

Bacteriophages produce highly evolved lytic enzymes, called endolysins, to lyse peptidoglycan and release their progeny from bacterial cells. Due to their potent lytic activity and specificity, the use of endolysins has gained increasing attention as a natural alternative to antibiotics. Since most endolysins from Gram-positive-bacterium-infecting phages have a modular structure, understanding the function of each domain is crucial to make effective endolysin-based therapeutics. Here, we report the functional and biochemical characterization of a Bacillus cereus phage endolysin, LysPBC2, which has an unusual spore binding domain and a cell wall binding domain. A single point mutation in the spore binding domain greatly enhanced the lytic activity of endolysin at the cost of reduced thermostability. This work contributes to the understanding of the role of each domain in LysPBC2 and will provide insight for the rational design of efficient antimicrobials or diagnostic tools for controlling B. cereus.
KEYWORDS: Bacillus cereus, antimicrobial agents, bacteriophages, endolysin, spore binding domain, spores
ABSTRACT
To control the spore-forming human pathogen Bacillus cereus, we isolated and characterized a novel endolysin, LysPBC2, from a newly isolated B. cereus phage, PBC2. Compared to the narrow host range of phage PBC2, LysPBC2 showed very broad lytic activity against all Bacillus, Listeria, and Clostridium species tested. In addition to a catalytic domain and a cell wall binding domain, LysPBC2 has a spore binding domain (SBD) partially overlapping its catalytic domain, which specifically binds to B. cereus spores but not to vegetative cells of B. cereus. Both immunogold electron microscopy and a binding assay indicated that the SBD binds the external region of the spore cortex layer. Several amino acid residues required for catalytic or spore binding activity of LysPBC2 were determined by mutagenesis studies. Interestingly, LysPBC2 derivatives with impaired spore binding activity showed an increased lytic activity against vegetative cells of B. cereus compared with that of wild-type LysPBC2. Further biochemical studies revealed that these LysPBC2 derivatives have lower thermal stability, suggesting a stabilizing role of SBD in LysPBC2 structure.
IMPORTANCE Bacteriophages produce highly evolved lytic enzymes, called endolysins, to lyse peptidoglycan and release their progeny from bacterial cells. Due to their potent lytic activity and specificity, the use of endolysins has gained increasing attention as a natural alternative to antibiotics. Since most endolysins from Gram-positive-bacterium-infecting phages have a modular structure, understanding the function of each domain is crucial to make effective endolysin-based therapeutics. Here, we report the functional and biochemical characterization of a Bacillus cereus phage endolysin, LysPBC2, which has an unusual spore binding domain and a cell wall binding domain. A single point mutation in the spore binding domain greatly enhanced the lytic activity of endolysin at the cost of reduced thermostability. This work contributes to the understanding of the role of each domain in LysPBC2 and will provide insight for the rational design of efficient antimicrobials or diagnostic tools for controlling B. cereus.
INTRODUCTION
Bacillus cereus is a Gram-positive spore-forming bacterium that is widespread in the natural environment. B. cereus belongs to the Bacillus cereus group consisting of genetically closely related species: B. cereus, B. thuringiensis, B. anthracis, B. mycoides, B. weihenstephanensis, B. pseudomycoides, and B. cytotoxicus (1). Under starvation or harsh environmental conditions, B. cereus can form spores to survive, and these spores are metabolically dormant and can withstand extremes of heat, desiccation, and many chemical agents (2). The outer portion of the spores consists of a cortex, a spore coat layer, and an exosporium. The cortex is the innermost layer; it is made of thick peptidoglycan and is responsible for maintaining the highly dehydrated state of the core (3). A proteinaceous spore coat encases the core and the cortex and is essential for protecting spores from various environmental stresses (2). Separated from the coat by an interspace, the exosporium is the loose-fitting outermost spore layer and is composed of an inner basal layer and an outer hair-like nap (4).
Since B. cereus produces various toxins, including enterotoxins, phospholipases, hemolysins, and an emetic toxin (cereulide), it has been frequently associated with severe local and systemic infections as well as food poisoning cases (5). The antibiotic treatment options, however, seem to be limited because B. cereus is generally resistant to beta-lactam antibiotics (5), and a number of B. cereus strains are increasingly reported as resistant to vancomycin (6), aminoglycosides (7), erythromycin (8), and fluoroquinolones (9). For these reasons, the demand for alternative strategies to combat B. cereus has grown.
Endolysins are bacteriophage-encoded peptidoglycan hydrolases required for host cell lysis and the release of phage progeny at the late stage of infection. Because endolysins are bactericidal enzymes with highly evolved specificities toward specific target bacteria, they have been considered novel antibacterial agents and have shown promising potential in therapy, disinfection, and diagnostics (10, 11). While most endolysins from phages that infect Gram-negative bacteria adopt a single-domain globular structure, typical endolysins from Gram-positive phages have a modular structure consisting of one or more enzymatic active domains (EADs) and a C-terminal cell wall binding domain (CBD) (12, 13). The EAD has conserved active sites and cleaves specific bonds within the peptidoglycan, whereas the CBD recognizes a highly specific ligand in the cell wall and targets the endolysin to its substrate (14). This modular nature has been extensively exploited by protein engineers to generate novel endolysins with optimized or new properties (15, 16).
Here, we characterized a novel broad-spectrum endolysin, LysPBC2, derived from a newly isolated B. cereus phage, PBC2, and constructed various LysPBC2 derivatives via domain deletion, enhanced green fluorescent protein (EGFP) fusion, and site-directed mutagenesis to determine the role of each domain of LysPBC2. Our findings may facilitate the development of effective biocontrol and detection agents for B. cereus.
RESULTS
Isolation of phage PBC2 and its endolysin.
The B. cereus phage PBC2 was isolated from sewage. PBC2 belongs to the Siphoviridae family (see Fig. S1A in the supplemental material) and showed high host specificity, producing plaques only against the ATCC 13061 strain of B. cereus out of the nine B. cereus strains tested (Table 1). The genome of PBC2 contains 168,689 bp, including 251 open reading frames (ORFs) and 17 tRNAs (see Fig. S2), and a comparative genomic analysis revealed that PBC2 and B. anthracis phage Tsamsa are closely related (Fig. S1B and C) (17). Genes encoding a putative holin (ORF195) and an endolysin (ORF197, N-acetylmuramoyl-l-alanine amidase) were found, suggesting that PBC2 may use these two proteins for host cell lysis. Since PBC2 features a very narrow host range and harbors several lysogeny cassettes (ORF209 and ORF241), we concluded PBC2 itself may not be suitable as a biocontrol agent and turned our attention to its endolysin, LysPBC2.
TABLE 1.
The host ranges of phage PBC2, LysPBC2, and LysPBC2_EAD and the binding activity of LysPBC2_CBD
| Species | Straina | Plaque |
Relative lytic activityb
|
LysPBC2_CBD binding | |
|---|---|---|---|---|---|
| PBC2 | LysPBC2 | LysPBC2_EAD | |||
| B. cereus group strains | |||||
| Bacillus cereus | ATCC 27348 | − | ++ | + | + |
| Bacillus cereus | ATCC 21768 | − | ++ | + | + |
| Bacillus cereus | ATCC 13061 | + | ++ | + | + |
| Bacillus cereus | ATCC 14579 | − | ++ | ++ | + |
| Bacillus cereus | ATCC 21772 | − | ++ | + | + |
| Bacillus cereus | ATCC 10876 | − | + | + | + |
| Bacillus cereus | KCTC 3674 | − | ++ | + | + |
| Bacillus cereus | ATCC 10987 | − | ++ | ++ | + |
| Bacillus cereus | KCTC 1094 | − | + | + | + |
| Bacillus thuringiensis | ATCC 10792 | − | ++ | + | + |
| Bacillus mycoides | ATCC 6462 | − | ++ | + | + |
| Other Gram-positive strains | |||||
| Bacillus subtilis | ATCC 23857 | − | ++ | ++ | − |
| Bacillus subtilis | ATCC 6051 | − | +++ | +++ | − |
| Bacillus megaterium | JCM 2506 | − | +++ | +++ | − |
| Bacillus circulans | JCM 2504 | − | +++ | +++ | − |
| Bacillus licheniformis | JCM 2505 | − | +++ | +++ | − |
| Bacillus pumilus | JCM 2508 | − | +++ | +++ | − |
| Listeria monocytogenes | ScottA | − | + | + | − |
| Listeria monocytogenes | EGDe | − | + | + | − |
| Clostridium perfringens | ATCC 13124 | − | ++ | + | − |
| Clostridium perfringens | NCCP 15911 | − | ++ | + | − |
| Staphylococcus aureus | ATCC 29213 | − | − | − | − |
| Staphylococcus epidermidis | ATCC 35983 | − | − | − | − |
| Enterococcus faecalis | ATCC 10100 | − | − | − | − |
| Lactococcus lactis | ATCC 11454 | − | − | − | − |
| Gram-negative strainsc | |||||
| Escherichia coli | MG 1655 | −/− | −/++ | −/++ | −/+ |
| Cronobacter sakazakii | ATCC 29544 | −/− | −/++ | −/++ | −/+ |
ATCC, American Type Culture Collection; KCTC, Korean Collection for Type Cultures; NCCP, National Culture Collection for Pathogens; JCM, Japan Collection of Microorganisms.
Relative lytic activity of endolysin was obtained by measuring the percent drop in OD at 600 nm in 6 min. −, no lysis; +, limited lysis; ++, medium lysis; +++, rapid lysis.
Gram-negative strains were pretreated with 0.1 M EDTA (before/after).
A conserved domain analysis revealed that LysPBC2 contains three domains, one N-terminal amidase_2 domain (PF01510) and two C-terminal bacterial Src homology 3 (SH3) domains (Fig. 1A). LysPBC2 shares overall 81% amino acid sequence identity with the endolysin of Tsamsa (17). In addition, LysPBC2 showed close relatedness to a B. anthracis prophage lambdaBa02 endolysin, PlyL (GenBank identifier [ID] AAP27798, 37% identity) (18), B. anthracis phage gamma endolysin, PlyG (GenBank ID AAM97149, 37% identity) (19), B. cereus phage TP21 endolysin, Ply21 (GenBank ID CAA72267, 33% identity) (20), and a B. cereus phage BPS13 endolysin, LysBPS13 (GenBank ID YP_006907567, 31% identity) (21) (Fig. 1B). Three Zn2+ binding residues (His31, His131, and Cys139) and two active-site residues (Glu92 and Lys137) were found to be conserved in the catalytic amidase_2 domain (18, 22, 23). Recombinantly produced LysPBC2 showed potent antimicrobial activity against B. cereus cells (see Fig. S3). In contrast to the narrow host range of PBC2, LysPBC2 showed a broad lytic spectrum and was able to lyse all B. cereus strains tested (Table 1). Several other Bacillus species tested, Listeria monocytogenes, Clostridium perfringens, and even EDTA-treated Gram-negative bacteria, were also sensitive to the action of LysPBC2. All of these LysPBC2-sensitive bacteria have the same peptidoglycan type (A1γ) (24), suggesting that LysPBC2 recognizes and cleaves the conserved moiety within the peptidoglycan subgroup A1γ. To confirm the binding selectivity of LysPBC2, we created EGFP-fused LysPBC2_CBD (residues 163 to 311 of LysPBC2) and tested its binding ability toward different bacteria. Interestingly, as shown in Table 1 and Fig. S4, LysPBC2_CBD showed specific binding to B. cereus group strains but not to other species, including B. subtilis, Listeria, and Clostridium strains, all of which are sensitive to the lytic action of LysPBC2. These unexpected results indicate that the lytic spectrum of LysPBC2 exceeds the binding range of the endolysin and that the specific cell wall components only present in B. cereus group strains may be required for the binding of PBC2_CBD. Lastly, to test the effect of CBD binding on the lytic activity of LysPBC2, we produced a LysPBC2_EAD (residues 1 to 165 of LysPBC2) and performed a turbidity reduction assay. The LysPBC2_EAD alone also showed lytic activity against a broad spectrum of bacteria, similarly to LysPBC2, but its activity against B. cereus was lower than that of full-length endolysin (Table 1; see also Fig. S5). These results suggest that CBD binding is not essential for lytic activity but is necessary for LysPBC2 to exert full lytic activity against B. cereus cells.
FIG 1.

Spore binding domain of LysPBC2. (A) Schematic diagram of the multimodular structure of LysPBC2. (B) Sequence alignment of LysPBC2-related endolysins. Conserved residues are shaded in gray (>60% identity) and black (>80% identity). *, conserved amidase catalytic residue. The brackets indicate SBD (blue) and SBD core (red) regions. (C) Binding capacities of EGFP only, EGFP-SBD, and EGFP-CBD for B. cereus spores and vegetative cells. (D) Spore binding selectivity of the SBD.
Spore binding domain of LysPBC2.
LysPBC2 showed overall sequence similarity to the B. anthracis phage endolysin PlyG (37% amino acid identity) (Fig. 1B). Previously, Yang et al. reported that PlyG has a separate 60-amino-acid domain, located mainly within the catalytic domain of PlyG (residues 106 to 165), that specifically binds to B. anthracis spores but not to vegetative cells (25). Because the EAD of LysPBC2 has significant amino acid sequence identity (57%) to the catalytic domain of PlyG, we assumed that LysPBC2 also has a spore binding domain (SBD). To confirm this hypothesis, two truncated fragments (residues 108 to 170 for SBD1 and residues 127 to 147 for the SBD core) of LysPBC2 were fused with EGFP and expressed in Escherichia coli (see Fig. S6A). Fluorescence microscopy analysis revealed that the EGFP-SBD1 fusion protein displayed a clear ability to bind to B. cereus spores, while only limited binding was observed with the EGFP-SBD core fusion protein (Fig. S6B). Thus, we refer to the SBD1 construct as the SBD of LysPBC2 (LysPBC2_SBD). LysPBC2_SBD discriminated between spores and the vegetative form of B. cereus, because it did not bind to vegetative cells (Fig. 1C). On the other hand, the LysPBC2_CBD labeled only vegetative cells and not the spores of B. cereus. A combined SBD and CBD (SCBD) fused with EGFP showed binding capacity to both spores and vegetative cells, although the vegetative cells were more strongly decorated by the fusion protein in the mixed samples (Fig. S6C). In addition, LysPBC2_SBD specifically binds to B. cereus spores, whereas spores of Bacillus subtilis, Bacillus licheniformis, and Bacillus megaterium did not react with LysPBC2_SBD (Fig. 1D). Altogether, these results validate the existence of separate domains within LysPBC2 for specific recognition of B. cereus spores and vegetative cells.
LysPBC2_SBD localizes to the spore cortex near the inner coat.
The earlier study reported that the exosporium of the spores may be the most probable binding target of PlyG_SBD (25). To test whether the exosporium is a binding target of LysPBC2_SBD, we used physical extraction (sonication) and/or chemical extraction (SDS, urea) methods to disrupt the exosporium layer (26, 27). A closer analysis of the ruthenium red-stained hair-like nap of the exosporium revealed that the sonicated spores had an almost intact nap layer, while the spores extracted with both SDS and urea exhibited greater disruption of the exosporium layer (Fig. 2A). Further experiments with the EGFP-SBDs showed that the SDS–urea-treated spores displayed much higher fluorescence intensity than the intact spores and sonicated spores (Fig. 2B). Quantitative fluorescence analysis confirmed this result; a >7-fold higher signal was observed with SDS–urea-treated spores than with intact spores (Fig. 2C). Our observations were contrary to the previous results for PlyG_SBD, which showed significantly reduced fluorescence intensity toward sonicated spores (25). Despite the substantial sequence homologies between LysPBC2_SBD and PlyG_SBD (46% identity), the contradictory results may be due in part to the different natures of the binding targets or strain differences (spores from B. cereus ATCC 13061 versus B. anthracis A16) or even to how the spores were prepared prior to sonication (26). To determine the binding target of the LysPBC2_SBD, immunogold electron microscopy was employed using rabbit anti-GFP antibody as the primary antibody and a secondary antibody that was reactive against the primary antibody and labeled with 10-nm gold nanoparticles. An average of 6 to 7 gold particles were found per spore, and the particles were generally observed in the external region of cortex layer of the B. cereus spores (Fig. 2D). We also tried to see chemically extracted spores by electron microscopy (EM), but we were only able to observe heavily deformed spores (data not shown). A further analysis of the controls, where the primary antibody was omitted, showed few gold particles scattered randomly over the spore core or outside the spores (Fig. 2E). These results, in combination with the fluorescence microscopy data, suggest that the binding target of LysPBC2_SBD is the spore external cortex region near the inner coat layers. In addition, our findings demonstrate a novelty of LysPBC2_SBD and may have implications for the development of B. cereus spore-specific diagnostic agents.
FIG 2.

The outermost layer of the cortex is the most probable binding target of the SBD. (A, top) TEM of untreated spores. Inset shows exosporium basal layer and hair-like nap. (Bottom) TEM of spores sonicated or treated with SDS and urea buffer. All panels stained with ruthenium red to visualize the heavily glycosylated exosporium nap. (B) Binding profiles of EGFP-SBD with B. cereus intact spores, sonicated spores, and SDS–urea-treated spores. (C) Relative fluorescence intensities of differently treated spores after incubation with EGFP-SBD and EGFP-only proteins. Immunogold electron micrographs of ultrathin sections of B. cereus spores were labeled with primary antibody (D) or without primary antibody (E). The arrows point to 10-nm gold nanoparticles. The scale bars represent 100 nm.
Single point mutations within the SBD enhance the lytic activity of LysPBC2.
A sequence alignment analysis and a structural model of LysPBC2 revealed that conserved residues in the SBD core region are located near active sites of several endolysins (Fig. 1B and 3A). This observation prompted us to identify important residues for spore binding and/or catalytic activity and to test if the alteration of spore binding modulates the lytic activity of endolysin. Derivatives of LysPBC2 having single amino acid replacement as shown on Fig. 2B were made by site-directed mutagenesis, and we evaluated their spore binding activity toward B. cereus spores using EGFP-fused SBD derivatives. Spore binding assays showed that all the tested SBD derivatives except a catalytic residue mutant (K137E) displayed decreased spore binding activity compared to that of the wild-type SBD (Fig. 3B). Specifically, charge inversion mutations (K129E, R142E, and R148E) significantly impaired the spore binding ability of SBD. These results suggest that these positively charged residues within SBD, not the catalytic residue K137, are involved in spore binding. Then, we measured the lytic activity against B. cereus vegetative cells using LysPBC2 derivatives containing the single point mutation within the SBD region. A turbidity reduction assay revealed that the catalytic residue mutant (K137E) of LysPBC2 did not have any lytic activity against vegetative B. cereus cells, but unexpectedly, the other SBD mutants of LysPBC2 showed increased lytic activity compared to that of the wild-type LysPBC2 (Fig. 3C), proposing an inhibitory effect of an intact SBD on the lytic activity of LysPBC2.
FIG 3.
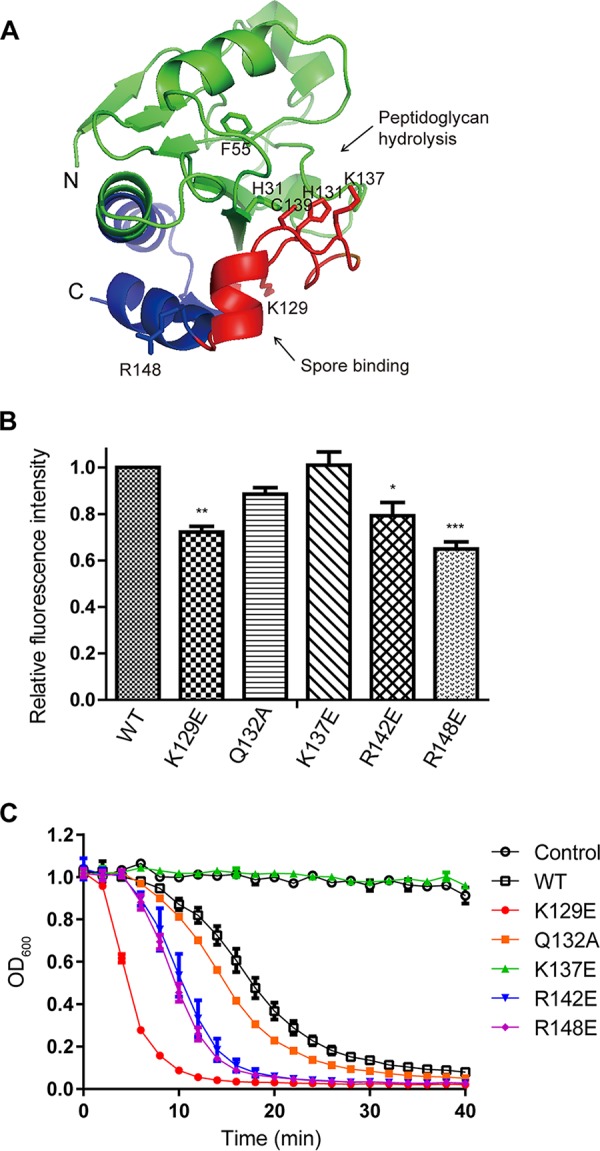
The effects of point mutations within SBD on the activity of LysPBC2. (A) Structural model of the catalytic domain of LysPBC2 (residues 1 to 158). The SBD region is represented in blue with the highly conserved core region (residues 127 to 147) in red. The N and C termini are marked N and C, respectively. Catalytic residues (H31, F55, H131, K137, and C139) and key residues for spore binding (K129 and R148) are shown as sticks. (B) Effects of point mutations within the SBD on spore binding. *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus the control (WT), determined by one-way ANOVA with Tukey’s post hoc test. (C) Effects of point mutations within the SBD on the lytic activity of LysPBC2 against vegetative cells.
Contribution of the SBD to the stability of LysPBC2.
The increase in lytic activity with SBD mutation led us to wonder why LysPBC2 harbors the intact SBD. We hypothesized that the single mutation within the conserved SBD region may cause a structural change and alter the stability of the endolysin. To see how the SBD region affects the stability of the endolysin, we performed thermal inactivation assays using a wild-type LysPBC2 and two LysPBC2 derivatives (K129E and R148E mutants) which showed enhanced lytic activities but significantly lower spore binding activities. As shown in Fig. 4, heat treatment at 60°C was sufficient to inactivate both K129E and R148E mutant enzymes, whereas the wild-type LysPBC2 retained substantial activity at 60°C, suggesting that the intact SBD contributes to the thermal stability of LysPBC2. It is worth noting that the lytic activity of the K129E mutant was lower than that of wild-type endolysin under all tested conditions, and this was contrary to the previous result shown in Fig. 3C. We attributed this discrepancy to the different reaction conditions; whereas the buffer used in thermal inactivation assay contained 200 mM NaCl, the turbidity reduction assay was carried out in the absence of NaCl. To confirm if the addition of NaCl decreased the lytic activity of the K129E mutant, we performed the turbidity reduction assay with various concentrations of NaCl. The lytic activity of wild-type LysPBC2 was relatively unaffected by NaCl concentrations between 0 and 200 mM, whereas the lytic activity of the K129E mutant was greatly reduced by the addition of the NaCl (see Fig. S7), suggesting that the K129 residue is involved in the NaCl sensitivity of LysPBC2. In conjunction with thermal inactivation data, these results propose a stabilizing role of the intact SBD in LysPBC2.
FIG 4.

Contribution of SBD to endolysin resistance to thermal inactivation. The residual lytic activity of WT, K129E, and R148E after heat treatment (10 min) was measured using turbidity reduction assays. The lytic activity of endolysin was measured at 12.5 min and was normalized to the activity displayed by the 20°C heat treatment group.
DISCUSSION
We have shown that the endolysin LysPBC2 has a broad lytic spectrum considering the narrow host range of phage PBC2. While PBC2 can only infect a single B. cereus strain (ATCC 13061) out of 11 B. cereus group strains, LysPBC2 lyses all Listeria, Clostridium, and Bacillus species tested. To the best of our knowledge, this is the first report of an endolysin that can kill three important human pathogens (B. cereus, L. monocytogenes, and C. perfringens) commonly associated with food poisoning outbreaks. The catalytic domain of LysPBC2 belongs to the N-acetylmuramoyl-l-alanine amidase family and shares high sequence similarity to those of other Bacillus phage endolysins, such as the endolysin of Tsamsa (identity, 89%), PlyL (identity, 58%), PlyG (identity, 57%), and Ply21 (identity, 51%). Despite the sequence similarities, LysPBC2 exhibited markedly different host ranges among the related endolysins. Previous studies have revealed that PlyG only kills B. anthracis and some rare B. cereus strains (19), while Ply21 and PlyL lyse B. cereus but have only detectable activities against B. subtilis cells (18). The Tsamsa endolysin also showed lytic activity specific for B. cereus group strains but not for B. subtilis and B. megaterium strains (17). Although LysPBC2 demonstrated strong activities toward all B. cereus strains tested, the Bacillus species other than the B. cereus group were found to be the most susceptible species to the action of LysPBC2. Considering that a number of Bacillus species, including B. subtilis, B. licheniformis, and B. pumilus, are increasingly implicated in food poisoning, spoilage, and systemic infections (28–30), the use of the broad-spectrum LysPBC2 could be beneficial for preventing diseases associated with Bacillus species.
Like other typical endolysins from phages infecting Gram-positive bacteria, LysPBC2 has a modular organization consisting of an N-terminal catalytic domain and a C-terminal CBD. The LysPBC2_CBD is composed of tandem repeated SH3 domains and has little sequence similarity to those of the related endolysins (PlyG, PlyL, and Ply21). It is interesting to note that LysPBC2_EAD alone has lytic activity and LysPBC2_CBD only bound to B. cereus group species, even though LysPBC2 has broad lytic activity against various Gram-positive bacteria, including Listeria, Clostridium, and Bacillus. This suggests that the LysPBC2_EAD alone has intrinsic binding activity toward conserved peptidoglycan (PG) moieties of target bacteria and lyses them. The contribution of the CBD on the lytic activity of LysPBC2 varies depending on the treated bacterial species. The removal of the CBD led to a decrease in lytic activity against B. cereus group and Clostridium strains, while it barely affected the activity toward B. subtilis group strains and even enhanced the activity against Listeria strains. As opposed to the previous studies showing that the CBD has an inhibitory effect on lytic activity when it is not bound to cognate target bacteria (18, 31), our results indicate that the effect of the CBD on lytic activity is variable. Based on our results, we suggest that each enzyme-sensitive bacterial species should be tested to determine the contribution of the CBD to the activity of an endolysin.
In addition to a catalytic domain and a CBD, LysPBC2 has a spore binding domain (SBD) that partially overlaps the catalytic domain, similar to the case of PlyG (25). Although both SBDs have significant sequence similarities (46% identity) and contain several conserved zinc-coordinating and catalytic residues (H131, C139, and K137 for LysPBC2), they showed quite different characteristics in terms of binding targets. It was shown that the PlyG_SBD specifically recognizes B. anthracis spores but not B. cereus spores, and the exosporium was proposed as the most probable binding site of the PlyG_SBD (25). On the other hand, we showed that the LysPBC2_SBD binds specifically to B. cereus spores and likely targets the boundary region between the spore’s cortex and coats. This finding is surprising, because it has been reported that the channels of the exosporium layer are too small to permit the diffusion of large proteins (32). We speculate that the EGFP-tagged LysPBC2_SBD (326 amino acids) was barely able to cross the exosporium channels (∼3.4 nm) (33, 34) and holes/pits (diameter of 5 to 7 nm) (35) of the outer spore coat of B. cereus at the concentrations we used. It is also possible that the loosely attached exosporium had been damaged during spore preparations (e.g., repeated centrifugation/wash cycles and freeze/thaw steps). We also found that the two conserved positively charged amino acids (K129 and R148) within the SBD are mainly involved in spore binding, and surprisingly, these two LysPBC2 derivatives (K129E and R148E) had increased lytic activity against vegetative cells of B. cereus under certain conditions. These results suggest a sophisticated regulatory potential of the SBD for the catalytic efficiency of LysPBC2. Subsequent biochemical studies revealed that these mutant endolysins have reduced thermal stability and salt tolerance, proposing a stabilizing role of the SBD in LysPBC2. Further biophysical and structural studies are necessary to elucidate how LysPBC2_SBD regulates enzymatic kinetics and how the SBD contributes to the endolysin’s stability.
Given that the genetic interchange between phages and bacterial hosts occurs naturally (36), PBC2 might have acquired the spore binding domain from the host sporulation proteins during evolution. A BLASTP analysis using the spore and cell wall binding domain (SCBD) of LysPBC2 as the query revealed a significant homology with many spore-lytic enzymes of B. cereus, supporting this speculation. Indeed, a number of germination-specific N-acetylmuramoyl-l-alanine amidases have been identified in B. cereus spores (37), and several of these major cortex-lytic enzymes are thought to be localized to the spore coat (38, 39). Then why does the PBC2 phage have an SBD in its genome? The exact biological role of the SBD is still unknown, but we speculate two possibilities since spores are refractory to phage infection and propagation. If the phage were to infect a B. cereus cell that is in the process of forming a spore, or if the vegetative cell begins sporulation during a phage infection, the endolysin may prevent the completion of the sporulation by degrading the forming spore cortex and/or hijacking the precursors of cortex peptidoglycan. In this case, the SBD can facilitate this process by localizing the endolysin on developing spores (presumably at the spore coat/cortex boundary region). Alternatively, the localization of LysPBC2 on the developing spores may be evolutionarily beneficial to both phage and host if the endolysin promotes spore germination under favorable environmental conditions. Recently, one study revealed that a Clostridium tyrobutyricum phage endolysin, CTP1L, can bind to clostridial spores (40). Although they did not find an SBD within CTP1L, it is intriguing to identify additional SBDs from phages infecting spore-forming bacteria.
In summary, we isolated and characterized a novel endolysin, LysPBC2. In addition to a catalytic domain and a cell wall binding domain, LysPBC2 has a spore binding domain. LysPBC2 derivatives with impaired spore binding demonstrated increased lytic activity at the expense of reduced stability. Since LysPBC2 showed strong lytic activities against a broad range of bacterial genera, such as Bacillus, Listeria, and Clostridium, and has a modular structure with distinct B. cereus group-specific cell or spore binding domains, our findings hold promise for the future development of effective biocontrol agents and detection tools.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this study are listed in Table 1. The B. cereus ATCC 13061 strain was used for the isolation and propagation of the PBC2 phage. All of the Bacillus strains and Gram-negative bacteria were grown in Luria-Bertani (LB) broth at 37°C. Listeria, Staphylococcus, and Clostridium strains were grown in brain heart infusion (BHI) broth at 37°C. All of the media were purchased from Difco (Sparks, MD) and used according to the manufacturer’s instructions.
Isolation and purification of the PBC2 phage.
A sewage sample was collected from the Seonam Water Reclamation Center in Seoul, South Korea. PBC2 was isolated and purified according to methods previously described (41). Briefly, a 25-ml sample was added to equal volumes of 2× LB broth and incubated with shaking at 37°C for 24 h. After centrifugation (10,000 × g for 10 min), the filtered supernatant was mixed with overnight cultures of a B. cereus host strain for initial propagation. Phages were isolated by the double-agar overlay method by picking plaques and eluting them in SM buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 10 mM MgSO4). For propagation and purification, phage particles were precipitated with polyethylene glycol 6000 (PEG 6000) (10%; Sigma) and 1 M sodium chloride. The precipitated phages were resuspended in SM buffer and purified by CsCl density gradient (step densities, 1.3, 1.45, 1.5, and 1.7 g/ml) ultracentrifugation at 78,500 × g for 2 h. Separated phages were dialyzed against SM buffer and stored at 4°C until further use.
Host range.
Host range studies were performed by using plaque-forming assays. Briefly, diluted phage stocks (10 μl) were spotted onto molten top LB agar (0.4%) containing bacterial cells grown overnight. The formation of a single plaque was verified after incubation at 37°C for 12 h.
Morphological analysis by TEM.
Purified PBC2 (1 × 1010 PFU/ml) was visualized by transmission electron microscopy (TEM) conducted as previously described (41). Based on the morphology, PBC2 was classified according to the guidelines of the International Committee on Taxonomy of Viruses.
Genome sequencing and in silico analysis.
Phenol-chloroform extraction was used to isolate the genomic DNA of PBC2 (42), and it was sequenced using the Genome Sequencer FLX system (Titanium serie; Macrogen, Seoul, South Korea). The high-quality filtered reads were assembled using GS De Novo Assembler (v. 2.6) and the Glimmer 3 (43), FgenesB (http://linux1.softberry.com/berry.phtml), and GeneMarkS (44) applications, and the software were used to predict open reading frames (ORFs). The ORFs were limited to those encoding proteins of more than 50 amino acids, and the prediction of ribosomal binding sites (RBS) of each ORF was conducted using RBSfinder (J. Craig Venter Institute, Rockville, MD, USA). The predicted ORFs were annotated based on the results from BLASTP (45), InterProScan (46), and the NCBI conserved domain database (47). tRNAs were predicted by using the tRNAscan-SE program (48). The phylogenetic tree was constructed with MEGA5 by the neighbor-joining method and bootstrap analysis (1,000 replicates) with p-distance values (49). The average nucleotide identity (ANI) was calculated with Kalign (50). Amino acid sequence alignments of the proteins were conducted using ClustalX2 (51). Secondary and tertiary structures of endolysins were predicted using Phyre2 and the ESyPred3D server (52, 53). Visual inspection of protein structural models was performed using PyMol software (54).
Production of recombinant proteins.
All plasmids used in this work are listed in Table 2. The gene fragments encoding endolysin (LysPBC2) and the catalytic domain of the endolysin (LysPBC2_EAD) were cloned into pET15b and pET28a (Novagen), respectively. For the enhanced green fluorescent protein (EGFP)-fused proteins, gene fragments encoding the CBD or SBD were digested with the corresponding restriction enzymes (TaKaRa Clontech, Kyoto, Japan) and subcloned into EGFP-containing pET28a (55). The site-directed mutants of LysPBC2 were generated using overlapping PCR with the primers listed in Table 3. The sequences were verified for all constructs. The cloned plasmid was transformed into E. coli BL21(DE3) (Invitrogen, Carlsbad, CA, USA). The expression and purification of recombinant proteins were performed as previously described with some modifications (41, 55). Freshly transformed cells were grown in LB at 37°C to an optical density at 600 nm (OD600) of 0.6 and induced with isopropyl-β-d-thiogalactoside (IPTG; 1 mM). The induced culture was shaken for 20 h at 18°C, centrifuged, and frozen at −20°C. After thawing, the cells were resuspended in a lysis buffer containing 200 mM NaCl and 50 mM Tris-Cl (pH 8.0) and disrupted by sonication (Sonifier 250; Branson, Danbury, CT, USA). After centrifugation (21,000 × g for 1 h at 4°C), the supernatant was filtered (0.20-μm pore size; Sartorius, Göttingen, Germany), and the soluble protein was purified by immobilized metal affinity chromatography (Poly-Prep chromatography column, catalog no. C731-1550; Bio-Rad, Hercules, CA, USA) using a nickel-nitrilotriacetic acid (Ni-NTA) Superflow column (Qiagen, Valencia, CA, USA). The purity of the proteins was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (see Fig. S8). The purified protein was stored at −80°C after buffer exchange to the storage buffer (50 mM Tris-Cl, 200 mM NaCl, and 30% glycerol; pH 8.0) using PD Miditrap G-25 (GE Healthcare, Waukesha, USA).
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Reference or source |
|---|---|---|
| pET15b | Ampr vector with a T7 promoter | Novagen |
| pET28a | Kanr vector with a T7 promoter | Novagen |
| pEGFP | Ampr egfp cassette vector | Clontech |
| pET28a-EGFP | EGFP gene cloned into NdeI-BamHI site of pET28a | 55 |
| pET15b-LysPBC2 | LysPBC2 gene cloned into NdeI-BamHI site of pET15b | This work |
| pET15b-LysPBC2_K129E | LysPBC2 (K129E) gene cloned into NdeI-BamHI site of pET15b | This work |
| pET15b-LysPBC2_Q132A | LysPBC2 (Q132A) gene cloned into NdeI-BamHI site of pET15b | This work |
| pET15b-LysPBC2_K137E | LysPBC2 (K137E) gene cloned into NdeI-BamHI site of pET15b | This work |
| pET15b-LysPBC2_R142E | LysPBC2 (R142E) gene cloned into NdeI-BamHI site of pET15b | This work |
| pET15b-LysPBC2_R148E | LysPBC2 (R148E) gene cloned into NdeI-BamHI site of pET15b | This work |
| pET28a-LysPBC2_EAD | EAD fragment (Met1-Ser165) of LysPBC2 cloned into NdeI-BamHI site of pET28a | This work |
| pET28a-EGFP-PBC2_CBD | CBD fragment (Gly163-Asp311) of LysPBC2 cloned into BamHI-HindIII site of pET28a-EGFP | This work |
| pET28a-EGFP-PBC2_SBD1 | SBD fragment (Ala108-Thr170) of LysPBC2 cloned into EcoRI-XhoI site of pET28a-EGFP | This work |
| pET28a-EGFP-PBC2_SBD-core | SBD fragment (Arg127-Gly147) of LysPBC2 cloned into EcoRI-XhoI site of pET28a-EGFP | This work |
TABLE 3.
Primers used in this study
| Primer | Sequence (5′→3′) |
|---|---|
| fNde_LysPBC2 | GCGCATATGGCTATTTCAGTAAGACAAAAATTGGTG |
| rBamH_LysPBC2 | GCGGGATCCTTAGTCTCTTACAAAACGAACGAATGAAGAGT |
| rBamH_LysPBC2_EAD | GCGGGATCCTTAGCTTCCTCCACCGTTTAATTCAGCTT |
| fBamH_LysPBC2_CBD | GCGGGATCCGGAGGAAGCACAGGTGGCGGTAC |
| rHind_LysPBC2_CBD | GCGAAGCTTTTAGTCTCTTACAAAACGAACGAATGAAGAGT |
| fEcoR_LysPBC2_SBD1 | GCGGAATTCGCTTTAGCTATCAAGTTCGTTGCACAAC |
| rXho_LysPBC2_SBD1 | GCGCTCGAGTTAAGTACCGCCACCTGTGCTTCCTC |
| fEcoR_LysPBC2_SBD-core | GCGGAATTCAGAGTTAAGAAGCACCAAGATTGGAGCG |
| rXho_LysPBC2_SBD-core | GCGCTCGAGTTATCCTTCATCAAGAATACGGTGTGGGC |
| f_LysPBC2_K129E | GGGGCATTGATAGAGTTGAAAAGCACCAAGATTGGAGCGG |
| r_LysPBC2_K129E | CCGCTCCAATCTTGGTGCTTTTCAACTCTATCAATGCCCC |
| f_LysPBC2_Q132A | GATAGAGTTAAGAAGCACGCGGATTGGAGCGGTAAATACTGC |
| r_LysPBC2_Q132A | GCAGTATTTACCGCTCCAATCCGCGTGCTTCTTAACTCTATC |
| f_LysPBC2_K137E | CAAGATTGGAGCGGTGAATACTGCCCACACC |
| r_LysPBC2_K137E | GGTGTGGGCAGTATTCACCGCTCCAATCTTG |
| f_LysPBC2_R142E | GCGGTAAATACTGCCCACACGAAATTCTTGATGAAGGACG |
| r_LysPBC2_R142E | CGTCCTTCATCAAGAATTTCGTGTGGGCAGTATTTACCGC |
| f_LysPBC2_R148E | CACCGTATTCTTGATGAAGGAGAATGGCAAGCTGTTAAAAATGC |
| r_LysPBC2_R148E | GCATTTTTAACAGCTTGCCATTCTCCTTCATCAAGAATACGGTG |
Lytic activity of endolysin.
The lytic activity of the endolysin was confirmed with a turbidity reduction assay as previously described (41) with some modifications. Exponentially growing bacterial cells were resuspended with the reaction buffer (20 mM Tris-Cl [pH 8.0]) to adjust the OD600 to approximately 1.0. Then, the purified endolysin was added to a final concentration of 0.4 μM, and OD600 values were monitored over time. For the lytic activity of the EADs, an equimolar concentration (0.4 μM) was used to take into account the differences in protein molecular weight. For Gram-negative bacteria, cells were pretreated with a buffer containing 20 mM Tris-Cl (pH 8.0) and 0.1 M EDTA for 5 min at room temperature (RT). Then, the cells were washed three times with reaction buffer to remove residual EDTA, and the endolysin was added. For the viable cell count assay, bacterial cells (B. cereus ATCC 13061) at the mid-exponential growth phase (OD600 of 0.5) were resuspended with the reaction buffer (20 mM Tris-Cl [pH 8.0]) to adjust the OD600 to 1.0 and were further diluted by 100-fold with the reaction buffer. Then, different concentrations of endolysin were added to the cells, which were then incubated for 1 h at 37°C with shaking. After incubation, the cells were harvested by centrifugation (17,000 × g for 1 min), and the supernatants were discarded to remove residual endolysins. The cells were serially diluted before plating them on BHI agar and incubating overnight at 30°C.
Cell binding assay with fluorescence microscopy.
The binding property of the EGFP-CBD fusion protein was examined as previously described (56). Briefly, 1 ml of exponentially grown bacterial cells was centrifuged (16,000 × g for 1 min) and resuspended in 1 ml of phosphate-buffered saline (PBS; GenDEPOT, Barker, NY, USA). Next, 100 μl of cells was incubated together with 0.4 μM GFP-CBD fusion protein at room temperature for 5 min. For Gram-negative strains, EDTA-pretreated cells were incubated with 4 μM GFP-CBD fusion protein for 10 min. The cells were washed twice with PBS and observed by epifluorescence microscopy (DE/Axio Imager A1 microscope; Carl Zeiss, Oberkochen, Germany) with a GFP filter (470/40 nm excitation, 495 nm dichroic, 525/50 nm emission).
Preparation of spores.
B. cereus spores were prepared using Difco sporulation medium (DSM) as described previously (57). In brief, cells were spread on DSM agar and incubated at 37°C for 3 to 5 days. The resulting bacterial lawns were scraped from the plates, resuspended in ice-cold deionized water, and harvested by centrifugation at 16,000 × g for 5 min at 4°C. The spore pellets were washed approximately ten times in ice-cold water by repeated centrifugation. Microscopy examination was conducted to check that the spores were >95% pure. Spores were stored at −20°C until further use. To remove the exosporium, physical and detergent methods were used (26, 27). For the detergent method, PBS-washed spores were subjected to boiling in chemical extraction buffer (62.5 mM Tris-Cl [pH 6.8], 10% glycerol, 2% SDS, 8 M urea, 2% β-mercaptoethanol) for 10 min. Spores were then harvested by centrifugation (21,000 × g for 1.5 min), and the spore pellets were washed two times in PBS. For the physical method, spores were sonicated (Vibra Cell; Sonics & Materials, USA) with an amplitude of 20% and 120 pulse cycles on ice (5 s on/9 s off). Then, the spores were pelleted by centrifugation (9,000 × g for 5 min) and washed two times with PBS.
Spore binding assays with SBD.
Spores in PBS solution were reacted with 1 µM of EGFP-tagged SBD (spore binding domain) protein at RT for 5 min. After washing three times with PBS by centrifugation (16, 000 × g for 1 min), the labeled spores were analyzed by either fluorescence microscopy (DE/Axio Imager A1 microscope; Carl Zeiss, Oberkochen, Germany) or a SpectraMax i3 multimode microplate reader (Molecular Devices, Sunnyvale, CA, USA) with excitation at 485 nm and emission at 535 nm. Statistical analysis was performed with GraphPad Prism version 5.01. Statistical significance was calculated with one-way analysis of variance (ANOVA) and Tukey’s post hoc test.
Electron microscopy.
Spore sections were prepared and viewed as previously described (26) with some modifications. Spores were fixed for 1 h at 37°C in 1 ml of 2% glutaraldehyde and 0.1 M sodium cacodylate buffer (pH 7.4) containing 0.1% ruthenium red (Electron Microscopy Sciences, Fort Washington, PA, USA). Spore pellets were then washed in 0.1 M sodium cacodylate buffer (pH 7.4) and postfixed for 3 h at room temperature in 1% osmium tetroxide (Electron Microscopy Sciences) in 0.1 M sodium cacodylate solution containing 0.1% ruthenium red. Spores were then washed twice in distilled water and further stained with 0.5% uranyl acetate for 30 min at 4°C. The spores were dehydrated in a graded ethanol series, including 30%, 50%, 70%, 90%, and 100% ethanol (three times), which was followed by two 15-min rinses in propylene oxide. The spores were infiltrated for 2 h in a 1:1 mixture of Spurr’s low-viscosity resin (Electron Microscopy Sciences) and propylene oxide and then overnight in 100% Spurr’s resin. The spores were infiltrated for 2 h in fresh Spurr’s resin and embedded again in fresh resin. This suspension was incubated at 70°C for 24 h to embed the spores in polymerized resin. Sections were cut at a 100-nm thickness (Ultramicrotome MT-X; RMC, Tucson, AZ, USA), placed on copper grids, and stained with 2% uranyl acetate for 7 min. The sections were then treated with Reynolds’ lead citrate (58) for 7 min and examined by TEM (JEM1010; JEOL, Japan) at 80 kV.
For immunogold electron microscopy (IEM), spores were reacted with 1 µM EGFP-SBD at 37°C for 30 min and washed three times with PBS. Spores were then fixed in a 2% glutaraldehyde and 2% paraformaldehyde PBS solution overnight at 4°C and postfixed for 1 h in 1% osmium tetroxide. After two 10-min washes in PBS, the spores were dehydrated in 70%, 80%, 90%, 95%, and (twice) 100% ethanol and infiltrated with and embedded in Embed 812 resin (Electron Microscopy Sciences). This suspension was incubated at 55°C for 48 h to embed the spores in polymerized resin. Ultrathin sections (70 nm) were collected on Formvar/carbon-coated nickel grids (Electron Microscopy Sciences) and treated with freshly prepared 4% sodium metaperiodate for 30 min. The sections were then blocked in TBST buffer (50 mM Tris-Cl [pH 7.5], 200 mM NaCl, 0.1% Triton X-100) containing 2% bovine serum albumin (BSA) for 30 min. The grids were incubated for 3 h in TBST buffer containing rabbit anti-GFP antibody (30 μg/ml, Abcam), washed six times with TBST, and blocked for 15 min in TBST containing 2% BSA. The grids were then incubated with 10-nm gold-conjugated goat anti-rabbit IgG H+L (10 μg/ml; Abcam) for 90 min. After a series of washes in TBST and distilled water, the sections were briefly stained with uranyl acetate and lead citrate before examination with a JSM-1200EX II (JEOL) microscope.
Thermal inactivation of enzymes.
The thermal inactivation assay was performed as previously described (59) with slight modifications. Briefly, the reaction buffer (50 mM Tris-Cl [pH 8.0], 200 mM NaCl) was equilibrated on a thermal cycler (C1000 Touch; Bio-Rad, Hercules, CA, USA) at temperatures ranging from 10°C to 80°C. Endolysin samples were then added to the heated buffers and incubated for an additional 10 min. After heat treatment, the enzymes were chilled on ice and aliquoted into wells of a 96-well microplate containing B. cereus ATCC 13061 cells, resulting in final LysPBC2 and B. cereus concentrations of 2 µg/ml and OD600 of 1.0, respectively. The turbidity reduction was measured every 5 min for 1 h at 37°C using a microplate reader (SpectraMax i3; Molecular Devices, Sunnyvale, CA, USA). Endolysin activity is presented as the maximum velocity (12.5 min) corresponding to the linear portion of the reduction curve. All data are expressed as the means ± standard deviations (SDs) from triplicate experiments and normalized to the lytic activity displayed by the 20°C heat treatment group. Description and statistical analysis of each curve were performed with GraphPad Prism version 5.01.
Salt sensitivity test.
B. cereus ATCC 13061 cells were resuspended in the reaction buffers (20 mM Tris-Cl [pH 8.0]) containing different NaCl concentrations ranging from 0 mM to 200 mM. Then, fresh enzymes (wild type [WT] and K129E) were added to each sample to achieve a final concentration of 2 µg/ml. Using a microplate reader, the turbidity was measured every 5 min for 1 h at 37°C.
Accession number(s).
The GenBank accession numbers of PBC2 and LysPBC2 are KT070867 and AKQ08512, respectively.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Basic Science Research Programs (NRF-2017R1A2A1A17069378) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning, and Korea Institute of Planning, and by IPET through the Agriculture, Food and Rural Affairs Research Center Support Program, funded by the Ministry of Agriculture, Food and Rural Affairs (MAFRA) (710012-03-1-SB110). This work was also supported by the BK21 Plus Program of the Department of Agricultural Biotechnology, Seoul National University, Seoul, South Korea.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02462-18.
REFERENCES
- 1.Guinebretiere MH, Auger S, Galleron N, Contzen M, De Sarrau B, De Buyser ML, Lamberet G, Fagerlund A, Granum PE, Lereclus D, De Vos P, Nguyen-The C, Sorokin A. 2013. Bacillus cytotoxicus sp. nov. is a novel thermotolerant species of the Bacillus cereus group occasionally associated with food poisoning. Int J Syst Evol Microbiol 63:31–40. doi: 10.1099/ijs.0.030627-0. [DOI] [PubMed] [Google Scholar]
- 2.Nicholson WL, Munakata N, Horneck G, Melosh HJ, Setlow P. 2000. Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol Mol Biol Rev 64:548–572. doi: 10.1128/MMBR.64.3.548-572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imamura D, Kuwana R, Takamatsu H, Watabe K. 2011. Proteins involved in formation of the outermost layer of Bacillus subtilis spores. J Bacteriol 193:4075–4080. doi: 10.1128/JB.05310-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fazzini MM, Schuch R, Fischetti VA. 2010. A novel spore protein, ExsM, regulates formation of the exosporium in Bacillus cereus and Bacillus anthracis and affects spore size and shape. J Bacteriol 192:4012–4021. doi: 10.1128/JB.00197-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bottone EJ. 2010. Bacillus cereus, a volatile human pathogen. Clin Microbiol Rev 23:382–398. doi: 10.1128/CMR.00073-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khera M, Pathengay A, Jindal A, Jalali S, Mathai A, Pappuru RR, Relhan N, Das T, Sharma S, Flynn HW. 2013. Vancomycin-resistant Gram-positive bacterial endophthalmitis: epidemiology, treatment options, and outcomes. J Ophthalmic Inflamm Infect 3:46. doi: 10.1186/1869-5760-3-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frenzel E, Kranzler M, Stark TD, Hofmann T, Ehling-Schulz M. 2015. The endospore-forming pathogen Bacillus cereus exploits a small colony variant-based diversification strategy in response to aminoglycoside exposure. mBio 6:e01172-15. doi: 10.1128/mBio.01172-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Citron DM, Appleman MD. 2006. In vitro activities of daptomycin, ciprofloxacin, and other antimicrobial agents against the cells and spores of clinical isolates of Bacillus species. J Clin Microbiol 44:3814–3818. doi: 10.1128/JCM.00881-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simm R, Vörös A, Ekman JV, Sødring M, Nes I, Kroeger JK, Saidijam M, Bettaney KE, Henderson PJF, Salkinoja-Salonen M, Kolstø A-B. 2012. BC4707 is a major facilitator superfamily multidrug resistance transport protein from Bacillus cereus implicated in fluoroquinolone tolerance. PLoS One 7:e36720. doi: 10.1371/journal.pone.0036720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischetti VA. 2008. Bacteriophage lysins as effective antibacterials. Curr Opin Microbiol 11:393–400. doi: 10.1016/j.mib.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fenton M, Ross P, McAuliffe O, O'Mahony J, Coffey A. 2010. Recombinant bacteriophage lysins as antibacterials. Bioeng Bugs 1:9–16. doi: 10.4161/bbug.1.1.9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loessner MJ. 2005. Bacteriophage endolysins–current state of research and applications. Curr Opin Microbiol 8:480–487. doi: 10.1016/j.mib.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Son B, Kong M, Ryu S. 2018. The auxiliary role of the amidase domain in cell wall binding and exolytic activity of staphylococcal phage endolysins. Viruses 10:E284. doi: 10.3390/v10060284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oliveira H, Melo LD, Santos SB, Nobrega FL, Ferreira EC, Cerca N, Azeredo J, Kluskens LD. 2013. Molecular aspects and comparative genomics of bacteriophage endolysins. J Virol 87:4558–4570. doi: 10.1128/JVI.03277-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerstmans H, Criel B, Briers Y. 2018. Synthetic biology of modular endolysins. Biotechnol Adv 36:624–640. doi: 10.1016/j.biotechadv.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 16.Sao-Jose C. 2018. Engineering of phage-derived lytic enzymes: improving their potential as antimicrobials. Antibiotics (Basel) 7:E29. doi: 10.3390/antibiotics7020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganz HH, Law C, Schmuki M, Eichenseher F, Calendar R, Loessner MJ, Getz WM, Korlach J, Beyer W, Klumpp J. 2014. Novel giant siphovirus from Bacillus anthracis features unusual genome characteristics. PLoS One 9:e85972. doi: 10.1371/journal.pone.0085972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Low LY, Yang C, Perego M, Osterman A, Liddington RC. 2005. Structure and lytic activity of a Bacillus anthracis prophage endolysin. J Biol Chem 280:35433–35439. doi: 10.1074/jbc.M502723200. [DOI] [PubMed] [Google Scholar]
- 19.Schuch R, Nelson D, Fischetti VA. 2002. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 418:884–889. doi: 10.1038/nature01026. [DOI] [PubMed] [Google Scholar]
- 20.Loessner MJ, Maier SK, Daubek-Puza H, Wendlinger G, Scherer S. 1997. Three Bacillus cereus bacteriophage endolysins are unrelated but reveal high homology to cell wall hydrolases from different bacilli. J Bacteriol 179:2845–2851. doi: 10.1128/jb.179.9.2845-2851.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park J, Yun J, Lim JA, Kang DH, Ryu S. 2012. Characterization of an endolysin, LysBPS13, from a Bacillus cereus bacteriophage. FEMS Microbiol Lett 332:76–83. doi: 10.1111/j.1574-6968.2012.02578.x. [DOI] [PubMed] [Google Scholar]
- 22.Kikkawa HS, Ueda T, Suzuki S, Yasuda J. 2008. Characterization of the catalytic activity of the gamma-phage lysin, PlyG, specific for Bacillus anthracis. FEMS Microbiol Lett 286:236–240. doi: 10.1111/j.1574-6968.2008.01280.x. [DOI] [PubMed] [Google Scholar]
- 23.Dziarski R, Gupta D. 2006. The peptidoglycan recognition proteins (PGRPs). Genome Biol 7:232. doi: 10.1186/gb-2006-7-8-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schleifer KH, Kandler O. 1972. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 36:407–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Wang DB, Dong Q, Zhang Z, Cui Z, Deng J, Yu J, Zhang XE, Wei H. 2012. Existence of separate domains in lysin PlyG for recognizing Bacillus anthracis spores and vegetative cells. Antimicrob Agents Chemother 56:5031–5039. doi: 10.1128/AAC.00891-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson BM, Binkley JM, Stewart GC. 2011. Current physical and SDS extraction methods do not efficiently remove exosporium proteins from Bacillus anthracis spores. J Microbiol Methods 85:143–148. doi: 10.1016/j.mimet.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang DB, Yang R, Zhang ZP, Bi LJ, You XY, Wei HP, Zhou YF, Yu Z, Zhang XE. 2009. Detection of B. anthracis spores and vegetative cells with the same monoclonal antibodies. PLoS One 4:e7810. doi: 10.1371/journal.pone.0007810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Logan NA. 2012. Bacillus and relatives in foodborne illness. J Appl Microbiol 112:417–429. doi: 10.1111/j.1365-2672.2011.05204.x. [DOI] [PubMed] [Google Scholar]
- 29.Celandroni F, Salvetti S, Gueye SA, Mazzantini D, Lupetti A, Senesi S, Ghelardi E. 2016. Identification and pathogenic potential of clinical Bacillus and Paenibacillus isolates. PLoS One 11:e0152831. doi: 10.1371/journal.pone.0152831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salkinoja-Salonen MS, Vuorio R, Andersson MA, Kampfer P, Andersson MC, Honkanen-Buzalski T, Scoging AC. 1999. Toxigenic strains of Bacillus licheniformis related to food poisoning. Appl Environ Microbiol 65:4637–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Low LY, Yang C, Perego M, Osterman A, Liddington R. 2011. Role of net charge on catalytic domain and influence of cell wall binding domain on bactericidal activity, specificity, and host range of phage lysins. J Biol Chem 286:34391–34403. doi: 10.1074/jbc.M111.244160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart GC. 2015. The exosporium layer of bacterial spores: a connection to the environment and the infected host. Microbiol Mol Biol Rev 79:437–457. doi: 10.1128/MMBR.00050-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kailas L, Terry C, Abbott N, Taylor R, Mullin N, Tzokov SB, Todd SJ, Wallace BA, Hobbs JK, Moir A, Bullough PA. 2011. Surface architecture of endospores of the Bacillus cereus/anthracis/thuringiensis family at the subnanometer scale. Proc Natl Acad Sci U S A 108:16014–16019. doi: 10.1073/pnas.1109419108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ball DA, Taylor R, Todd SJ, Redmond C, Couture-Tosi E, Sylvestre P, Moir A, Bullough PA. 2008. Structure of the exosporium and sublayers of spores of the Bacillus cereus family revealed by electron crystallography. Mol Microbiol 68:947–958. doi: 10.1111/j.1365-2958.2008.06206.x. [DOI] [PubMed] [Google Scholar]
- 35.Plomp M, Leighton TJ, Wheeler KE, Malkin AJ. 2005. Architecture and high-resolution structure of Bacillus thuringiensis and Bacillus cereus spore coat surfaces. Langmuir 21:7892–7898. doi: 10.1021/la050412r. [DOI] [PubMed] [Google Scholar]
- 36.Hendrix RW. 2002. Bacteriophages: evolution of the majority. Theor Popul Biol 61:471–480. doi: 10.1006/tpbi.2002.1590. [DOI] [PubMed] [Google Scholar]
- 37.Moriyama R, Fukuoka H, Miyata S, Kudoh S, Hattori A, Kozuka S, Yasuda Y, Tochikubo K, Makino S. 1999. Expression of a germination-specific amidase, SleB, of bacilli in the forespore compartment of sporulating cells and its localization on the exterior side of the cortex in dormant spores. J Bacteriol 181:2373–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bagyan I, Setlow P. 2002. Localization of the cortex lytic enzyme CwlJ in spores of Bacillus subtilis. J Bacteriol 184:1219–1224. doi: 10.1128/jb.184.4.1219-1224.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chirakkal H, O’Rourke M, Atrih A, Foster SJ, Moir A. 2002. Analysis of spore cortex lytic enzymes and related proteins in Bacillus subtilis endospore germination. Microbiology 148:2383–2392. doi: 10.1099/00221287-148-8-2383. [DOI] [PubMed] [Google Scholar]
- 40.Gómez-Torres N, Dunne M, Garde S, Meijers R, Narbad A, Ávila M, Mayer MJ. 2018. Development of a specific fluorescent phage endolysin for in situ detection of Clostridium species associated with cheese spoilage. Microb Biotechnol 11:332–345. doi: 10.1111/1751-7915.12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong M, Ryu S. 2015. Bacteriophage PBC1 and its endolysin as an antimicrobial agent against Bacillus cereus. Appl Environ Microbiol 81:2274–2283. doi: 10.1128/AEM.03485-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 43.Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 46.Zdobnov EM, Apweiler R. 2001. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 47.Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, Hao L, He S, Hurwitz DI, Jackson JD, Ke Z, Krylov D, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Thanki N, Yamashita RA, Yin JJ, Zhang D, Bryant SH. 2007. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res 35:D237–D240. doi: 10.1093/nar/gkl951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar S, Nei M, Dudley J, Tamura K. 2008. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 9:299–306. doi: 10.1093/bib/bbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lassmann T, Sonnhammer EL. 2005. Kalign–an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics 6:298. doi: 10.1186/1471-2105-6-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 52.Lambert C, Leonard N, De Bolle X, Depiereux E. 2002. ESyPred3D: prediction of proteins 3D structures. Bioinformatics 18:1250–1256. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 53.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 54.DeLano WL. 2005. The case for open-source software in drug discovery. Drug Discov Today 10:213–217. doi: 10.1016/S1359-6446(04)03363-X. [DOI] [PubMed] [Google Scholar]
- 55.Kong M, Sim J, Kang T, Nguyen HH, Park HK, Chung BH, Ryu S. 2015. A novel and highly specific phage endolysin cell wall binding domain for detection of Bacillus cereus. Eur Biophys J 44:437–446. doi: 10.1007/s00249-015-1044-7. [DOI] [PubMed] [Google Scholar]
- 56.Kong M, Shin JH, Heu S, Park JK, Ryu S. 2017. Lateral flow assay-based bacterial detection using engineered cell wall binding domains of a phage endolysin. Biosens Bioelectron 96:173–177. doi: 10.1016/j.bios.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 57.Harwood CR, Cutting SM. 1990. Molecular biological methods for Bacillus. John Wiley & Sons Ltd., Chichester, United Kingdom. [Google Scholar]
- 58.Reynolds ES. 1963. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol 17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heselpoth RD, Owens JM, Nelson DC. 2015. Quantitative analysis of the thermal stability of the gamma phage endolysin PlyG: a biophysical and kinetic approach to assaying therapeutic potential. Virology 477:125–132. doi: 10.1016/j.virol.2014.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.