

Abstract
Background/Aims:
Timely review of research protocols by Institutional Review Boards leads to more rapid initiation of clinical trials, which is critical to expeditious translation from bench to bedside. This observational study examined the impact of a single Institutional Review Board (sIRB) on time and efforts required to initiate clinical trials by the National Institute of Child Health and Human Development Cooperative Reproductive Medicine Network.
Methods:
Collection of data from the same six main clinical sites for three current clinical trials and two past clinical trials, including time from IRB submission to approval, pages submitted, consent form length, number of required attachments, other regulatory requirements, order of review at central or local sites, and language in documents at individual participating sites. Results from two past clinical trials were also included.
Results:
While time required for actual IRB submission’s review and initial approval was reduced with use of a sIRB for multicenter trials (from a mean of 66.7 days to 24.0 days), total time was increased (to a mean of 111.2 or 123.3 days). In addition to sIRB approval, all institutions required local approval of some components (commonly consent language and use of local language), which varied considerably. The sIRB relied on local institutions for adding or removing personnel, conflict of interest review, and auditing of activities.
Conclusions:
A sIRB reduced time for initial review and approval of protocols and informed consents, although time for the entire process was increased, as individual institutions retained oversight of components of required regulatory review. In order to best achieve the National Institute of Health’s goals for improved efficiency in initiation and conduct of multisite clinical research, greater coordination with local IRBs is key to streamlining and accelerating initiation of multisite clinical research.
Keywords: Institutional review board, multi-center studies, single IRB
Introduction
In June 2016, the National Institute of Health (NIH) provided guidance directing single Institutional Review Board (IRB) review of NIH multicenter studies for United States/domestic institutions. Specifically, NIH sought to improve efficiency of IRB review of multicenter studies, reduce time needed for initiation of studies, allow for consistency of ethical review, and reduce regulatory burden on investigators1 (and staff) and administrators, thereby reducing cost of research. This was to be accomplished by converting the former prevailing approach for multicenter studies where each participating institution would have IRB review done at that institution, to a model where a single IRB would serve as the IRB of record for all participating institutions. This concept was based on prior guidance2, 3 and widespread recognition of the considerable time and effort required to initiate human subject research. Of note, while previously the term “Central IRB” was used to describe models of streamlined review for multiple clinical sites conducting the same trial, the term “Single IRB” (sIRB) is now more commonly used, and will be the term employed throughout this article.
Consumption of resources is compounded in multicenter clinical research studies by redundancy of review, namely, same regulatory preparation and institutional review is required at each participating site. To address this compounding of time and effort, the National Cancer Institute (NCI) established a Central Institutional Review Board that could provide IRB regulatory oversight for clinical sites (and institutions) willing to cede oversight to the NCI Central Institutional Review Boards.4 Such an approach may also reduce variability of decisions of IRBs at different institutions and improve consistency in informed consent documents.5–10 However, there has been hesitancy by many medical schools and medical centers/hospitals to use sIRBs.2, 11, 12 Stated concerns included adequacy of review, uncertainty that sIRB would recognize and/or address local considerations, concern with expertise of sIRB members, and concern for local institutional liability following central review.13 Nonetheless, the Office of Human Research Protection provided guidance supportive of use of sIRBs, and now many national entities encourage or recommend use of sIRB, including the NIH14–16 (which made use of a single IRB mandatory for NIH funded multisite trials beginning January, 2018).
While use of a sIRB addresses one of the barriers to prompt study initiation, regulatory approval required to initiate clinical trials is not limited to IRB approval. Other university/medical center/hospital requirements include determination of qualifications of study team members, oversight, reporting and management of conflicts of interest of investigators and coordinators, review of biosafety, radiation safety, and chemical safety issues, data storage, cost coverage analysis, financial review, as well as review of plans for privacy protection and Health Insurance Portability and Accountability Act compliance. Additionally, institutional compliance officers, privacy officers, and legal offices may provide guidance for specific required wording for multiple sections of the informed consent document, which may vary by institution and/or by State. Surprisingly, there has been little consideration of consistency of these additional requirements, and specifically how these obligatory items are addressed in multicenter protocols that use a single IRB. Implementation of processes to promote use of sIRBs for multicenter trials have not routinely addressed responsibility for these additional areas of regulatory oversight of human subject research, specifically whether it should be the purview of the sIRB or local institutional IRBs. Consideration is also lacking for the sequence of review for items for which local institutions are unwilling to relinquish review, and for which the sIRB is not providing review and oversight. This lack of coordination could ultimately increase time required to study initiation.
To address these issues, we consider the time required to achieve IRB approval for the National Institute of Child Health and Human Development Cooperative Reproductive Medicine Network studies when reviewed by local or sIRBs. To our knowledge this was the first sIRB for a National Institute of Child Health and Human Development trial/network, and thus provides an important touchstone for future such endeavors. In this report, first, we sought to examine the time required for local IRB approval of studies for which a sIRB would be the IRB of record. Second, we sought to determine the variability in time required for local institution initiation of clinical trials, including determination of which areas of management the local site would relinquish (and central site would accept) with regards to conduct of human subject research. Third, we sought to compare length of time required for IRB approval to the Reproductive Medicine Network’s prior report examining IRB approval that utilized local IRBs,17 and to each other.
Methods
Regulatory approval characteristics were collected from principal participating institutions of the Reproductive Medicine Network for three ongoing randomized clinical trials: 1) Optimal Treatment for Women with a Persisting Pregnancy of Unknown Location – Randomized Clinical Trial of Women at Risk for an Ectopic Pregnancy: Active Treatment versus Expectant Management (No Treatment) (ACTorNOT; NCT02152696); 2) Improving Reproductive Fitness through Pretreatment with Lifestyle Modification in Obese Women with Unexplained Infertility (FIT-PLESE; NCT02432209); and 3) Males, Antioxidants, and Infertility (MOXI; NCT02421887) Trial. Data collection was initiated in August 2016 with clarifications through June 2017. The principal participating institutions represented six of sixteen clinical sites for the ACTorNOT protocol, six of 10 for FIT-PLESE, and six of 10 for MOXI. The additional sites joined the protocols after initiation of the study, and are not included in this analysis. The additional sites used a combination of the sIRB and local IRBs (if mandated by their local institution). Metrics for these studies were compared to prior Reproductive Medicine Network studies, which were included in an earlier study of the IRB review process,17 for the Pregnancy in Polycystic Ovary Syndrome (PPCOS) I and II studies. These metrics included the time from submission to approval, the total number of pages in the IRB submission, the number of pages in the consent form, and the number of attachments.
For MOXI and FIT-PLESE studies, the IRB of a single participating institution (University of Pennsylvania) was the sIRB of record for all participating primary Reproductive Medicine Network clinical sites. Each clinical site had to meet the regulatory requirements of the sIRB, as well as the additional requirements of their own institution. The ACTorNOT protocol was reviewed by IRBs at each individual institution. For all protocols, multiple characteristics regarding the regulatory approval process were collected. These included total length of time from IRB submission to approval, number of pages of the submission and consent, and time required for other institutional approval processes such as Biosafety review, Health Insurance Portability and Accountability Act and Privacy Board Review, and conflict of interest disclosures. Additionally, for protocols reviewed by the sIRB, each clinical site was asked to identify additional local institutional requirements for each of these components, as well as whether the local institution required changes in wording of the consent to meet local requirements. This included specification of which sections of the consent form required local review/alterations. Total time from initial submission to the sIRB to final approval at each clinical (relying) site included 1) length of time for initial sIRB review, 2) time for submission to sIRB for relying site, 3) time for submission to the local IRB, 4) time for transmission of approved protocol and consent from the sIRB to the clinical site, 5) revision of documents by each clinical site for sIRB submission, and 6) time waiting to submit to the sIRB. The latter was necessary because the sIRB would only allow one amendment at a time to be undergoing review (including addition of additional relying sites). The above data were collected from six main clinical sites, all of which participated in all of the three current studies. For information purposes, results from two previous studies conducted in the Reproductive Medicine Network were also included.17 The full name for these two previous trials are: Treatment of Infertility in Women with Polycystic Ovary Syndrome (PPCOS I); Pregnancy in Polycystic Ovary Syndrome II (PPCOS II).
As with the Reproductive Medicine Network’s prior IRB-related study,17 calculations of page lengths did not include the page numbers of the actual Reproductive Medicine Network protocol, case report forms, or investigative drug brochures. Items included in page counts were institutionally required protocol summary form, consent form (with Health Insurance Portability and Accountability Act and Privacy verbiage), attachments, disclosures, advertisements, and any other IRB forms requested at time of initial IRB review.
Statistical analysis
Statistical analysis was performed comparing the protocol reviewed solely by each clinical site’s institution’s IRB with protocols reviewed by the sIRB (FIT-PLESE or MOXI) utilizing the paired Student’s T-test. The main outcome for comparison is the mean IRB approval time. Other items for comparison include total number of pages of IRB submission, length of the consent forms, and numbers of attachments required. Data are expressed as mean + SD. Statistical significance was defined as p<0.05.
Results
The time to select and establish a sIRB that would be utilized by the Reproductive Medicine Network was nine months. Once the sIRB was established, the duration of time for all agreements to be signed with clinical site institutions was one to six months. Length of time from IRB submission to approval at individual local IRBs for the ACTorNOT protocol was 66.7+32.7 days (Table 1). In comparison, time duration from IRB submission to initial approval at the sIRB for FIT-PLESE and MOXI protocols was 24 calendar days each. In addition to time for initial sIRB approval, time required for local site submissions to the sIRB and clinical site IRB and the intervening time to the clinical site’s institutional approval was 111.2 + 22.2 days for FIT-PLESE (range 82 to 137 days, p=0.007 when compared to that for ACTorNOT) (Table 1) with two sites requiring more than one submission to the clinical site’s IRB. For the MOXI protocol, total time till clinical site IRB approval was 123.3 + 41.9 days (range 82 to 193 days, p=0.031 when compared to that for ACTorNOT) (Table 1) with three of the sites requiring more than one local clinical site submission. The total number of pages for two protocols that underwent sIRB review (and involved administration of either medications or nutraceuticals to promote live births), MOXI was within the range of reasonable uncertainty (NS), while being slightly less for FIT-PLESE (p=0.038) compared to number of pages for the protocol (ACTorNOT) which underwent solely local review. Of note, the number of pages of consent forms for FIT-PLESE (p<0.001) and MOXI (p=0.011) were longer when compared to that for ACTorNOT study. No significant difference was found in the number of attachments required for submission to local clinical site IRBs for ACTorNOT when compared to that for submission to sIRB for FIT-PLESE (p=0.50) or MOX (p=0.40) study (Table 1).
Table 1.
Time from IRB submission to approval and characterization of IRB submissions from RMN institutions relying on a cIRB.
| Protocol | n | Submission to Approval (days) | IRB Submission (total pages)a | Consent Form (pages) | Number of Attachments | |||
|---|---|---|---|---|---|---|---|---|
| Initial IRB Review |
Subsequent Central IRBc |
Local IRBd | Total Dayse | |||||
| ACT or NOT | 6 | 66.7±32.7 | 67.5±27.0 | 10.2±02.0 | 07.6±07.3 | |||
| FIT-PLESE | 6 | 24 | 6.0±1.8 | 11.8±10.1 | 111.2±22.2f | 48.8±16.6g | 24.2±05.0f | 08.3±07.3 |
| MOXI | 6 | 24 | 8.2±5.6 | 11.7±12.9 | 123.3±41.9g | 44.8±07.4 | 15.2±01.5g | 09.2±06.9 |
| PPCOS Ib | 4 | 67.0±09.9 | 40.8±22.4 | 11.3±07.4 | 03.5±04.4 | |||
| PPCOS IIb | 7 | 81.7±50.6 | 96.4±44.2 | 24.3±04.5 | 11.6±06.4 | |||
– Excludes the RMN study protocol, Case Report Forms, and any Investigational Drug Brochure
– From the RMNs prior report on IRBs, Ref 18
– Days for approval of individual relying sites at central IRB d
– Days for local approval
– Days from initial central IRB submission to final IRB approval for local site, including days for between submissions including report generation, dissemination of IRB determinations, incorporation of any modifications, and time until submission (such as review of prior submission related to other items).
– p<0.01 when compared to that for ACTorNOT
– p<0.05 when compared to that for ACTorNOT
For institutions that relied on another institution as the sIRB, variation was observed as to specific topics required for review by the local IRB in addition to sIRB approval. Each of the clinical site institutions required local review of the protocol (Table 2), with four allowing central review initially and two requiring clinical site review first. In addition, the clinical site’s institutions also very commonly required clinical site review (either initially or after central review, n=3) for protocol and consent. Additionally, there was considerable variability in the requirements for sIRB and clinical site review of the subject injury language, subject payment language, Health Insurance Portability and Accountability Act language, adding or removing personnel, and study advertising (whether by print, radio-TV, and internet) (Table 2). The sIRB did not review qualifications of the clinical site institutions investigators and coordinators participating in the trial, addition or removal of study personnel, or conflicts of interest of study personnel.
Table 2.
Requirements for local approval and order of approval for IRB protocols and consents
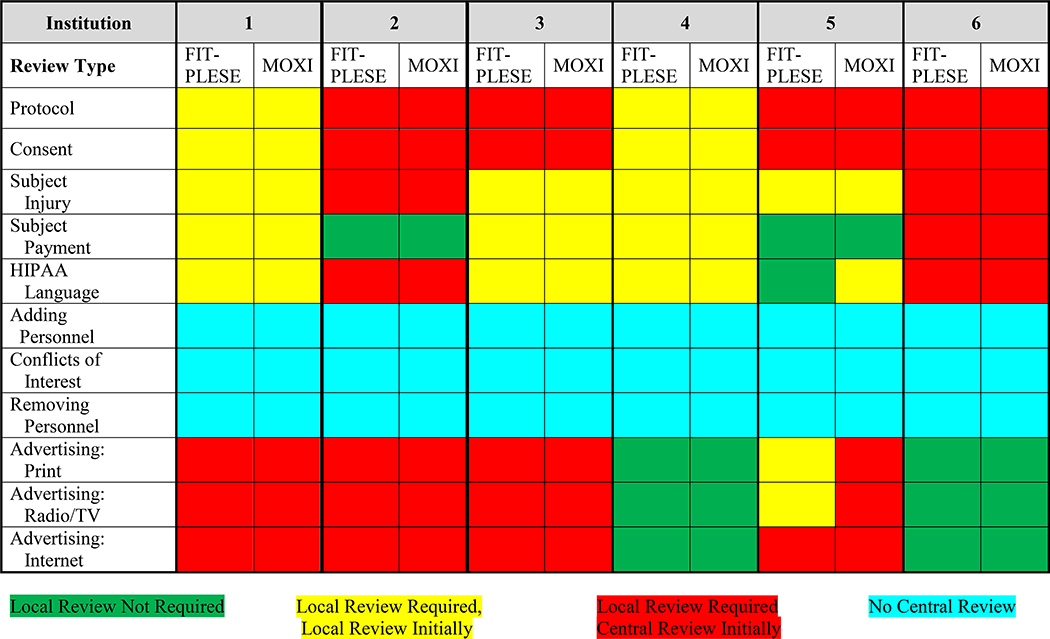 |
Variation among the clinical site institutions also existed for local consent language content related to subject injury, indemnification, Health Insurance Portability and Accountability Act, privacy, and subject compensation, both as to whether it was required, as well as timing of clinical site review (Table 3). Finally, variation was observed regarding whether additional clinical site institutional reviews were required for approval in addition to sIRB approval before study initiation for items including HIPPA, biosafety, radiation safety, chemical safety, and medical center/hospital review (Table 4). Considerable variation also existed in time for reviews and approvals for protocol amendments by both single and clinical site IRBs. In part, this duration was related to timing of initial approval to subsequent submission, review, and approval. Of note, individual sites frequently required additional time for local IRB amendment submissions.
Table 3.
Local Institution Requirements for Use of Local Language
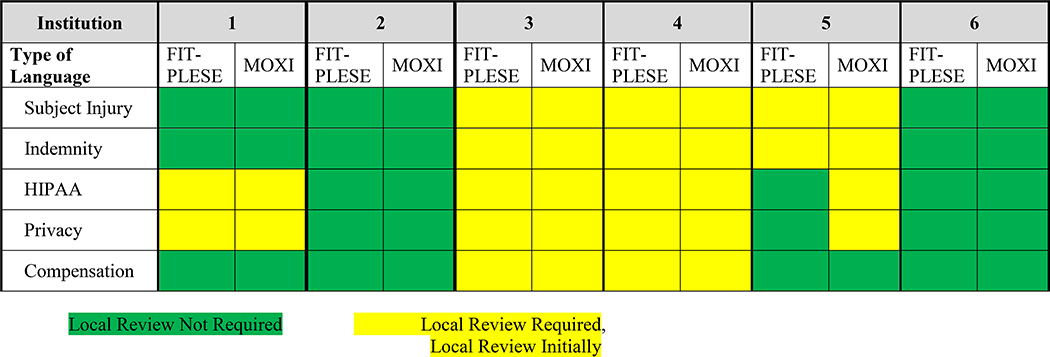 |
Table 4.
Local Approvals Required in Addition to cIRB Approval
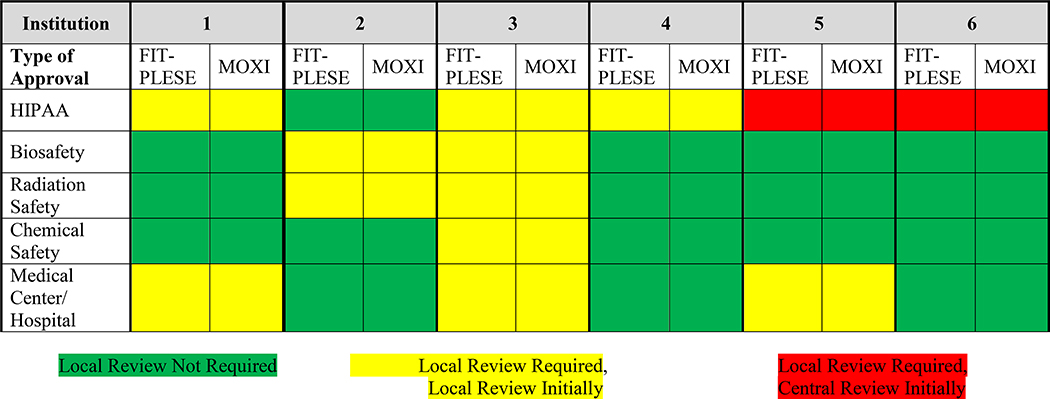 |
Discussion
Rights and welfare of human subjects participating in clinical trials is of paramount importance! Also important is timeliness of study initiation and conduct, so as to hasten adoption of new medications, devices, and therapeutic approaches to improve human health. Time to study initiation impacts expenses incurred for study start up, including time required for review by IRB staff, IRB members, as well as the effort of study team staff who prepare regulatory and study initiation paperwork.
Prior reports have identified great variability in requests arising from IRB protocol review.5–10 These evaluations can include requests for clarification of, or changes to, the protocol, additional safety testing, alterations in patient population included, changes in inclusion or exclusion criteria, and alteration of wording of consent form. For multicenter protocols, variation in requests from different local IRBs typically results in further delays while differences are adjudicated through the coordinating center. At an extreme, such variability in requests has the potential to result in variations in the conduct of the study, making it difficult to assure compatibility of data collected across all study sites. Consequently, a challenge has been posed that multiple IRB reviews may be counterproductive by increasing burdensomeness18 or detracting from the likelihood of maintaining ethical standards,19 in addition to the generally recognized issue of redundancy.
Thus use of a central (or single) IRB for a multicenter clinical trial could theoretically provide many potential advantages. In our experience comparing our prior reports (Table 1) and current studies reviewed by local and sIRBs, use of sIRBs resulted in a reduction in time for initial IRB review, but a total increase in time to initiation of study because of need for sequential review by local and sIRBs, as was required by each institution.
Importantly, there was considerable variation in local institution requirements to rely upon the sIRB, with local sites continuing to require submission of protocol and consent for review, as well as review for subject injury, subject payment, Health Insurance Portability and Accountability Act language, and advertising. Local sites were responsible for adding or removing personnel, and conflict of interest review. Additionally, several institutions required local review by the medical center/hospital and by Biosafety, Radiation Safety, and/or Chemical Safety committees. Distribution of which of these items was required is depicted in Tables 2, 3, and 4.
Of particular interest, while most institutions allowed use of sIRB for Health Insurance Portability and Accountability Act and Privacy Board oversight, additional local input was still required for many components of human subject research initiation before local institutional “release” of the study for initiation at that site. Specifically, many institutions required use of local institutional Health Insurance Portability and Accountability Act language be incorporated into the document, thus requiring alteration to consent form language initially approved by sIRB. This was also the case for subject injury language and indemnity clauses. In these situations, approval of amendments containing these modifications was required by the sIRB, further prolonging time to study initiation at that clinical site. Of note, because of logistics of submission of amendments to the sIRB with their requirement of a limit of one amendment at a time, an initial amendment from one institution delayed amendments from other sites.
Moreover, delays occurred while compiling needed modifications from all sites, so that all could be submitted simultaneously for sIRB review. It was also the case that when Continuing Review of the protocol was due, all amendments requested by sites, including recruitment materials, were delayed until after approval had been given for Continuing Review.
Up till now, despite purported advantages of use of a sIRB, acceptance of use of sIRBs remained limited.20–22 In part, this initial hesitancy to accept sIRBs may have reflected concern for institutional exposure as related to Federal human subject research oversight bodies. This concern was considerably ameliorated following statements by the US Office of Human Research Protection, the Food and Drug Administration, and the Department of Health and Human Services supporting use of sIRBs.3, 23, 24 In part, this may be achievable by generalized use of IRB Reliance Agreements such as the one recently developed by the National Center for Advancing Translational Science.25 Acceptance of use of sIRBs has been further promoted by issuance of NIH RFAs, which have encouraged use of sIRBs, such as the Request For Proposal for the Reproductive Medicine Network, which strongly encouraged applicants to identify whether their institution would allow use of a sIRB, and more recently requirement for use of a sIRB for NIH funded multicenter studies, scheduled for beginning January 2018.
Further refinements of considerations for use of a sIRB are needed to achieve the goal of improving efficiency and expediting human subject research, by additionally addressing function and language expectations of individual participating sites. Future considerations will also need to consider expenses of the sIRB, which are overseeing trials not only at their site [for which they may receive indirect (facilities and administration) dollars], but also for time, efforts, and infrastructure needed for overseeing human subject research by other participating sites.
The use of a sIRB, which is also the IRB of one of the clinical sites in the network, has potential for cost savings compared to use of a commercial IRB; however, institutional IRBs often do not have much experience acting as a sIRB and do not necessarily have systems in place to handle so many relying sites. Additionally, the Data Coordinating Center for multicenter clinical trials will now have an additional responsibility that will need to be considered when planning personnel and budgetary requirements of being the coordinating center. This is especially the case for continuing review, as instead of sites each handling their own local submission, the Data Coordinating Center assembles all pertinent information across all sites.
There are several limitations of this study. First, observations represent the initial experience of a single multicenter network, which had previously utilized only local IRBs. Second, the experiences were based on central (single) IRB practices, policies, and approaches of one unique institutional IRB, which served as the single IRB of record; experiences may have varied with a different sIRB. Third, since the time of trial initiation and clinical sites collecting the data for the two prior trials were different from those of the current three trials, no formal comparison was performed using data from these two trials. Last, as would be the case with local IRB submissions, our observations are based on experiences with individuals who prepared our IRB transactions (correspondence, submissions, stipulation responses, and dissemination of IRB correspondence) and individuals from the sIRB who handled our submissions.
In summary, implementation of a single (central) IRB for clinical studies of the NIH/National Institute of Child Health and Human Development Cooperative Reproductive Medicine Network was associated with a shorter initial time for IRB review and approval, as compared to prior Network studies as well as a concurrent study initiated by the same Network sites utilizing local IRBs. However, total time to implementation of clinical trials was increased when considering all aspects of the submission and review process at both central and local levels. This observation suggests that achieving increased efficiency for conducting multisite clinical research may begin with central IRB approval, but more comprehensive guidance from NIH leading to enhanced coordination with local IRBs will likely be required to achieve the goal to streamline and accelerate the startup process.
Acknowledgements
The authors would like to acknowledge the advice of Louis De Paolo, PhD for his constructive guidance and comments to this manuscript.
Funding:
This work was supported by National Institutes of Health (NIH)/Eunice Kennedy Shriver National Institute of Child Health and Human Development Grants U10 HD39005 (to M.P.D.), U10 HD27049 (to C.C.), U10 HD38992 (to R.S.L.), U10 HD077680 (to K.R.H.), U10 HD077844 (to A.Z.S.), U10 HD077841 (to M.C.) U10 HD055925 (to H.Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Child Health and Human Development or NIH.
Footnotes
Declaration of Conflicting Interests
Conflicts of Interest include Dr. Michael P. Diamond – NIH Funding, AbbVie and ObsEva Funding, Board of Directors and Stockholder for Advanced Reproductive Care; Dr. Esther Eisenberg – NIH Employee; Dr. Hao Huang – No conflicts; Dr. Christos Coutifaris – NIH Funding; Dr. Richard Legro – Consultant for Ogeda, Millendo, Kindex, Fractyl and Bayer; Ferring funding; Dr. Karl R. Hansen – Yale University/ Reproductive Medicine Network/National Institute of Child Health and Human Development, Roche Diagnostics and Ferring International Pharmascience Center US funding; Dr. Anne Steiner – No conflicts; Dr. Marcelle I. Cedars – No conflicts; Dr. Kurt Barnhart – No conflicts; Tracey Ziolek – No conflicts; Tracey Thomas – No conflicts; Dr. Kate Maurer – No conflicst; Dr. Stephen A. Krawetz – No conflicts; Dr. Robert Wild – No conflicts; Dr. J. C. Trussell – No conflicts; Dr. Nanette Santoro – No conflicts; Dr. Heping Zhang – NIH Funding.
References
- 1.The National Institutes of Health. Final NIH policy on the use of a single institutional review board for multi-site research. June 21, 2016, https://grants.nih.gov/grants/guide/notice-files/NOT-OD-16-094.html (2016, accessed 27 July 2018).
- 2.McNeil C Central IRBs: why are some institutions reluctant to sign on? J Natl Cancer Inst 2005; 97: 953–955. [DOI] [PubMed] [Google Scholar]
- 3.The Office of the Secretary, HHS and the Food and Drug Administration, HHS. Human subjects research protections: enhancing protections for research subjects and reducing burden, delay, and ambiguity for investigators In: Department of Health and Human Services (ed) Federal Register, 2011, pp. 44512–44531. [Google Scholar]
- 4.Christian MC, Goldberg JL, Killen J, et al. A central institutional review board for multi-institutional trials. N Engl J Med 2002; 346: 1405–1408. [DOI] [PubMed] [Google Scholar]
- 5.Silverman H, Hull SC and Sugarman J. Variability among institutional review boards’ decisions within the context of a multicenter trial. Crit Care Med 2001; 29: 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randal J Growing pains: central review board project still developing. J Natl Cancer Inst 2003; 95: 636–637. [DOI] [PubMed] [Google Scholar]
- 7.Greene SM and Geiger AM. A review finds that multicenter studies face substantial challenges but strategies exist to achieve Institutional Review Board approval. J Clin Epidemiol 2006; 59: 784–790. [DOI] [PubMed] [Google Scholar]
- 8.Greene SM, Geiger AM, Harris EL, et al. Impact of IRB requirements on a multicenter survey of prophylactic mastectomy outcomes. Ann Epidemiol 2006; 16: 275–278. [DOI] [PubMed] [Google Scholar]
- 9.Helfand BT, Mongiu AK, Roehrborn CG, et al. Variation in institutional review board responses to a standard protocol for a multicenter randomized, controlled surgical trial. J Urol 2009; 181: 2674–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abramovici A, Salazar A, Edvalson T, et al. Review of multicenter studies by multiple institutional review boards: characteristics and outcomes for perinatal studies implemented by a multicenter network. Am J Obstet Gynecol 2015; 212: 110 e1–e6. [DOI] [PubMed] [Google Scholar]
- 11.Loh ED and Meyer RE. Medical schools’ attitudes and perceptions regarding the use of central institutional review boards. Acad Med 2004; 79: 644–651. [DOI] [PubMed] [Google Scholar]
- 12.Check DK, Weinfurt KP, Dombeck CB, et al. Use of central institutional review boards for multicenter clinical trials in the United States: a review of the literature. Clin Trials 2013; 10: 560–567. [DOI] [PubMed] [Google Scholar]
- 13.Ervin AM, Taylor HA and Ehrhardt S. NIH policy on single-IRB review - A new era in multicenter studies. N Engl J Med 2016; 375: 2315–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaufmann P and O’Rourke PP. Central institutional review board review for an academic trial network. Acad Med 2015; 90: 321–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hudson KL, Lauer MS and Collins FS. Toward a new era of trust and transparency in clinical trials. JAMA 2016; 316: 1353–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordon VM, Culp MA and Wolinetz CD. Final NIH policy on the use of a single institutional review board for multisite research. Clin Transl Sci 2017; 10: 130–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlaff WD, Zhang H, Diamond MP, et al. Increasing burden of institutional review in multicenter clinical trials of infertility: the Reproductive Medicine Network experience with the Pregnancy in Polycystic Ovary Syndrome (PPCOS) I and II studies. Fertil Steril 2011; 96: 15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silberman G and Kahn KL. Burdens on research imposed by institutional review boards: the state of the evidence and its implications for regulatory reform. Milbank Q 2011; 89: 599–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menikoff J The paradoxical problem with multiple-IRB review. N Engl J Med 2010; 363: 1591–1593. [DOI] [PubMed] [Google Scholar]
- 20.Klitzman R How local IRBs view central IRBs in the US. BMC Med Ethics 2011; 12: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mascette AM, Bernard GR, Dimichele D, et al. Are central institutional review boards the solution? The National Heart, Lung, and Blood Institute Working Group’s report on optimizing the IRB process. Acad Med 2012; 87: 1710–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flynn KE, Hahn CL, Kramer JM, et al. Using central IRBs for multicenter clinical trials in the United States. PloS One 2013; 8: e54999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menikoff J OHRP Correspondence. 2010.
- 24.US Food and Drug Administration. Guidance for industry: using a centralized IRB review process in multicenter clinical trials, 2006. [Google Scholar]
- 25.National Center for Advancing Translational Sciences. NCATS SMART IRB Reliance Authorization Agreement.