences compared to the XBP1u-transfected sample (P < 0.05) as determined by a two-tailed Student t test. NS, not significant.
To test whether the XBP1es protein has transactivating properties similar to those of XBP1s on ERSE-containing promoters, XBP1es and XBP1s cDNAs were constructed from the mouse XBP1u cDNA (49) by site-directed mutagenesis. The three constructs (XBP1u, XBP1s, and XBP1es) and a plasmid control encoding eGFP were transfected in HEK293 cells, and XBP1 proteins were detected by Western blotting using an anti-XBP1 antibody raised against amino acids 76 to 263 of the protein. A protein product of the expected size and of similar intensity was detected in cells transfected with XBP1s and XBP1es cDNAs (Fig. 9A). In cells transfected with the cDNA encoding XBP1u, no protein product was visible most probably due to the instability of XBP1u (3).
MA104 cells were then cotransfected with reporter plasmids encoding the firefly luciferase gene under the control of the XBP1, GRP78 or GRP94 promoters (50). A reporter plasmid with the firefly luciferase gene under the control of the CHOP promoter (50), which is not targeted by XBP1 (51) but is induced after prolonged stress, was used as a negative control. To standardize the assay, the reporter renilla luciferase under the control of the cytomegalovirus (CMV) promoter was included in all transfections. As illustrated in Fig. 9B, XBP1u had only very limited trans-acting activity on the GRP78, GRP94, and XBP1 promoters and no activity on the CHOP promoter. Conversely, XBP1es and XBP1s both transactivated the GRP78, GRP94, and XBP1 promoters to similar levels but had no effect on the CHOP promoter. Thus, the XBP1es protein was functional and, at first approximation, had transactivation properties similar to XBP1s transcription factor.
DISCUSSION
We have shown that during infection with several rotavirus strains, a new form of XBP1 RNA is synthesized that corresponds to an RNA lacking exon 4 and potentially encodes a functional XBP1 transcription factor. We correlated XBP1 exon skipping with the early induction of the nuclear localization of PABPC. Using classical and reverse genetics, we identified NSP3 and, more precisely, its eIF4G-binding domain as the trigger of XBP1 exon skipping.
The nuclear localization of PABPC requires the release of the bound poly(A) RNA and the subsequent unmasking of nuclear import signals within its RNA recognition motifs (52). Since the interaction of PABPC and eIF4G is allosterically regulated by poly(A) binding to PABPC (53), it is probable that eviction of PABPC from eIF4G by NSP3 interaction with eIF4G (32) also disrupts the PABPC-poly(A) interaction and thus unmasks the PABPC nuclear import signal. The difference in the kinetics of nuclear localization of PABPC upon rotavirus infection may originate from a difference in the strength of the eIF4G-NSP3 interaction and hence the amino acid sequence of the NSP3 eIF4G-binding domain. This mechanism can explain the segregation of the ES phenotype according to the T genotype of the viral strain and the eIF4G-binding domain of NSP3.
The nuclear roles of PABPC are not well known (43). PABPC is not required for mRNA export from the nucleus (54) but interacts with pre-RNA (44), and an increased level of PABPC in the nucleus results in hyperadenylation and nuclear retention of transcripts (55), probably by interfering with the functions of nuclear PABP. Our observations that the splicing of cellular genes can be altered by rotavirus infection are thus consistent with alteration of the nuclear functioning by PABPC1 localization in the nucleus. Relocalization of PABPC into the nucleus have been observed in stressed cells after heat shock (56) or UV irradiation (54), and in both cases extensive changes in RNA splicing have been observed (57). Several other viral infections induce PABPC relocalization (58–60). In the case of HSV1 infection, nuclear RNA export was affected, and profound modifications of the splicing of cellular genes were observed (61). In the case of orthoreovirus, another double-stranded RNA (dsRNA) virus, infection perturbs RNA splicing (62, 63), but PABPC localization has not been studied. Our observation that the splicing of the NCOA1 RNA is affected by rotavirus infection regardless of the ES phenotype might indicate that rotavirus infection is more perturbative for RNA splicing than reported here. In this regard, it is interesting that the nuclear localization of the RNA processing factor sam68 is modified during rotavirus infection (36) and that the localization of several cellular RNA binding proteins, including hnRNPs (64), is perturbed during rotavirus infection (47, 48). A thorough study of RNA splicing during rotavirus infection using deep sequencing is required to enlighten this point.
However, skipping of exon 4 of XBP1 is observed only with some strains of rotavirus. The biological significance that only some strains exhibit this effect on XBP1 splicing is still puzzling since this correlates neither with the animal or human origin of the virus nor with the capacity to replicate outside the intestines (65), which has also been linked to the origin of gene 7 (SA11 replication is restricted to the intestine but not RRV, and this capacity is genetically linked to gene 7; however, SA11 and RRV are both ES– viruses). Nor does this finding correlate with the kinetics of cell protein synthesis shutoff (see Fig. 4 in reference 26): cell protein synthesis is halved after 4 h with RF, but only 6 h after infection with RRV, whereas gene 7 monoreassortants have intermediate phenotypes.
We only correlated the ES phenotype with the kinetics of PABPC nuclear accumulation that itself correlates with the eIF4G-binding domain of the NSP3 protein. Whether this property provides an advantage for viral replication in vivo, requires a more in depth comparison of the replication of ES– or ES+ viruses.
Taking into account the prominent role of XBP1 in stress and immune responses, one cannot totally discard the hypothesis of a role of XBP1 exon skipping as a specific cell response to infection or to nuclear localization of PABPC, especially since the XBP1es protein is a functional transcription factor.
The XBP1es protein is probably not highly expressed in rotavirus-infected cells because its RNA is detected at 6 h after infection, when the cellular protein synthesis shutoff induced by rotavirus is already fully efficient (26, 36). Furthermore, our fractionation experiments showed that most of the XBP1es RNA remains in the nucleus, where translation is not efficient (66).
However, it cannot be totally excluded that a small amount of the XBP1es transcription factor is synthesized before completion of translation shutoff. The expression of the XBP1es protein, which has transactivation properties similar to those of XBP1s, may be a viral mechanism to favor gene expression of proviral factors. Consistent with this idea, rotavirus is dependent on HSP chaperones (67–69), and activation of the IRE1/XBP1 arm of the UPR early in flavivirus infection reduces virus cytotoxicity (70). We have shown here that the XBP1es protein could transactivate the chaperone GRP94 and GRP78 promoters similarly to the XBP1s protein. However, studies of cellular gene induction during rotavirus infection are not consistent: GRP78 and GRP94 mRNA and protein levels decrease in cells infected with the OSU (ES+) strain (20) but increase during SA11 (ES–) infection (71).
Studies have uncovered a critical role of XBP1 in mammalian host defenses and the innate immune response (14–16, 72). For example, some Toll-like receptors specifically activate the IRE1α branch (but not the PERK or ATF6 branches) of the UPR and its downstream target XBP1 (15, 16). XBP1 is also involved in the transcription stimulation of the beta interferon gene (17, 18). Exon skipping in XBP1 could be another means of inducing the synthesis of a functional XBP1 transcription factor in the course of a cellular response to infection. Relevant to this hypothesis, alternative splicing has been shown to regulate host antiviral responses (73, 74), and pathways linking splicing regulation with the antiviral response are now considered emerging mechanisms of evasion in viral infection (72, 74–77).
XBP1 is ubiquitously expressed (78) and is involved in diverse human diseases such as cancer (76, 79), neurodegeneration (80), obesity (81), and inflammatory diseases (82). Regulation of XBP1 activities in an IRE1α-independent manner by the regulation of exon skipping might take place in physiological conditions other than rotavirus infection. Indeed, similar XBP1 exon skipping was found to occur at the gastrula and early neurula stages of Xenopus development (83). Moreover, cDNAs with the same exon skipping are present in EST libraries from mice (GenBank BQ884979, 948 bp) and adult swine bone marrow cells (GenBank HX206384, 628 bp and with a 5′ untranslated region), showing that XBP1 exon skipping could probably occur in physiological conditions other than rotavirus infection.
MATERIALS AND METHODS
Cells and viruses.
Embryonic rhesus monkey kidney MA104 cells were maintained in Eagle minimum essential medium (EMEM) supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. COS7 cells were maintained in Dulbecco modified EMEM.
The bovine RF and UK, human WA, porcine OSU, and simian RRV and SA11-4F strains of group A rotavirus were used to infect MA104 cells. Virus titers were determined by counting immunoperoxidase/carbazole-stained infected cells in 96-well plates 24 h after infection with a 5-fold dilution of the viral inoculum.
Infections were performed at an MOI of 10 PFU/cell in EMEM in the presence of trypsin (0.44 μg/ml) and antibiotics but without serum.
To avoid stress induced by the culture medium replacement, the cells were washed with fresh serum-free medium the day before use, and a small volume of the overnight medium was left on the cell before proceeding to infection. Mock infections were made by adding trypsin to the overnight medium.
Plasmid construction and mutagenesis.
The XBP1s and XBP1es cDNAs were obtained by site-directed mutagenesis using PFU polymerase and DpnI digestion on the mouse XBP1u cDNA cloned into pCMV2 (49). Mutations were confirmed by nucleotide sequencing of the whole coding sequences.
To minimize structural perturbations in the chimeric NSP3 proteins, the chimeras were engineered using the KpnI site preexisting in the RF gene and positioned in regions encoding identical amino acid sequences in the RF and RRV genes. A silent T-to-C mutation was introduced by site-directed mutagenesis at position 769 of the RRV gene 7 cDNA cloned into the pRiboz vector (84) to create a KpnI site. The KpnI-EcoRI fragment of the RRV plasmid was then exchanged with the KpnI-EcoRI fragment of the RF plasmid, leading to chimeric RRV/RF genes 07 cloned into pRiboz. The pRiboz RF-Kp-RRV07 plasmid thus encodes a chimeric NSP3 protein with amino acids 1 to 247 from RF and amino acids 248 to 313 from RRV NSP3, and the pRiboz RRV-Kp-RF07 plasmid encodes a chimeric NSP3 protein with amino acids 1 to 247 from RRV and amino acids 248 to 313 from RF NSP3.
Cell RNA purification, RT-PCR, and RT-qPCR.
Total RNA from cells was purified with an RNeasy RNA isolation kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol and quantified using a NanoDrop ND-1000 spectrophotometer. Then, 1 μg of total RNA was used for reverse transcription with SuperScript II reverse transcriptase (42°C, 50 min, 200 U) and 10 μM random oligonucleotides or oligo(dT) (when indicated) primers. The PCR conditions were as follows: denaturation at 94°C for 30 s, hybridization at 50°C for 30 s, and elongation at 72°C for 30 s, with 35 cycles for XBP1 and 25 cycles for GAPDH. The primers used for PCR were chosen from the Macaca mulatta (rhesus monkey) genome using Primer 7 software and are indicated in Table 1. To specifically quantify the XBP1es isoform, a sense primer spanning the exon 3-to-5 junction sequence (Fig. 2B) (11 nucleotides on each side) and a reverse primer positioned in exon 5 (PCR product, 169 bp) were used.
A XBP1es qPCR was performed using 5 μl of a 1:5 dilution of reverse-transcribed RNA (1:15 for GAPDH) with MESA green qPCR MasterMix Plus (Eurogentec) with a MxPro3000 (Stratagene) apparatus and Comparative Quantification software. The efficiency of the primers (Table 1) was >98% (26). Results were analyzed according to the 2–ΔΔCT method with GAPDH mRNA as an endogenous reference (85) since the GAPDH threshold cycle (CT) values are not significantly modified up to 9 h after infection with either of the two strains used (26).
dsRNA extraction and RT-PCR.
Viral dsRNA was purified from 0.4 ml of infected cell culture medium by TRIzol (Life Technologies) or RNAzol (Sigma-Aldrich) RT, and 200 ng of RNA was analyzed by electrophoresis for 24 h on a 20-cm 10% polyacrylamide gel in Tris-glycine-SDS buffer (86). RNA was detected by silver staining using a Silver Stain Plus kit from Bio-Rad. To determine the parental origin of some genes, RT-PCR with strain-specific primers was used. dsRNA (1 μg in 10 μl) with random primers (5 μM) was denatured by incubation in boiling water and reverse transcribed (50 min at 42°C) using 200 U of Moloney murine leukemia virus reverse transcriptase (20 μl, final volume). PCR was conducted on 1 μl of the reverse transcriptase reaction in a final volume of 50 μl with 200 nM concentrations of each primer and 0.2 U of Taq polymerase. The following PCR conditions were used: 35 cycles of denaturation at 95°C for 0.5 min, annealing at 40°C for 1 min, and elongation at 72°C for 1 min.
Cell transfection, Western blotting, and luciferase assays.
DNA was introduced into MA104 cells by lipofection using Lipofectamine 2000 according to the supplier’s instructions (Life Technologies). Cells were recovered at 36 h posttransfection and assayed with the Dual-Glo luciferase assay system (Life Technologies). HEK293 cell lysates were used to detect XBP1 proteins with a rabbit polyclonal antibody from Santa-Cruz (SC-7160). Western blots were visualized using an OdysseyFC imager and Image Studio software (LI-COR).
Rotavirus reassortment and reverse genetics.
Monoreassortants between the RF and RRV and the RF and SA11-4F strains of rotavirus were selected from the progeny viruses obtained after mixed infection at an MOI of 10 for each parent. Some monoreassortants were obtained by back-crossing a multireassortant with one of the parental viruses. The progeny viruses were plaque purified and amplified, and the parental origin of the genome segments was determined by the electrophoretic mobility of the genomic RNA on 10% polyacrylamide gels after silver staining. Genes 1, 2, 3, 7, 8, 9, and 10 whose parental origin could not be unambiguously assigned were further analyzed by RT-PCR using specific primers for the gene of each parent. Monoreassortant candidates were then plaque purified twice and reanalyzed after amplification (data are available upon request).
For rotavirus reverse genetics, the protocol and plasmids described by Kanai et al. (45) were used. The plasmid pT7 SA NSP3 was replaced by the chimeric constructs in pRiboz as described above.
Immunofluorescence staining and microscopy.
MA104 cells (105) seeded on 12-mm glass coverslips were infected with different rotavirus strains at an MOI of 10 and incubated at 37°C for various times. The cells were fixed with 4% paraformaldehyde at room temperature and processed as described in Harb et al. (35). The fixed cells were incubated with the primary antibodies (rabbit polyclonal antibody against rotavirus NSP2 [1:100] [87] and 10E10 monoclonal antibody specific for PABP-C1 [Santa Cruz, sc-32318; 1:100]) for 1 h and then with Alexa Fluor 488-conjugated goat anti-mouse IgG and Alexa Fluor 546-conjugated goat anti-rabbit IgG (Molecular Probes, Inc., Eugene, OR) at a dilution of 1:1,000. Before being mounted on glass slides with ProLong Antifade (Molecular Probes), the cells were incubated with DAPI (4′,6′-diamidino-2-phenylindole) for 10 min. The cells were visualized with a Zeiss Axio-Imager microscope (20× objective; Zeiss AG, Gottingen, Germany). Images were captured using the same acquisition parameters. Mock-infected cells were examined first and used to set the background before imaging the infected samples. For immunofluorescence quantification, three fields were randomly selected for each time point postpostinfection, and cytoplasmic and nuclear PABPC fluorescence levels were quantified using the Cell Profiler pipeline (88) and processed using ImageJ (http://rsb.info.nih.gov/ij/index.html). The nuclear and cytoplasmic compartments were delimited by DAPI and NSP2 fluorescence, respectively.
Nuclear and cytoplasmic extracts.
Nuclear and cytoplasmic extracts of infected and mock-infected cells were prepared as previously described (89).
ACKNOWLEDGMENTS
This study was supported by the Institut National de la Recherche Agronomique (INRA) and the Centre National de la Recherche Scientifique (CNRS). M.G. was supported by a Ph.D. thesis fellowship from Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche. P.V., A.C., C.L., and D.P. are staff members of the INRA. M.D. is a staff member of University of Evry Val d’Essones.
Reporter plasmids with GRP78, GRP94, CHOP, or XBP1 promoters were kindly provided by Kazutoshi Mori (Kyoto University, Japan), and the plasmid encoding the full-length mouse XBP1 cDNA was kindly provided by Ling Qi (Cornell University, Ithaca, NY).
REFERENCES
- 1.Hetz C, Martinon F, Rodriguez D, Glimcher LH. 2011. The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol Rev 91:1219–1243. doi: 10.1152/physrev.00001.2011. [DOI] [PubMed] [Google Scholar]
- 2.Walter P, Ron D. 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 4.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 5.Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, Heindl K, Hoffmann T, Busslinger M, Martinez J. 2014. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J 33:2922–2936. doi: 10.15252/embj.201490332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosmaczewski SG, Edwards TJ, Han SM, Eckwahl MJ, Meyer BI, Peach S, Hesselberth JR, Wolin SL, Hammarlund M. 2014. The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep 15:1278–1285. doi: 10.15252/embr.201439531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu Y, Liang FX, Wang X. 2014. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol Cell 55:758–770. doi: 10.1016/j.molcel.2014.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. 2004. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem 136:343–350. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- 9.Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR, 3rd, Su AI, Kelly JW, Wiseman RL. 2013. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 3:1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. 2007. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev Cell 13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 11.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. 2004. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 12.Sriburi R, Jackowski S, Mori K, Brewer JW. 2004. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol 167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. 2003. A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell 4:265–271. doi: 10.1016/S1534-5807(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 14.Bettigole SE, Glimcher LH. 2015. Endoplasmic reticulum stress in immunity. Annu Rev Immunol 33:107–138. doi: 10.1146/annurev-immunol-032414-112116. [DOI] [PubMed] [Google Scholar]
- 15.Martinon F, Chen X, Lee AH, Glimcher LH. 2010. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinon F, Glimcher LH. 2011. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr Opin Immunol 23:35–40. doi: 10.1016/j.coi.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. 2008. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-β induction via X-box binding protein 1. Eur J Immunol 38:1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng L, Liu YP, Sha H, Chen H, Qi L, Smith JA. 2010. XBP-1 couples endoplasmic reticulum stress to augmented IFN-beta induction via a cis-acting enhancer in macrophages. J Immunol 185:2324–2330. doi: 10.4049/jimmunol.0903052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tate JE, Burton AH, Boschi-Pinto C, Parashar UD, World Health Organization-Coordinated Global Rotavirus Surveillance Network. 2016. Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000-2013. Clin Infect Dis 62:S96–S105. doi: 10.1093/cid/civ1013. [DOI] [PubMed] [Google Scholar]
- 20.Zambrano JL, Ettayebi K, Maaty WS, Faunce NR, Bothner B, Hardy ME. 2011. Rotavirus infection activates the UPR but modulates its activity. Virol J 8:359. doi: 10.1186/1743-422X-8-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trujillo-Alonso V, Maruri-Avidal L, Arias CF, López S. 2011. Rotavirus infection induces the unfolded protein response of the cell and controls it through the nonstructural protein NSP3. J Virol 85:12594–12604. doi: 10.1128/JVI.05620-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubio RM, Mora SI, Romero P, Arias CF, Lopez S. 2013. Rotavirus prevents the expression of host responses by blocking the nucleocytoplasmic transport of polyadenylated mRNAs. J Virol 87:6336–6345. doi: 10.1128/JVI.00361-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montero H, Rojas M, Arias CF, Lopez S. 2008. Rotavirus infection induces the phosphorylation of eIF2α but prevents the formation of stress granules. J Virol 82:1496–1504. doi: 10.1128/JVI.01779-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gratia M, Vende P, Charpilienne A, Baron HC, Laroche C, Sarot E, Pyronnet S, Duarte M, Poncet D. 2016. Challenging the roles of NSP3 and untranslated regions in rotavirus mRNA translation. PLoS One 11:e0145998. doi: 10.1371/journal.pone.0145998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montero H, Arias CF, Lopez S. 2006. Rotavirus nonstructural protein NSP3 is not required for viral protein synthesis. J Virol 80:9031–9038. doi: 10.1128/JVI.00437-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gratia M, Sarot E, Vende P, Charpilienne A, Baron CH, Duarte M, Pyronnet S, Poncet D. 2015. Rotavirus NSP3 is a translational surrogate of the poly(A) binding protein-poly(A) complex. J Virol 89:8773–8782. doi: 10.1128/JVI.01402-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deo RC, Groft CM, Rajashankar KR, Burley SK. 2002. Recognition of the rotavirus mRNA 3’ consensus by an asymmetric NSP3 homodimer. Cell 108:71–81. doi: 10.1016/S0092-8674(01)00632-8. [DOI] [PubMed] [Google Scholar]
- 28.Poncet D, Aponte C, Cohen J. 1993. Rotavirus protein NSP3 (NS34) is bound to the 3’ end consensus sequence of viral mRNAs in infected cells. J Virol 67:3159–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poncet D, Laurent S, Cohen J. 1994. Four nucleotides are the minimal requirement for RNA recognition by rotavirus nonstructural protein NSP3. EMBO J 13:4165–4173. doi: 10.1002/j.1460-2075.1994.tb06734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Groft CM, Burley SK. 2002. Recognition of eIF4G by rotavirus NSP3 reveals a basis for mRNA circularization. Mol Cell 9:1273–1283. doi: 10.1016/S1097-2765(02)00555-5. [DOI] [PubMed] [Google Scholar]
- 31.Piron M, Delaunay T, Grosclaude J, Poncet D. 1999. Identification of the RNA-binding, dimerization, and eIF4GI-binding domains of rotavirus nonstructural protein NSP3. J Virol 73:5411–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piron M, Vende P, Cohen J, Poncet D. 1998. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J 17:5811–5821. doi: 10.1093/emboj/17.19.5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vende P, Piron M, Castagne N, Poncet D. 2000. Efficient translation of rotavirus mRNA requires simultaneous interaction of NSP3 with the eukaryotic translation initiation factor eIF4G and the mRNA 3′ end. J Virol 74:7064–7071. doi: 10.1128/JVI.74.15.7064-7071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chizhikov V, Patton JT. 2000. A four-nucleotide translation enhancer in the 3′-terminal consensus sequence of the nonpolyadenylated mRNAs of rotavirus. RNA. RNA 6:814–825. doi: 10.1017/S1355838200992264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harb M, Becker MM, Vitour D, Baron CH, Vende P, Brown SC, Bolte S, Arold ST, Poncet D. 2008. Nuclear localization of cytoplasmic poly(A)-binding protein upon rotavirus infection involves the interaction of NSP3 with eIF4G and RoXaN. J Virol 82:11283–11293. doi: 10.1128/JVI.00872-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green VA, Pelkmans L. 2016. A systems survey of progressive host-cell reorganization during rotavirus infection. Cell Host Microbe 20:107–120. doi: 10.1016/j.chom.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 37.Shang J, Lehrman MA. 2004. Discordance of UPR signaling by ATF6 and Ire1p-XBP1 with levels of target transcripts. Biochem Biophys Res Commun 317:390–396. doi: 10.1016/j.bbrc.2004.03.058. [DOI] [PubMed] [Google Scholar]
- 38.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, Koong AC. 2011. Identification of an Ire1α endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matthijnssens J, Ciarlet M, Rahman M, Attoui H, Banyai K, Estes MK, Gentsch JR, Iturriza-Gomara M, Kirkwood CD, Martella V, Mertens PP, Nakagomi O, Patton JT, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Desselberger U, Van Ranst M. 2008. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch Virol 153:1621–1629. doi: 10.1007/s00705-008-0155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matthijnssens J, Ciarlet M, Heiman E, Arijs I, Delbeke T, McDonald SM, Palombo EA, Iturriza-Gomara M, Maes P, Patton JT, Rahman M, Van Ranst M. 2008. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J Virol 82:3204–3219. doi: 10.1128/JVI.02257-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matthijnssens J, Theuns S. 2015. Minutes of the 7th Rotavirus Classification Working Group meeting. Rotavirus Classification Working Group, Leuven, Belgium: https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/minutes-of-the-7th-rcwg-meeting. [Google Scholar]
- 42.Gilbertson S, Federspiel JD, Hartenian E, Cristea IM, Glaunsinger B. 2018. Changes in mRNA abundance drive shuttling of RNA binding proteins, linking cytoplasmic RNA degradation to transcription. eLife 7:e37663. doi: 10.7554/eLife.37663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burgess HM, Gray NK. 2012. An integrated model for the nucleocytoplasmic transport of cytoplasmic poly(A)-binding proteins. Commun Integr Biol 5:243–247. doi: 10.4161/cib.19347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hosoda N, Lejeune F, Maquat LE. 2006. Evidence that poly(A) binding protein C1 binds nuclear pre-mRNA poly(A) tails. Mol Cell Biol 26:3085–3097. doi: 10.1128/MCB.26.8.3085-3097.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanai Y, Komoto S, Kawagishi T, Nouda R, Nagasawa N, Onishi M, Matsuura Y, Taniguchi K, Kobayashi T. 2017. Entirely plasmid-based reverse genetics system for rotaviruses. Proc Natl Acad Sci U S A 114:2349–2354. doi: 10.1073/pnas.1618424114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sorek R, Shemesh R, Cohen Y, Basechess O, Ast G, Shamir R. 2004. A non-EST-based method for exon-skipping prediction. Genome Res 14:1617–1623. doi: 10.1101/gr.2572604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhowmick R, Mukherjee A, Patra U, Chawla-Sarkar M. 2015. Rotavirus disrupts cytoplasmic P bodies during infection. Virus Res 210:344–354. doi: 10.1016/j.virusres.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Oceguera A, Peralta AV, Martínez-Delgado G, Arias CF, López S. 2018. Rotavirus RNAs sponge host cell RNA binding proteins and interfere with their subcellular localization. Virology 525:96–105. doi: 10.1016/j.virol.2018.09.013. [DOI] [PubMed] [Google Scholar]
- 49.Chen H, Qi L. 2010. SUMO modification regulates the transcriptional activity of XBP1. Biochem J 429:95–102. doi: 10.1042/BJ20100193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. 2000. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol 20:6755–6767. doi: 10.1128/MCB.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee AH, Iwakoshi NN, Glimcher LH. 2003. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar GR, Shum L, Glaunsinger BA. 2011. Importin alpha-mediated nuclear import of cytoplasmic poly(A) binding protein occurs as a direct consequence of cytoplasmic mRNA depletion. Mol Cell Biol 31:3113–3125. doi: 10.1128/MCB.05402-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Safaee N, Kozlov G, Noronha AM, Xie J, Wilds CJ, Gehring K. 2012. Interdomain allostery promotes assembly of the poly(A) mRNA complex with PABP and eIF4G. Mol Cell 48:375–386. doi: 10.1016/j.molcel.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 54.Burgess HM, Richardson WA, Anderson RC, Salaun C, Graham SV, Gray NK. 2011. Nuclear relocalization of cytoplasmic poly(A)-binding proteins PABP1 and PABP4 in response to UV irradiation reveals mRNA-dependent export of metazoan PABPs. J Cell Sci 124:3344–3355. doi: 10.1242/jcs.087692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar GR, Glaunsinger BA. 2010. Nuclear import of cytoplasmic poly(A) binding protein restricts gene expression via hyperadenylation and nuclear retention of mRNA. Mol Cell Biol 30:4996–5008. doi: 10.1128/MCB.00600-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma S, Bhattacharjee RB, Bag J. 2009. Expression of poly(A)-binding protein is upregulated during recovery from heat shock in HeLa cells. FEBS J 276:552–570. doi: 10.1111/j.1742-4658.2008.06803.x. [DOI] [PubMed] [Google Scholar]
- 57.Pai AA, Luca F. 2018. Environmental influences on RNA processing: biochemical, molecular and genetic regulators of cellular response. Wiley Interdiscip Rev RNA 2018:e1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blakqori G, van Knippenberg I, Elliott RM. 2009. Bunyamwera orthobunyavirus S-segment untranslated regions mediate poly(A) tail-independent translation. J Virol 83:3637–3646. doi: 10.1128/JVI.02201-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walsh D, Mathews MB, Mohr I. 2013. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb Perspect Biol 5:a012351. doi: 10.1101/cshperspect.a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salaun C, MacDonald AI, Larralde O, Howard L, Lochtie K, Burgess HM, Brook M, Malik P, Gray NK, Graham SV. 2010. Poly(A)-binding protein 1 partially relocalizes to the nucleus during herpes simplex virus type 1 infection in an ICP27-independent manner and does not inhibit virus replication. J Virol 84:8539–8548. doi: 10.1128/JVI.00668-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu B, Li X, Huo Y, Yu Y, Zhang Q, Chen G, Zhang Y, Fraser NW, Wu D, Zhou J. 2016. Cellular responses to HSV-1 infection are linked to specific types of alterations in the host transcriptome. Sci Rep 6:28075. doi: 10.1038/srep28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boudreault S, Martenon-Brodeur C, Caron M, Garant JM, Tremblay MP, Armero VE, Durand M, Lapointe E, Thibault P, Tremblay-Letourneau M, Perreault JP, Scott MS, Lemay G, Bisaillon M. 2016. Global profiling of the cellular alternative RNA splicing landscape during virus-host interactions. PLoS One 11:e0161914. doi: 10.1371/journal.pone.0161914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rivera-Serrano EE, Fritch EJ, Scholl EH, Sherry B. 2017. A cytoplasmic RNA virus alters the function of the cell splicing protein SRSF2. J Virol 91:e02488-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dhillon P, Tandra VN, Chorghade SG, Namsa ND, Sahoo L, Rao CD. 2018. Cytoplasmic relocalization and colocalization with viroplasms of host cell proteins, and their role in rotavirus infection. J Virol 92:e00612-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mossel EC, Ramig RF. 2002. Rotavirus genome segment 7 (NSP3) is a determinant of extraintestinal spread in the neonatal mouse. J Virol 76:6502–6509. doi: 10.1128/JVI.76.13.6502-6509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dahlberg J, Lund E. 2012. Nuclear translation or nuclear peptidyl transferase? Nucleus 3:320–321. doi: 10.4161/nucl.20754. [DOI] [PubMed] [Google Scholar]
- 67.Dutta D, Bagchi P, Chatterjee A, Nayak MK, Mukherjee A, Chattopadhyay S, Nagashima S, Kobayashi N, Komoto S, Taniguchi K, Chawla-Sarkar M. 2009. The molecular chaperone heat shock protein-90 positively regulates rotavirus infection. Virology 391:325–333. doi: 10.1016/j.virol.2009.06.044. [DOI] [PubMed] [Google Scholar]
- 68.Dutta D, Chattopadhyay S, Bagchi P, Halder UC, Nandi S, Mukherjee A, Kobayashi N, Taniguchi K, Chawla-Sarkar M. 2011. Active participation of cellular chaperone Hsp90 in regulating the function of rotavirus nonstructural protein 3 (NSP3). J Biol Chem 286:20065–20077. doi: 10.1074/jbc.M111.231878. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Maruri-Avidal L, López S, Arias CF. 2008. Endoplasmic reticulum chaperones are involved in the morphogenesis of rotavirus infectious particles. J Virol 82:5368–5380. doi: 10.1128/JVI.02751-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu CY, Hsu YW, Liao CL, Lin YL. 2006. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J Virol 80:11868–11880. doi: 10.1128/JVI.00879-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu A, Bellamy AR, Taylor JA. 1998. BiP (GRP78) and endoplasmin (GRP94) are induced following rotavirus infection and bind transiently to an endoplasmic reticulum-localized virion component. J Virol 72:9865–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith JA. 2014. A new paradigm: innate immune sensing of viruses via the unfolded protein response. Front Microbiol 5:222. doi: 10.3389/fmicb.2014.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lad SP, Yang G, Scott DA, Chao T-H, Correia J. d S, de la Torre JC, Li E. 2008. Identification of MAVS splicing variants that interfere with RIGI/MAVS pathway signaling. Mol Immunol 45:2277–2287. doi: 10.1016/j.molimm.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 74.Zhu C, Xiao F, Hong J, Wang K, Liu X, Cai D, Fusco DN, Zhao L, Jeong SW, Brisac C, Chusri P, Schaefer EA, Zhao H, Peng LF, Lin W, Chung RT. 2015. EFTUD2 is a novel innate immune regulator restricting hepatitis C virus infection through the RIG-I/MDA5 pathway. J Virol 89:6608–6618. doi: 10.1128/JVI.00364-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Almstead LL, Sarnow P. 2007. Inhibition of U snRNP assembly by a virus-encoded proteinase. Genes Dev 21:1086–1097. doi: 10.1101/gad.1535607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu YC, Kuo RL, Lin JY, Huang PN, Huang Y, Liu H, Arnold JJ, Chen SJ, Wang RY, Cameron CE, Shih SR. 2014. Cytoplasmic viral RNA-dependent RNA polymerase disrupts the intracellular splicing machinery by entering the nucleus and interfering with Prp8. PLoS Pathog 10:e1004199. doi: 10.1371/journal.ppat.1004199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.De Maio FA, Risso G, Iglesias NG, Shah P, Pozzi B, Gebhard LG, Mammi P, Mancini E, Yanovsky MJ, Andino R, Krogan N, Srebrow A, Gamarnik AV. 2016. The dengue virus NS5 protein intrudes in the cellular spliceosome and modulates splicing. PLoS Pathog 12:e1005841. doi: 10.1371/journal.ppat.1005841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takayanagi S, Fukuda R, Takeuchi Y, Tsukada S, Yoshida K. 2013. Gene regulatory network of unfolded protein response genes in endoplasmic reticulum stress. Cell Stress Chaperones 18:11–23. doi: 10.1007/s12192-012-0351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vincenz L, Jager R, O’Dwyer M, Samali A. 2013. Endoplasmic reticulum stress and the unfolded protein response: targeting the Achilles heel of multiple myeloma. Mol Cancer Ther 12:831–843. doi: 10.1158/1535-7163.MCT-12-0782. [DOI] [PubMed] [Google Scholar]
- 80.Dunys J, Duplan E, Checler F. 2014. The transcription factor X-box binding protein-1 in neurodegenerative diseases. Mol Neurodegener 9:35. doi: 10.1186/1750-1326-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sha H, He Y, Yang L, Qi L. 2011. Stressed out about obesity: IRE1α-XBP1 in metabolic disorders. Trends Endocrinol Metab 22:374–381. doi: 10.1016/j.tem.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, Blumberg RS. 2008. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cao Y, Knochel S, Oswald F, Donow C, Zhao H, Knochel W. 2006. XBP1 forms a regulatory loop with BMP-4 and suppresses mesodermal and neural differentiation in Xenopus embryos. Mech Dev 123:84–96. doi: 10.1016/j.mod.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 84.Troupin C, Dehee A, Schnuriger A, Vende P, Poncet D, Garbarg-Chenon A. 2010. Rearranged genomic RNA segments offer a new approach to the reverse genetics of rotaviruses. J Virol 84:6711–6719. doi: 10.1128/JVI.00547-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 86.Gombold JL, Estes MK, Ramig RF. 1985. Assignment of simian rotavirus SA11 temperature-sensitive mutant groups B and E to genome segments. Virology 143:309–320. doi: 10.1016/0042-6822(85)90118-7. [DOI] [PubMed] [Google Scholar]
- 87.Martin D, Duarte M, Lepault J, Poncet D. 2010. Sequestration of free tubulin molecules by the viral protein NSP2 induces microtubule depolymerization during rotavirus infection. J Virol 84:2522–2532. doi: 10.1128/JVI.01883-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barbier J, Dutertre M, Bittencourt D, Sanchez G, Gratadou L, de la Grange P, Auboeuf D. 2007. Regulation of H-ras splice variant expression by cross talk between the p53 and nonsense-mediated mRNA decay pathways. Mol Cell Biol 27:7315–7333. doi: 10.1128/MCB.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]