

HBV and its hepadnavirus relatives infect a wide range of vertebrates, with a long infectious history (hundreds of millions of years). Such a long history generally allows adaptive mutations in hosts to escape from infection while simultaneously allowing adaptive mutations in viruses to overcome host barriers. However, there is no published molecular evidence for such a coevolutionary arms race between hepadnaviruses and hosts. In the present study, we performed coevolutionary phylogenetic analysis between hepadnaviruses and the sodium taurocholate cotransporting polypeptide (NTCP), an HBV receptor, combined with virological experimental assays for investigating the biological significance of NTCP sequence variation. Our data provide the first molecular evidence supporting that HBV-related hepadnaviruses drive adaptive evolution in the NTCP sequence, including a mechanistic explanation of how NTCP mutations determine host viral susceptibility. Our novel insights enhance our understanding of how hepadnaviruses evolved with their hosts, permitting the acquisition of strong species specificity.
KEYWORDS: HBV, NTCP, coevolution, dN/dS, entry, evolution, hepadnavirus, positive selection, receptor, transporter
ABSTRACT
Hepatitis B virus (HBV) and its hepadnavirus relatives infect a wide range of vertebrates, from fish to human. Hepadnaviruses and their hosts have a long history of acquiring adaptive mutations. However, there are no reports providing direct molecular evidence for such a coevolutionary “arms race” between hepadnaviruses and their hosts. Here, we present evidence suggesting that the adaptive evolution of the sodium taurocholate cotransporting polypeptide (NTCP), an HBV receptor, has been influenced by virus infection. Evolutionary analysis of the NTCP-encoding genes from 20 mammals showed that most NTCP residues are highly conserved among species, exhibiting evolution under negative selection (dN/dS ratio [ratio of nonsynonymous to synonymous evolutionary changes] of <1); this observation implies that the evolution of NTCP is restricted by maintaining its original protein function. However, 0.7% of NTCP amino acid residues exhibit rapid evolution under positive selection (dN/dS ratio of >1). Notably, a substitution at amino acid (aa) 158, a positively selected residue, converting the human NTCP to a monkey-type sequence abrogated the capacity to support HBV infection; conversely, a substitution at this residue converting the monkey Ntcp to the human sequence was sufficient to confer HBV susceptibility. Together, these observations suggested a close association of the aa 158 positive selection with the pressure by virus infection. Moreover, the aa 158 sequence determined attachment of the HBV envelope protein to the host cell, demonstrating the mechanism whereby HBV infection would create positive selection at this NTCP residue. In summary, we provide the first evidence in agreement with the function of hepadnavirus as a driver for inducing adaptive mutation in host receptor.
IMPORTANCE HBV and its hepadnavirus relatives infect a wide range of vertebrates, with a long infectious history (hundreds of millions of years). Such a long history generally allows adaptive mutations in hosts to escape from infection while simultaneously allowing adaptive mutations in viruses to overcome host barriers. However, there is no published molecular evidence for such a coevolutionary arms race between hepadnaviruses and hosts. In the present study, we performed coevolutionary phylogenetic analysis between hepadnaviruses and the sodium taurocholate cotransporting polypeptide (NTCP), an HBV receptor, combined with virological experimental assays for investigating the biological significance of NTCP sequence variation. Our data provide the first molecular evidence supporting that HBV-related hepadnaviruses drive adaptive evolution in the NTCP sequence, including a mechanistic explanation of how NTCP mutations determine host viral susceptibility. Our novel insights enhance our understanding of how hepadnaviruses evolved with their hosts, permitting the acquisition of strong species specificity.
INTRODUCTION
Over 250 million people are chronically infected worldwide with hepatitis B virus (HBV), and hepatitis B resulted in 887,000 deaths in 2015 (1–3). HBV and its hepadnavirus relatives infect a wide range of vertebrates, including fish, amphibians, reptiles, birds, and mammals (4–12). Phylogenetic analyses suggest that the coevolutionary history of hepadnaviruses and their hosts spans hundreds of millions of years (4, 8, 9, 11). During the long-term coevolution of virus and host, host proteins that interact directly with a virus accumulate adaptive mutations to permit escape from virus infection, while viruses acquire mutations that overcome host barriers (13). In the course of this coevolutionary “arms race,” viruses continuously exert selective pressures on their host, thereby inducing multiple adaptive mutations in the host genome (14). One of the best-known examples of such coevolution is embodied by the conflict between (host-encoded) restriction factors and their (virus-encoded) viral antagonists (13). Host restriction factors inhibit viral replication at different steps during virus proliferation in the host. Conversely, viruses encode factors that antagonize the function of host restriction factors and thereby circumvent the host’s attempt to eliminate viruses (15). During the course of long-term viral selective pressures, some host restriction factors evolve rapidly, a process that is evidenced as a ratio of nonsynonymous to synonymous mutations in the host gene (the dN/dS ratio) that exceeds 1 (termed positive selection) (16). For example, host restriction factors against human immunodeficiency virus type 1 (HIV-1), including tripartite motif-containing protein 5-alpha (TRIM5α) (17), apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3 G (APOBEC3G) (18), bone marrow stromal antigen 2 (BST2; also known as tetherin, CD317, and HM1.24) (19–22), and SAM domain and HD domain 1 (SAMHD1) (23, 24), have been reported to exhibit rapid evolution (dN/dS ratio of >1), likely due to the selective pressure exerted by HIV-1 infection. Regarding the coevolution of hepadnaviruses and host restriction factors, Abdul et al. recently reported an evolutionary analysis of an HBV restriction factor, the Structural Maintenance of Chromosomes 5/6 (Smc5/6) complex (25), a complex originally identified based on its housekeeping function in genomic stability (26). However, Abdul et al. did not detect a clear signature of positive selection that was suggested to be induced by hepadnavirus infection. In contrast, Enard et al. reported that host proteins interacting with viruses with a long history display higher rates of adaptive mutations (14); those authors showed that host proteins reported to interact with HBV exhibited a strong signature of adaptation during coevolution with viruses, which was at a degree similar to that seen for HIV-1-interacting host proteins. However, molecules subject to such a selective pressure by hepadnaviruses have not (to our knowledge) been identified to date.
Hepadnaviruses infect their hosts in a highly species-specific manner; for instance, HBV can infect only humans, chimpanzees, and treeshrews, but not monkeys, including both Old World and New World monkeys (27). The sodium taurocholate cotransporting polypeptide (NTCP; also designated solute carrier family 10A1 [SLC10A1]) was recently identified as a host factor that functions as an HBV entry receptor. NTCP, which originally was characterized as a hepatic transporter for the uptake of bile acids by hepatocytes, binds to the HBV envelope protein, notably to the preS1 region, thereby mediating viral entry into the host cells (28). Among host factors involved in HBV proliferation processes (29–31), NTCP has been suggested to be a key determinant of the species specificity of HBV, as primary monkey hepatocytes can support the replication of intracellular HBV but not the entry of the virus into host cells (32), and complementation of the monkey cells with human NTCP (hNTCP) permits HBV entry and thereby the whole infection cycle both in cell culture and in vivo (33, 34). These results indicate that the inability of monkey Ntcp to support HBV infection serves as the species barrier preventing HBV infection in monkey. However, the evolutionary relationship between NTCP sequences in different species and susceptibility to hepadnavirus infection has not been analyzed previously. Virus entry receptors generally have their own original function in cellular physiology. Thus, their sequences typically are conserved during evolution to maintain their functional profile, showing a dN/dS ratio of <1 that indicates negative selection (35). Indeed, transferrin receptor (TfR1), which serves as an entry receptor for arenaviruses and mouse mammary tumor virus (36), exhibits such evolutionarily negative selection across their entire sequences. Interestingly, however, a small percentage of positions in the TfR1 coding sequence is rapidly evolving under positive selection, and these sites were shown to correspond to a virus-binding surface (36). In another example, the Niemann-Pick disease, type C1 (NPC1), a receptor for filovirus, shows a similar evolutionary property (37). These data strongly suggest that virus infection serves as a pressure for the evolution of viral entry receptors. In addition, positive selection sites on virus entry receptors can provide critical information, permitting the identification of the receptor domains that interface with the virus. However, to our knowledge, there have been no reports of such an evolutionary analysis for a hepadnavirus receptor.
In this study, we focused on the phylogenetic analyses of hepadnaviruses and NTCP based on molecular evolutionary analyses to detect positive selection sites and investigated the biological significance of NTCP sequence changes using virological experimental assays in our cell culture model (38). Evolutionary analyses of the NTCPs from 20 mammalian species revealed that NTCP was highly conserved among these mammals but, interestingly, identified several sites that were subject to positive selection. Among these positive selection sites, the amino acid at NTCP residue 158 (aa 158) determined the HBV host species specificity. Our virological experiments showed that a single substitution converting aa 158 of hNTCP to the corresponding amino acid of cynomolgus macaque Ntcp completely negated the protein’s ability to mediate viral infection, whereas a cynomolgus macaque Ntcp protein mutated to carry the hNTCP aa 158 rendered macaque Ntcp susceptible to the virus, suggesting that this site regulates viral susceptibility. Moreover, aa 158 of NTCP was shown to mediate viral attachment to the host cell surface. In a manner analogous to that of TfR1 and NPC1, these data suggest that NTCP receives an evolutionary pressure by virus infection, which provides the first evidence on the molecular level for coevolution between hepadnaviruses and hosts.
(This article was submitted to an online preprint archive [39].)
RESULTS
A small portion of sites in NTCP evolve under positive selection in mammals.
We performed maximum likelihood (ML)-based molecular evolutionary analyses (Fig. 1) using 20 NTCP coding sequences from mammalian species, including human, monkeys, bats, rabbit, rat, and mouse (Table 1). To detect positive selection sites in mammalian NTCPs, two pairs of site models were conducted using PAML, ver. 4.8 (40, 41). The differences between neutral models and selection models (M1a versus M2a, respectively, or M7 versus M8, respectively) were significant (P < 0.01 or P < 0.005, respectively), suggesting that NTCP genes have been evolving under positive selection in mammals (Table 2). The M2a analysis estimated that 66.2% (p0 = 0.662) of the codons were under negative selection with a dN/dS ratio of (ω0) 0.086; 33.1% (p1 = 0.331) were under neutral evolution, and 0.7% (p2 = 0.007) were under positive selection with a dN/dS ratio (ω2) of 4.526. Thus, only a small number of sites in mammalian NTCPs was under positive selection. The same data set also was analyzed to detect positive selection sites by fixed effects likelihood (FEL), random effects likelihood (REL) (42), and mixed effects model of evolution (MEME) (43), and 27 positively selected sites were inferred by at least one of the four evolutionary analyses, as summarized in Tables 2 and 3.
FIG 1.

ML tree of the 20 NTCP sequences analyzed in this study. An ML tree of 20 mammalian full-length NTCP sequences was reconstructed using PhyML 3.0. Bootstrap values from 1,000 replicates are indicated at each node. The scale bar indicates the number of nucleotide substitutions per site. The corresponding GenBank accession numbers are listed in Table 1. The indicated amino acid sequences correspond to the amino acids at residues 157 and 158 in each NTCP species.
TABLE 1.
GenBank accession numbers of mammalian NTCP sequences used in this study
| ID no. | Accession no. | Scientific name | Common namea | Order |
|---|---|---|---|---|
| 1 | NM_003049.3 | Homo sapiens | Human | Primates (hominoids) |
| 2 | XM_510035.5 | Pan troglodytes | Chimpanzee | |
| 3 | XM_003824101.2 | Pan paniscus | Bonobo | |
| 4 | XM_002824890.2 | Pongo abelii | Sumatran orangutan | |
| 5 | NM_001283323.1 | Macaca fascicularis | Cynomolgus macaque | Primates (Old World monkeys) |
| 6 | KT382283.1 | Papio hamadryas | Hamadryas baboon | |
| 7 | KT382281.1 | Chlorocebus aethiops | Grivet | |
| 8 | KT326157.1 | Semnopithecus sp. | Gray langur | |
| 9 | KT382285.1 | Callithrix jacchus | Marmoset | Primates (New World monkeys) |
| 10 | KT382284.1 | Saguinus oedipus | Cotton-top tamarin | |
| 11 | KR153327.1 | Saimiri sciureus | Squirrel monkey | |
| 12 | XM_006091030.2 | Myotis lucifugus | Little brown bat | Chiroptera |
| 13 | XM_005877950.2 | Myotis brandtii | Brandt's bat | |
| 14 | XM_008138900.1 | Eptesicus fuscus | Big brown bat | |
| 15 | XM_006915181.2 | Pteropus alecto | Black flying fox | |
| 16 | JQ608471.1 | Tupaia belangeri | Northern treeshrew | Scandentia |
| 17 | NM_001082768.1 | Oryctolagus cuniculus | European rabbit | Lagomorpha |
| 18 | XM_015490579.1 | Marmota marmota | Alpine marmot | Rodentia |
| 19 | NM_017047.1 | Rattus norvegicus | Brown rat | |
| 20 | NM_001177561.1 | Mus musculus | House mouse |
The common name of each mammals is identical to that shown in Fig. 1.
TABLE 2.
Log likelihood values and estimated parameters under models in PAMLa
| Model | lnL | 2ΔlnL | P value | Estimated parameter(s) | Positive selection sites |
|---|---|---|---|---|---|
| M0 | −5,972.703 | ω = 0.344 | |||
| M1a | −5,868.144 | p0 = 0.667, p1 = 0.333 ω0 = 0.085, ω1 = 1 | |||
| M2a | −5,863.300 | 9.687 | <0.01 | p0 = 0.662, p1 = 0.331, p2 = 0.007 ω0 = 0.086, ω1 = 1, ω2 = 4.526 | K157, A351 |
| M7 | −5,868.087 | p = 0.237, q = 0.430 | |||
| M8 | −5,862.086 | 12.002 | <0.005 | p0 = 0.991, p = 0.252, q = 0.469 p1 = 0.009, ω = 3.784 | K157, A351 |
Two pairs of site models with CODEML implemented in the PAML v 4.8 were performed. The first pair involves M1a (neutral, 2 site classes, ω > 1 not allowed) versus M2a (selection, 3 site classes, ω > 1 allowed), and the second pair consists of M7 (neutral, 10 site classes, ω > 1 not allowed) versus M8 (selection, 11 site classes, ω > 1 allowed). lnL, log likelihood.
TABLE 3.
Positive selection sites inferred under FEL, REL, and MEMEc
| Sitea | FEL |
REL |
MEME |
|||
|---|---|---|---|---|---|---|
| dN/dS | P valueb | dN/dS | Bayes factorb | ω+ | P valueb | |
| 25 | 3.03 | 0.056 | 0.32 | 9.987 | >100 | 0.034 |
| 62 | 0.575 | 0.278 | −0.094 | 6.955 | >100 | 0.087 |
| 107 | 2.268 | 0.045 | 0.31 | 12.91 | >100 | 0.062 |
| 129 | 2.834 | 0.014 | 1.08 | 37.539 | >100 | 0.022 |
| 142 | 1.127 | 0.156 | −0.018 | 8.682 | >100 | 0 |
| 157 | 4.646 | 0.004 | 1.843 | 2,566.71 | >100 | 0.007 |
| 158 | 1.311 | 0.3 | −0.158 | 4.877 | >100 | 0.05 |
| 160 | 0.959 | 0.437 | −0.21 | 4.018 | >100 | 0.061 |
| 165 | 1.673 | 0.095 | 0.06 | 9.433 | >100 | 0.071 |
| 166 | 1.152 | 0.067 | 0.188 | 23.465 | >100 | 0.088 |
| 175 | 2.486 | 0.051 | 0.475 | 13.608 | >100 | 0.064 |
| 184 | −1.037 | 0.275 | −0.783 | 0.55 | >100 | 0.038 |
| 198 | 1.592 | 0.099 | 0.031 | 8.999 | >100 | 0.11 |
| 207 | 1.491 | 0.087 | 0.044 | 10.293 | >100 | 0.11 |
| 210 | 0.044 | 0.966 | −0.286 | 3.225 | >100 | 0.047 |
| 223 | 2.671 | 0.051 | 0.22 | 7.959 | >100 | 0.001 |
| 248 | 1.896 | 0.03 | 0.275 | 19.36 | >100 | 0.043 |
| 308 | −0.797 | 0.634 | −0.401 | 1.487 | 25.428 | 0.095 |
| 330 | 1.321 | 0.085 | 0.104 | 13.859 | >100 | 0.11 |
| 333 | 0.883 | 0.094 | 0.175 | 23.698 | >100 | 0.047 |
| 334 | 1.05 | 0.098 | 0.146 | 18.083 | >100 | 0.122 |
| 339 | 1.723 | 0.044 | 0.172 | 15.24 | >100 | 0.062 |
| 342 | 1.03 | 0.169 | −0.022 | 8.713 | >100 | 0.077 |
| 347 | 1.956 | 0.212 | −0.044 | 4.988 | >100 | 0.001 |
| 348 | 0.719 | 0.699 | −0.166 | 2.831 | >100 | 0.008 |
| 351 | 1.961 | 0.705 | 1.346 | 743.714 | >100 | 0.003 |
| 352 | 0.739 | 0.5 | −0.187 | 3.923 | 25.945 | 0.048 |
Codons varying between human (NM_003049.3) and cynomolgus macaque (NM_001283323.1) NTCPs are underlined.
The default significant cutoff values were used and shown in boldface: P value of <0.1 for FEL, Bayes factor of >50 for REL, and P value of <0.1 for MEME.
FEL, REL, and MEME were employed to detect the positive selection sites through the DATAMONKEY webserver.
Single positive selection site of NTCP is a key determinant of HBV susceptibility.
To date, hepadnaviruses have been found in multiple hominoids, including chimpanzees and orangutans (44). On the other hand, no HBV or hepadnavirus infections have been reported in Old World monkeys, even though this family is the closest to the hominoids (Fig. 1). (Although a unique case of HBV genotype D infection was reported in Macaca fascicularis [cynomolgus macaques] from Mauritius [45], another recent paper showed that these macaques exhibited no evidence of current or prior HBV infection [34]). Since Old World monkeys are the closest species to human that are regarded to be nonsusceptible to HBV or hepadnaviruses, we focused on these two lineages for the comparison among NTCP genes. Notably, there are 13 amino acid differences between the human NTCP (NM_003049.3; hNTCP) and the cynomolgus macaque Ntcp (NM_001283323.1; Macaca fascicularis, Old World monkey; mNtcp). Evolutionary analysis comparing the corresponding gene sequences identified five sites (codons 142, 157, 158, 160, and 165) in the NTCP open reading frame (ORF) that are under positive selection (Table 3 and Fig. 2).
FIG 2.
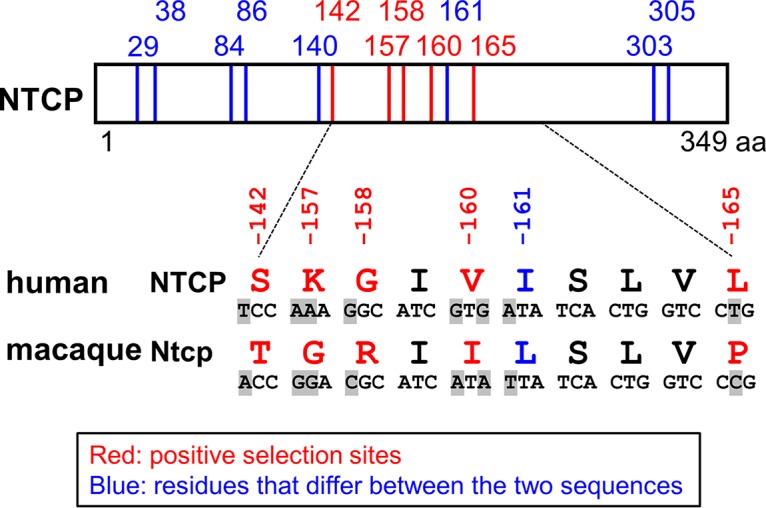
Comparison between human and macaque NTCPs. Schematic representation of the primary amino acid and nucleotide sequence of NTCP. The 13 amino acid positions at which human (NM_003049.3) and cynomolgus macaque (NM_001283323.1) sequences differ are indicated by the numbers above the box showing the NTCP protein and the vertical lines (in red and blue). Among these 13 positions, five residues (indicated in red) were inferred to be under positive selection in the 20 mammalian sequences evaluated in this study. The amino acid sequences at residues 142 and 157 to 165 are compared between human and cynomolgus macaques, and their corresponding nucleotide differences are also highlighted in gray, as shown under the box. The indicated numbers correspond to the amino acid positions in human NTCP.
We next addressed whether the amino acids encoded by these sites are involved in susceptibility to HBV infection. We prepared HepG2 cells transiently expressing hNTCP or a series of single-mutant variants in which a single amino acid was replaced by the corresponding macaque residue (S142T, K157G, G158R, V160I, and L165P) (Fig. 3A). Similarly, we produced HepG2 cells expressing a variant of hNTCP in which aa 157 to 165 were replaced by the corresponding macaque sequence [designated hNTCP(m157-165)]; this substitution has been reported to abrogate NTCP’s receptor function (28) (Fig. 3A). At 24 h after transfection, protein expression of NTCP or its variants was confirmed by immunoblotting of cell lysates. As shown in Fig. 3A, the mutant hNTCPs were expressed at levels equivalent to that of wild-type hNTCP, except for hNTCP-L165P, which had much lower protein expression (Fig. 3A). To further validate whether each of the NTCP variants maintained its functional properties as a bile acid transporter, we also measured NTCP-mediated uptake of [3H]taurocholic acid in these cells. All NTCP variants, including hNTCP-L165P, possessed the capacity to take up bile acid, retaining at least 40% of the wild-type activity and providing activity 6- to 24-fold greater than that of the background (assessed under sodium-free conditions) (Fig. 3B). Thus, all of the hNTCP variant-expressing HepG2 cell lines in this experiment retained the significant NTCP functions; however, hNTCP-G158R showed a relatively low activity of bile acid uptake, having approximately 40% of that of the wild type. The impact of a mutation in aa 158 on bile acid uptake was further examined in Fig. 7 and is discussed below (see the last paragraph of Results).
FIG 3.
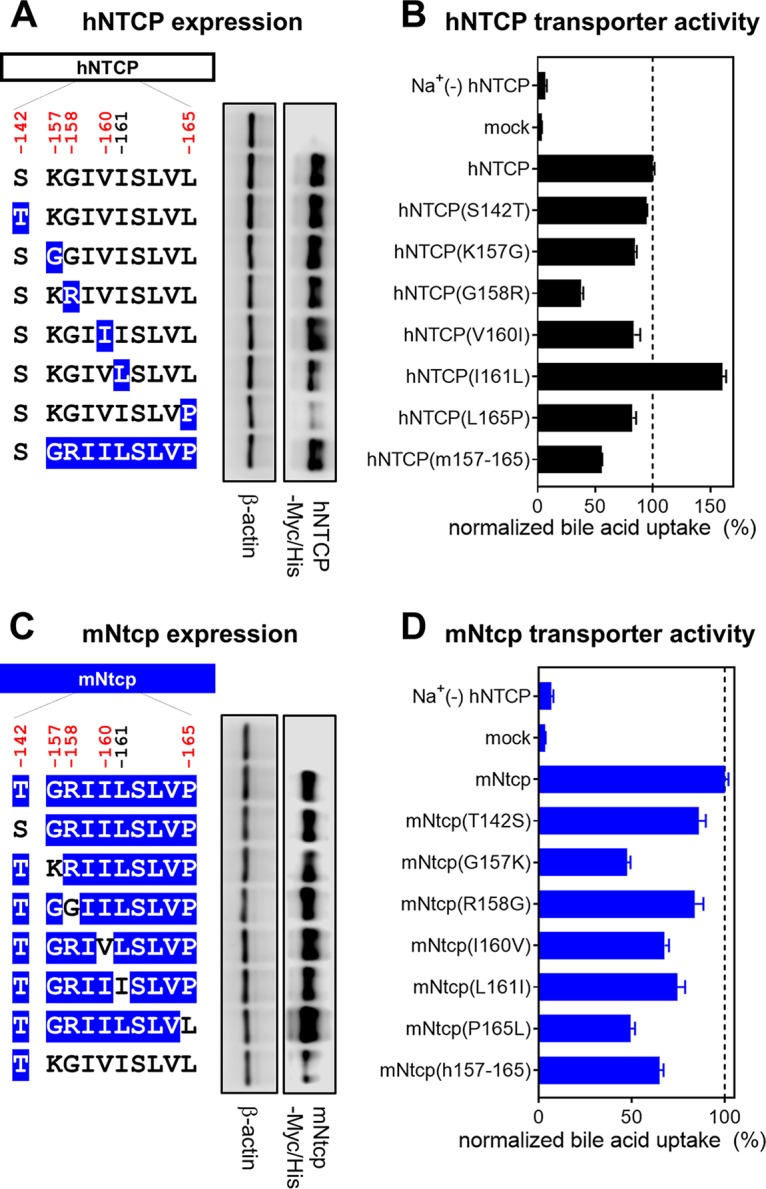
NTCP expression and transporter activity. (A and C, left) Schematic representation of NTCP variants. Black and blue boxes/amino acids indicate those from human and macaque, respectively. The constructs shown in panels A and C are variants in which hNTCP (black) and mNtcp (blue) were used as backbones. Amino acid positions under positive selection are shown in red at the top of the scheme. (Right) Western blotting was used to assess the expression level of Myc/His-tagged NTCP or its variants in HepG2 cells transfected with the respective construct. Actin was used as an internal control. (B and D) Transporter activities of NTCP in these cells were measured by transporter assay using [3H]taurocholic acid as a substrate in either sodium-free buffer (to determine background level) or sodium-containing buffer. Normalized transporter activities are shown and were calculated by dividing the detected radioactivity values by NTCP expression levels determined by immunofluorescence analysis. Results are presented as means ± SEM (n = 3).
FIG 7.
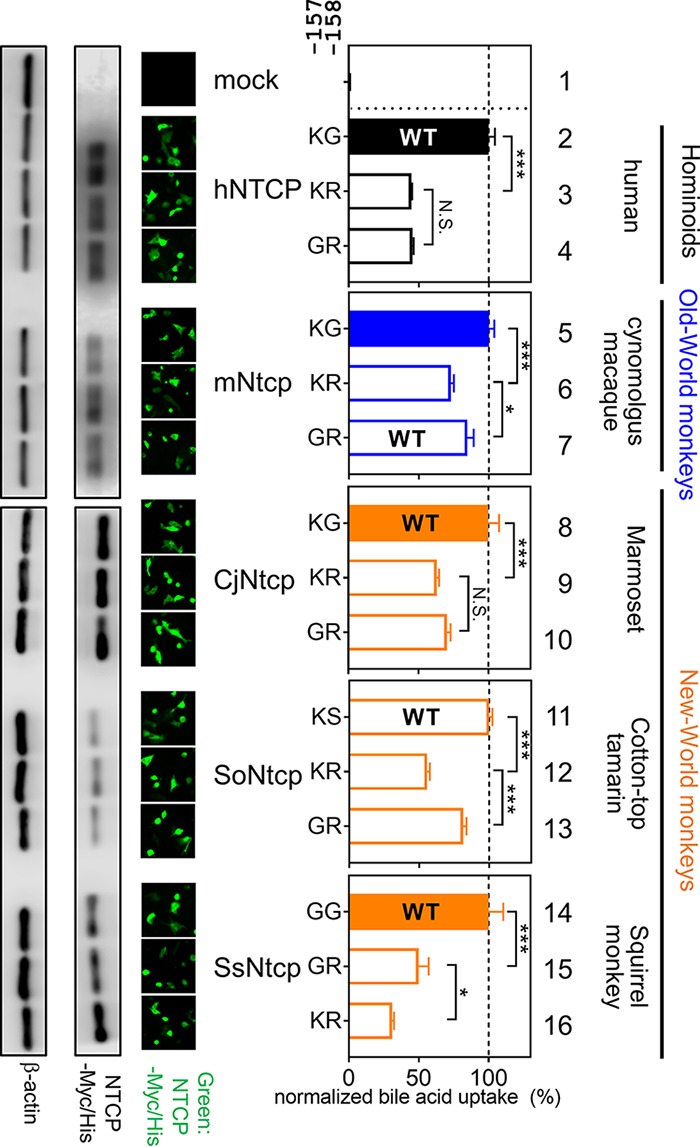
Impact of mutations at aa 157 and 158 in NTCP for bile acid uptake function. (Left) Expression levels of Myc/His-tagged NTCP or its variants in HepG2 cells transfected with each construct were examined by Western blot and immunofluorescence analyses (green) as described for Fig. 3 and 4. (Middle) Two amino acids indicate the sequences at aa 157 and 158 of each NTCP variant. (Right) Transporter activities for each NTCP variant were measured by transporter assay, as done for Fig. 3. Normalized transporter activities, calculated by dividing the transporter activity by NTCP expression levels, are shown as described for Fig. 3. Closed and open bars in the graph indicate HBV-susceptible and nonsusceptible NTCP, respectively. The wild-type NTCP in each species is also shown as WT. Results are presented as means ± SEM (n = 3). One, two, and three asterisks indicate P < 0.05, P < 0.01, and P < 0.001, respectively.
We then analyzed the ability of these cell lines to support HBV infection. Specifically, an HBV infection assay was performed (per Materials and Methods) (38) to evaluate the cells’ susceptibility to HBV infection by detecting intracellular hepatitis B core antigen (HBcAg) at 13 days postinoculation by immunofluorescence analysis (Fig. 4A, right, red, DMSO [dimethyl sulfoxide]). The detected levels of HBcAg were normalized to the expression level of NTCP in the respective line, permitting assessment of each NTCP’s ability to support HBV infection (Fig. 4A, center graph). In addition, to confirm that the observed fluorescence signals were derived from HBV infection (and did not reflect nonspecific background), the HBV infection assay was also performed in parallel in the presence of Myrcludex B (Myr-B), an HBV entry inhibitor (46), a condition that should abrogate the specific signal derived from HBV infection (Fig. 4A, right, Myr-B). As shown in Fig. 4A, HBV infection was observed in the lines expressing wild-type hNTCP but not in that expressing hNTCP(m157-165) (Fig. 4A, lanes 2 and 9), consistent with a previous report (28). HBV infection was supported by all but one of the NTCP variants, hNTCP(S142T), hNTCP(K157G), hNTCP(V160I), hNTCP(I161L), and hNTCP(L165P) (Fig. 4A, lanes 3, 4, 6, 7, and 8). In contrast, the level of HBV infection in hNTCP(G158R)-expressing cells was similar to the background level (Fig. 4A, lanes 1 and 5), suggesting that aa 158 was unique among the examined sites in determining HBV susceptibility.
FIG 4.
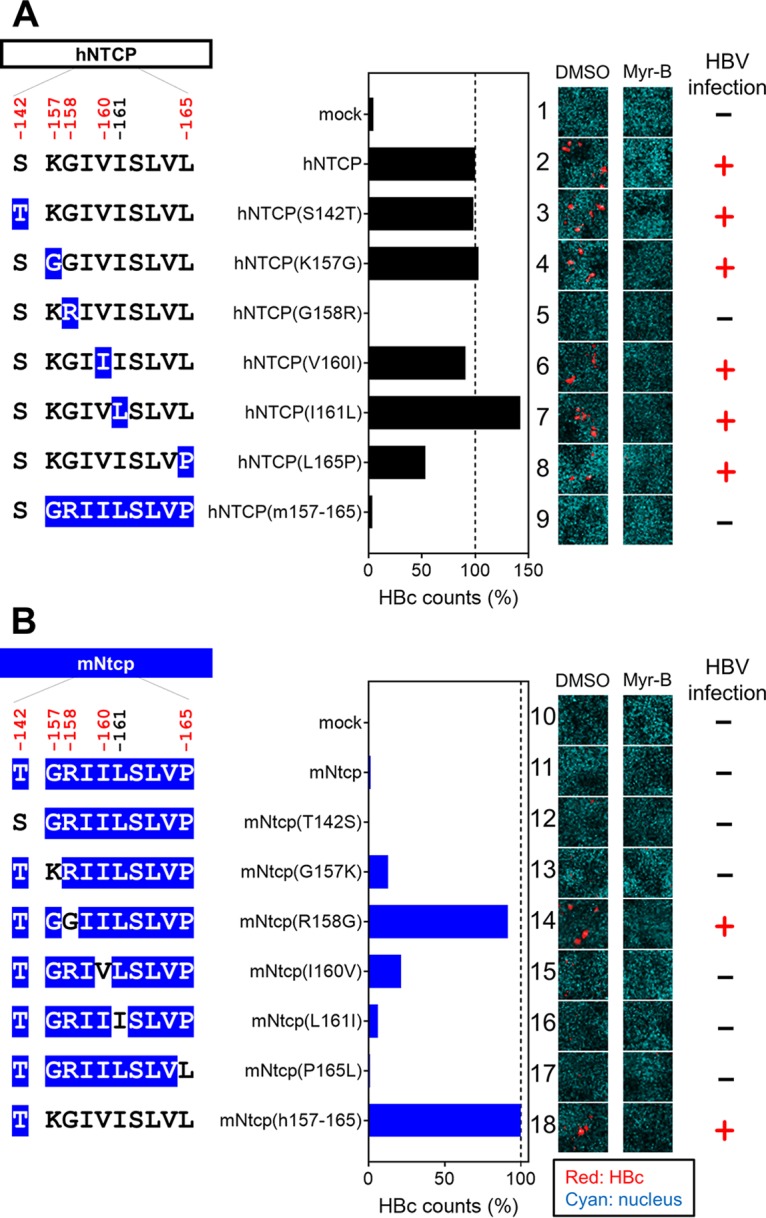
Single positive selection site on NTCP, at aa 158, is a key determinant for supporting HBV infection. (Left) Schematic representation of the wild type and variants of hNTCP (A) and mNtcp (B) (black, human-type residues; blue, macaque-type residues). (Middle) HBV susceptibility in HepG2 cells expressing each NTCP construct was determined by detecting HBc-positive cells; the detected value was normalized to the NTCP expression level in the same cell line. (Right) HBc (red) and the nucleus (cyan) were detected in the HBV infection assay by immunofluorescence analysis in the presence or absence of Myr-B, an HBV entry inhibitor. On the right, the inability (−) or ability (+) to support HBV infection (without Myr-B) for each construct is summarized.
We then performed a complementary experiment using the mNtcp backbone and substituting single amino acids with the human residues for the corresponding sites; the resulting mutant mNtcps were examined to determine whether any of the amino acid substitutions endowed cells with viral entry. Using the same methods as those used for Fig. 3A and B, we confirmed the protein expression as well as the transporter activity of the wild-type and mutant mNtcps. Notably, all of the mNtcp derivatives were expressed in HepG2 cells and provided functional bile acid uptake (to levels at least 50% of that of the wild type, with activity 7- to 13-fold higher than the background level) (Fig. 3C and D). The HBV infection assay indicated that the wild-type mNtcp did not support HBV infection (Fig. 4B, lane 11), while the substitution of the human aa 157 to 165 in mNtcp [mNtcp(h157-165)] permitted infection (Fig. 4B, lane 18), as previously reported (28). Among the single-amino-acid changes, only the substitution to the human sequence at aa 158 (R158G) was sufficient to endow mNtcp with HBV receptor function equivalent to that of mNtcp(h157-165) (Fig. 4B, lane 14). Thus, a single positive selection site in NTCP, corresponding to aa 158, was a key determinant for HBV susceptibility.
The positive selection site of NTCP is critical for HBV attachment.
NTCP is involved in the specific attachment of HBV to the host cell surface through binding to the preS1 region of the large surface protein (LHB) of HBV (47). To explore the impact of the NTCP substitutions on the interaction with HBV preS1, we examined the attachment of a fluorescence-labeled preS1 peptide (6-carboxytetramethylrhodamine-labeled preS1 peptide, or TAMRA-preS1) to target cells expressing the wild-type NTCP or its variants (Fig. 5A and B, right, red, DMSO). To confirm the specificity of the observed TAMRA-preS1 signals, the preS1 binding assay was (as above) performed in parallel in the presence of Myr-B, an attachment inhibitor (46) (Fig. 5A and B, right, Myr-B). Expression in HepG2 cells of variants based on hNTCP or mNtcp backbones was confirmed by immunofluorescence analysis (Fig. 5A and B, right, green). As shown in Fig. 5A, while expression of hNTCP supported preS1 attachment, replacement of aa 158 in hNTCP with the macaque-type residue [hNTCP(G158R)] completely abrogated the ability to bind to the preS1 peptide (Fig. 5A, lane 5). On the other hand, replacement of aa 158 in mNtcp with the human residue [mNtcp(R158G)] endowed mNtcp with preS1 binding activity (Fig. 5B, lane 14). Notably, although the preS1 binding activity of mNtcp(R158G) was still approximately 40% of that of mNtcp(h157-165), this activity was significantly higher than that of wild-type mNtcp (Fig. 5B, lane 11), and this result correlated well with the profile of the HBV infection (Fig. 4).
FIG 5.

Single positive selection site on NTCP is critical for HBV envelope binding. (Left) Schematic representation of hNTCP (A) or mNtcp (B) variants (black, human-type residues; blue, macaque-type residues). (Middle) preS1-cell binding mediated by NTCP mutants. (Right) The relative amounts of preS1-cell binding were determined by counting preS1-attached cells (red), and the values were normalized to NTCP expression levels (green) in the respective cell lines. preS1-cell binding was examined by preS1 binding assay (red) either in the presence or absence of Myr-B. Expression levels of Myc/His-tagged NTCP were simultaneously visualized by immunofluorescence analysis (green).
Hepatitis D virus (HDV) virions harbor an envelope identical or very similar to that of HBV particles, entering cells in a manner similar to that employed by HBV (48). Consistent with the above-described results obtained in the HBV infection and preS1-binding assays, HDV susceptibility in hNTCP-expressing cells was abrogated by the G158R substitution in hNTCP (Fig. 6A, lane 5), whereas the R158G substitution in mNtcp endowed cells with susceptibility to HDV infection (Fig. 6B, lane 14). Taken together, these observations suggested that a single position on NTCP, at aa 158, determined the susceptibility to HBV/HDV infection by facilitating viral attachment.
FIG 6.
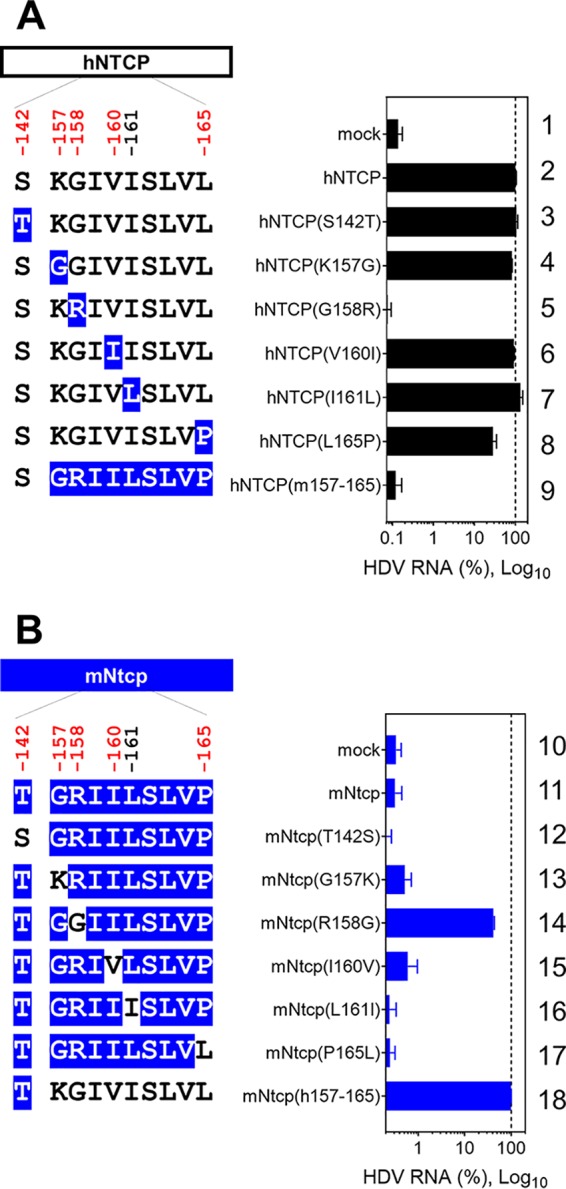
Single positive selection site in NTCP determines HDV susceptibility. (Left) Schematic representation of hNTCP (A) or mNtcp (B) variants (black, human-type residues; blue, macaque-type residues). (Right) HDV infection assay for determining the susceptibility to HDV infection of HepG2 cells expressing each NTCP. Intracellular HDV RNA was normalized by the NTCP expression level in the respective cell line. Results are presented as means ± SEM (n = 3).
Impact of mutations at aa 157 and 158 in NTCP for bile acid uptake function.
Thus, aa 158 was shown to be a key determinant of HBV’s host species specificity. However, our biochemical experiment showed the reduced bile acid uptake activity of hNTCP(G158R) (approximately 40% of that of the wild type) (Fig. 3B). Therefore, we hypothesized that arginine at aa 158 in NTCP acquires the nonsusceptibility to HBV infection on one hand but is disadvantageous for the function for bile acid uptake on the other hand (termed hypothesis A). In fact, all hominoids and two out of three New World monkeys still possess glycine at aa 158 (Fig. 1), despite the disadvantage of its susceptibility to virus infection. Interestingly, moreover, the amino acid at aa 157 in all the Old World monkey NTCPs is also changed to glycine, in contrast to that of all the hominoids and two out of three New World monkey species, which possess lysine/arginine at aa 157, raising a further hypothesis that the 157G substitution compensates for the impaired transporter activity caused by the G158R substitution (mentioned as hypothesis B). This may explain why aa 157 was identified as a strong positive selection site: the 158R substitution acquires HBV nonsusceptibility but reduces the bile acid uptake activity (hypothesis A), and the additional substitution 157G rescues the reduced activity of bile acid uptake (hypothesis B). We addressed the above two hypotheses by conducting the bile acid transporter assay using variants with substitution at aa 157 and 158 of NTCPs from hominoids, Old World monkey species, and New World monkey species (marmoset [KT382285.1, Callithrix jacchus, CjNtcp], cotton-top tamarin [KT382284.1, Saguinus oedipus, SoNtcp], and squirrel monkey [KR153327.1, Saimiri sciureus, SsNtcp]) used as backbone NTCPs. As a result, transporter assay using aa 158 mutants of NTCP derived from five species entirely supports hypothesis A: bile acid uptake was significantly reduced by alteration from their original amino acid (mostly glycine) to arginine at aa 158 in NTCP from human (Fig. 7, KG to KR, lanes 2 and 3), New World monkey species (Fig. 7, CjNtcp, KG to KR, lanes 8 and 9; SoNtcp, KS to KR, lanes 11 and 12; and SsNtcp, GG to GR, lanes 14 and 15), and from glycine to arginine at aa 158 in the Old World monkey mNtcp backbone (Fig. 7, KG to KR in lanes 5 and 6). All of these results are in agreement with hypothesis A that 158R in NTCP is disadvantageous for NTCP’s original function for bile acid uptake. On the other hand, in the experiments with aa 157 substitutions to address hypothesis B, replacement of lysine with glycine at aa 157 significantly rescued the reduced transporter activities of mNtcp, SoNtcp, and SsNtcp (Fig. 7, mNtcp, KR to GR, lanes 6 and 7; SoNtcp, KR to GR, lanes 12 and 13; SsNtcp, KR to GR, lanes 16 and 15), while K157G substitution did not show significant recovery of the bile acid uptake by hNTCP or CjNtcp (Fig. 7, hNTCP, KR to GR in lanes 3 and 4; CjNtcp, KR to GR in lanes 9 and 10). Thus, hypothesis B, that the substitution at aa 157 compensates for the impaired bile acid uptake function caused by G158R substitution, was not necessarily generalized for all the NTCP species examined. However, the KR variants in aa 157 to 158 showed the lowest transporter activities in all of the NTCP backbones tested in this study (Fig. 7, lanes 3, 6, 9, 12, and 16), which suggests the disadvantage of the double basic residues KR at aa 157 and 158 for maintaining NTCP’s functionality for bile acid transport, serving as a possible driving force to swap G from aa 158 to aa 157. Thus, the transporter assay suggests the impact of the substitution at aa 157 and 158 in NTCP on bile acid uptake activity, although the overall difference is moderate.
DISCUSSION
In this study, we employed four evolutionary analyses to detect positive selection sites of NTCP by using NTCP sequences derived from 20 mammalian species. A PAML site model (M2a) revealed that NTCP is highly conserved in mammals (across 66.2% of the codons), indicating that NTCP is critical for (or advantageous to) host viability. This is in agreement with a previous report showing that NTCP-knockout mice could exhibit body weight loss and elevated serum bile acid levels (49). While the majority of NTCP sequence was subjected to negative selection, positive selection sites were inferred from different methods: aa 157 was detected as a positive selection site by all four evolutionary analyses, while other residues were inferred as positive selection sites by specific methods: aa 158 was detected as a positively selected site only by the MEME method. This might be because three of the other evolutionary methods preferably detect sites subject to pervasive positive selection throughout its evolutionary history, and it is often difficult to identify the episodic positively selected sites that affect a small subset of lineages among all the analyzed lineages (43). The MEME method is improved to infer both episodic and pervasive positive selection at the level of an individual site, and its advantage enables us to identify aa 158 as a positive selection site. The branch site model implemented in PAML v4.8 (50) did not detect aa 158 as a positively selected site in the Old World monkeys’ lineage, which could be explained by PAML branch site models requiring a priori assumptions, sometimes leading to poor statistical performance (51). In conclusion, Nozawa et al. states that the limitation of identifying positive selection from real sequence data using these evolutionary methods has to be complemented by experimental confirmation, e.g., site-directed mutagenesis, to understand adaptive evolution (52). In this point of view, we employed a strategy combining phylogenetic and evolutionary analyses with virological and biological experimental assays as discussed below.
To verify the biological significance of the inferred positive selection sites in NTCP, we focused on a comparison of the protein between human and the Old World monkey lineage. Notably, the latter family is phylogenetically the closest to human, but Old World monkeys are not susceptible to HBV infection (44), rendering these simians the best counterpart for comparison. Interestingly, our infection experiments suggested that a single positively selected site inferred in the present study, aa 158, is a key determinant of HBV’s host species specificity. The consistent role of aa 158 was reported by a very recent paper (53). This observation is supported by the fact that NTCPs that carry the human-type sequence at aa 158 (e.g., NTCP of chimpanzee, orangutan, New World monkey, treeshrew, and mouse) (Fig. 1) is known to interact with HBV preS1 (33, 53–55) (although these species, except for chimpanzee and treeshrew, are not susceptible to HBV infection because of unknown restriction mechanism in postentry steps [53, 55]). Given that the crystal structure of NTCP has not been solved, the exact mechanism whereby aa 158 facilitates hepadnavirus entry remains unknown. However, our data suggest that this residue is a primary mediator of the interaction between the preS1 region of the HBV envelope protein and the cell surface (Fig. 5). This observation would be analogous to the analyses of two other virus receptors, TfR1 and NPC1, which showed overall negative selection, with a subset of positive selection sites in the virus-binding interface as a signature of a coevolutionary relationship of these receptors with virus infection (36, 37).
To date, most of the relevant studies have reported that error-prone RNA viruses and lethal viruses (e.g., HIV and filovirus) exert selective pressure on their host proteins (17–24, 37). On the other hand, if a virus is not pathogenic to the host, adaptive mutations in host proteins would not be expected to occur (13). In most cases, HBV infection in adult human is controlled by immunity; only in a minority of cases does chronic infection occur for developing chronic liver disease, and a limited number of cases develop fulminant hepatitis (56). Therefore, it has been unclear whether hepadnavirus could serve as a driver of adaptive evolution in host proteins. However, hepadnavirus infection in malnourished wildlife species, which can be immunocompromised, may result in higher morbidity and mortality (57). In addition, an analysis by Enard et al. of the evolutionary patterns of ∼9,900 proteins conserved in mammalian genomes revealed that host proteins that have been in contact with viruses for extended evolutionary periods in a wide range of lineages display higher rates of adaptation to viruses; among these proteins, those that interact with HBV proteins had the strongest signature of adaptation, suggesting the coevolution of host factors and hepadnaviruses (14). However, Enard et al. did not provide a molecular analysis showing the detailed gene sites that were under positive selection by virus infection. Therefore, the present study provides the first clear evidence for positive selection (in NTCP) mediated by hepadnaviruses. We hypothesize that the predecessors of Old World monkeys were infected with pathogenic hepadnaviruses that imposed strong selective pressures; while these ancient Old World monkeys declined or became extinct, their descendants (the extant Old World monkeys) presumably evolved to escape hepadnavirus infection by accruing a mutation at NTCP aa 158. Indeed, examples of endangerment or extinction of species caused by virulent virus infection have been reported (58–60). Further investigation is needed to determine whether any hepadnavirus exists in extant Old World monkeys, because the number of primate species surveyed for hepadnavirus infection has been limited (44). However, all known Old World monkey NTCPs carry 158R (i.e., HBV-nonsusceptible NTCP) (53), consistent with the apparent absence of hepadnavirus in Old World monkeys. Simian immunodeficiency virus (SIV) is nonpathogenic in its natural hosts, while its virus relatives are highly pathogenic to rhesus macaque, an experimental host, and human, who only recently have become hosts (61, 62). These observations support the idea that a virus recently acquired from different species possesses highly pathogenic properties and may impose selective genetic pressure on the new host; subsequent host adaptation over the course of a long history of infection would render such a virus less pathogenic to the new host (63). Interestingly, hepadnaviruses have an extremely long history of infection of their respective hosts. The present study demonstrates that the host species specificity of a virus can be determined by a single positively selected site in a host receptor, as evidenced by a comparison of HBV susceptibility between human and Old World monkeys and correlation with the sequence of the NTCP gene. Our biochemical experiment, however, showed that arginine at aa 158 (i.e., HBV-nonsusceptible residue) reduced the NTCP’s original function for bile acid uptake. This observation may explain why most primates still possess glycine at aa 158, despite its disadvantage of susceptibility to HBV infection. Interestingly, acquiring an additional mutation at aa 157 from lysine to glycine partially rescued the reduced bile acid transporter function of 158R in the mNtcp, SoNtcp, and SsNtcp backbones. This compensatory function of aa 157 mutation may be the basis of the inference of aa 157 as a positively selected site across the four evolutionary analyses used in this study. This point should be further examined in the future. Although there is a possibility that viruses or factors other than hepadnavirus also exerted selective pressure on NTCP, we provide the first evidence that at least hepadnaviruses can be a driver for inducing adaptive mutations in NTCP.
Surprisingly, our study implies that the HBV cross-species barrier is secured by a single-amino-acid change in the virus receptor. Several studies have reported that a single mutation on a virus envelope protein determines species specificity (64, 65), but our insight is unique in showing that a single mutation in a host receptor plays a key role in determining the host specificity. It is not known whether all hepadnaviruses (or which hepadnaviruses) use NTCP as their entry receptor. However, tent-making bat hepadnavirus (5) and New World monkey hepadnaviruses (53) are reported to interact with NTCP, suggesting that NTCP functions as a receptor for a wide range of hepadnaviruses (at least from bat to human). The present analysis suggests that HBV-related hepadnaviruses drive an adaptive evolution in their hosts’ proteins. Our novel insights are expected to improve our understanding of how hepadnaviruses coevolved with their hosts over evolutionary intervals, thereby gaining strong host species specificity.
MATERIALS AND METHODS
Molecular evolutionary analyses.
Twenty mammalian full-length NTCP protein sequences were collected from GenBank, as listed in Table 1, and aligned by using MUSCLE implemented in MEGA7 (66). The resulting alignment was verified manually at the amino acid level. A maximum likelihood (ML) tree of the 20 NTCP sequences was reconstructed using PhyML 3.0 (67) with 1,000 bootstrap resamplings and was used for further analysis (Fig. 1). The best-fitting substitution model was determined using Smart Model Selection (SMS) (68) implemented in PhyML 3.0. The Akaike information criterion (AIC) selected TN93 + G as the best-fit model. To test positive selection or the presence of sites with ω (dN/dS) of >1 in the evolution of NTCP, we performed two pairs of site models (69) with CODEML implemented in PAML v 4.8 (40, 41): the first pair employed M1a (neutral, 2 site classes, ω > 1 not allowed) versus M2a (selection, 3 site classes, ω > 1 allowed), and the second pair consisted of M7 (neutral, 10 site classes, ω > 1 not allowed) versus M8 (selection, 11 site classes, ω > 1 allowed). FEL, REL (42), and MEME (43) also were employed to detect positive selection sites through the DATAMONKEY webserver (70). HKY85 was determined as the best-fitting substitution model based on the model selection tool in DATAMONKEY. The following default significant cutoff values were used: P value of <0.1 for FEL, Bayes factor of >50 for REL, and P value of <0.1 for MEME. We further analyzed positive selection in the Old World monkey lineage by the branch site model (model A) (50) implemented in PAML v4.8.
Plasmids and cells.
Plasmids were constructed to permit expression of hNTCP, mNtcp (Macaca fascicularis), CjNtcp (Callithrix jacchus), SoNtcp (Saguinus oedipus), and SsNtcp (Saimiri sciureus) tagged with Myc/His at the C terminus. Specifically, the open reading frame encoding hNTCP (NM_003049.3), mNtcp (NM_001283323.1), CjNtcp (KT382285.1), SoNtcp (KT382284.1), and SsNtcp (KR153327.1) was inserted between the KpnI and XbaI sites of the pEF4-Myc/His A vector (Thermo Fisher Scientific, Waltham, MA). Individual mutant constructs were constructed using oligonucleotide-directed mutagenesis (71).
HepG2 cells were cultured as described previously (38). To generate NTCP-expressing cells, HepG2 cells were transfected with each NTCP expression plasmid using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer's protocol. At 24 h posttransfection, the cells were trypsinized and transferred into 96-well plates; the replated cells then were subjected to preS1 binding assay or virus infection assay (performed as described below) at 48 h posttransfection.
Western blotting.
Cells were treated first with lysing buffer (1% NP-40, 150 mM NaCl, 50 mM Tris-HCl, pH 7.5) and then with 250 U peptide-N-glycosidase F (PNGase F) to digest N-linked oligosaccharides from glycoproteins. NTCP expression was examined by immunoblotting using monoclonal anti-c-Myc antibody (Santa Cruz Biotechnology, Dallas, TX) at a 1:3,000 dilution, as described previously (56). Beta-actin was detected as a loading control by using monoclonal anti-β-actin antibody (Sigma, St. Louis, MO) at a 1:10,000 dilution. The chemiluminescence signals were scanned with C-DiGit (LI-COR Biosciences, Lincoln, NE) and analyzed with Image Studio Lite, ver. 5.2 (LI-COR Biosciences).
NTCP transporter assay.
NTCP transporter activity was measured by incubating cells with [3H]taurocholic acid at 37°C for 15 min in a sodium-containing buffer to allow substrate uptake into the cells. After washing to remove free [3H]taurocholic acid, cells were lysed and intracellular radioactivity was measured using an LSC-6100 liquid scintillation counter (Hitachi-Aloka Medical, Tokyo, Japan) as described previously (71). To measure the background signal, the same assay was performed in a sodium-free buffer in which the NTCP transporter does not function. To evaluate the transporter activity of equivalent amounts of NTCP protein (normalized NTCP transporter activity), we divided the value for bile acid uptake by the level of NTCP expression determined by immunofluorescence analysis. Results are presented as means ± standard errors of the means (SEM) (n = 3).
HBV preparation and infection.
HBV inocula were derived from the culture supernatant of Hep38.7-Tet cells (genotype D); these inocula were prepared as described previously (72). For the HBV infection assay, HBV was infected into recipient cells at 12,000 genome equivalents (GEq)/cell in the presence of 4% polyethylene glycol (PEG) 8000 for 16 h, followed by washing (to remove free virus) and culturing the cells for an additional 12 days, according to the previously described protocol (73). Indirect immunofluorescence analysis for detection of HBc was conducted to evaluate HBV infection; this technique was performed essentially as described previously (74). Briefly, after fixation with 4% paraformaldehyde and permeabilization with 0.3% Triton X-100, the cells were treated with a polyclonal rabbit anti-HBV core antibody (Thermo Fisher Scientific) at a 1:100 dilution and then with an Alexa Fluor 594 (Thermo Fisher Scientific)-conjugated donkey anti-rabbit IgG secondary antibody at a 1:500 dilution; nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) at a 1:5,000 dilution. Stained cells were photographed using a BZ-X710 microscope (Keyence, Osaka, Japan), and the images were analyzed with BZ-X Analyzer 1.3.1.1 (Keyence). The relative number of HBV-infected cells was determined by counting HBc-positive cells using the Hybrid Cell Count module of the Keyence analyzer, and the values were normalized for the NTCP expression levels determined by immunofluorescence analyses.
preS1 binding assay.
To examine binding between the preS1 region in the large surface protein of HBV (LHBs) and host cells, cells were exposed to 20 nM 6-carboxytetramethylrhodamine-labeled preS1 peptide (TAMRA-preS1) at 37°C for 30 min; unbound peptide then was removed by washing. To simultaneously detect Myc/His-tagged NTCP, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X-100, and the tagged NTCP was detected with a monoclonal anti-c-Myc antibody at a 1:1,200 dilution and with an Alexa Fluor 488-conjugated goat anti-mouse IgG secondary antibody (Thermo Fisher Scientific) at a 1:1,000 dilution. Fluorescence was quantified as described above.
HDV preparation and infection.
HDV was prepared from culture supernatants of Huh-7 cells transfected with pSVLD3 (kindly provided by John Taylor at the Fox Chase Cancer Center) and pT7HB2.7 as described previously (75, 76). The cells were inoculated with HDV at 15 GEq/cell in 5% PEG 8000 for 16 h, followed by washing (to remove free virus) and culturing of the cells for an additional 6 days. Total intracellular RNA was extracted and reverse transcribed using a TaqMan Gene Expression Cells-to-CT kit (Thermo Fisher Scientific) according to the manufacturer's protocol. Intracellular HDV RNA was quantified by real-time reverse transcription-PCR (RT-PCR) as described previously (77). Results are presented as means ± SEM (n = 3).
Statistics.
GraphPad Prism 7.03 was used for the statistical tests. Multiple comparisons within respective species were performed using one-way analysis of variance; posttest comparisons, performed only if P values were <0.05, were performed using Tukey’s multiple comparisons.
ACKNOWLEDGMENTS
HDV expression plasmids were kindly provided by John Taylor at the Fox Chase Cancer Center. This study was supported by KAKENHI (JP16J40232, JP16K19145, JP17H04085, JP66KT0111, and JP17K15708), the JST CREST program, the Japan Agency for Medical Research and Development, AMED (JP18fk0310114j0002, JP18fk0310101j1002, JP18fk0310103j0002, JP18fk0310103j0202, JP18fm0208019j0002, JP18fm0208019h0202, JP18fk0210036j0001, and JP18fk0210009j0003), the Takeda Science Foundation, the MSD Life Science Foundation, and the Pharmacological Research Foundation, Tokyo. We have no conflicts of interest to declare.
REFERENCES
- 1.World Health Organization. 2017. Global hepatitis report, 2017. World Health Organization, Geneva, Switzerland: http://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/. [Google Scholar]
- 2.Alter H, Block T, Brown N, Brownstein A, Brosgart C, Chang KM, Chen PJ, Chisari FV, Cohen C, El-Serag H, Feld J, Gish R, Glenn J, Greten T, Guo H, Guo JT, Hoshida Y, Hu J, Kowdley KV, Li W, Liang J, Locarnini S, Lok AS, Mason W, McMahon B, Mehta A, Perrillo R, Revill P, Rice CM, Rinaudo J, Schinazi R, Seeger C, Shetty K, Tavis J, Zoulim F. 2018. A research agenda for curing chronic hepatitis B virus infection. Hepatology 67:1127–1131. doi: 10.1002/hep.29509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuen MF, Ahn SH, Chen DS, Chen PJ, Dusheiko GM, Hou JL, Maddrey WC, Mizokami M, Seto WK, Zoulim F, Lai CL. 2016. Chronic hepatitis B virus infection: disease revisit and management recommendations. J Clin Gastroenterol 50:286–294. doi: 10.1097/MCG.0000000000000478. [DOI] [PubMed] [Google Scholar]
- 4.Dill JA, Camus AC, Leary JH, Di Giallonardo F, Holmes EC, Ng TF. 2016. Distinct viral lineages from fish and amphibians reveal the complex evolutionary history of hepadnaviruses. J Virol 90:7920–7933. doi: 10.1128/JVI.00832-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drexler JF, Geipel A, Konig A, Corman VM, van Riel D, Leijten LM, Bremer CM, Rasche A, Cottontail VM, Maganga GD, Schlegel M, Muller MA, Adam A, Klose SM, Carneiro AJ, Stocker A, Franke CR, Gloza-Rausch F, Geyer J, Annan A, Adu-Sarkodie Y, Oppong S, Binger T, Vallo P, Tschapka M, Ulrich RG, Gerlich WH, Leroy E, Kuiken T, Glebe D, Drosten C. 2013. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc Natl Acad Sci U S A 110:16151–16156. doi: 10.1073/pnas.1308049110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hahn CM, Iwanowicz LR, Cornman RS, Conway CM, Winton JR, Blazer VS. 2015. Characterization of a novel hepadnavirus in the white sucker (Catostomus commersonii) from the Great Lakes region of the United States. J Virol 89:11801–11811. doi: 10.1128/JVI.01278-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mason WS, Seal G, Summers J. 1980. Virus of Pekin ducks with structural and biological relatedness to human hepatitis B virus. J Virol 36:829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suh A, Brosius J, Schmitz J, Kriegs JO. 2013. The genome of a Mesozoic paleovirus reveals the evolution of hepatitis B viruses. Nat Commun 4:1791. doi: 10.1038/ncomms2798. [DOI] [PubMed] [Google Scholar]
- 9.Suh A, Weber CC, Kehlmaier C, Braun EL, Green RE, Fritz U, Ray DA, Ellegren H. 2014. Early mesozoic coexistence of amniotes and hepadnaviridae. PLoS Genet 10:e1004559. doi: 10.1371/journal.pgen.1004559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Summers J, Smolec JM, Snyder R. 1978. A virus similar to human hepatitis B virus associated with hepatitis and hepatoma in woodchucks. Proc Natl Acad Sci U S A 75:4533–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lauber C, Seitz S, Mattei S, Suh A, Beck J, Herstein J, Borold J, Salzburger W, Kaderali L, Briggs JAG, Bartenschlager R. 2017. Deciphering the origin and evolution of hepatitis B viruses by means of a family of non-enveloped fish viruses. Cell Host Microbe 22:387–399. doi: 10.1016/j.chom.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Carvalho Dominguez Souza BF, Konig A, Rasche A, de Oliveira Carneiro I, Stephan N, Corman VM, Roppert PL, Goldmann N, Kepper R, Muller SF, Volker C, de Souza AJS, Gomes-Gouvea MS, Moreira-Soto A, Stocker A, Nassal M, Franke CR, Rebello Pinho JR, Soares M, Geyer J, Lemey P, Drosten C, Netto EM, Glebe D, Drexler JF. 2018. A novel hepatitis B virus species discovered in capuchin monkeys sheds new light on the evolution of primate hepadnaviruses. J Hepatol 68:1114–1122. doi: 10.1016/j.jhep.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 13.Duggal NK, Emerman M. 2012. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol 12:687–695. doi: 10.1038/nri3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enard D, Cai L, Gwennap C, Petrov DA. 2016. Viruses are a dominant driver of protein adaptation in mammals. Elife 5:e12469. doi: 10.7554/eLife.12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kluge SF, Sauter D, Kirchhoff F. 2015. SnapShot: antiviral restriction factors. Cell 163:774–774. doi: 10.1016/j.cell.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Yang Z. 2014. Molecular evolution: a statistical approach. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 17.Sawyer SL, Wu LI, Emerman M, Malik HS. 2005. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A 102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sawyer SL, Emerman M, Malik HS. 2004. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol 2:E275. doi: 10.1371/journal.pbio.0020275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta RK, Hue S, Schaller T, Verschoor E, Pillay D, Towers GJ. 2009. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog 5:e1000443. doi: 10.1371/journal.ppat.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi T, Takeuchi JS, Ren F, Matsuda K, Sato K, Kimura Y, Misawa N, Yoshikawa R, Nakano Y, Yamada E, Tanaka H, Hirsch VM, Koyanagi Y. 2014. Characterization of red-capped mangabey tetherin: implication for the co-evolution of primates and their lentiviruses. Sci Rep 4:5529. doi: 10.1038/srep05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNatt MW, Zang T, Hatziioannou T, Bartlett M, Fofana IB, Johnson WE, Neil SJ, Bieniasz PD. 2009. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog 5:e1000300. doi: 10.1371/journal.ppat.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeuchi JS, Ren F, Yoshikawa R, Yamada E, Nakano Y, Kobayashi T, Matsuda K, Izumi T, Misawa N, Shintaku Y, Wetzel KS, Collman RG, Tanaka H, Hirsch VM, Koyanagi Y, Sato K. 2015. Coevolutionary dynamics between tribe Cercopithecini tetherins and their lentiviruses. Sci Rep 5:16021. doi: 10.1038/srep16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim ES, Fregoso OI, McCoy CO, Matsen FA, Malik HS, Emerman M. 2012. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe 11:194–204. doi: 10.1016/j.chom.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyerson NR, Rowley PA, Swan CH, Le DT, Wilkerson GK, Sawyer SL. 2014. Positive selection of primate genes that promote HIV-1 replication. Virology 454-455:291–298. doi: 10.1016/j.virol.2014.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdul F, Filleton F, Gerossier L, Paturel A, Hall J, Strubin M, Etienne L. 2018. Smc5/6 antagonism by HBx is an evolutionarily conserved function of hepatitis B virus infection in mammals. J Virol 92:e00769-18. doi: 10.1128/JVI.00769-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X, Wu Y, Yu Y, Xiong Y, Su L. 2016. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep 16:2846–2854. doi: 10.1016/j.celrep.2016.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrera A, Guerra B, Lee H, Lanford RE. 2004. Analysis of host range phenotypes of primate hepadnaviruses by in vitro infections of hepatitis D virus pseudotypes. J Virol 78:5233–5243. doi: 10.1128/JVI.78.10.5233-5243.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen DH, Ludgate L, Hu J. 2008. Hepatitis B virus-cell interactions and pathogenesis. J Cell Physiol 216:289–294. doi: 10.1002/jcp.21416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gehring A, Bertoletti A, Tavis JE. 2014. Host factor-targeted hepatitis B virus therapies. Intervirology 57:158–162. doi: 10.1159/000360938. [DOI] [PubMed] [Google Scholar]
- 31.Block TM, Guo H, Guo JT. 2007. Molecular virology of hepatitis B virus for clinicians. Clin Liver Dis 11:685–706. doi: 10.1016/j.cld.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lucifora J, Vincent IE, Berthillon P, Dupinay T, Michelet M, Protzer U, Zoulim F, Durantel D, Trepo C, Chemin I. 2010. Hepatitis B virus replication in primary macaque hepatocytes: crossing the species barrier toward a new small primate model. Hepatology 51:1954–1960. doi: 10.1002/hep.23602. [DOI] [PubMed] [Google Scholar]
- 33.Lempp FA, Wiedtke E, Qu B, Roques P, Chemin I, Vondran FWR, Le Grand R, Grimm D, Urban S. 2017. Sodium taurocholate cotransporting polypeptide is the limiting host factor of hepatitis B virus infection in macaque and pig hepatocytes. Hepatology 66:703–716. doi: 10.1002/hep.29112. [DOI] [PubMed] [Google Scholar]
- 34.Burwitz BJ, Wettengel JM, Muck-Hausl MA, Ringelhan M, Ko C, Festag MM, Hammond KB, Northrup M, Bimber BN, Jacob T, Reed JS, Norris R, Park B, Moller-Tank S, Esser K, Greene JM, Wu HL, Abdulhaqq S, Webb G, Sutton WF, Klug A, Swanson T, Legasse AW, Vu TQ, Asokan A, Haigwood NL, Protzer U, Sacha JB. 2017. Hepatocytic expression of human sodium-taurocholate cotransporting polypeptide enables hepatitis B virus infection of macaques. Nat Commun 8:2146. doi: 10.1038/s41467-017-01953-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coffin JM. 2013. Virions at the gates: receptors and the host-virus arms race. PLoS Biol 11:e1001574. doi: 10.1371/journal.pbio.1001574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demogines A, Abraham J, Choe H, Farzan M, Sawyer SL. 2013. Dual host-virus arms races shape an essential housekeeping protein. PLoS Biol 11:e1001571. doi: 10.1371/journal.pbio.1001571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng M, Ndungo E, Kaczmarek ME, Herbert AS, Binger T, Kuehne AI, Jangra RK, Hawkins JA, Gifford RJ, Biswas R, Demogines A, James RM, Yu M, Brummelkamp TR, Drosten C, Wang LF, Kuhn JH, Muller MA, Dye JM, Sawyer SL, Chandran K. 2015. Filovirus receptor NPC1 contributes to species-specific patterns of ebolavirus susceptibility in bats. Elife 4:e11785. doi: 10.7554/eLife.11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iwamoto M, Watashi K, Tsukuda S, Aly HH, Fukasawa M, Fujimoto A, Suzuki R, Aizaki H, Ito T, Koiwai O, Kusuhara H, Wakita T. 2014. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem Biophys Res Commun 443:808–813. doi: 10.1016/j.bbrc.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 39.Takeuchi JS, Fukano K, Iwamoto M, Tsukuda S, Suzuki R, Aizaki H, Muramatsu M, Wakita T, Sureau C, Watashi K. 2018. A single adaptive mutation in sodium taurocholate cotransporting polypeptide induced by hepadnaviruses determines virus species specificity. bioRxiv 10.1101/391490. [DOI] [PMC free article] [PubMed]
- 40.Xu B, Yang Z. 2013. PAMLX: a graphical user interface for PAML. Mol Biol Evol 30:2723–2724. doi: 10.1093/molbev/mst179. [DOI] [PubMed] [Google Scholar]
- 41.Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 42.Kosakovsky Pond SL, Frost SD. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 43.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764. doi: 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Souza BF, Drexler JF, Lima RS, Rosário MO, Netto EM. 2014. Theories about evolutionary origins of human hepatitis B virus in primates and humans. Braz J Infect Dis 18:535–543. doi: 10.1016/j.bjid.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dupinay T, Gheit T, Roques P, Cova L, Chevallier-Queyron P, Tasahsu S-I, Le Grand R, Simon F, Cordier G, Wakrim L, Benjelloun S, Trépo C, Chemin I. 2013. Discovery of naturally occurring transmissible chronic hepatitis B virus infection among Macaca fascicularis from Mauritius Island. Hepatology 58:1610–1620. doi: 10.1002/hep.26428. [DOI] [PubMed] [Google Scholar]
- 46.Petersen J, Dandri M, Mier W, Lutgehetmann M, Volz T, von Weizsacker F, Haberkorn U, Fischer L, Pollok JM, Erbes B, Seitz S, Urban S. 2008. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat Biotechnol 26:335–341. doi: 10.1038/nbt1389. [DOI] [PubMed] [Google Scholar]
- 47.Yan H, Peng B, Liu Y, Xu G, He W, Ren B, Jing Z, Sui J, Li W. 2014. Viral entry of hepatitis B and D viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J Virol 88:3273–3284. doi: 10.1128/JVI.03478-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor JM. 2013. Virus entry mediated by hepatitis B virus envelope proteins. World J Gastroenterol 19:6730–6734. doi: 10.3748/wjg.v19.i40.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Slijepcevic D, Kaufman C, Wichers CG, Gilglioni EH, Lempp FA, Duijst S, de Waart DR, Elferink RP, Mier W, Stieger B, Beuers U, Urban S, van de Graaf SF. 2015. Impaired uptake of conjugated bile acids and hepatitis b virus pres1-binding in na(+)-taurocholate cotransporting polypeptide knockout mice. Hepatology 62:207–219. doi: 10.1002/hep.27694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Z, dos Reis M. 2011. Statistical properties of the branch-site test of positive selection. Mol Biol Evol 28:1217–1228. doi: 10.1093/molbev/msq303. [DOI] [PubMed] [Google Scholar]
- 51.Kosakovsky Pond SL, Murrell B, Fourment M, Frost SD, Delport W, Scheffler K. 2011. A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol 28:3033–3043. doi: 10.1093/molbev/msr125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nozawa M, Suzuki Y, Nei M. 2009. Reliabilities of identifying positive selection by the branch-site and the site-prediction methods. Proc Natl Acad Sci U S A 106:6700–6705. doi: 10.1073/pnas.0901855106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muller SF, Konig A, Doring B, Glebe D, Geyer J. 2018. Characterisation of the hepatitis B virus cross-species transmission pattern via Na+/taurocholate co-transporting polypeptides from 11 New World and Old World primate species. PLoS One 13:e0199200. doi: 10.1371/journal.pone.0199200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhong G, Yan H, Wang H, He W, Jing Z, Qi Y, Fu L, Gao Z, Huang Y, Xu G, Feng X, Sui J, Li W. 2013. Sodium taurocholate cotransporting polypeptide mediates woolly monkey hepatitis B virus infection of Tupaia hepatocytes. J Virol 87:7176–7184. doi: 10.1128/JVI.03533-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan H, Peng B, He W, Zhong G, Qi Y, Ren B, Gao Z, Jing Z, Song M, Xu G, Sui J, Li W. 2013. Molecular determinants of hepatitis B and D virus entry restriction in mouse sodium taurocholate cotransporting polypeptide. J Virol 87:7977–7991. doi: 10.1128/JVI.03540-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee WM. 1993. Acute liver failure. N Engl J Med 329:1862–1872. doi: 10.1056/NEJM199312163292508. [DOI] [PubMed] [Google Scholar]
- 57.Becker DJ, Streicker DG, Altizer S. 2015. Linking anthropogenic resources to wildlife-pathogen dynamics: a review and meta-analysis. Ecol Lett 18:483–495. doi: 10.1111/ele.12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daszak P, Berger L, Cunningham AA, Hyatt AD, Green DE, Speare R. 1999. Emerging infectious diseases and amphibian population declines. Emerg Infect Dis 5:735–748. doi: 10.3201/eid0506.990601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tarlinton RE, Meers J, Young PR. 2006. Retroviral invasion of the koala genome. Nature 442:79–81. doi: 10.1038/nature04841. [DOI] [PubMed] [Google Scholar]
- 60.McCarthy AJ, Shaw MA, Goodman SJ. 2007. Pathogen evolution and disease emergence in carnivores. Proc Biol Sci 274:3165–3174. doi: 10.1098/rspb.2007.0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Compton AA, Malik HS, Emerman M. 2013. Host gene evolution traces the evolutionary history of ancient primate lentiviruses. Philos Trans R Soc Lond B Biol Sci 368:20120496. doi: 10.1098/rstb.2012.0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silvestri G, Paiardini M, Pandrea I, Lederman MM, Sodora DL. 2007. Understanding the benign nature of SIV infection in natural hosts. J Clin Investig 117:3148–3154. doi: 10.1172/JCI33034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arien KK, Vanham G, Arts EJ. 2007. Is HIV-1 evolving to a less virulent form in humans? Nat Rev Microbiol 5:141–151. doi: 10.1038/nrmicro1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Vries RP, Tzarum N, Peng W, Thompson AJ, Ambepitiya Wickramasinghe IN, de la Pena ATT, van Breemen MJ, Bouwman KM, Zhu X, McBride R, Yu W, Sanders RW, Verheije MH, Wilson IA, Paulson JC. 2017. A single mutation in Taiwanese H6N1 influenza hemagglutinin switches binding to human-type receptors. EMBO Mol Med 9:1314–1325. doi: 10.15252/emmm.201707726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Teng Q, Xu D, Shen W, Liu Q, Rong G, Li X, Yan L, Yang J, Chen H, Yu H, Ma W, Li Z. 2016. A single mutation at position 190 in hemagglutinin enhances binding affinity for human type sialic acid receptor and replication of H9N2 avian influenza virus in mice. J Virol 90:9806–9825. doi: 10.1128/JVI.01141-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 68.Lefort V, Longueville JE, Gascuel O. 2017. SMS: smart model selection in PhyML. Mol Biol Evol 34:2422–2424. doi: 10.1093/molbev/msx149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nielsen R, Yang Z. 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shimura S, Watashi K, Fukano K, Peel M, Sluder A, Kawai F, Iwamoto M, Tsukuda S, Takeuchi JS, Miyake T, Sugiyama M, Ogasawara Y, Park SY, Tanaka Y, Kusuhara H, Mizokami M, Sureau C, Wakita T. 2017. Cyclosporin derivatives inhibit hepatitis B virus entry without interfering with NTCP transporter activity. J Hepatol 66:685–692. doi: 10.1016/j.jhep.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ogura N, Watashi K, Noguchi T, Wakita T. 2014. Formation of covalently closed circular DNA in Hep38.7-Tet cells, a tetracycline inducible hepatitis B virus expression cell line. Biochem Biophys Res Commun 452:315–321. doi: 10.1016/j.bbrc.2014.08.029. [DOI] [PubMed] [Google Scholar]
- 73.Watashi K, Liang G, Iwamoto M, Marusawa H, Uchida N, Daito T, Kitamura K, Muramatsu M, Ohashi H, Kiyohara T, Suzuki R, Li J, Tong S, Tanaka Y, Murata K, Aizaki H, Wakita T. 2013. Interleukin-1 and tumor necrosis factor-alpha trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID). J Biol Chem 288:31715–31727. doi: 10.1074/jbc.M113.501122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsukuda S, Watashi K, Hojima T, Isogawa M, Iwamoto M, Omagari K, Suzuki R, Aizaki H, Kojima S, Sugiyama M, Saito A, Tanaka Y, Mizokami M, Sureau C, Wakita T. 2017. A new class of hepatitis B and D virus entry inhibitors, proanthocyanidin and its analogs, that directly act on the viral large surface proteins. Hepatology 65:1104–1116. doi: 10.1002/hep.28952. [DOI] [PubMed] [Google Scholar]
- 75.Kuo MY, Chao M, Taylor J. 1989. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: role of delta antigen. J Virol 63:1945–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sureau C, Guerra B, Lee H. 1994. The middle hepatitis B virus envelope protein is not necessary for infectivity of hepatitis delta virus. J Virol 68:4063–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gudima S, He Y, Meier A, Chang J, Chen R, Jarnik M, Nicolas E, Bruss V, Taylor J. 2007. Assembly of hepatitis delta virus: particle characterization, including the ability to infect primary human hepatocytes. J Virol 81:3608–3617. doi: 10.1128/JVI.02277-06. [DOI] [PMC free article] [PubMed] [Google Scholar]