

It is generally accepted that the antiviral effects of vaccine-induced classical CD8+ T-cell responses against human immunodeficiency virus (HIV) are limited to partial reductions in viremia after the establishment of productive infection. Here we show that rhesus macaques (RMs) vaccinated with Vif and Nef acquired simian immunodeficiency virus (SIV) infection at a lower (albeit not statistically significant) rate than control RMs following repeated intrarectal challenges with a pathogenic SIV clone. All animals in the present experiment expressed the elite control-associated major histocompatibility complex class I (MHC-I) molecule Mamu-B*08 that binds immunodominant epitopes in Vif and Nef. Though preliminary, these results provide tantalizing evidence that the protective efficacy of vaccine-elicited CD8+ T cells may be greater than previously thought. Future studies should examine if vaccine-induced cellular immunity can prevent systemic viral replication in RMs that do not express MHC-I alleles associated with elite control of SIV infection.
KEYWORDS: human immunodeficiency virus, simian immunodeficiency virus, vaccines
ABSTRACT
Approximately 50% of rhesus macaques (RMs) expressing the major histocompatibility complex class I (MHC-I) allele Mamu-B*08 spontaneously control chronic-phase viremia after infection with the pathogenic simian immunodeficiency virus mac239 (SIVmac239) clone. CD8+ T-cell responses in these animals are focused on immunodominant Mamu-B*08-restricted SIV epitopes in Vif and Nef, and prophylactic vaccination with these epitopes increases the incidence of elite control in SIVmac239-infected Mamu-B*08-positive (Mamu-B*08+) RMs. Here we evaluated if robust vaccine-elicited CD8+ T-cell responses against Vif and Nef can prevent systemic infection in Mamu-B*08+ RMs following mucosal SIV challenges. Ten Mamu-B*08+ RMs were vaccinated with a heterologous prime/boost/boost regimen encoding Vif and Nef, while six sham-vaccinated MHC-I-matched RMs served as the controls for this experiment. Vaccine-induced CD8+ T cells against Mamu-B*08-restricted SIV epitopes reached high frequencies in blood but were present at lower levels in lymph node and gut biopsy specimens. Following repeated intrarectal challenges with SIVmac239, all control RMs became infected by the sixth SIV exposure. By comparison, four vaccinees were still uninfected after six challenges, and three of them remained aviremic after 3 or 4 additional challenges. The rate of SIV acquisition in the vaccinees was numerically lower (albeit not statistically significantly) than that in the controls. However, peak viremia was significantly reduced in infected vaccinees compared to control animals. We found no T-cell markers that distinguished vaccinees that acquired SIV infection from those that did not. Additional studies will be needed to validate these findings and determine if cellular immunity can be harnessed to prevent the establishment of productive immunodeficiency virus infection.
IMPORTANCE It is generally accepted that the antiviral effects of vaccine-induced classical CD8+ T-cell responses against human immunodeficiency virus (HIV) are limited to partial reductions in viremia after the establishment of productive infection. Here we show that rhesus macaques (RMs) vaccinated with Vif and Nef acquired simian immunodeficiency virus (SIV) infection at a lower (albeit not statistically significant) rate than control RMs following repeated intrarectal challenges with a pathogenic SIV clone. All animals in the present experiment expressed the elite control-associated major histocompatibility complex class I (MHC-I) molecule Mamu-B*08 that binds immunodominant epitopes in Vif and Nef. Though preliminary, these results provide tantalizing evidence that the protective efficacy of vaccine-elicited CD8+ T cells may be greater than previously thought. Future studies should examine if vaccine-induced cellular immunity can prevent systemic viral replication in RMs that do not express MHC-I alleles associated with elite control of SIV infection.
INTRODUCTION
The ultimate goal of a prophylactic human immunodeficiency virus (HIV) vaccine is to block acquisition of infection. While induction of broadly neutralizing antibodies (bnAbs) remains our best hope to achieve this kind of protection, engendering bnAbs by vaccination has been exceedingly difficult due to structural features of the Env glycoprotein and its high variability among circulating isolates (1). Notwithstanding these barriers, it is well established that the passive transfer of monoclonal HIV-specific bnAbs can afford complete protection against mucosal challenge with simian/HIV (SHIV) chimeras in rhesus macaques (RMs) (2). Importantly, mounting evidence indicates that virus neutralization at the mucosal portal of entry is not required for the protection afforded by monoclonal bnAb treatment (3, 4). Indeed, in order to characterize the anatomic sites of bnAb-mediated protection, Liu et al. have recently treated female RMs with a fully protective dose of the bnAb PGT121 and subsequently challenged them intravaginally with SHIV-SF162P3 (4). The authors reported that within the first week after transmission, the virus had disseminated to distal tissues and undergone a few rounds of replication before the initial foci of infected cells decayed or were cleared by bnAb-mediated antiviral mechanisms. Notably, during these first few days after infection, bnAb-treated RMs remained aviremic and failed to develop adaptive T-cell responses against the virus. These results, together with those by Hessell et al. showing that early treatment with monoclonal bnAbs halts SHIV infection in infant RMs (3), indicate that even in cases of monoclonal bnAb-mediated protection of mucosally acquired immunodeficiency virus infection, the transmitted/founder virus can still spread to distant tissues from the site of transmission. However, because of bnAb interception or the spontaneous decay of virus-infected cells, the nascent infection cannot spread beyond the initial foci of infected cells.
Cellular immune responses have also been a major focus of HIV vaccine development efforts since compelling evidence implicated major histocompatibility complex class I (MHC-I)-restricted CD8+ T cells in the virologic control of lentivirus replication (5–8). Because CD8+ T-cell activation depends on the recognition of cognate peptide–MHC-I complexes on the surface of target cells (9), a condition that might be satisfied only after infection has been initiated, it is generally accepted that vaccine-elicited cellular immunity alone cannot prevent HIV infection. In this regard, the expectation for vaccine-induced, HIV-specific CD8+ T cells has been to decrease viral replication after acquisition of infection in hopes to delay progression to AIDS and reduce the chances of transmission (10). Although this endpoint of HIV vaccine efficacy has not yet been achieved in clinical trials, vaccine-elicited T-cell responses have been repeatedly shown to lower simian immunodeficiency virus (SIV) viremia in RMs (11–18), demonstrating the feasibility of engendering anti-lentiviral T-cell immunity by vaccination.
Mounting evidence suggests that vaccine-elicited T cells can afford more than partial control of immunodeficiency virus replication. For example, live-attenuated SIV vaccines are extremely effective in nonhuman primates, often resulting in sterilizing immunity against challenge with sequence-matched pathogenic strains of SIV (19). Although protection in these cases also involves humoral immune responses (20), given the homology of Env sequences between the vaccine and challenge viruses, the magnitude of lymph node (LN)-based antiviral T cells alone can predict the efficacy of live-attenuated SIV vaccination (21). The recent evaluation of rhesus cytomegalovirus (RhCMV)-vectored SIV vaccines also indicates that T-cell immunity can dramatically alter the course of SIV infection (22–24). As a member of the Herpesviridae family (25), RhCMV persistently infects RMs, thereby providing chronic low-level antigen exposure. This type of immune stimulation favors the generation of effector memory T cells (TEM) that recirculate through extralymphoid tissues and that are endowed with immediate antiviral activity (26). Remarkably, approximately half of RhCMV-vaccinated RMs manifest complete control of viral replication shortly after SIVmac239 infection (22–24). Except for occasional viral load (VL) blips in the ensuing weeks, these successful vaccinees remain aviremic in the chronic phase and ultimately clear SIVmac239 infection (24). The 68-1 RhCMV strain utilized in these experiments is noteworthy in that it induces broadly targeted CD8+ TEM responses that recognize epitopes presented by MHC-II and the nonclassical MHC-I molecule Mamu-E (27, 28). Moreover, even though env was included in the vaccine regimen, vaccination with 68-1 RhCMV env did not elicit anti-Env antibodies, reinforcing the conclusion that vaccine-induced, nonclassical CD8+ TEM responses are responsible for protection. Importantly, 68-1 RhCMV vaccinees harbored high frequencies of SIV-specific TEM at key sites of virus entry and amplification (23), a property that might have facilitated the interception of SIV-infected cells in the first stages of infection. Collectively, these vaccine studies indicate that lentiviral infections are vulnerable to T-cell-mediated immunity early after transmission.
Elite control of HIV infection is defined as spontaneous suppression of chronic-phase viremia in the absence of antiretroviral therapy (8). Consistent with a crucial role of CD8+ T-cell responses in this phenotype, some MHC-I alleles (e.g., HLA-B*27 and HLA-B*57) are enriched in cohorts of elite controllers (ECs) and the expressed MHC-I molecules present immunodominant HIV epitopes (6, 8, 29). Similar to human ECs, expression of certain RM MHC-I alleles is also associated with elite control of SIV infection (30–32). For example, approximately 50% of SIVmac239-infected Mamu-B*08-positive (Mamu-B*08+) RMs manifest some measure of control of viral replication in the chronic phase (30). Due to the virulence and high replicative capacity of SIVmac239, EC status in the macaque model is defined as having a set point VL of less than 1,000 viral RNA (vRNA) copies/ml of plasma. Interestingly, even though HLA-B*27 and Mamu-B*08 differ considerably in their amino acid sequences, these MHC-I molecules share similar peptide binding motifs, characterized by the requirement for Arg in position 2 as the primary anchor and major overlaps in the residues occupying other secondary anchor positions (33). Virologic control in Mamu-B*08+ RMs is largely dependent on CD8+ T cells directed against three immunodominant Mamu-B*08-restricted epitopes: Vif RL8, Vif RL9, and Nef RL10 (34, 35). Indeed, we have shown that prophylactic vaccination with these three epitopes significantly increases the incidence of elite control in Mamu-B*08+ RMs after high-dose intrarectal (IR) challenges with SIVmac239 (36). Of note, the immunization protocol utilized in that study, a recombinant yellow fever virus 17D (rYF17D) prime followed by a recombinant adenovirus type 5 (rAd5) boost, resulted in low levels of SIV-specific CD8+ T cells at the time of SIV challenge. Curiously, we have recently reported that focusing Mamu-B*08-restricted CD8+ T cells on the Nef RL10 epitope did not improve containment of SIVmac239 infection (37), suggesting that elite control in Mamu-B*08+ RMs might depend on the concerted action of CD8+ T-cell responses targeting the three immunodominant epitopes.
Here we explored the hypothesis that vaccine-induced classical MHC-I-restricted CD8+ T-cell responses can prevent systemic SIV replication in RMs rectally challenged with SIVmac239. In order to increase the chances of a successful outcome, only Mamu-B*08+ RMs were utilized in this experiment due to their propensity for mounting efficacious CD8+ T-cell responses against SIV. It is important to emphasize that expression of Mamu-B*08 does not affect susceptibility to SIV infection but affects only postacquisition control of chronic viremia. Since recurrent antigen exposure is also a key determinant of antiviral T-cell immunity (38, 39), SIVmac239 genes encoding Mamu-B*08-restricted epitopes were delivered by sequential immunizations with rAd5, recombinant vesicular stomatitis virus (rVSV), and recombinant rhesus monkey rhadinovirus (rRRV) vectors. The last vector is a herpesvirus that establishes persistent infection in RMs (40, 41). A single inoculation of rRRV vectors encoding SIV Env, Gag, and a Rev-Tat-Nef fusion has been shown to significantly decrease viral replication in RMs following intravenous challenge with SIVmac239 (42). Unlike 68-1 RhCMV, though, rRRV vaccination results in TEM-biased CD8+ T-cell responses restricted by classical MHC-I molecules (17, 42, 43). Here we report the efficacy of SIV-specific T-cell responses induced by the aforementioned rAd5/rVSV/rRRV regimen in Mamu-B*08+ RMs subjected to repeated IR challenges with SIVmac239.
RESULTS
Sixteen Mamu-B*08+ RMs of Indian origin were selected for this study (Table 1). Ten animals were vaccinated with SIVmac239 gene inserts encoding the immunodominant Mamu-B*08-restricted epitopes Vif RL8 (amino acids [aa] 172 to 179), Vif RL9 (aa 123 to 131), and Nef RL10 (aa 137 to 146) (Fig. 1). These SIV inserts were delivered by an rAd5 priming vector, followed by boosts with rVSV and then rRRV vectors (Fig. 1). It should be noted that some of the vaccine vectors employed in this study encoded segments of vif and nef or full-length vif and nef fused with other genes, such as tat and rev (see Materials and Methods). As a result, the rAd5/rVSV/rRRV vaccine regimen also elicited cellular immune responses against Tat and Rev in some of the animals. Additionally, the nef insert delivered by the rVSV and rRRV vectors also included the subdominant Mamu-B*08-restricted epitope Nef RL9b (aa 246 to 254). The remaining six monkeys were sham immunized with empty vectors or vectors containing irrelevant inserts and served as the controls for this experiment (Fig. 1).
TABLE 1.
Animal characteristics
| Experimental group | Animal identifier | Relevant MHC-I allele(s) | Age (yr)a | Sex |
|---|---|---|---|---|
| Vaccinees | r08009 | Mamu-A*01, -B*08, -B*17 | 6 | Male |
| rhBB35 | Mamu-B*08 | 9.9 | Female | |
| r08024 | Mamu-A*01, -B*08 | 5.9 | Male | |
| r08038 | Mamu-A*01, -B*08 | 5.8 | Male | |
| r10038 | Mamu-B*08 | 4 | Female | |
| r10051 | Mamu-B*08 | 3.9 | Female | |
| r01065 | Mamu-B*08 | 12.7 | Female | |
| r09041 | Mamu-B*08 | 5 | Male | |
| r10091 | Mamu-B*08 | 3.5 | Male | |
| r10072 | Mamu-B*08 | 3.7 | Male | |
| Controls | r11063 | Mamu-A*01, -B*08 | 2.7 | Female |
| r10055 | Mamu-A*01, -B*08 | 3.8 | Male | |
| r00008 | Mamu-A*02, -B*08 | 14.3 | Female | |
| r07011 | Mamu-B*08 | 6.7 | Male | |
| r02075 | Mamu-B*08 | 11.9 | Male | |
| rhBH87 | Mamu-B*08 | 2.4 | Male |
Animal age at beginning of study.
FIG 1.

Experimental design. Ten Mamu-B*08+ RMs were vaccinated with an rAd5/rVSV/rRRV regimen expressing the immunodominant Mamu-B*08-restricted epitopes Vif RL8, Vif RL9, and Nef RL10. Six Mamu-B*08+ RMs were sham vaccinated and served as the controls. At week 41, vaccine efficacy was assessed by subjecting all monkeys to repeated IR challenges with a marginal dose of SIVmac239 (200 TCID50) every 2 weeks. The results for four vaccinees are color coded in blue in this figure and subsequent ones because they resisted greater than six IR challenges with SIVmac239.
We monitored the kinetics of vaccine-induced SIV-specific CD8+ T-cell responses in blood by fluorochrome-labeled MHC-I–peptide tetramer staining. This analysis revealed a robust expansion of CD8+ T cells against all three immunodominant Mamu-B*08-restricted epitopes, especially after the rVSV and rRRV boosts (Fig. 2A to C). Low to modest levels of CD8+ T cells directed against the subdominant Nef RL9b epitope were also detected in a subset of animals (Fig. 2D). Overall, there was considerable variation in the magnitude of Vif RL8-, Vif RL9-, and Nef RL10-specific CD8+ T cells among the 10 vaccinees. As a reference, however, the combined frequency of these responses at the time of the first SIV challenge was still significantly higher than that achieved in our previous Mamu-B*08 trial (P = 0.007 from Student's t test; Fig. 2E), where a poorly immunogenic rYF17D/rAd5 regimen encoding the aforementioned immunodominant epitopes increased the incidence of elite control after SIVmac239 infection (36).
FIG 2.

Kinetics of vaccine-induced CD8+ T-cell responses targeting Mamu-B*08-restricted SIV epitopes. Fluorochrome-labeled Mamu-B*08 tetramers folded with peptides corresponding to SIV epitopes were used to track vaccine-elicited SIV-specific CD8+ T cells in PBMC. (A to D) The percentages of live tetramer+ CD8+ T cells specific for Vif RL8 (aa 172 to 179) (A), Vif RL9 (aa 123 to 131) (B), Nef RL10 (aa 137 to 146) (C), and Nef RL9b (aa 246 to 254) (D) are shown at multiple time points throughout the vaccine phase. The times of each vaccination (black dotted lines) and the day of the first IR SIVmac239 challenge (red solid line) are indicated in each graph. (E) Comparison of the total magnitude of vaccine-induced CD8+ T cells against Vif RL8, Vif RL9, and Nef RL10 at the time of the first SIV challenge between vaccinees in the present experiment and those in our previous Mamu-B*08 SIV vaccine trial (36). The P value was determined by Student's t test after log transformation. Lines represent means, and each symbol denotes one vaccinee. The vaccinees in the present experiment that resisted greater than 6 IR SIV challenges (r10038, r10091, r09041, and r08009) are color coded in blue. The remaining six vaccinees that acquired SIV infection and manifested partial or no control of viral replication are indicated by black symbols.
Because the goal of the rAd5/rVSV/rRRV vaccine regimen described here was to induce SIV-specific TEM responses, we characterized the memory phenotype of tetramer-positive (tetramer+) CD8+ T cells at week 37 after rAd5 prime. Memory CD8+ T cells in RMs can be classified as three subsets based on the surface expression of CD28 and CCR7: central memory (TCM; CD28+ CCR7+), transitional memory (TEM1; CD28+ CCR7−), and terminally differentiated effector memory (TEM2; CD28− CCR7−) (44). We noticed high animal-to-animal variability in the frequencies of these subsets within the same and across different tetramer+ CD8+ T-cell populations (Fig. 3). Nevertheless, the TEM2 phenotype predominated within the three SIV epitope-specific CD8+ T cells, although in the case of Mamu-B*08/Vif RL8 tetramer+ cells, the mean frequencies of cells displaying the TEM2 and TCM signatures were not statistically different (P = 0.188 from a post hoc Fisher least-significant-difference test; Fig. 3A). We also characterized expression of granzyme B (Gzm B) within tetramer+ CD8+ T cells, as the granule exocytosis pathway has been reported to be the primary mechanism by which HIV-specific CD8+ T cells lyse HIV-infected CD4+ T cells (45–47). We found that the proportion of tetramer+ CD8+ T cells that expressed Gzm B directly ex vivo also varied between the animals (Fig. 3A to C), but the mean frequencies of Gzm B-expressing cells were not statistically different among the three tetramer+ populations (P = 0.6 from repeated-measurement analysis of variance [ANOVA]).
FIG 3.

Memory phenotype of vaccine-induced CD8+ T cells in PBMC. RM memory T cells can be classified into three subsets based on the differential expression of the cell surface molecules CD28 and CCR7: fully differentiated effector memory (TEM2; CD28− CCR7−), transitional memory (TEM1; CD28+ CCR7−), and central memory (TCM; CD28+ CCR7+). Based on the expression of these two markers, the memory phenotype of vaccine-induced CD8+ T cells against Vif RL8 (A), Vif RL9 (B), and Nef RL10 (C) in PBMC was delineated at week 37 after rAd5 prime. The panels on the left show the percentages of MHC-I tetramer+ CD8+ T cells that display each of the three memory signatures. This flow cytometric analysis also evaluated the intracellular expression of the cytotoxicity-associated molecule granzyme B (Gzm B) by each MHC-I tetramer+ CD8+ T-cell population, as shown in the panels on the right. Lines represent means. The P values were determined by repeated-measurement ANOVA. Vaccinees are color coded as described in the legend of Fig. 2. Data from monkey r08038 were not available for this analysis.
To determine if vaccine-induced CD8+ T cells were present at relevant sites of SIV replication, we searched for tetramer+ CD8+ T cells in disaggregated lymphocytes from colorectal and peripheral LN biopsy specimens obtained at week 37. This analysis revealed that CD8+ T cells targeting the three epitopes were present in LN (Fig. 4) and, in the case of Vif RL9 and Nef RL10, at significantly lower frequencies than their blood counterparts (Fig. 4B and C). The colorectal biopsy specimens also harbored vaccine-elicited CD8+ T cells, albeit at levels even lower than those found in LN (Fig. 4A to C). Overall, vaccine-elicited CD8+ T cells against the three epitopes reached their highest frequencies in peripheral blood mononuclear cells (PBMC), followed by LN and then the gut (Fig. 4D).
FIG 4.

Tissue distribution of vaccine-induced CD8+ T cells against Mamu-B*08-restricted SIV epitopes. (A to C) The frequencies of vaccine-elicited CD8+ T cells against Vif RL8 (A), Vif RL9 (B), and Nef RL10 (C) were quantified in PBMC and disaggregated lymphocyte suspensions obtained from lymph node (LN) and gut (colon plus rectal) biopsy specimens by fluorochrome-labeled MHC-I tetramer staining. (D) The combined frequencies of Vif RL8-, Vif RL9-, and Nef RL10-specific CD8+ T cells in each tissue are shown. The samples for this analysis were harvested at week 37 after rAd5 prime. Lines represent means. The P values were determined by repeated-measurement ANOVA. Vaccinees are color coded as described in the legend of Fig. 2.
We also carried out intracellular cytokine staining (ICS) assays at week 41 to determine the total magnitude of vaccine-induced SIV-specific T-cell responses at the time of the first SIV challenge. Vif and Nef were the primary targets of vaccine-induced CD8+ T cells, followed by Rev and Tat (Fig. 5A). CD4+ T-cell responses were below the detection limits in all animals, except for animals r08024 and r10051 (Fig. 5B). Because the ability of CD8+ T cells to perform multiple antiviral functions upon stimulation has been proposed as a marker of effective HIV-specific CD8+ T-cell responses (48), we also used ICS to characterize the functional profile of vaccine-induced CD8+ T-cell responses against Vif RL8, Vif RL9, and Nef RL10. This analysis revealed that the majority of CD8+ T cells targeting these immunodominant epitopes were capable of elaborating two or three functions, which consisted of degranulation, as measured by CD107a upregulation, and production of gamma interferon (IFN-γ) and/or tumor necrosis factor alpha (TNF-α) (Fig. 5C and D).
FIG 5.

Total magnitude and functional profile of vaccine-induced SIV-specific T-cell responses at the time of the first SIV challenge. These analyses were carried out in PBMC collected at study week 41. (A and B) CD8+ and CD4+ T-cell responses were measured by ICS using pools of SIVmac239 peptides (15-mers overlapping by 11 amino acids). The percentages of responding CD8+ (A) and CD4+ (B) T cells were calculated by adding the frequencies of positive responses producing any combination of three immunological functions (IFN-γ, TNF-α, and CD107a). Each panel shows the magnitude of T-cell responses against Vif, Nef, and both Rev and Tat and the sum of T-cell responses against these proteins. The P values were determined by repeated-measurement ANOVA. (C to E) Functional profile of vaccine-induced CD8+ T cells directed against Vif RL8, Vif RL9, and Nef RL10. ICS was used to evaluate the ability of vaccine-induced CD8+ T cells to degranulate (based on CD107a upregulation) and/or produce the cytokines IFN-γ and TNF-α upon stimulation. The antigen stimuli for this assay consisted of peptides corresponding to the Mamu-B*08-restricted epitopes Vif RL8 (C), Vif RL9 (D), and Nef RL10 (E). The combinations of functions tested are shown below each panel. The P values were determined by repeated-measurement ANOVA. Lines represent means. Vaccinees are color coded as described in the legend of Fig. 2.
Vaccine efficacy was assessed at week 41 by subjecting both control and vaccinated macaques to repeated IR challenges with a marginal dose (200 50% tissue culture infective doses [TCID50]) of SIVmac239. In order to pinpoint the time of infection, vaccinated and control animals were challenged every 2 weeks and VLs were evaluated 7 and 10 days after each exposure (Fig. 6A). Monkeys that remained aviremic on both days 7 and 10 were rechallenged on day 14. Animals exhibiting positive VLs on either one of these occasions were considered to be infected and were no longer challenged.
FIG 6.

Acquisition of SIV infection in vaccinees and control animals. (A) Challenge scheme. Macaques were exposed to SIV on day 0 and subsequently bled on days 7 and 10. Plasma collected on days 7 and 10 was assayed for the presence of SIV RNA, and a decision was made as to whether or not to challenge the animals on day 14. Macaques that remained aviremic on both days 7 and 10 were rechallenged, whereas monkeys with a positive VL on either of these days were not rechallenged. (B) Kinetics of SIVmac239 acquisition in vaccinated and control macaques. Individual animals in the vaccine and control groups are depicted along with the IR SIVmac239 exposures that caused (filled circles) or did not cause (empty circles) productive infection. Although macaque r08009 likely acquired infection after the 7th SIV exposure, it ended up being challenged 9 times (see the text for details). Macaque r10091 skipped challenge 7 but was rechallenged two additional times (see the text for details). (C) Survival analysis of SIV-infected vaccinated and control animals. The P value was determined by the Cox proportional hazard model (hazard ratio, 0.59; 95% confidence interval, 0.19 to 1.85).
All control monkeys acquired SIV infection by the 6th IR challenge (Fig. 6B). By comparison, four vaccinees (r08009, r10038, r09041, and r10091) remained uninfected after the 6th exposure (Fig. 6B). Although the infection rate for the vaccinated group was numerically lower than that of the control group, this difference was not statistically significant (P = 0.363; Fig. 6C). We kept challenging the uninfected vaccinees to evaluate the extent to which they were resistant to SIV infection (Fig. 6B).
Of note, vaccinee r10091 had a VL of 1,000 vRNA copies/ml 7 days after the 6th exposure, but no virus was detected in its day 10 plasma sample. Because this pattern resembled the abrupt and early control of SIV replication manifested by macaques prophylactically vaccinated with 68-1 RhCMV-based vectors (22, 23), r10091 was not subjected to a 7th IR challenge (Fig. 6B). Instead, we bled this animal in the following week and carried out an ICS assay. This analysis revealed no signs of secondary CD8+ T-cell responses against vaccine-encoded antigens (Vif, Nef, Rev, and Tat; Fig. 7) or primary CD8+ T-cell responses against SIV proteins that were not included in the vaccine (Gag, Env, Vpr, and Vpx; Fig. 7). In support of this, the VL blip detected 7 days after the 6th challenge was not confirmed in a replicate plasma sample from that same time point, indicating that the initial VL measurement was a false positive. As a result, we resumed challenging r10091 in the next cycle, which corresponded to the 8th exposure for vaccinees r10038 and r09041 (Fig. 7B).
FIG 7.
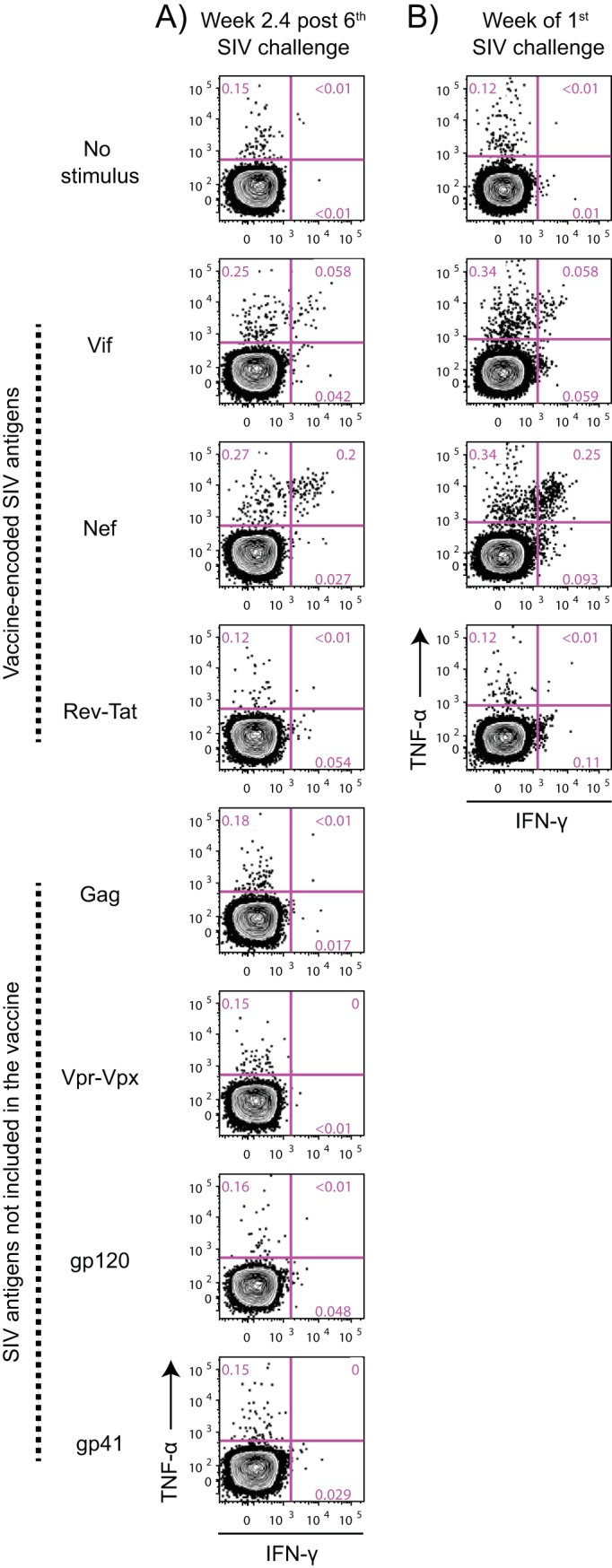
No signs of anamnestic expansion of vaccine-induced T-cell responses or de novo induction of SIV-specific T-cell responses in r10091 after the 6th SIV exposure. (A) SIV-specific CD8+ T-cell responses at week 2.4 after challenge 6. The antigen stimuli for this assay consisted of pools of SIV peptides corresponding to vaccine-encoded SIV proteins (Vif, Nef, and Rev-Tat) or antigens that were not included in the vaccine (Gag, gp120, gp41, and Vpr-Vpx). The results are shown as contour plots displaying the percentages of CD8+ T cells producing IFN-γ and/or TNF-α in the absence of stimulation (no stimulus) or in response to the aforementioned SIV peptide pools. (B) Baseline (week of the 1st SIV challenge) levels of vaccine-induced CD8+ T-cell responses against Vif, Nef, and Rev-Tat are shown as a reference. In all panels, the cells were gated on live CD14− CD16− CD20− CD3+ CD4− CD8+ lymphocytes.
Even though monkey r08009 became infected after the 7th exposure, we were initially unaware of this due to a false-negative result based on a technical VL assay issue, with the result corrected upon repeat testing. Consequently, this animal ended up being challenged two additional times (Fig. 6B). As for the remaining vaccinees, they remained aviremic until the 9th (r10091) or 10th (r10038 and r09041) exposures (Fig. 6B), at which point we halted the challenges in order to explore potential mechanisms of SIV resistance.
Half of the animals in the control group (three of the six) became ECs after infection (Fig. 8A), a rate that is consistent with our previous report on the outcome of SIVmac239 infection in unvaccinated Mamu-B*08+ macaques (30). By comparison, five of the seven (71%) SIVmac239-infected vaccinees controlled chronic-phase viremia to <1,000 vRNA copies/ml (Fig. 8B to F). This incidence of ECs among vaccinees was not significantly different from that in the control group (P = 0.59 from Fisher’s exact test). Of note, animal r08009 had a second surge of viremia at week 7 postinfection (p.i.) (Fig. 8B), likely as a consequence of the additional IR SIV challenges that this animal was subjected to after it became infected at the 7th exposure (Fig. 6B). After curbing the second burst of viral replication, r08009 experienced a transient increase in viremia at week 20 p.i. but then reasserted control of viral replication (Fig. 8B). Despite these unusual VL kinetics, r08009 was still among the infected vaccinees with the best control of SIVmac239 replication (Fig. 8B). Although monkey r10072 had low VLs of approximately 1,000 vRNA copies/ml early in the chronic phase, it progressively lost control of viral replication at later time points (Fig. 8G). Monkey r10051, on the other hand, never experienced VLs below 1,000 vRNA copies/ml during the course of SIVmac239 infection (Fig. 8H). Overall, vaccine-induced T-cell responses resulted in a modest, albeit statistically significant, reduction in peak VLs compared to those in the control group (P = 0.014 from Student's t test; Fig. 8I).
FIG 8.

Plasma virus concentrations after SIVmac239 infection. The viral loads of animals that acquired SIV infection were log transformed and correspond to the number of vRNA copies per milliliter of plasma. (A to H) Viral load traces for animals in the control group (A) or individual vaccinees that became infected (B to H). The dotted and dashed lines are for reference only and indicate VLs of 103 and 106 vRNA copies/ml of plasma, respectively. The two unintentional SIV challenges delivered to r08009 are indicated by vertical solid red lines in panel B. (I) Comparison of peak viral loads between SIV-infected vaccinees and control animals. The P value was calculated using Student's t test after log transformation. Lines correspond to geometric means, and each symbol denotes an animal. Monkey r08009 is color coded in blue because it manifested stringent control of chronic-phase viremia.
The fact that four vaccinees (r08009, r10038, r10091, and r09041; 40% of the vaccinated group) resisted 6 or more IR SIV challenges was unexpected, considering the overall rate of SIVmac239 acquisition in control monkeys subjected to the same IR SIVmac239 challenge regimen employed here. Of the 60 SIV-naive RMs (including the 6 contemporaneous control animals) that have been challenged with the same dose of the same SIVmac239 stock used here, only 11 (18%) have remained uninfected after the 6th IR exposure. This observation led us to hypothesize that vaccine-induced T-cell responses were responsible for this effect. To explore this possibility, we first assessed whether vaccine-elicited SIV-specific CD8+ T cells in the four animals that resisted ≥6 IR SIV challenges shared any unique immunological attributes that could distinguish them from the other vaccinees that acquired SIV infection at earlier challenges. We found that neither memory phenotype (Fig. 3), tissue distribution (Fig. 4), nor the functional profile (Fig. 5C to E) of the vaccine-induced SIV epitope-specific CD8+ T cells distinguished the four vaccinees that resisted SIV infection from their group counterparts that acquired infection after the 6th SIV exposure (Table 2). Furthermore, the total magnitude of vaccine-elicited CD8+ T-cell responses against Vif, Nef, Rev, and Tat did not predict the rate of SIV acquisition in vaccinees (Fig. 5A; Table 2).
TABLE 2.
Lack of associations between parameters of vaccine-induced CD8+ T-cell responses and acquisition of SIV infectiona
| Immunological variable | SIV target | Tissue | Time point (study wk) | Hazard ratio (95% confidence interval) | P value |
|---|---|---|---|---|---|
| Frequency of Vif RL8-specific CD8+ TEM2 responsesb | Vif RL8 | PBMC | 37 | 0.00 (0.00–12.88) | 0.173 |
| Frequency of Vif RL9-specific CD8+ TEM2 responsesb | Vif RL9 | PBMC | 37 | 0.02 (0.00–248.68) | 0.400 |
| Frequency of Nef RL10-specific CD8+ TEM2 responsesb | Nef RL10 | PBMC | 37 | 2.53 (0.36–17.70) | 0.350 |
| Frequency of CD8+ T cells in PBMCc | Vif RL8 + Vif RL9 + Nef RL10 | PBMC | 37 | 1.29 (0.37–4.52) | 0.695 |
| Frequency of CD8+ T cells in LNc | Vif RL8 + Vif RL9 + Nef RL10 | LN biopsy specimens | 37 | 2.17 (0.49–9.70) | 0.310 |
| Frequency of CD8+ T cells in gutc | Vif RL8 + Vif RL9 + Nef RL10 | Colorectal biopsy specimens | 37 | 12.66 (0.02–9,756.33) | 0.454 |
| Frequency of polyfunctional CD8+ T cellsd | Vif RL8 + Vif RL9 + Nef RL10 | PBMC | 41 | 0.04 (0.00–20.78) | 0.304 |
| Total frequency of SIV-specific CD8+ T cellse | Vif, Nef, Rev, and Tat | PBMC | 41 | 0.98 (0.23–4.11) | 0.981 |
To appropriately account for censoring (three animals never acquired SIV infection), the Cox proportional hazard model was used for this analysis.
Calculated as the fraction of tetramer+ CD8+ T cells in PBMC that expressed the TEM2 memory phenotype.
Based on the data shown in Fig. 4D.
Calculated as the fraction of SIV epitope-specific CD8+ T cells capable of degranulating (as measured by CD107a upregulation) and producing both IFN-γ and TNF-α (based on the data in Fig. 5C to E).
Based on the data shown in Fig. 5A.
Next, we assessed whether r10038, r10091, and r09041 developed T-cell responses against SIV proteins that were not included in the vaccine after being repeatedly challenged with SIVmac239. Monkey r08009 was not included in this analysis because it became infected after the 7th SIV exposure. The premise for this analysis was that, over the course of these serial SIV exposures, enough cells could have become transiently infected in vivo to prime de novo SIV-specific T-cell responses. Along these lines, Hansen et al. have reported that 68-1 RhCMV/SIV vaccinees that manifest early control of viral replication after SIVmac239 infection develop de novo T-cell responses against antigens that were not included in the vaccine (23). We therefore searched for T-cell responses against Gag, Pol, Vpr, Vpx, and Env in the three SIV-exposed aviremic vaccinees 4 days prior to the 10th IR SIV challenge. This analysis revealed no evidence that the three animals developed primary SIV-specific T cells or that they mounted anamnestic T-cell responses against vaccine-encoded antigens (Fig. 9A). Of note, an SIV gp140 enzyme-linked immunosorbent assay carried out 4 days before (r10038 and r10091) and 2 weeks after (r09041) the 10th IR SIV exposure also showed that the three SIV-exposed aviremic vaccinees were Env seronegative.
FIG 9.

No evidence for de novo induction of SIV-specific CD8+ T-cell responses or SIV rebound following CD8α depletion in vaccinees that resisted repeated IR exposures to SIV. (A) Macaques r10038, r10091, and r09041 were bled 4 days prior to the 10th IR SIV challenge, and an ICS assay was carried out in PBMC. The antigen stimuli for this assay consisted of peptides corresponding to SIV antigens that were included in the vaccine (Vif, Nef, and Rev-Tat) as well as proteins that were not included in the vaccine (Gag, Pol, Vpr, Vpx, and Env). The percentages of responding CD8+ T cells displayed in the panel were calculated by adding the background-subtracted frequencies of positive responses producing any combination of IFN-γ, TNF-α, and CD107a. Lines represent means, and each symbol corresponds to one vaccinee. (B to D) To evaluate whether r10038 and r09041 harbored replication-competent SIV, these animals were treated with a single i.v. infusion of 50 mg/kg of a CD8α-depleting MAb 39 weeks after the 10th IR SIVmac239 challenge. Monkey r10091 was not subjected to this procedure because it had to be euthanized prior to it. The absolute counts of CD8+ T cells (CD3+ CD8α+) and NK cells (CD3− CD8α+ CD16+) per microliter of blood after the CD8α depletion are shown in panels B and C, respectively. (D) Viral loads after CD8α depletion. The VLs were log transformed and correspond to the number of vRNA copies per milliliter of plasma. The dash-dot line indicates the limit of detection (LOD) of the VL assay (15 vRNA copies/ml of plasma).
Whitney et al. have shown that even if antiretroviral therapy (ART) is initiated in RMs 3 days after IR SIV challenge and subsequently maintained for 6 months, SIV rebound still occurred upon treatment interruption (49). Critically, these animals remained aviremic for the duration of the ART regimen, during which time no SIV-specific immune responses were detected (49). In light of these data, we investigated whether the SIV-exposed aviremic vaccinees in the present experiment harbored replication-competent virus that was being kept under control by their vaccine-induced CD8+ T cells. Our approach was to deplete CD8+ lymphocytes by the infusion of an anti-CD8α monoclonal antibody and monitor the treated animals for the development of viremia in the ensuing weeks. Of note, monkey r10091 developed unmanageable self-injurious behavior and had to be euthanized prior to this procedure. As a result, only vaccinees r10038 and r09041 were subjected to the CD8α depletion. Treatment with the anti-CD8α antibody occurred 39 weeks after the 10th IR SIV exposure and transiently eliminated both circulating CD8+ T cells and NK cells in r10038 and r09041 (Fig. 9B and C). However, the CD8α depletion did not result in SIV rebound (Fig. 9D), indicating that both animals lacked replication-competent SIV. Thus, it is unclear why r10091, r10038, and r09041 resisted 9 to 10 IR challenges with SIVmac239, whereas the majority of vaccinated and all control animals became infected by the 6th IR SIV challenge.
DISCUSSION
Because Mamu-B*08+ RMs are predisposed to developing effective CD8+ T-cell responses against SIV, these animals constitute a potentially useful resource to study T-cell-mediated containment of immunodeficiency virus replication. With this rationale in mind, the goal of the present study was to evaluate the protective efficacy of a heterologous prime/boost/boost vaccine regimen encoding immunodominant SIV epitopes in Mamu-B*08+ monkeys. We show that six repeated IR challenges with SIVmac239 resulted in productive infection in 6/6 control animals compared to 6/10 vaccinees. This difference in SIV acquisition was not, however, statistically significant. After 3 to 4 additional challenges, one of the vaccinees became infected but the other three remained aviremic following the 9th or 10th IR SIV exposure. This outcome is intriguing and potentially interesting but must be interpreted with caution, given the small number of animals utilized in the present experiment. Mucosal transmission of immunodeficiency viruses is a poorly understood process that can be shaped by both selective and stochastic processes (50). Moreover, several factors might impact the outcome of IR challenge studies, including mucosal integrity (51), local inflammation (52), the gut microbiome (53, 54), and the amount of intraluminal feces present at the time of virus exposure (55). Thus, we cannot rule out the possibility that the delayed (although statistically not significant) infection rate observed in the vaccine group was due to factors unrelated to vaccine-elicited T-cell immunity.
Nevertheless, it is conceivable that the resistance to productive SIV infection observed in the three vaccinees that remained aviremic after 9 to 10 IR challenges was vaccine mediated. Indeed, mounting evidence suggests that lentivirus-specific CD8+ T cells can do more than reduce viremia in productively infected individuals. For instance, a recent study has shown that adoptively transferring large quantities of SIV-specific CD8+ T cells into RMs 3 days after a high-dose IR challenge with SIV can reduce the number of transmitted/founder viruses in infected animals (56). Although this approach did not prevent the establishment of productive infection, it lends support to the notion that high frequencies of vaccine-elicited CD8+ T cells situated at relevant sites of virus replication can intercept a nascent infection before it becomes systemic. Along these lines, Xu et al. have recently evaluated the efficacy of a mucosal T-cell-based vaccine encoding SIV tat, vif, rev, and vpr in RMs (57). Following repeated IR challenges with SIVmac251, the authors observed that the vaccinees became infected at a lower rate than the control animals. Although the P value for this difference (0.08) was slightly above the nominal significance level of 0.05, it is noteworthy that 2/6 vaccinees compared to 0/6 controls remained aviremic after 10 IR exposures to SIVmac251. In contrast to our findings in the three Mamu-B*08+ vaccinees that resisted 9 to 10 IR challenges with SIVmac239, the two aviremic vaccinees in the study by Xu and colleagues developed primary T-cell responses against Gag (an antigen that was not included in the vaccine) and anamnestic T-cell responses against vaccine-encoded SIV antigens (57). Together, these observations indicate that these two animals became infected but that, perhaps through the action of their vaccine-induced T cells, the nascent SIV infection could not spread beyond the initial foci of infected cells.
Assuming that CD8+ T-cell immunity was responsible for the SIV challenge outcomes reported here and by Xu and colleagues (57), where did vaccine-induced SIV-specific CD8+ T cells intercept the first cells that became infected? Considering the rapidity with which SIV can traverse the rectal mucosa and disseminate to local and distant lymphoid structures (58), T-cell-mediated suppression of viral replication might have occurred at multiple anatomic sites. Consistent with this notion, the ability of 50% of 68-1 RhCMV/SIV vaccinees to control viral replication shortly after SIVmac239 infection is presumably due to the presence of potent SIV-specific TEM responses at both effector and secondary lymphoid tissues at the time of SIV exposure (23, 24). These vaccine-induced TEM responses are thought to restrict viral spread beyond a relatively small and short-lived population of initially infected cells, with clearance or spontaneous decay of this population over time (23, 24, 59). Of note, vaccine-induced CD8+ T cells directed against Mamu-B*08-restricted epitopes were also detected in colorectal and LN biopsy specimens of all vaccinees in the present study prior to SIV challenge. However, the frequencies of these responses were low in some of the animals and did not distinguish between vaccinees that acquired SIV infection and those that did not. Thus, despite the hint that vaccine-induced CD8+ T cells prevented systemic SIV replication in the present experiment, larger monkey trials will be needed to validate this hypothesis.
MATERIALS AND METHODS
Research animals and ethics statement.
Sixteen RMs expressing the Mamu-B*08 MHC-I allele were used. The details regarding animal welfare described herein are either similar or identical to those published in one of our previous experiments (18). The Indian-origin RMs (Macaca mulatta) utilized in this study were housed at the Wisconsin National Primate Research Center (WNPRC). All animals were cared for in accordance with the guidelines of the Weatherall report (66) and the principles described in the National Research Council’s Guide for the Care and Use of Laboratory Animals (60) under a protocol approved by the University of Wisconsin Graduate School Animal Care and Use Committee. Furthermore, the RMs used in this study were managed according to the animal husbandry program of WNPRC, which aims at providing consistent and excellent care to nonhuman primates at the center. This program is employed by the Colony Management Unit and is based on the laws, regulations, and guidelines promulgated by the United States Department of Agriculture, Institute for Laboratory Animal Research (e.g., Guide for the Care and Use of Laboratory Animals, 8th ed. [60]), Public Health Service, National Research Council, Centers for Disease Control and Prevention, and the Association for Assessment and Accreditation of Laboratory Animal Care International. The nutritional plan utilized by the WNPRC is based on recommendations published by the National Research Council. Specifically, RMs were fed twice daily with the 2050 Teklad Global 20% protein primate diet, and food intake was closely monitored by animal research technicians. This diet was also supplemented with a variety of fruits, vegetables, and other edible objects as part of the environmental enrichment program established by the Behavioral Management Unit. Paired/grouped animals exhibiting stereotypical and/or incompatible behaviors were reported to the behavioral management staff and managed accordingly. All primary enclosures (i.e., stationary cages, mobile racks, and pens) and animal rooms were cleaned daily with water and sanitized at least once every 2 weeks. Lights were on a 12-h-light, 12-h-dark diurnal schedule. Vaccinations were performed under anesthesia (ketamine administered at 5 to 12 mg/kg of body weight, depending on the animal), and all efforts were made to minimize suffering. Euthanasia was performed at the end of the study or whenever an animal experienced conditions deemed distressful by one of the veterinarians at WNPRC. All euthanasias were performed in accordance with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association and consisted of an intravenous (i.v.) overdose (greater than or equal to 50 mg/kg or to effect) of sodium pentobarbital or equivalent, as approved by a clinical veterinarian, preceded by ketamine (at least 15 mg/kg body weight) given by the intramuscular (i.m.) route. Additional animal information, including MHC-I alleles, age at the beginning of study, and sex, is shown in Table 1.
In vivo CD8α depletion.
CD8α-expressing lymphocytes were transiently depleted using the complementarity-determining region-grafted rhesus IgG1 CD8α-specific monoclonal antibody (MAb) MT807R1, which was provided by the NIH Nonhuman Primate Reagent Resource (Boston, MA). Monkeys r10038 and r09041 received a single i.v. injection of 50 mg/kg of body weight of MT807R1. The frequencies of lymphocyte subsets before and after the MT807R1 injection were monitored as described previously (17). The absolute counts of CD8+ T cells and NK cells were calculated by multiplying their respective frequencies in flow cytometric assays by the absolute number of white blood cells obtained from matching complete blood counts.
Vaccinations.
The vaccinated RMs were primed with two rAd5 vectors produced by Viraquest, Inc. One construct expressed a vif minigene corresponding to Vif aa 1 to 110, while the other expressed a nef minigene corresponding to Nef aa 45 to 210. The immunogenicity of these rAd5 vectors expressing vif and nef minigenes is described elsewhere (16). These vif and nef minigenes were codon optimized for expression in mammalian cells. The control animals were sham vaccinated with an empty Ad5 vector lacking any inserts, which was provided by the International AIDS Vaccine Initiative. A dose of 1011 viral particles of each rAd5 vector was administered intramuscularly to the left and right shoulders.
The rVSV boost occurred at study week 18. One rVSV vector expressing a fusion of the SIVmac239 Nef, Tat, and Vif proteins was administered intramuscularly to the RMs in the vaccine group. The control animals were sham vaccinated with an rVSV construct encoding the malaria CSP antigen. These rVSV constructs were based on a modified attenuated virus strain developed by Profectus Bioscience (61). Profectus Biosciences also provided the rVSV vectors for this study. A dose of 107 PFU of each rVSV vector was administered intramuscularly to the thighs.
The rRRV boost occurred at study week 25. The RMs in the vaccine group received a mixture of three rRRV vectors, each containing the full-length open reading frames of SIVmac239 vif or nef or a fusion of rev-tat-nef. The vif and nef genes were codon optimized for expression in mammalian cells, whereas the rev-tat-nef fusion gene retained its original codon usage. In order to prevent Nef-mediated downregulation of MHC-I molecules, both nef- and rev-tat-nef-expressing rRRV constructs lacked the nucleotides corresponding to Nef aa 239 to 240 (62). The control RMs were sham vaccinated with a rRRV vector encoding enhanced fluorescent green protein. One milliliter of phosphate-buffered saline (PBS) containing 7.1 × 107 genome copies of each of the appropriate rRRV vectors was administered via both the i.v. and IR routes. Details about the generation of these rRRV vectors have been described previously (42, 63).
SIVmac239 challenges.
At 16 weeks after the rRRV boost, all vaccinated and control RMs were subjected to repeated IR inoculations of 200 TCID50 (4.8 × 105 vRNA copies) of the same SIVmac239 stock described in our recent study (17). These IR challenges occurred every 2 weeks. Plasma VLs were assessed 7 and 10 days after each exposure. Once an animal experienced a positive VL at either one of these time points, it was no longer challenged. Only RMs that remained aviremic at both time points were rechallenged on day 14.
SIV RNA viral load measurements.
VLs were measured using 0.5 ml of EDTA-anticoagulated rhesus macaque plasma based on a modification of a previously published procedure (64). Total RNA was extracted from plasma samples using Qiagen DSP virus/pathogen midikits on a QIAsymphony XP laboratory automation instrument platform. Six replicate two-step reverse transcription-PCRs were performed per sample using a randomly primed reverse transcription reaction, followed by 45 cycles of PCR using the following primers and probe: forward primer SGAG21 [5′-GTCTGCGTCAT(dP)TGGTGCATTC-3′], reverse primer SGAG22 [5′-CACTAG(dK)TGTCTCTGCACTAT(dP)TGTTTTG-3′], and probe PSGAG23 [5′-FAM-CTTC(dP)TCAGT(dK)TGTTTCACTTTCTCTTCTGCG-BHQ1-3′], where FAM is 6-carboxyfluorescein and BHQ1 is black hole quencher 1]. The limit of reliable quantitation on an input volume of 0.5 ml of plasma was 15 vRNA copies/ml.
Sample processing.
We isolated peripheral blood mononuclear cells (PBMC) from EDTA-treated blood by Ficoll-Paque Plus (GE Health Sciences) density centrifugation. Red blood cells were lysed by treating PBMC with ACK buffer (H2O containing 8.3 g/liter of NH4Cl, 1.0 g/liter KHCO3, and 0.42 g/liter Na2EDTA) for 5 min. Cells were subsequently washed in R10 medium (RPMI 1640 medium supplemented with GlutaMAX [Life Technologies], 10% fetal bovine serum, and 1% antibiotic/antimycotic) and then resuspended at various concentrations depending on the application. LN and gut biopsy specimens were obtained as described previously (36). To process the LN biopsy specimens, we placed the tissue specimen in 100-μm cell strainers and gently pushed against the nylon mesh using a 3-ml syringe plunger until the tissue was disrupted. We counted the ensuing cell suspension and then used it in immunological assays. Rectal and colon biopsy specimens were obtained from the same animal and pooled during the lymphocyte isolation process. These samples were washed once in R10 medium and then resuspended in collagenase medium (R10 medium with 0.5 mg/ml of type II collagenase from Clostridium histolyticum [Sigma-Aldrich]). After rocking the cells at 80 rpm for 30 min at 37°C on a tabletop shaker, the supernatant was collected and washed twice with R10 medium. This digestion cycle was repeated two additional times. The cells collected after each digestion cycle were then pooled and layered on top of a 40% to 90% Percoll gradient. After a 30-min centrifugation at 450 × g, purified gut lymphocytes were collected from the interface of the 40% and 90% Percoll layers. The cells were then washed once with R10 medium, counted, and, finally, used in immunological assays. Of note, all tissues described above were harvested at WNPRC and subsequently refrigerated to 4°C until they were shipped overnight to the University of Miami in temperature-controlled packages. Blood draws intended for VL measurements were an exception. In those cases, blood was processed shortly after phlebotomy and plasma aliquots were immediately frozen at −80°C.
Quantification of MHC-I tetramer+ CD8+ T cells in PBMC.
The tetramer staining assays performed as part of the time course analysis of vaccine-induced SIV-specific CD8+ T cells were done by labeling approximately 800,000 PBMC with titrated amounts of fluorochrome-conjugated MHC-I tetramers at room temperature for 45 min, as described previously (65). The cells were then stained with fluorochrome-labeled MAbs directed against the surface molecules CD3 (clone SP34-2), CD8α (clone RPA-T8), CD14 (clone M5E2), CD16 (clone 3G8), and CD20 (clone 2H7) for 25 min. This step also included an amine-reactive dye (ARD; LIVE/DEAD Fixable Aqua Dead cell stain; Life Technologies). The cells were then washed with wash buffer (Dulbecco's PBS with 0.1% bovine serum albumin and 0.45 g/liter NaN3) and fixed with PBS containing 2% paraformaldehyde (PFA) for 20 min at 4°C. The cells were washed one more time and then stored at 4°C until acquisition.
In order the determine the memory phenotype of tetramer+ CD8+ T cells, PBMC were labeled with the appropriate MHC-I tetramer reagents and then stained with the same MAbs against CD8α, CD14, CD16, and CD20 described above, with the addition of MAbs against CD28 (clone 28.2) and CCR7 (clone 150503). ARD Aqua was used in this step as well. The cells were then washed with wash buffer and fixed with PBS containing 2% PFA for 20 min at 4°C. Next, the cells were permeabilized by treatment with perm buffer (1× BD FACS Lysing Solution 2 [Beckton Dickinson] and 0.05% Tween 20 [Sigma-Aldrich]) for 10 min. The cells were then washed once and stained with the aforementioned CD3 MAb and a granzyme B-specific MAb (clone GB12). After a 30-min incubation in the dark at room temperature, the cells were washed and stored at 4°C until acquisition. All samples were acquired using FACSDIVA (version 6) software on a Special Order Research Product BD LSR-II apparatus equipped with a 50-mW 405-nm violet, a 100-mW 488-nm blue, and a 30-mW 635-nm red laser.
We used FlowJo (version 9.6) software (TreeStar, Inc.) to analyze the data. First, we gated on diagonally clustered singlets by plotting forward scatter height (FSC-H) versus forward scatter area (FSC-A) and then side scatter height (SSC-H) versus side scatter area (SSC-A). Next, we created a time gate that included only those events that were recorded within the 5th and 90th percentiles and then gated on dump channel-negative CD3+ cells. At this stage, we delineated the lymphocyte population based on its FSC-A and SSC-A properties and subsequently gated on CD8+ cells. After outlining tetramer+ cells, we conducted our memory phenotyping analysis within this gate. Cells stained with fluorochrome-labeled MAbs of the same isotypes as the anti-granzyme B, anti-CD28, and anti-CCR7 MAbs guided the identification of the memory subsets within the tetramer+ population. Based on this gating strategy, all tetramer frequencies reported in this report correspond to the percentages of live CD3+ CD8+ tetramer+ lymphocytes.
ICS assay.
PBMC were stimulated with the appropriate pools of SIV peptides in R10 medium containing costimulatory MAbs against CD28 and CD49d for 9 h at 37°C in a 5.0% CO2 incubator. A phycoerythrin-conjugated MAb specific for CD107a was also included in the assay. To inhibit protein transport, brefeldin A (BioLegend, Inc.) and GolgiStop (BD Biosciences) were added to all tubes 1 h into the incubation period. The antigen stimuli for the intracellular cytokine staining (ICS) assays performed during the vaccine phase consisted of pools of SIVmac239 peptides (15-mers overlapping by 11 aa), as indicated in Fig. 7 and 9. In some cases, two peptide pools were combined in the same test. Peptides corresponding to Mamu-B*08-restricted SIV epitopes were also used as the antigen stimuli in ICS assays. The final assay concentration of each peptide was 10.0 μM. We employed the same steps outlined above to stain molecules on the surface of cells, fix them with 2% paraformaldehyde, and permeabilize them with Perm Buffer. The surface staining MAb cocktail included the same MAbs against CD14, CD16, CD20, and CD8α described above, as well as ARD Aqua, with the addition of an anti-CD4 (clone OKT4) MAb. After permeabilization, the cells were incubated with MAbs against CD3 (clone SP34-2; BD Biosciences), IFN-γ (clone 4S.B3; BioLegend, Inc.), TNF-α (clone Mab11; BD Biosciences), and CD69 (clone FN50; BioLegend, Inc.) for 1 h in the dark at room temperature After this incubation was completed, the cells were washed and subsequently stored at 4°C until acquisition. We analyzed the data by gating first on live CD14− CD16− CD20− CD3+ lymphocytes and then on cells expressing either CD4 or CD8 but not both markers. We then conducted functional analyses within these two compartments. Cells were considered positive for IFN-γ, TNF-α, or CD107a only if they coexpressed these molecules with CD69, a marker of recent activation. Once the appropriate gates were created, we employed the Boolean gate platform to generate a full array of possible combinations, equating to 8 response patterns when testing three functions (23 = 8). Leukocyte activation cocktail (LAC; BD Pharmingen)-stimulated cells stained with fluorochrome-labeled MAbs of the same isotypes as those against IFN-γ, TNF-α, and CD107a guided the identification of positive populations. We used two criteria to determine if the responses were positive. First, the frequency of events in each Boolean gate had to be at least 2-fold higher than their corresponding values in background-subtracted negative-control tests. Second, the Boolean gates for each response had to contain ≥10 events. The magnitude of responding CD4+ or CD8+ T cells was calculated by adding the frequencies of positive responses producing any combination of IFN-γ, TNF-α, and CD107a. Background subtraction and calculation of the frequencies of responding cells were performed with Microsoft Excel software.
Statistics.
The Cox proportional hazard model was used to determine whether the rAd5/rVSV/rRRV vaccine regimen evaluated here affected acquisition of SIV infection and to examine the association between infection rate and vaccine-induced T-cell responses. For these analyses, the number of challenges until productive infection was used as the time scale. To account for right skewness and inconsistent variances, all the T-cell response data were log transformed after adding a constant value (0.1). Student's t test was used after checking significant differences in variance by the F test (i) to compare the vaccine-induced CD8+ T-cell responses between the current study and our previous study (Fig. 2E) and (ii) to compare peak VLs between vaccinees and control animals (Fig. 8E). Repeated-measurement ANOVA was used to (i) test the difference in Gzm B expression between different epitopes (Fig. 3) and (ii) test the difference in the CD8+ T-cell responses between different memory T cells (Fig. 3), different SIV proteins (Fig. 5A and B), and different anatomical sites (Fig. 4). All P values reported in this paper are two-tailed.
ACKNOWLEDGMENTS
We thank Teresa Maidana Giret for confirming the MHC-I genotype of the monkeys in this study; Leydi Guzman for administrative assistance; Xiwei Chen and Stephanie Dickinson for assistance with statistical analyses; all members of the Immunology Services Unit at WNPRC, Kelli Oswald, Rebecca Shoemaker, Randy Fast, Mary Lopez, and Marina Kemelman for excellent technical support; and Eric Peterson and Kristin Crosno for professional animal care. We thank John Eldridge and Profectus Biosciences for providing rVSV-SIV vaccines based on their attenuated rVSV vector platform.
This work was funded by Public Health Service grants R01 AI108421 (to D.I.W.) and R37 AI052056 (to D.I.W.) from the National Institute of Allergy and Infectious Diseases. Partial support came from federal funds from the Office of Research Infrastructure Programs (P51 OD011106) and the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E (to J.D.L.). We also acknowledge the Miami Center for AIDS Research (P30 AI073961) for its support. The International AIDS Vaccine Initiative’s work is made possible by generous support from many donors, including The Bill & Melinda Gates Foundation, the Ministry of Foreign Affairs of Denmark, Irish Aid, the Ministry of Finance of Japan, the Ministry of Foreign Affairs of the Netherlands, the Norwegian Agency for Development Cooperation (NORAD), the United Kingdom Department for International Development (DFID), and the United States Agency for International Development (USAID). The full list of IAVI donors is available at www.iavi.org.
The contents are the responsibility of the International AIDS Vaccine Initiative and do not necessarily reflect the views of USAID or the United States government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Burton DR, Hangartner L. 2016. Broadly neutralizing antibodies to HIV and their role in vaccine design. Annu Rev Immunol 34:635–659. doi: 10.1146/annurev-immunol-041015-055515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hessell AJ, Malherbe DC, Haigwood NL. 2018. Passive and active antibody studies in primates to inform HIV vaccines. Expert Rev Vaccines 17:127–144. doi: 10.1080/14760584.2018.1425619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hessell AJ, Jaworski JP, Epson E, Matsuda K, Pandey S, Kahl C, Reed J, Sutton WF, Hammond KB, Cheever TA, Barnette PT, Legasse AW, Planer S, Stanton JJ, Pegu A, Chen X, Wang K, Siess D, Burke D, Park BS, Axthelm MK, Lewis A, Hirsch VM, Graham BS, Mascola JR, Sacha JB, Haigwood NL. 2016. Early short-term treatment with neutralizing human monoclonal antibodies halts SHIV infection in infant macaques. Nat Med 22:362–368. doi: 10.1038/nm.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu J, Ghneim K, Sok D, Bosche WJ, Li Y, Chipriano E, Berkemeier B, Oswald K, Borducchi E, Cabral C, Peter L, Brinkman A, Shetty M, Jimenez J, Mondesir J, Lee B, Giglio P, Chandrashekar A, Abbink P, Colantonio A, Gittens C, Baker C, Wagner W, Lewis MG, Li W, Sekaly RP, Lifson JD, Burton DR, Barouch DH. 2016. Antibody-mediated protection against SHIV challenge includes systemic clearance of distal virus. Science 353:1045–1049. doi: 10.1126/science.aag0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen TM, Altfeld M, Geer SC, Kalife ET, Moore C, O'Sullivan KM, DeSouza I, Feeney ME, Eldridge RL, Maier EL, Kaufmann DE, Lahaie MP, Reyor L, Tanzi G, Johnston MN, Brander C, Draenert R, Rockstroh JK, Jessen H, Rosenberg ES, Mallal SA, Walker BD. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J Virol 79:13239–13249. doi: 10.1128/JVI.79.21.13239-13249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goulder PJ, Walker BD. 2012. HIV and HLA class I: an evolving relationship. Immunity 37:426–440. doi: 10.1016/j.immuni.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 8.Walker BD, Yu XG. 2013. Unravelling the mechanisms of durable control of HIV-1. Nat Rev Immunol 13:487–498. doi: 10.1038/nri3478. [DOI] [PubMed] [Google Scholar]
- 9.Parham P. 2009. Antigen recognition by T lymphocytes, p 125–154. In Foltin J, Masson S, Ghezzi K, Engels A, Lawrence E, Jeffcock E (ed), The immune system, 3rd ed Garland Science, Taylor & Francis Group, LLC, New York, NY. [Google Scholar]
- 10.Watkins DI, Burton DR, Kallas EG, Moore JP, Koff WC. 2008. Nonhuman primate models and the failure of the Merck HIV-1 vaccine in humans. Nat Med 14:617–621. doi: 10.1038/nm.f.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hel Z, Nacsa J, Tryniszewska E, Tsai WP, Parks RW, Montefiori DC, Felber BK, Tartaglia J, Pavlakis GN, Franchini G. 2002. Containment of simian immunodeficiency virus infection in vaccinated macaques: correlation with the magnitude of virus-specific pre- and postchallenge CD4+ and CD8+ T cell responses. J Immunol 169:4778–4787. doi: 10.4049/jimmunol.169.9.4778. [DOI] [PubMed] [Google Scholar]
- 12.Hel Z, Tsai WP, Tryniszewska E, Nacsa J, Markham PD, Lewis MG, Pavlakis GN, Felber BK, Tartaglia J, Franchini G. 2006. Improved vaccine protection from simian AIDS by the addition of nonstructural simian immunodeficiency virus genes. J Immunol 176:85–96. doi: 10.4049/jimmunol.176.1.85. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, O'Brien KL, Lynch DM, Simmons NL, La Porte A, Riggs AM, Abbink P, Coffey RT, Grandpre LE, Seaman MS, Landucci G, Forthal DN, Montefiori DC, Carville A, Mansfield KG, Havenga MJ, Pau MG, Goudsmit J, Barouch DH. 2009. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature 457:87–91. doi: 10.1038/nature07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson NA, Reed J, Napoe GS, Piaskowski S, Szymanski A, Furlott J, Gonzalez EJ, Yant LJ, Maness NJ, May GE, Soma T, Reynolds MR, Rakasz E, Rudersdorf R, McDermott AB, O'Connor DH, Friedrich TC, Allison DB, Patki A, Picker LJ, Burton DR, Lin J, Huang L, Patel D, Heindecker G, Fan J, Citron M, Horton M, Wang F, Liang X, Shiver JW, Casimiro DR, Watkins DI. 2006. Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239. J Virol 80:5875–5885. doi: 10.1128/JVI.00171-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson NA, Keele BF, Reed JS, Piaskowski SM, MacNair CE, Bett AJ, Liang X, Wang F, Thoryk E, Heidecker GJ, Citron MP, Huang L, Lin J, Vitelli S, Ahn CD, Kaizu M, Maness NJ, Reynolds MR, Friedrich TC, Loffredo JT, Rakasz EG, Erickson S, Allison DB, Piatak MJ, Lifson JD, Shiver JW, Casimiro DR, Shaw GM, Hahn BH, Watkins DI. 2009. Vaccine-induced cellular responses control simian immunodeficiency virus replication after heterologous challenge. J Virol 83:6508–6521. doi: 10.1128/JVI.00272-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martins MA, Wilson NA, Piaskowski SM, Weisgrau KL, Furlott JR, Bonaldo MC, Veloso de Santana MG, Rudersdorf RA, Rakasz EG, Keating KD, Chiuchiolo MJ, Piatak MJ, Allison DB, Parks CL, Galler R, Lifson JD, Watkins DI. 2014. Vaccination with Gag, Vif, and Nef gene fragments affords partial control of viral replication after mucosal challenge with SIVmac239. J Virol 88:7493–7516. doi: 10.1128/JVI.00601-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martins MA, Tully DC, Shin YC, Gonzalez-Nieto L, Weisgrau KL, Bean DJ, Gadgil R, Gutman MJ, Domingues A, Maxwell HS, Magnani DM, Ricciardi M, Pedreño-Lopez N, Bailey V, Cruz MA, Lima NS, Bonaldo MC, Altman JD, Rakasz E, Capuano S, Reimann KA, Piatak M, Lifson JD, Desrosiers RC, Allen TM, Watkins DI. 2017. Rare control of SIVmac239 infection in a vaccinated rhesus macaque. AIDS Res Hum Retroviruses 33:843–858. doi: 10.1089/AID.2017.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martins MA, Shin YC, Gonzalez-Nieto L, Domingues A, Gutman MJ, Maxwell HS, Castro I, Magnani DM, Ricciardi M, Pedreño-Lopez N, Bailey V, Betancourt D, Altman JD, Pauthner M, Burton DR, von Bredow B, Evans DT, Yuan M, Parks CL, Ejima K, Allison DB, Rakasz E, Barber GN, Capuano S, Lifson JD, Desrosiers RC, Watkins DI. 2017. Vaccine-induced immune responses against both Gag and Env improve control of simian immunodeficiency virus replication in rectally challenged rhesus macaques. PLoS Pathog 13:e1006529. doi: 10.1371/journal.ppat.1006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koff WC, Johnson PR, Watkins DI, Burton DR, Lifson JD, Hasenkrug KJ, McDermott AB, Schultz A, Zamb TJ, Boyle R, Desrosiers RC. 2006. HIV vaccine design: insights from live attenuated SIV vaccines. Nat Immunol 7:19–23. doi: 10.1038/ni1296. [DOI] [PubMed] [Google Scholar]
- 20.Manrique J, Piatak M, Lauer W, Johnson W, Mansfield K, Lifson J, Desrosiers R. 2013. Influence of mismatch of Env sequences on vaccine protection by live attenuated simian immunodeficiency virus. J Virol 87:7246–7254. doi: 10.1128/JVI.00798-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukazawa Y, Park H, Cameron MJ, Lefebvre F, Lum R, Coombes N, Mahyari E, Hagen SI, Bae JY, Reyes MD, Swanson T, Legasse AW, Sylwester A, Hansen SG, Smith AT, Stafova P, Shoemaker R, Li Y, Oswald K, Axthelm MK, McDermott A, Ferrari G, Montefiori DC, Edlefsen PT, Piatak MJ, Lifson JD, Sekaly RP, Picker LJ. 2012. Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat Med 18:1673–1681. doi: 10.1038/nm.2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, Legasse AW, Axthelm MK, Oswald K, Trubey CM, Piatak MJ, Lifson JD, Nelson JA, Jarvis MA, Picker LJ. 2009. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med 15:293–299. doi: 10.1038/nm.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak MJ, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen SG, Piatak MJ, Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson JA, Fruh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, Picker LJ. 2013. Immune clearance of highly pathogenic SIV infection. Nature 502:100–104. doi: 10.1038/nature12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pellett PE, Roizman B. 2013. Herpesviridae, p 1802–1819. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 26.Masopust D, Picker LJ. 2012. Hidden memories: frontline memory T cells and early pathogen interception. J Immunol 188:5811–5817. doi: 10.4049/jimmunol.1102695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, Gilbride RM, Lewis MS, Gilliam AN, Ventura AB, Malouli D, Xu G, Richards R, Whizin N, Reed JS, Hammond KB, Fischer M, Turner JM, Legasse AW, Axthelm MK, Edlefsen PT, Nelson JA, Lifson JD, Fruh K, Picker LJ. 2013. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340:1237874. doi: 10.1126/science.1237874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen SG, Wu HL, Burwitz BJ, Hughes CM, Hammond KB, Ventura AB, Reed JS, Gilbride RM, Ainslie E, Morrow DW, Ford JC, Selseth AN, Pathak R, Malouli D, Legasse AW, Axthelm MK, Nelson JA, Gillespie GM, Walters LC, Brackenridge S, Sharpe HR, Lopez CA, Fruh K, Korber BT, McMichael AJ, Gnanakaran S, Sacha JB, Picker LJ. 2016. Broadly targeted CD8(+) T cell responses restricted by major histocompatibility complex E. Science 351:714–720. doi: 10.1126/science.aac9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goulder PJ, Watkins DI. 2008. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol 8:619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loffredo JT, Maxwell J, Qi Y, Glidden CE, Borchardt GJ, Soma T, Bean AT, Beal DR, Wilson NA, Rehrauer WM, Lifson JD, Carrington M, Watkins DI. 2007. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J Virol 81:8827–8832. doi: 10.1128/JVI.00895-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nomura T, Yamamoto H, Shiino T, Takahashi N, Nakane T, Iwamoto N, Ishii H, Tsukamoto T, Kawada M, Matsuoka S, Takeda A, Terahara K, Tsunetsugu-Yokota Y, Iwata-Yoshikawa N, Hasegawa H, Sata T, Naruse TK, Kimura A, Matano T. 2012. Association of major histocompatibility complex class I haplotypes with disease progression after simian immunodeficiency virus challenge in Burmese rhesus macaques. J Virol 86:6481–6490. doi: 10.1128/JVI.07077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yant LJ, Friedrich TC, Johnson RC, May GE, Maness NJ, Enz AM, Lifson JD, O'Connor DH, Carrington M, Watkins DI. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol 80:5074–5077. doi: 10.1128/JVI.80.10.5074-5077.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loffredo JT, Sidney J, Bean AT, Beal DR, Bardet W, Wahl A, Hawkins OE, Piaskowski S, Wilson NA, Hildebrand WH, Watkins DI, Sette A. 2009. Two MHC class I molecules associated with elite control of immunodeficiency virus replication, Mamu-B*08 and HLA-B*2705, bind peptides with sequence similarity. J Immunol 182:7763–7775. doi: 10.4049/jimmunol.0900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loffredo JT, Friedrich TC, Leon EJ, Stephany JJ, Rodrigues DS, Spencer SP, Bean AT, Beal DR, Burwitz BJ, Rudersdorf RA, Wallace LT, Piaskowski SM, May GE, Sidney J, Gostick E, Wilson NA, Price DA, Kallas EG, Piontkivska H, Hughes AL, Sette A, Watkins DI. 2007. CD8+ T cells from SIV elite controller macaques recognize Mamu-B*08-bound epitopes and select for widespread viral variation. PLoS One 2:e1152. doi: 10.1371/journal.pone.0001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loffredo JT, Bean AT, Beal DR, Leon EJ, May GE, Piaskowski SM, Furlott JR, Reed J, Musani SK, Rakasz EG, Friedrich TC, Wilson NA, Allison DB, Watkins DI. 2008. Patterns of CD8+ immunodominance may influence the ability of Mamu-B*08-positive macaques to naturally control simian immunodeficiency virus SIVmac239 replication. J Virol 82:1723–1738. doi: 10.1128/JVI.02084-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mudd PA, Martins MA, Ericsen AJ, Tully DC, Power KA, Bean AT, Piaskowski SM, Duan L, Seese A, Gladden AD, Weisgrau KL, Furlott JR, Kim YI, Veloso de Santana MG, Rakasz E, Capuano S, Wilson NA, Bonaldo MC, Galler R, Allison DB, Piatak MJ, Haase AT, Lifson JD, Allen TM, Watkins DI. 2012. Vaccine-induced CD8+ T cells control AIDS virus replication. Nature 491:129–133. doi: 10.1038/nature11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martins MA, Tully DC, Cruz MA, Power KA, Veloso de Santana MG, Bean DJ, Ogilvie CB, Gadgil R, Lima NS, Magnani DM, Ejima K, Allison DB, Piatak MJ, Altman JD, Parks CL, Rakasz EG, Capuano S, Galler R, Bonaldo MC, Lifson JD, Allen TM, Watkins DI. 2015. Vaccine-induced simian immunodeficiency virus-specific CD8+ T-cell responses focused on a single Nef epitope select for escape variants shortly after infection. J Virol 89:10802–10820. doi: 10.1128/JVI.01440-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masopust D, Ha SJ, Vezys V, Ahmed R. 2006. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol 177:831–839. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- 39.Picker LJ, Hansen SG, Lifson JD. 2012. New paradigms for HIV/AIDS vaccine development. Annu Rev Med 63:95–111. doi: 10.1146/annurev-med-042010-085643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Damania B, Desrosiers RC. 2001. Simian homologues of human herpesvirus 8. Philos Trans R Soc Lond B Biol Sci 356:535–543. doi: 10.1098/rstb.2000.0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desrosiers RC, Sasseville VG, Czajak SC, Zhang X, Mansfield KG, Kaur A, Johnson RP, Lackner AA, Jung JU. 1997. A herpesvirus of rhesus monkeys related to the human Kaposi’s sarcoma-associated herpesvirus. J Virol 71:9764–9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bilello JP, Manrique JM, Shin YC, Lauer W, Li W, Lifson JD, Mansfield KG, Johnson RP, Desrosiers RC. 2011. Vaccine protection against simian immunodeficiency virus in monkeys using recombinant gamma-2 herpesvirus. J Virol 85:12708–12720. doi: 10.1128/JVI.00865-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shin YC, Bischof GF, Lauer WA, Gonzalez-Nieto L, Rakasz EG, Hendricks GM, Watkins DI, Martins MA, Desrosiers RC. 2018. A recombinant herpesviral vector containing a near-full-length SIVmac239 genome produces SIV particles and elicits immune responses to all nine SIV gene products. PLoS Pathog 14:e1007143. doi: 10.1371/journal.ppat.1007143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Picker LJ, Reed-Inderbitzin EF, Hagen SI, Edgar JB, Hansen SG, Legasse A, Planer S, Piatak MJ, Lifson JD, Maino VC, Axthelm MK, Villinger F. 2006. IL-15 induces CD4 effector memory T cell production and tissue emigration in nonhuman primates. J Clin Invest 116:1514–1524. doi: 10.1172/JCI27564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, Kovacs CM, Rodriguez B, Sieg SF, Teixeira-Johnson L, Gudonis D, Goepfert PA, Lederman MM, Frank I, Makedonas G, Kaul R, Walker BD, Betts MR. 2010. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog 6:e1000917. doi: 10.1371/journal.ppat.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, Rood JE, Berkley AM, Sacha JB, Cogliano-Shutta NA, Lloyd M, Roby G, Kwan R, McLaughlin M, Stallings S, Rehm C, O'Shea MA, Mican JA, Packard BZ, Komoriya A, Palmer S, Wiegand AP, Maldarelli F, Coffin JM, Mellors JW, Hallahan CW, Follman DA, Connors M. 2008. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 29:1009–1021. doi: 10.1016/j.immuni.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shankar P, Xu Z, Lieberman J. 1999. Viral-specific cytotoxic T lymphocytes lyse human immunodeficiency virus-infected primary T lymphocytes by the granule exocytosis pathway. Blood 94:3084–3093. [PubMed] [Google Scholar]
- 48.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whitney JB, Hill AL, Sanisetty S, Penaloza-MacMaster P, Liu J, Shetty M, Parenteau L, Cabral C, Shields J, Blackmore S, Smith JY, Brinkman AL, Peter LE, Mathew SI, Smith KM, Borducchi EN, Rosenbloom DI, Lewis MG, Hattersley J, Li B, Hesselgesser J, Geleziunas R, Robb ML, Kim JH, Michael NL, Barouch DH. 2014. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 512:74–77. doi: 10.1038/nature13594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keele BF, Estes JD. 2011. Barriers to mucosal transmission of immunodeficiency viruses. Blood 118:839–846. doi: 10.1182/blood-2010-12-325860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hladik F, McElrath MJ. 2008. Setting the stage: host invasion by HIV. Nat Rev Immunol 8:447–457. doi: 10.1038/nri2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Passmore JA, Jaspan HB, Masson L. 2016. Genital inflammation, immune activation and risk of sexual HIV acquisition. Curr Opin HIV AIDS 11:156–162. doi: 10.1097/COH.0000000000000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Noguera-Julian M, Rocafort M, Guillén Y, Rivera J, Casadellà M, Nowak P, Hildebrand F, Zeller G, Parera M, Bellido R, Rodríguez C, Carrillo J, Mothe B, Coll J, Bravo I, Estany C, Herrero C, Saz J, Sirera G, Torrela A, Navarro J, Crespo M, Brander C, Negredo E, Blanco J, Guarner F, Calle ML, Bork P, Sönnerborg A, Clotet B, Paredes R. 2016. Gut microbiota linked to sexual preference and HIV infection. EBioMedicine 5:135–146. doi: 10.1016/j.ebiom.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sui Y, Dzutsev A, Venzon D, Frey B, Thovarai V, Trinchieri G, Berzofsky JA. 2018. Influence of gut microbiome on mucosal immune activation and SHIV viral transmission in naive macaques. Mucosal Immunol 11:1219–1229. doi: 10.1038/s41385-018-0029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smedley J, Turkbey B, Bernardo ML, Del Prete GQ, Estes JD, Griffiths GL, Kobayashi H, Choyke PL, Lifson JD, Keele BF. 2014. Tracking the luminal exposure and lymphatic drainage pathways of intravaginal and intrarectal inocula used in nonhuman primate models of HIV transmission. PLoS One 9:e92830. doi: 10.1371/journal.pone.0092830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ayala VI, Trivett MT, Barsov EV, Jain S, Piatak M, Trubey CM, Alvord WG, Chertova E, Roser JD, Smedley J, Komin A, Keele BF, Ohlen C, Ott DE. 2016. Adoptive transfer of engineered rhesus simian immunodeficiency virus-specific CD8+ T cells reduces the number of transmitted/founder viruses established in rhesus macaques. J Virol 90:9942–9952. doi: 10.1128/JVI.01522-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu H, Andersson AM, Ragonnaud E, Boilesen D, Tolver A, Jensen BA, Blanchard JL, Nicosia A, Folgori A, Colloca S, Cortese R, Thomsen AR, Christensen JP, Veazey RS, Holst PJ. 2017. Mucosal vaccination with heterologous viral vectored vaccine targeting subdominant SIV accessory antigens strongly inhibits early viral replication. EBioMedicine 18:204–215. doi: 10.1016/j.ebiom.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ribeiro Dos Santos P, Rancez M, Prétet J-L, Michel-Salzat A, Messent V, Bogdanova A, Couëdel-Courteille A, Souil E, Cheynier R, Butor C. 2011. Rapid dissemination of SIV follows multisite entry after rectal inoculation. PLoS One 6:e19493. doi: 10.1371/journal.pone.0019493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okoye AA, Hansen SG, Vaidya M, Fukazawa Y, Park H, Duell DM, Lum R, Hughes CM, Ventura AB, Ainslie E, Ford JC, Morrow D, Gilbride RM, Legasse AW, Hesselgesser J, Geleziunas R, Li Y, Oswald K, Shoemaker R, Fast R, Bosche WJ, Borate BR, Edlefsen PT, Axthelm MK, Picker LJ, Lifson JD. 2018. Early antiretroviral therapy limits SIV reservoir establishment to delay or prevent post-treatment viral rebound. Nat Med 24:1430–1440. doi: 10.1038/s41591-018-0130-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 61.Fuchs JD, Frank I, Elizaga ML, Allen M, Frahm N, Kochar N, Li S, Edupuganti S, Kalams SA, Tomaras GD, Sheets R, Pensiero M, Tremblay MA, Higgins TJ, Latham T, Egan MA, Clarke DK, Eldridge JH, Mulligan M, Rouphael N, Estep S, Rybczyk K, Dunbar D, Buchbinder S, Wagner T, Isbell R, Chinnell V, Bae J, Escamilla G, Tseng J, Fair R, Ramirez S, Broder G, Briesemeister L, Ferrara A. 2015. First-in-human evaluation of the safety and immunogenicity of a recombinant vesicular stomatitis virus human immunodeficiency virus-1 gag vaccine (HVTN 090). Open Forum Infect Dis 2:ofv082. doi: 10.1093/ofid/ofv082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Swigut T, Alexander L, Morgan J, Lifson J, Mansfield KG, Lang S, Johnson RP, Skowronski J, Desrosiers R. 2004. Impact of Nef-mediated downregulation of major histocompatibility complex class I on immune response to simian immunodeficiency virus. J Virol 78:13335–13344. doi: 10.1128/JVI.78.23.13335-13344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bilello JP, Morgan JS, Damania B, Lang SM, Desrosiers RC. 2006. A genetic system for rhesus monkey rhadinovirus: use of recombinant virus to quantitate antibody-mediated neutralization. J Virol 80:1549–1562. doi: 10.1128/JVI.80.3.1549-1562.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H, Wang S, Kong R, Ding W, Lee FH, Parker Z, Kim E, Learn GH, Hahn P, Policicchio B, Brocca-Cofano E, Deleage C, Hao X, Chuang GY, Gorman J, Gardner M, Lewis MG, Hatziioannou T, Santra S, Apetrei C, Pandrea I, Alam SM, Liao HX, Shen X, Tomaras GD, Farzan M, Chertova E, Keele BF, Estes JD, Lifson JD, Doms RW, Montefiori DC, Haynes BF, Sodroski JG, Kwong PD, Hahn BH, Shaw GM. 2016. Envelope residue 375 substitutions in simian-human immunodeficiency viruses enhance CD4 binding and replication in rhesus macaques. Proc Natl Acad Sci U S A 113:E3413–E3422. doi: 10.1073/pnas.1606636113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gonzalez-Nieto L, Domingues A, Ricciardi M, Gutman MJ, Maxwell HS, Pedreño-Lopez N, Bailey V, Magnani DM, Martins MA. 2016. Analysis of simian immunodeficiency virus-specific CD8+ T-cells in rhesus macaques by peptide-MHC-I tetramer staining. J Vis Exp 2016:54881. doi: 10.3791/54881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weatherall DJ. 2006. The use of non-human primates in research. https://mrc.ukri.org/documents/pdf/the-use-of-non-human-primates-in-research/.