

SUMMARY
Spermatogenesis is a complex and dynamic cellular differentiation process critical to male reproduction and sustained by spermatogonial stem cells (SSCs). Although patterns of gene expression have been described for aggregates of certain spermato- genic cell types, the full continuum of gene expression patterns underlying ongoing spermatogenesis in steady state was previously unclear. Here, we catalog single-cell transcriptomes for >62,000 individual spermatogenic cells from immature (postnatal day 6) and adult male mice and adult men. This allowed us to resolve SSC and progenitor spermatogonia, elucidate the full range of gene expression changes during male meiosis and spermiogenesis, and derive unique gene expression signatures for multiple mouse and human spermatogenic cell types and/or subtypes. These transcriptome datasets provide an information-rich resource for studies of SSCs, male meiosis, testicular cancer, male infertility, or contraceptive development, as well as a gene expression roadmap to be emulated in efforts to achieve spermatogenesis in vitro.
In Brief
Hermann et al. present single-cell transcriptomes from >62,000 individual spermatogenic cells from immature and adult male mice and adult men. Their analysis facilitates resolution of SSCs and progenitor spermatogonia, elucidates the full range of gene expression changes during male meiosis and spermiogenesis, and derives unique gene expression signatures for eleven mouse and human spermatogenic cell types.
Graphical Abstract
INTRODUCTION
Male fertility relies upon proper germ cell proliferation and differentiation within the seminiferous epithelium of the testis to facilitate the production of >85 million sperm per day by a normal man (Johnson et al., 1980). Steady-state spermatogenesis is driven by dynamic coordination between germ (spermatogenic) and supporting somatic cell types beginning with a critical balance between self-renewal of spermatogonial stem cells (SSCs) and initiation of their differentiation to both sustain the SSC pool while simultaneously generating progenitor spermatogonia that initiate spermatogenic differentiation to ultimately form testicular spermatozoa (de Rooij, 2017). Previous studies of gene expression patterns during spermatogenesis have relied largely on analyses of bulk RNA from aggregates of multiple spermatogenic cell types that, at least in mice, were often isolated from developing testes during the first wave of spermatogenesis (Shima et al., 2004; Laiho et al., 2013). However, it is unclear to what extent transcriptomes of spermatogenic cells from the distinct first-wave reflect those during steady-state adult spermatogenesis. Moreover, although informative, these past studies were unable to comprehensively characterize gene expression in less prevalent spermatogenic cell types or describe the extent of heterogeneity among populations of each spermatogenic cell type.
Single-cell mRNA profiling can comprehensively define the transcriptomes of a cell lineage while also delineating the extent of cellular heterogeneity and/or the existence of rare subpopulations (Wang and Navin, 2015). Initial forays into single-cell mRNA characterization of postnatal male germ cells largely focused on isolated populations of mouse and human spermatogonia (von Kopylow et al., 2016; Song et al., 2016; Guo et al., 2017; Neuhaus et al., 2017). One recent study described heterogeneity in gene expression patterns among 2,500 adult mouse spermatogenic cells from a total of two mice (Lukassen et al., 2018), and another examined transcriptomes of 1,204 manually picked spermatogenic cells isolated from the first wave of retinoic acid (RA)-synchronized mouse spermatogenesis (Chen et al., 2018). However, no previous single-cell RNA-seq study has (1) directly correlated spermatogo-nial single-cell transcriptome data with functional assessments of SSC fate based on the spermatogonial transplantation assay, (2) provided in-depth and unbiased analyses of all spermatogenic cell types in steady state, or (3) compared singlecell transcriptomes in equivalent immature and mature mouse spermatogenic cell types or in adult mouse and adult human spermatogenic cell types.
We used two methods of single-cell RNA-seq (10x Genomics and Fluidigm C1) to identify comprehensive gene expression patterns in >62,000 individual cells from the seminiferous epithelium in (1) the immature mouse testis, (2) the adult mouse testis, and (3) the adult human testis. These data were validated by protein immunostaining and independent RNA expression analyses using testis tissue, mixed seminiferous tubule cells, and enriched populations of spermatogonia, spermatocytes, and spermatids. The resulting sequence data were analyzed using unbiased approaches followed by retrospective correlation with transcriptomes of spermatogonial subtypes of known distinct functional capacities (Helsel and Oatley, 2017). Results reveal a conserved yet dynamic continuum of gene expression patterns across the full spectrum of spermatogenic development, as well as heterogeneity indicative of spermatogenic cell subtypes engaged in distinct biological pathways or functions. In addition, we have derived unique gene expression signatures that can be used to distinguish the presence of each spermato-genic cell type or subtype within samples of whole testis tissue from mice or men.
RESULTS
We used 10x Genomics analysis (greater throughput) validated by supplementary Fluidigm C1 single-cell RNA-seq analysis (greater depth) to evaluate gene expression heterogeneity throughout the complete spermatogenic lineage in 62,141 mouse and human spermatogenic cells. We first examined sper-matogenic cells derived from suspensions of seminiferous tubule cells obtained without prospective selection and then resolved datasets representing spermatogonia, spermatocytes, and spermatids in greater depth. Unsupervised cell clustering in concert with cell ordering in pseudotime facilitated profiling of the developmental kinetics of gene expression changes during spermatogenesis. Deduction of cell type/subtype identities was based on known spermatogenic cell-type-specific marker genes and/or a marker transgene (Id4-Egfp), as well as by comparison with results from parallel analyses of sorted cell types recovered by fluorescence-activated cell sorting (FACS) or Sta- Put gravity sedimentation (Bellvé et al., 1977b).
Single-Cell Transcriptomes of the Complete Cohort of Steady-State Spermatogenic Cells
We first used 10x Genomics analysis to profile transcriptomes of 4,651 and 7,134 spermatogenic cells from mice and men, respectively (Figure 1). Results were highly consistent (correlation coefficients of 0.97–0.99) among analyses of triplicate cell samples from each species (Figures S1E-S1G), with 99% droplet capture of single cells (Figures S1E-S1G). Unsupervised, unbiased clustering projected onto t-distributed stochastic neighbor embedding (tSNE) analysis plots revealed a heterogeneous distribution of multiple cell clusters representing the complete spermatogenic lineage in each species, with only minor contribution from testicular somatic cells, which we identified on the basis of somatic cell markers (Figures 1A-1E; Table S1). We identified 14 clusters of unselected spermatogenic cells in both the mouse (Figures 1A and 1C; Table S1) and human (Figures 1B and 1D; Table S1). We identified cell type(s) represented in each cluster, including major spermatogenic cell types, spermatogonia, spermatocytes, plus subtypes of each major cell type by cell-type-specific gene expression (Figures 1C-1E, S1C, and S1D) and validated a subset of these assignments with congruent protein immunolocalization patterns (Figures S1H and S1I). Among genes expressed during spermatogenesis, 9,400 of 28,625 and 7,031 of 20,939 were expressed throughout human and mouse spermatogenesis, respectively, with the remaining genes showing spermatogenic cell-type specificity. All of our single-cell gene expression data are publicly accessible in six GEO datasets plus 9 queryable Loupe Cell Browser files archived via Mendeley Data (Key Resources Table).
Figure 1. 10x Genomics Profiling of Unselected Adult Mouse and Human Spermatogenic Cells Reveals the Extent of Gene Expression Heterogeneity during Steady-State Spermatogenesis.
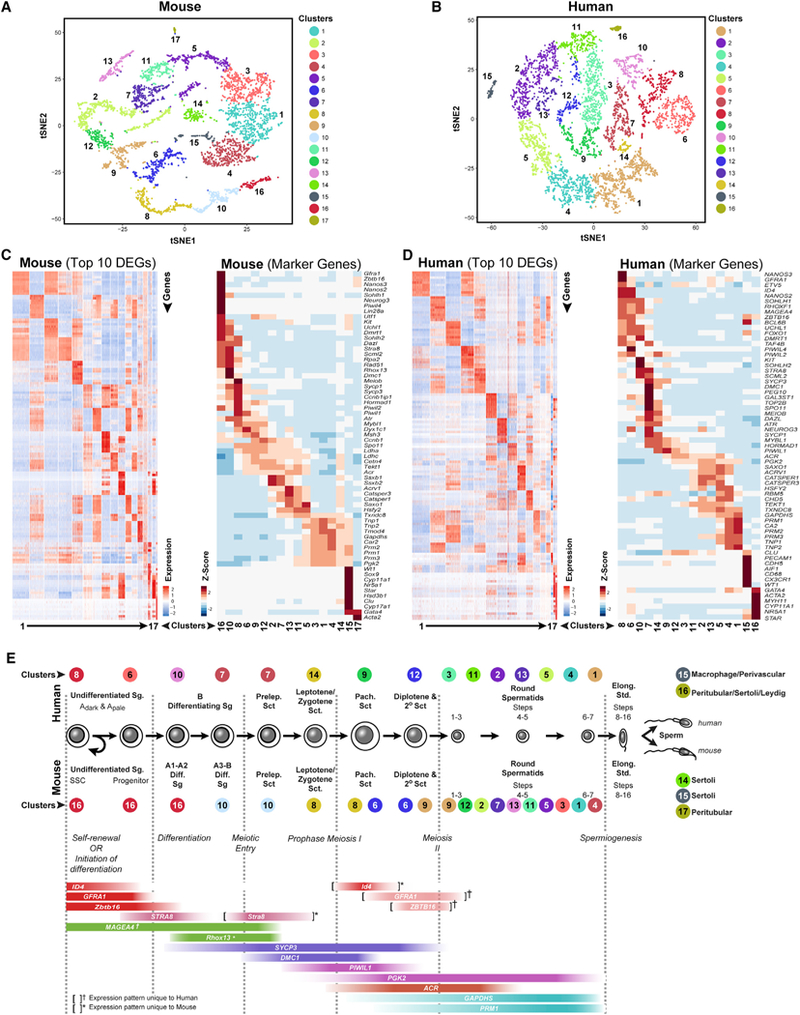
(A and B) tSNE plots show 10x Genomics profiling of unselected spermatogenic cells from (A) mouse testes and (B) human testes. Unbiased cell clusters are distinguished by color according to the key.
(C and D) Heatmaps show the top 10significantly differentially expressed genes (DEGs) between each cell cluster (left) and expression of key cell-type-specific markers (right) for (C) mouse and (D) human spermatogenic cells. Gene lists can be found in Table S1.
(E) Identification of cell clusters expressing the noted marker genes allowed clusters to be aligned with specific spermatogenic cell types (*mouse- or †human-specific expression patterns).
Heterogeneity among Adult Spermatogonia in Mice and Men
Cells from two clusters of mouse and four clusters of human spermatogenic cells expressed known spermatogonial genes (Gfra1, Kit, Nanos3, Rhox13, Sall4, and Zbtb16; Figure 1). When extracted and re-analyzed in isolation, these clusters were further resolved, ultimately yieldingfive and 10distinct clusters of human and mouse unselected mouse spermatogonia, respectively (Figures 2A and 2C; Table S1), distinguished by differentially expressed genes (DEGs) (Figures 2B and 2D; Table S1).
Figure 2. Adult Spermatogonia Are Heterogeneous in Mice and Men.
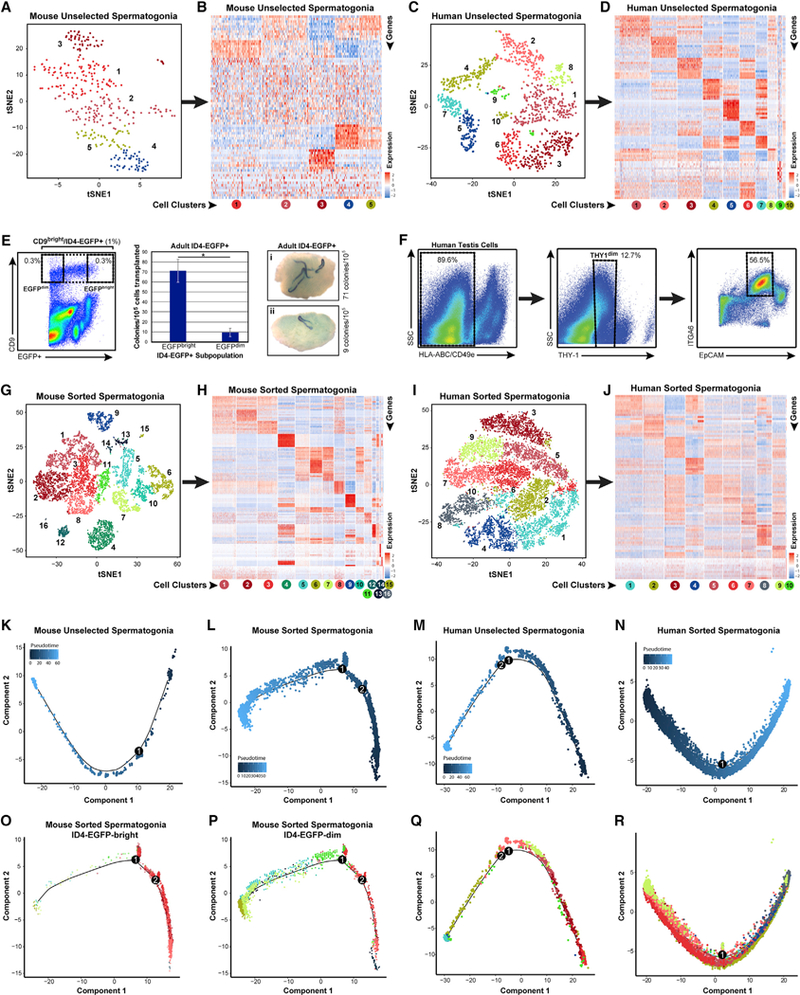
(A and C) Clusters of steady-state spermatogenic cells (Figures 1A and 1B) containing spermatogonia (mouse clusters 10and 16; human clusters 6,7, 8, and 10) were isolated and re-analyzed. tSNE plots show unbiased re-clustering of unselected spermatogonia from (A) mouse or (C) human testes (colors distinguish clusters).
(B and D) Heatmaps show the top 10 significantly differentially expressed genes (DEGs) between each cell cluster for (B) mouse and (D) human unselected spermatogonia (circles below heatmaps are colored and numbered by tSNE cluster). Gene lists can be found in Table S1.
(E and F) We also profiled sorted spermatogonia from (E) adult Id4-Egfp mouse testes (CD9bright/EGFP+, 1% of unsorted, and CD9bright/EGFPbright or CD9bright/EGFPdim subpopulations, each 0.3% of unsorted) and (F) adult human testes (HLA-ABCnegative, CD49enegative, THY1dim, ITGA6+, and EpCAMdim; ~6.4% of unsorted). Transplant of adult mouse EGFPbright/CD9bright versus EGFPdim/CD9bright spermatogonia shows 7.5-fold greater colonization activity of EGFPbright versus EGFPdim cells (*Student’s t test p < 0.02), demonstrating functional SSC enrichment and depletion, respectively.
(G and I) Additional tSNE plots show unbiased clustering of sorted adult spermatogonia from (G) mouse and (I) human testes (colors distinguish clusters).
(H and J) Heatmaps show the top 10 significantly DEGs between each cell cluster for sorted (H) mouse and (J) human spermatogonia.
(K-R) Pseudotime trajectories of (K-N) unselected and sorted spermatogonia in which cells are ordered from beginning (darkest blue color) to end (lightest blue) according tothe legend or (M), (N), (P), and (R), in which cells are colored asthey appear by cluster in the corresponding tSNE plots. Branch points in the singlecell trajectories are noted by black numbered circles. Spermatogonial clusters included in this trajectory analysis included (K) mouse unselected spermato- gonia—all clusters; (L, O, and P) mouse sorted spermatogonia—clusters 1–3, 5–8,10,11,13, and 14; (M and Q) human unselected spermatogonia—all clusters; and (N and R) human sorted spermatogonia—all clusters.
(L, O, and P) For mouse sorted spermatogonia (L) that were analyzed and displayed in pseudotime together, we also retrospectively displayed the two input subpopulations separately from the same pseudotime trajectory in (O) ID4-EGFPbright spermatogonia and (P) ID4-EGFPdim spermatogonia.
To correlate spermatogonial phenotype with function in the adult testis, we devised a spermatogonial isolation strategy using mice carrying an Id4-eGfp transgene (Chan et al., 2014). In immature mice, transplantable SSC capacity segregates nearly exclusively to the ID4-EGFP+ fraction (Chan et al., 2014), with maximum enrichment for transplantable SSCs achievable by sorting the most epifluorescent (ID4-EGFPbright) cells (Helsel et al., 2017). We used FACS to select cells that were CD9bright (to distinguish spermatogonia from spermatocytes) and ID4-EGFPbright and ID4-EGFPdim (to distinguish transplantable SSCs from progenitors; Figures 2E and S2A) as an effective means to identify and recover steady- state SSCs from the adult mouse testis. To recover spermatogonia from the adult human testis, we utilized markers previously shown by xenotransplantation to selectively enrich for human spermatogonia with colonization potential (Figures 2F and S2B; Dovey et al., 2013). Sorted adult mouse (6,945 cells) and human (11,104 cells) spermatogonia were profiled by 10x Genomics analysis, which yielded 14 mouse and 10 human clusters of spermatogonia (Figures 2G, 2I, S2C, and S2D; Table S1), each distinguished by DEGs (Figures 2H and 2J; Table S1). Parallel Fluidigm C1 single-cell RNA-seq data validated these 10x Genomics results (Figures S2E-S2N). Unbiased dynamic cell trajectory analyses of pseudotime order with both unselected and sorted spermatogonia from both mice and men (Figures 2K-2N) yielded developmental continua lacking major branching points (Figures 2K-2R).
Developmental Ordering of Spermatogonial Clusters
Pseudotime profiles were scrutinized based on genes known to be expressed by all spermatogonia (Ddx4) and genes that distinguish undifferentiated spermatogonia (Gfra1, Id4, and Nanos2) from differentiating spermatogonia (Dmrt1, Kit, Nanos3, and Stra8; Figures 3A-3D and S3A-S3D). Expression of genes specific to undifferentiated spermatogonia or differentiating spermatogonia was skewed preferentially toward the beginning and end of the trajectories, respectively, for both unselected and sorted spermatogonia from both mice and men (Figures 3A-3D and S3A-S3D). Surprisingly, although Stra8 levels increased as a function of the transition from undifferentiated to differentiating spermatogonia in mice (Figures 3A and 3B), very little STRA8 mRNA was detected among either undifferentiated or differentiating human spermatogonia (Figures 3C, 3D, and S3E-S3H).
Figure 3. Single-Cell Spermatogonial Trajectories Reveal Biological Transitions Coinciding with SSC Self-Renewal and Initiation of Differentiation in Pseudotime.
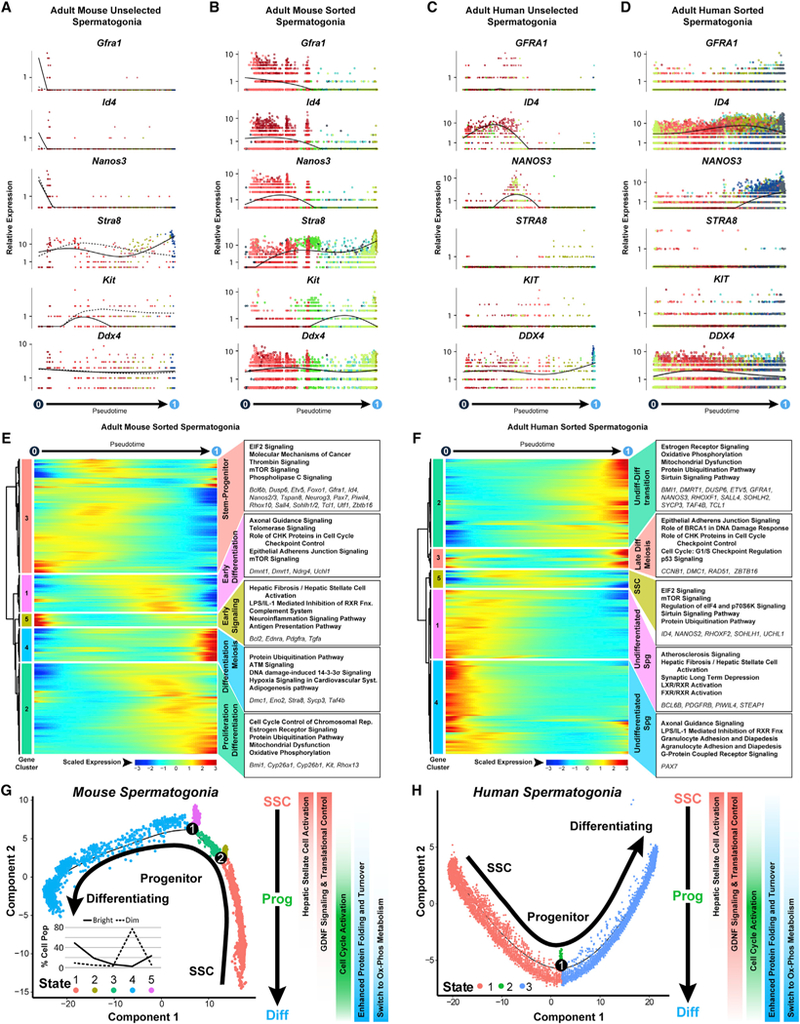
(A-D) Expression patterns of key landmark genes over pseudotime among (A) adult mouse unselected spermatogonia, (B) adult mouse sorted spermatogonia.
(C) adult human unselected spermatogonia, and (D) adult human sorted spermatogonia. Cells are colored according to the cluster colors in the corresponding tSNE plots(Figures 2A, 2C, 2G, and 2I) and ordered according tothe pseudotime plots (Figures 2K-2N). Pseudotime(scaled,0to 1)is indicated beloweach gene plot column.
(E and F) Clusters of genes that were differentially expressed across pseudotime from (E) adult mouse sorted spermatogonial and (F) adult human sorted spermatogonial datasets are shown as heatmaps according to expression color code noted at the bottom (see Table S2). Dendrograms show hierarchical relationship between gene clusters. The top five over-represented biological pathways from Ingenuity Pathway Analysis of each cluster are noted at the right in bold (see Table S3), and key genes are italicized.
(G and H) Trajectories (identical to Figures 2L and 2N) from (G) adult mouse sorted spermatogonia and (H) adult human sorted spermatogonia are colored by cell state and illustrate biologicallysignificant differences acrossthistrajectory, which is summarized tothe right ofeach plot. Inset graph in (G) showsthe distribution (percentage) ofthe sorted ID4-EGFPbright spermatogonia and ID4-EGFPdim spermatogonia as shown in Figures 2O and 2P, among the noted five spermatogonial states.
ID4-EGFPbright and ID4-EGFPdim mouse spermatogonia were inversely localized non-randomly among undifferentiated sper- matogonial clusters near the beginning and end of the pseudotime trajectory, respectively (Figures 2E, 2O, and 2P). Therefore, on the basis of (1) pseudotime ordering, (2) marker gene expression, and (3) transplantation competence, this mouse spermato-gonial trajectory appears to accurately depict the normal kinetics of spermatogonial development. Further, the striking similarity in gene expression patterns between mouse and human spermatogonial trajectories (Figures 3A-3D and S3A-S3D) lends credence to the extrapolation of these mouse results to human spermatogonial development. Among 14,668 orthologous genes expressed in SSC-enriched mouse spermatogonia in cluster 3 (Figure 2A) and SSC-enriched human spermatogonia in cluster 1 (Figure 2C), only 286 (1.9%) were differentially expressed (log fold-change [FC] > 1; p < 0.01; Table S4). Pathway analyses suggested that human SSCs express higher levels of genes involved in mRNA transport, surveillance, and degradation, and mouse SSCs express higher levels of genes involved in oxidative phosphorylation and proteasome function (Table S4).
Signaling and Metabolic Drivers of the Stem Cell and Differentiating Spermatogonial Fates
The prototypical SSC gene signature (Gfra1, Id4, Etv5, Nanos2, Pax7, Tspan8, Rhox10, and Zbtb16) was found early in pseudotime in gene cluster 3, along with expression of a panel of novel genes (Dusp6, Epha2, Ptpn13, Pvr, and Tcl1; Figure 3E; Tables S2 and S3). DUSP6 is a dual-specificity phosphatase that regulates mitogen-activated protein (MAP) kinase activity (Caunt and Keyse, 2013), PTPN13 is a phosphatase known to impede kinase cascade activity and influence cellular metabolic and proliferative states (Gurzov et al., 2015), and TCL1 is an AKT co-stimulator (Pekarsky et al., 2000), suggesting regulation of intracellular signaling pathways may play a key role in SSC function. Other genes that exhibited maximal expression at the early-to-mid- point of the pseudotime trajectory included those involved in control of translation (Eif4e, Eif4ebp1, Pabpc1, and Rptor; EIF2 signaling; mTOR signaling) and regulation of intracellular cell signaling pathways (F2r, Gnaq, Plce1, Ppp1cb, and Shc1; PLC signaling; Thrombin signaling; Figure 3E). Human spermatogonia gene cluster 5, which is also centered at the mid-point of the pseudotime trajectory, similarly showed expression of characteristic SSC genes encoding proteins that play roles in translational control (EIF4B, MLST8, and PABPC1; EIF2 signaling; mTOR signaling; Figure 3F), glycolysis (ALDOA, ENO3, PFKL, and TPI1), and known SSC genes (ID4 and NANOS2; Figure 3F),consistent with previous reports (Guo et al., 2017). However, other known human SSC genes (ETV5 and GFRA1) were expressed later in pseudotime coincident with expression of characteristic spermatogonial differentiation genes (DMRT1, NANOS3, and SOHLH2; Figures 3F and S3E-S3K).
Surprisingly, in the pseudotime trajectories representing spermatogonial development in both mice and men, a distinct cluster of genes (mouse cluster 5; human cluster 1) was expressed prior to the prototypical SSC gene signature (Figures 3E, 3F, S3J, and S3K). Genes involved in the hepatic stellate cell activation pathway (Bcl2, Ednra, Klf6, Pdgfra, and Tgfa) were upregulated in mouse unselected and sorted spermatogonia gene clusters 4 (Table S3) and 5 (Figure 3E; Table S3) in human unselected and sorted spermatogonia gene clusters 1 (Table S3) and 1 (Figure 3F; Table S3), respectively, and in Fluidigm-C1-sorted human spermatogonial gene cluster 2 (Figure S3K; Table S3). Activation of this pathway renders target cells responsive to inductive cytokine signaling (Tsuchida and Friedman, 2017), suggesting that the transition from quiescent SSCs to proliferative differentiating spermatogonia may involve responsiveness to cytokines. Transcripts encoding a master, growth-suppressing transcriptional regulator of this pathway, KLF6 (Ghiassi- Nejad et al., 2013), were significantly elevated in putative SSCs from all of our adult human and mouse spermatogonial datasets (Figures S3L-S3O) and in batch RNA sequencing (RNA-seq) results from SSC-enriched TSPAN8high spermatogonia (Mutoji et al., 2016) and SSC-enriched ID4-EGFPbright spermatogonia (Helsel et al., 2017) from the immature mouse testis.
Initiation of spermatogonial differentiation in mice was evident at the midpoint of the pseudotime trajectory when gene clusters 1 and 2 sequentially became activated (Figure 3E). This included elevated expression of genes involved in cell cycle activation and control (Cdc6, Cdc25e, Cdk1, Chek2, Rfc1, and Tp53) and spermatogonial differentiation (Dmrt1, Kit, Rhox13, and Stra8; Zhou et al., 2008; Busada et al., 2015; Figure 3E; Tables S2 and S3). Mouse and human spermatogonia that were approaching the end of the pseudotime trajectory (mouse gene cluster 2 and human gene cluster 2) expressed genes involved in mitochondrial function and oxidative phosphorylation (Atpf1, Cox5a, Cyc1, Ndufa1, and Surf1; Figures 3E and 3F), indicative of a metabolic shift away from glycolysis to support proliferation and differentiation. Late in the spermatogonial pseudotime trajectories (mouse clusters 2 and 4; human clusters 2 and 3), meiotic gene expression is initiated (Dmc1, Rad51, and Sycp3; Figures 3E, 3F, S3J, and S3K). Although the proteins encoded by these genes do not appear until the primary spermatocyte stage, transcription of these genes during the late spermatogonia phase predisposes the rapid availability of these mRNAs as these cells transition to spermatocytes. Finally, expression of genes involved in protein folding and turnover, including multiple DnaJ heat shock protein family (DNAJ) and heat shock protein (HSP) chaperones, as well as genes encoding proteasome subunits and ubiquitin-conjugating enzymes coincided with spermatogonial differentiation (Figures 3E and 3F; Tables S2 and S3).
Taken together, our single-cell-derived transcriptome data provide a more detailed and higher resolution account of gene expression patterns involved in spermatogonial development than previously available, including those that distinguish selfrenewal of SSCs and differentiation of progenitor spermatogonia (Figures 3G and 3H), indicating the SSC state is characterized by a unique signaling and transcriptional regulatory environment that is lost upon transition to an intermediary progenitor state. Conversely, as progenitors initiated differentiation, they acquired enhanced proliferative capacity by activating and/or elevating cell cycle gene expression, while simultaneously undergoing major metabolic shifts to oxidative phosphorylation and activation of protein translation, folding, and turnover.
Progression through Meiosis Is Characterized by Dynamic Changes to Metabolism and Signaling
Mouse and human steady-state spermatogenic cell clusters (Figures 1A and 1B) expressing spermatocyte genes (Dmc1, Hormad1, Mybl1, and Spo11; mouse clusters 6, 8, 9, and 10; human clusters 7, 9,12, and 14) were further resolved into 9 independent clusters of spermatocytes based on DEG patterns in each species (Figures 4A-4D; Table S1). Mouse and human spermatocytes expressed 15,684 orthologous genes with only 404 (2.6%) showing differential expression between species (log fold change > 1; p < 0.01; Table S4), indicative of higher levels of mRNA transport and degradation in human spermatocytes and higher levels of oxidative phosphorylation and ribosome function in mouse spermatocytes (Table S4). Adult pachytene spermatocytes enriched by StaPut could be resolved into 10 and 13 clusters for mouse and human, respectively (Figures S4A-S4D; Table S1). Unselected spermatocytes (Figures 4E and 4F) and StaPut-enriched pachytene spermatocytes (Figures S4D and S4E) were separately used for pseudotime trajectory analysis, which yielded distinct cell clusters that emerged sequentially in pseudotime and were, therefore, representative of distinct meiotic stages. We detected expression of multiple genes involved in processes unique to spermatocytes, including meiotic sex chromosome inactivation (MSCI) (Handel and Schi-menti, 2010), homolog synapsis (Sycp3), and meiotic recombination (Meiob, Mlh3, Rad51, Rpa2, and Spo11; Figures 4G-4J and S4F-S4I; Tables S2 and S3). We chronicled active expression of autosomal genes known to regulate MSCI (Atr [Royo et al., 2013] and Hormad1 [Wojtasz et al., 2012]), and inactivation of sex-linked genes (Pgk1, Scml2, and Zfy1) by MSCI (Wang et al., 2005) while confirming expression of autosomal mRNAs (Acr, Ldhc, and Pgk2) transcribed during meiosis, but not translated until the postmeiotic spermatid stage (Wang et al., 2005; Figures 4G-4J and S4F-S4I; Tables S2 and S3). Unselected spermatocytes reflected the entire range of first meiotic prophase, including preleptotene, leptotene, and zygotene spermatocytes, characterized by expression of recombination and synaptonemal complex genes (Mlh1, Rad51, Sycp1, Sycp3, and Top2b) and pre-MSCI expression of sex-linked genes (Figures 4G-4J and S4F-S4I), which appear to represent steady- state pre-pachynema spermatocyte transcriptomes.
Figure 4. Meiotic Progression among Steady-State Spermatocytes Is Characterized by Regulation of Dynamic Metabolic Transitions and Protein Turnover.
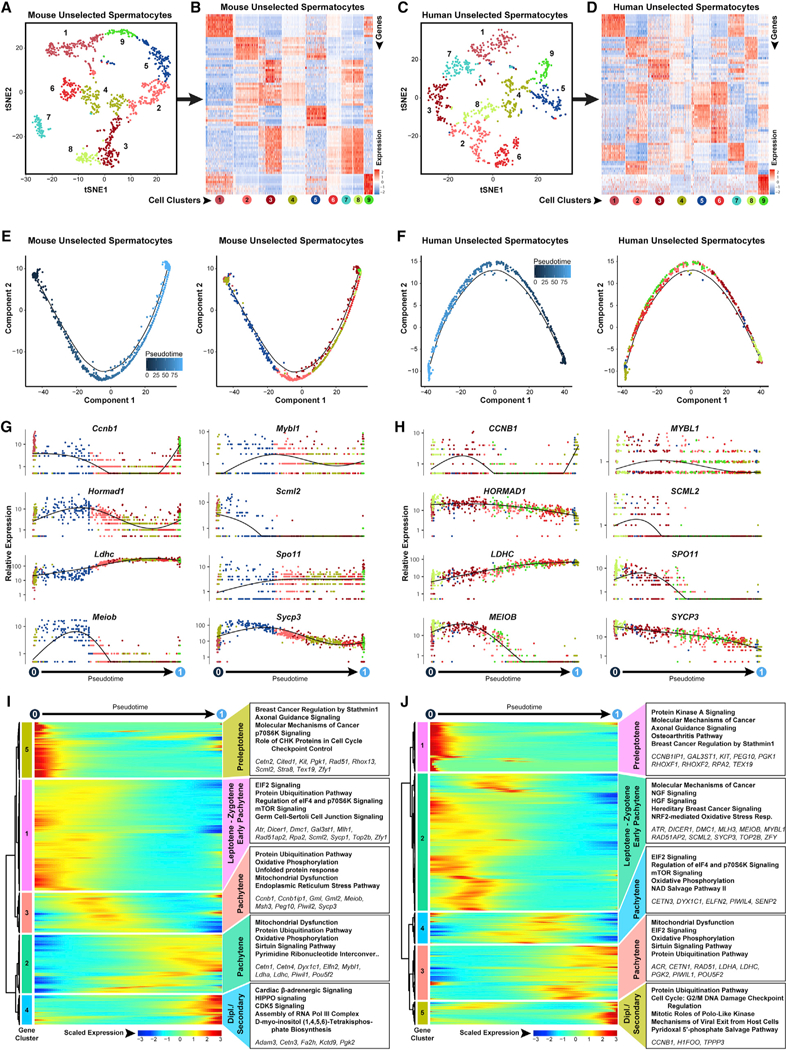
Clusters of steady-state spermatogenic cellsthat contained spermatocytes (Figure 1 Amouse clusters 6,8,9, and 10; Figure 1B human clusters 7, 9,12, and 14) were extracted and re-analyzed here.
(A and C) The tSNE plots show unbiased re-clustering of unselected spermatocytes from (A) mouse testes and (C) human testes, with color distinguishing clusters.
(B and D) Heatmaps show the top 10 significantly differentially expressed genes (DEGs) between each cell cluster for (B) mouse and (D) human unselected spermatocytes. Gene lists can be found in Table S1. Colors and numbering of circles below heatmaps match the cell clusters in the corresponding tSNE plot.
(E and F) Single-cell transcriptomes from (E) mouse unselected spermatocytes and (F) human unselected spermatocyteswere subsequently used for unbiased dynamic lineage analysis producing cell trajectories ordered in pseudotime (left) and cells colored according to tSNE cell cluster (right).
(G and H) Expression levels (vertical axis) of key genes among (G) mouse unselected spermatocytes and (H) human unselected spermatocytes ordered in pseudotime (cell coloring is according to tSNE clusters from A and C, respectively).
(I and J) Heatmaps show hierarchical relationship between clusters of genes that were differentially expressed across pseudotime from (I) mouse unselected spermatocytes and (J) human unselected spermatocytes (scaled expression according to legend; see Table S2). The top five over-represented biological pathways from GO analyses of each cluster are noted at the right in bold (see Table S3), and key genes are italicized
Expression of genes involved in spermatogonial differentiation (Kit, Rhox13, and Stra8, mouse gene cluster 5; KIT, RHOXF1, and RHOXF2, human gene cluster 1) was still evident at the pre- leptotene stage (Figures 4I and 4J). The sequential leptonema and zygonema stages (mouse gene cluster 1 and human gene cluster 2) were characterized by expression of genes involved in signaling pathway regulation, protein turnover and translational regulation, and growth factor signaling (NGFand HGF; Figures 4I and 4J; Tables S2 and S3). Maximal expression of Mybl1, a master transcriptional regulator of meiosis-related genes, occurred coincident with the transition to pachynema (Figures 4G and 4H; Bolcun-Filas et al., 2011).
Suppression of X-linked genes (Scml2 and Pgk1) and enhanced expression of genes involved in oxidative phosphorylation, mitochondrial function, and protein ubiquitination were observed among both unselected and StaPut-purified pachytene spermatocytes (mouse gene cluster 3; human gene cluster 4; Figures 4I, 4J, S4H, and S4I; Tables S2 and S3). Post-pachytene mouse spermatocytes, including both primary spermatocytes in diplonema and secondary spermatocytes, expressed higher levels of genes involved in HIPPO and CDK5 signaling pathways, and their human counterparts exhibited elevated levels of G2/M DNA damage checkpoint regulation and Polo-like kinase signaling (Figures 4I and 4J). Components of the p-adrenergic signaling pathway were upregulated in these post-pachytene spermatocytes in both species (Figures 4I and 4J). Finally, immu- nostaining for RHCG in mouse spermatocytes and round spermatids (Figure S4J) validated our single-cell measurements of Rhcg (Liu et al., 2000).
The Unique Haploid Transcriptome Facilitates Spermiogenesis
During spermatid morphogenesis (spermiogenesis), ongoing transcription occurs only in round spermatids due to subsequent genome condensation (Ward, 2010). Spermatid-containing clusters from the mouse and human steady-state spermatogenic cell datasets (Figures 1A and 1B) were identified based on expression of known spermatid-specific genes (Prm1, Prm2, Tnp1, Tnp2, and Catsper1 ; mouse clusters 1–5, 7,9, and 11–13; human clusters 1–5, 11, and 13). Unbiased analyses yielded 12 and 13 distinct clusters of unselected mouse and human spermatids, respectively (Figures 5A and 5C; Table S1), each distinguishable on the basis of DEG patterns (Figures 5B and 5D; Table S1). Between transcriptomes of unselected mouse and human spermatids, which together expressed 16,016 orthologous genes, only 632 (3.9%) were differentially expressed (log fold change > 1; p < 0.01; Table S4), with genes involved in ribosome function and oxidative phosphorylation upregulated in human spermatids and those involved in glycolysis, gluconeogenesis, and glucagon signaling upregulated in mouse spermatids. Mouse and human StaPut-enriched round spermatids were resolved into 11 and 15 clusters, respectively (Figures S5A and S5C).
Figure 5. Dynamic Transcriptome Changes during Spermiogenesis Precede Production of Functional Spermatozoa.
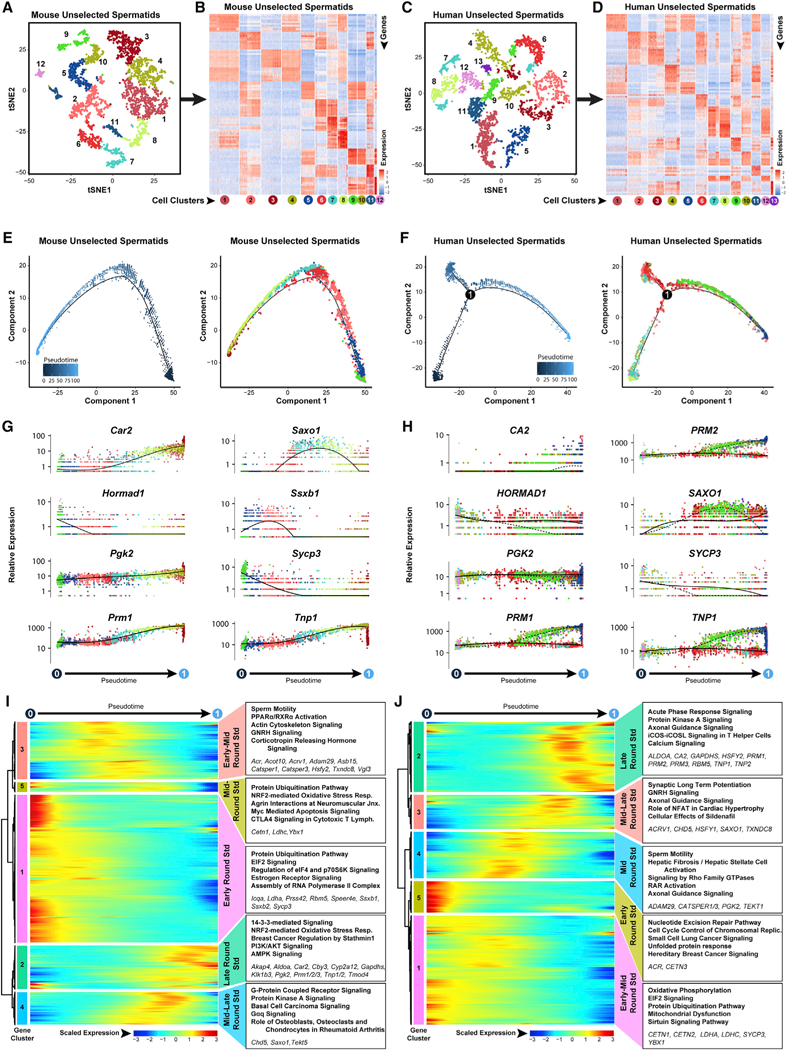
Spermatid-containing clustersfrom steady-state spermatogenic cells (Figure 1A, mouse clusters 1,2,3,4,5,7,9,11,12, and 13; Figure 1B, human clusters 1,2,3,4,5, 11, and 13) were extracted and re-analyzed here.
(A and C) These unselected spermatids were re-clustered, and the tSNE plots are shown for (A) mouse testes and (C) human testes, with color distinguishing clusters.
(B and D) Heatmaps show the top 10 significantly DEGs between each cell cluster for (B) mouse and (D) human unselected spermatids. Gene lists can befound in Table S1. Colors and numbering of circles below heatmaps match the corresponding tSNE plot.
(E and F) Single-cell transcriptomes from (E) mouse unselected spermatids and (F) human unselected spermatocytes were subsequently used for unbiased dynamic lineage analysis producing cell trajectories with spermatids ordered in pseudotime (left) and colored according to tSNE cell cluster (right).
(G and H) Expression levels (vertical axis) of key genes among (G) mouse unselected spermatids and (H) human unselected spermatids ordered in pseudotime (cell coloring is according to tSNE clusters from A and C, respectively).
(I and J) Heatmaps show a hierarchical relationship between clusters of genes that were differentially expressed across pseudotime from (I) mouse unselected spermatidsand (J) human unselected spermatids (scaled expression according to legend; see Table S2). The top five over-represented biological pathways from GO analyses of each gene cluster are noted at the right in bold (see Table S3), and key genes are italicized.
Spermatid cell clusters overlapped little in pseudotime, suggesting they denote distinct stages during spermiogenesis (Figures 5E, 5F, S5B, and S5D). Moreover, pseudotime trajectories of unselected spermatids closely aligned with StaPut-sorted round spermatids, in which elongating spermatids and residual bodies are absent (Bellvé et al., 1977b). Detection of latent meiotic gene expression (Hormad1 and Sycp3) among the earliest round spermatids coincided with meiotic exit and progression into spermiogenesis (Figures 5G, 5H, S5E, and S5F), as did reactivation of certain sex-linked mRNAs in early round spermatids (Ssxb1, Ssxb2, and Hsfy2; Turner et al., 2006; Namekawa et al., 2006; Figures 5G, 5H, S5E, and S5F). Subsequently, de novo activation of genes required for acquisition of sperm motility (Car2 and CA2; Wandernoth et al., 2015), as well as post- meiotic activation of transition protein (Tnp1 and Tnp2) and protamine (Prm1 and Prm2) genes required for sperm DNA packaging became evident at the midpoint of the pseudotime trajectory (Figures 5G-5J and S5E-S5H). In both mouse and human spermatids, genes encoding ion channels, second messenger enzymes, and kinases involved in sperm motility (mouse gene cluster 3; human gene cluster 4) were maximally expressed at the midpoint of round spermatid development (Figures 5I and 5J; Tables S2 and S3). Although initiated in primary spermatocytes, expression of sperm-specific glycolytic isozymes (Aldoa, Gapdhs, Ldhc, and Pgk2) peaks during the latter half of round spermatid development in mouse spermatid gene cluster 4 and human spermatid clusters 2 and 4, along with genes involved protein kinase A (PKA) signaling, calcium signaling, and G-protein-coupled receptor (GPCR) signaling required for acquisition of sperm motility (Figures 5I and 5J; Tables S2 and S3). Lastly, immunostaining for ACTL7B in mouse and human round spermatids (Figures S5I and S5J) validated our pseudotime expression profile of Actl7b/ACTL7B and previous Actl7b promoter-driven EGFP transgene expression (Hisano et al., 2003).
In mice, we identified specific cohorts of genes that appeared as waves with maximal expression during narrow windows relatively early during round spermatid pseudotime (Iqca, Prss42, and Speer4e; mouse gene cluster 1), slightly later during spermatid development (Acot10, Asb15, Saxo1, and Vgl3; mouse gene clusters 3 or 4), or late in spermatid pseudotime (Cby3, Cyp2a12, Klk1b3, and Tmod4; mouse gene cluster 2) and similar gene groups in human spermatids (Figures 5I, 5J, S5I, and S5J). Both unselected and StaPut-enriched human, but not mouse, round spermatids exhibited a marked bifurcation in the pseudotime trajectory indicative of two subgroups with distinct tran- scriptomes late during spermatid development (Figures 5F and S5F). One subgroup of human spermatids elevates or maintains expression of key known spermatid genes (CA2, PRM1, PRM2, and TNP1), whereas the other inexplicably fails to elevate and even shows decreases in levels of these same mRNAs.
Spermatogonial Gene Expression Programs in the Immature Mouse Testis Are Distinct from Those in the Adult Mouse Testis
It is generally accepted that the first wave of rodent spermatogenesis emanates from non-self-renewing spermatogonia or pros- permatogonia and is distinct from steady-state spermatogenesis sustained by a regulated balance between SSC self-renewal and initiation of differentiation (Geyer, 2017). This suggests that key differences should be discernable in spermatogonial transcrip- tomes at postnatal day 6 (P6) and adult ages. From P6 testes, 10x Genomics analysis of 3,466 unselected testicular cells and 9,628 sorted ID4-EGFPbright/ID4-EGFPdim spermatogonia that are highly enriched for or depleted of transplantable SSCs, respectively (Helsel et al., 2017), resolved into 11 and 12 cell clusters, respectively (Figures 6A-6D and S6A-S6C; Table S1). These clusters exhibited a small but consistent group of differentially expressed genes along with a large group of similarly expressed genes (Figures 6A-6D and S6A-S6C; Table S1), validated by Fluidigm C1 single-cell RNA-seq (data not shown). As with our previous adult spermatogonial analyses (Figures 2 and 3), parallel pseudotime analysis of the Ddx4+ (spermatogonia) subset of unselected P6 testis cells (cluster 5; Figure 6E) and sorted P6 spermatogonia (Figure 6F), distinguished ID4-EGFPbright and ID4-EGFPdim subpopulations, which skewed to the beginning and end of the trajectory, respectively (Figures 6G and 6H), as did pseudotime profiles of known markers distinguishing undifferentiated spermatogonia (Gfra1, Id4, Piwil4, and Rhox10) from differentiating spermatogonia (Dmrt1, Kit, Nanos3, Rhox13, and Stra8; Figures 6I, 6J, S6D, and S6E).
Figure 6. Neonatal Mouse Spermatogonia Exhibit Unique Developmental Characteristics during the First Wave of Spermatogenesis.
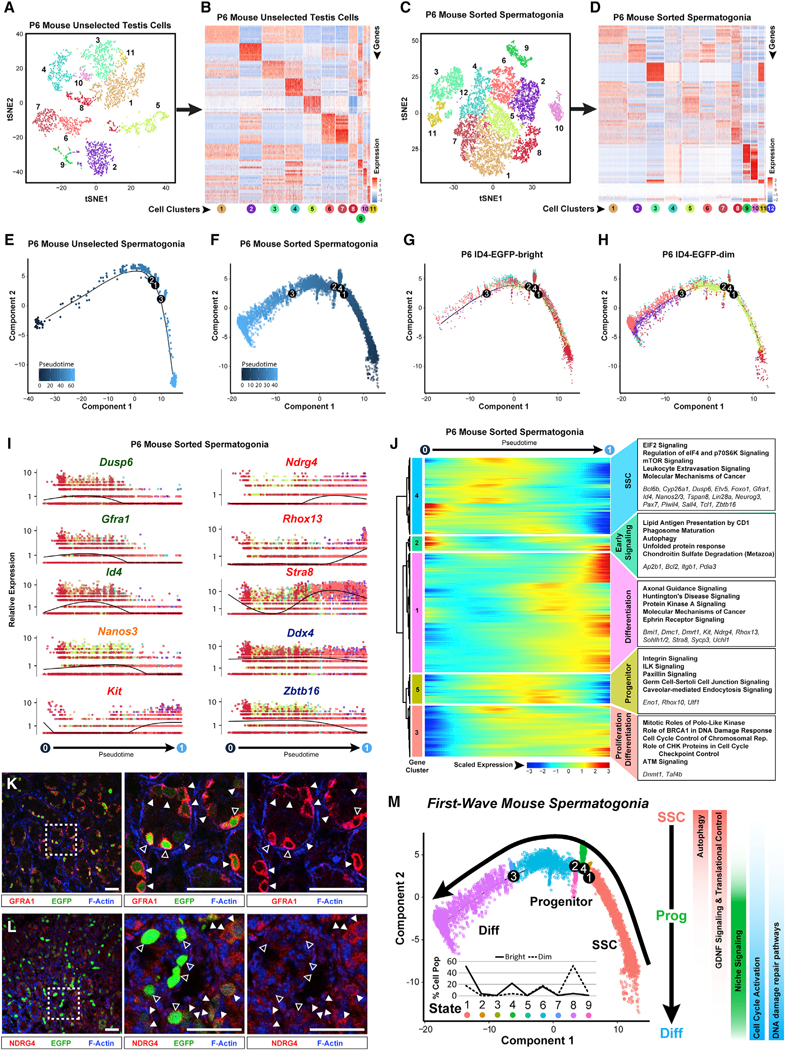
Unselected testis cells (containing a mixture of spermatogonia and testicular somatic cells) and sorted spermatogonia from P6 mouse testes were profiled by 10x Genomics analysis.
(A and C) Resulting tSNE plots show unbiased clustering of (A) P6 mouse unselected testiscellsand (C) sorted P6 sorted spermatogonia, with colordistinguishing clusters.
(B and D) Heatmaps show the top 10 significantly DEGs between each cell cluster for (B) P6 mouse unselected testis cells and (D) P6 mouse sorted spermatogonia. Gene lists can be found in Table S1. Colors and numbering of circles below heatmaps match the corresponding tSNE plot.
(E and F) Clusters of cells containing spermatogonia from (E) unselected P6 mouse testis cells (clutster 5 from A and B) or (F) sorted P6 mouse spermatogonia (clusters 1–9 from C and D) were subsequently used for unbiased dynamic lineage analysis producing cell trajectories with cells ordered in pseudotime.
(G and H) Retrospectively, (G) ID4-EGFPbr’9ht and (H) ID4-EGFPdim subpopulations contained within the sorted spermatogonia trajectory were displayed on the trajectory in isolation.
(I) Expression levels (vertical axis) of key genes among P6 sorted spermatogonia ordered in pseudotime (cell coloring is according to tSNE clusters from C). Similar plots for P6 mouse unselected spermatogonia are shown in Figure S6D.
(J) The heatmap shows hierarchical relationship between clusters of genesthat were differentially expressed across pseudotimefrom P6 sorted spermatogonia (scaled expression according to legend; see Table S2). The top five over-represented biological pathways from GO analyses ofeach cluster are noted at the right in bold (see Table S3), and key genes are italicized.
(K and L) Red immunostaining for (K) GFRA1 or (L) NDRG4 is shown together with green ID4-EGFP epifluorescence and blue F-actin counterstain (phalloidin) in sections of P6 Id4-Gfp testes (bar represents 50 μm; open arrowheads represent EGFPbright; solid arrowheads represent EGFPdim).
(M) The P6 spermatogonial trajectory (identical to Figure 6F) is colored by cell state and illustrates biologically significant differences across pseudotime and significant pathways are summarized to the right. Inset graph shows the distribution (percentage of the sorted ID4-EGFPbright spermatogonia and ID4-EGFPdim spermatogonia) as shown in (G) and (H), among the noted nine spermatogonial states.
Prior to expression of the prototypical SSC gene signature in P6 spermatogonia, we detected expression of a unique set of genes annotated for autophagy, phagosome maturation, and unfolded protein response (gene cluster 2; Figure 6I). Autophagy has been implicated in germ cell survival in the ovary prior to primordial follicle assembly (Gawriluk et al., 2011) and self-renewal of hematopoietic stem cells (Ho et al., 2017), and niche-driven autophagy is essential for proliferative expansion of germline stem cells in C. elegans (Ames et al., 2017). Expression of these genes in undifferentiated spermatogonia at P6, but not in adults, suggests potential unique involvement in establishing the foundational population of steady-state SSCs.
Expression of DNA repair and genome integrity gene pathways (BRCA1 DNA damage response; ATM signaling) was also greater in undifferentiated spermatogonia at P6 than in the adult testis (Figure 6J), which may reflect greater proliferation of spermatogonia in the immature testis. Reciprocally, expression of genes involved in mitochondrial function and oxidative phosphorylation was elevated in adult spermatogonia (Figure 6J), likely reflecting ongoing self-renewal of SSCs during steady-state spermatogenesis. Activation of genes involved in cell cycle control and replication (Ccnb1, Cdc20, Cdk1, Mcm5, and Pcna) was common to the transition from undifferentiated to differentiating spermatogonia at both P6 and adult stages (Figure 6J; Tables S2 and S3).
Analysis of a pooled dataset representing single-cell transcrip- tomes from immature and adult mouse spermatogonia resolved two clusters containing predominantly ID4-EGFPbright spermatogonia at P6 (clusters 3 and 5) but only a single cluster composed mostly of ID4-EGFPbright spermatogonia in the adult (cluster 2; Figure S6G; Table S1). The single cluster of adult ID4-EGFPbright spermatogonia was similar to one of the two clusters of P6 ID4-EGFPbright spermatogonia (cluster 5), with both showing enriched expression of genes involved in RNA metabolism and ribosome or ribonucleoprotein (RNP) biogenesis (Figure S6H). The other cluster of P6 ID4-EGFPbright spermatogonia (cluster 3) was unique to the immature testis and featured an overabundance of genes involved in cell cycle regulation, proliferation, and morphogenesis (Figure S6H). Of these clusters, only cell clusters 2 and 5 expressed the gene encoding TSPAN8, a marker which we previously demonstrated enriches for SSCs (Mutoji etal., 2016), suggesting that the foundational SSCs maintained until adulthood are a subpopulation of ID4-EGFPbright spermatogonia at P6 (Figure S6G). Thus, undifferentiated spermatogonia present in the immature testis are notably more heterogeneous than those in the adult testis. This is consistent with our suggestion that subsets of fetal prospermatogonia and/or neonatal undifferentiated spermatogonia adopt distinct epigenetic programming that predisposes their fate to form either the foundational SSC pool that ultimately sustains steady-state spermatogenesis or the non-self-renewing subset of spermatogonia that gives rise to the unique first wave of spermatogenesis (McCarrey, 2017).
Our analysis of single-cell transcriptomes directly reflects differential mRNA levels throughout spermatogenesis but does not detect the effects of post-transcriptional regulation of gene expression. To begin to investigate the extent of synchrony, or lack thereof, between gene expression at the RNA and protein levels, respectively, and to further validate our single-cell RNA sequencing (scRNA-seq) results, we performed immunostaining to localize the products of representative DEGs in sections of immature testes. We found that there was greater synchrony between upregulation of specific mRNAs and their encoded proteins (e.g., Ndrg4/NDRG4) than between downregulation of specific mRNAs and their encoded proteins (Gfra1 /GFRA1 and Dusp6/DUSP6), with the latter likely reflecting differential stabilities of encoded proteins following the reduction or loss of mRNA from each corresponding gene (Figures 6K, 6L, and S6C). Taken together, our single-cell transcriptome profiling of P6 mouse spermatogonia indicates that transitions among spermatogonial states in the immature testis reflect both the establishment of the foundational SSC pool as well as initiation of the first wave of spermatogenesis (Figure 6M).
qRT-PCR Analysis of Differentially Expressed Genes Validates the Single-Cell Transcriptomes and Provides Convenient Cell-type-Specific Gene Expression Signatures
As an independent validation of the continuum of spermatogenic cell types captured in our single-cell transcriptome data, and as an example of the utility of these datasets, we performed qRT- PCR to confirm the successive appearance of spermatogenic cell-type-specific gene expression signatures detectable in whole testis tissue across the first wave of spermatogenesis. We collected a daily series of testis tissue from mice at ages P6-P30, in which the sequential emergence of specific spermatogenic cell types is well characterized (Bellvé et al., 1977a; Figure 7). We identified simple, 3-gene expression signatures detectable by qRT-PCR and unique to each of 11 spermatogenic cell types or subtypes but absent from testicular somatic cells (Figure S7) and showed that the appearance of these signatures is consistent with the sequential emergence of each cell type during the first wave of spermatogenesis (Figures 7A-7C). These results provide a useful diagnostic tool for detection of specific spermatogenic cell types or subtypes within a testicular sample or biopsy. We also tested expression of a similar panel of cell-type-specific gene expression signatures in a series of 29 adult human testicular tissue biopsies with histologically confirmed complete spermatogenesis, as well as in StaPut-en- riched aggregates of human spermatogonia, spermatocytes, and spermatids (Figures 7D, 7E, and S7). Expression of all 11 spermatogenic cell-type-specific 3-gene signatures was detectable in the appropriate sorted cell population and each testis biopsy specimen (Figures 7D and 7E). These results demonstrate the utility and specificity of these expression signatures as a simple, rapid, and cost-effective yet highly specific means to assess or diagnose the presence or absence of any spermato-genic cell type in any sample of mouse or human testis tissue.
Figure 7. Validation of Single-Cell Transcriptomes and Derivation of Spermatogenic Cell-type-Specific Gene Expression Signatures.
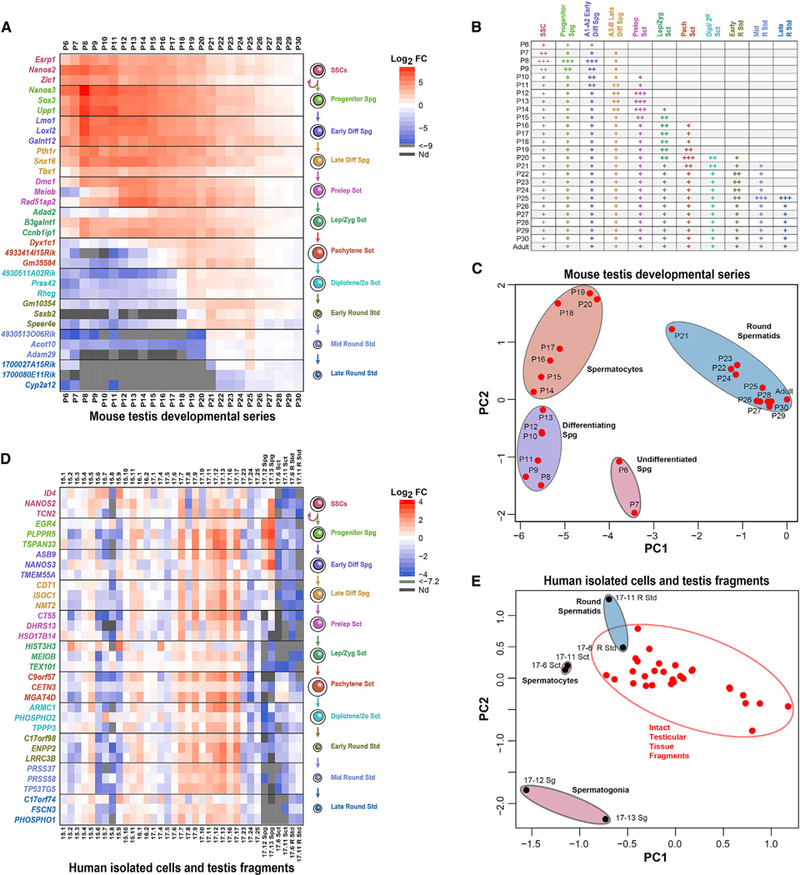
Germ-cell-specific genes that were significantly differentially expressed in pseudotime among spermatogonia, spermatocytes, and spermatids in both mice and men were tested for their ability to recognize specific spermatogenic cell types in complex mixtures using qRT-PCR.
(A) Log2 fold change values for qRT-PCR detection of 33 cell-type-specific signature genes in mouse testes at ages P6-P30 (made relative to levels in adult mouse testes).
(B) Detection of cell types in mouse testes by postnatal age (+++, robust; ++, moderate; +, detectable).
(C) Principal-component analysis (PCA) plot projects the qRT-PCR results from the 33-gene mouse spermatogenic cell gene expression panel.
(D) Log2 fold change values for qRT-PCR detection of 33 cell-type-specific signature genes in testis tissue fragments from 29 men or aggregate populations of isolated human spermatogonia, spermatocytes, or spermatids (relative to the gene-specific average from all 29 human tissue fragments).
(E) PCA plot of qRT-PCR results from the 33-gene human spermatogenic cell gene expression panel.
DISCUSSION
We used two different single-cell RNA-seq methods, 10x Genomics and Fluidigm C1, to perform a comprehensive analysis of gene expression during spermatogenesis in mice and humans. Our analysis of >62,000 individual spermatogenic cells from the immature and adult mouse testis and from the adult human testis afforded maximum resolution of gene expression patterns associated with the full continuum of cells within the mammalian spermatogenic cell lineage, including relatively transient, and hence rare, cell types for which little or no gene expression data were previously reported. We discerned 11 different, successive gene expression profiles corresponding to known cell types or subtypes within the spermatogenic lineage, from SSCs through haploid round spermatids, in both the mouse and human. In addition to confirming that spermatogenesis represents a continual progression of ever-changing gene expression profiles, our single-cell data have revealed more extensive heterogeneity among spermatogenic cell types or subtypes than previously reported.
We detected expression of a number of meiosis-related genes in spermatocytes, including the mouse and human homologs of Brdt, Dmrtb1, Fancm, Hspb1, Mcm8, Mcm9, Mei1, Meiob, Sycp2, Tex11, and Tex15, all of which manifest a maturation arrest male infertility phenotype when ablated in the mouse (Yat- senko et al., 2015). These potential sites of mutations in infertile men presenting with non-obstructive azoospermia (Flannigan and Schlegel, 2017) suggest our data may further inform ongoing efforts to identify novel genetic drivers of otherwise idiopathic male infertility. We also detected substantial variation in levels of transcripts from several recombination-related genes (Brca2, Spo11, RAD51, and RPA2), which may be related to the previously reported variation in levels of meiotic recombination among individual spermatocytes (Koehler et al., 2002). Loss of the meiotic program, reactivation of certain sex-linked genes as MSCI is replaced by postmeiotic sex chromatin (Turner et al., 2006), and gain of specific transcripts known to participate in chromatin repackaging during spermiogenesis (e.g., transition proteins and protamines) were all reflected among the transcrip- tomes evident in spermatids.
Heterogeneity among individual mouse spermatogonia allowed us to correlate different gene expression signatures with functionally distinct subpopulations of spermatogonia, including transplant-validated SSCs, progenitors, and differentiating spermatogonia. Although no similar functional readout is available for human spermatogonia, our ability to identify orthologous gene expression patterns conserved in subpopulations of functionally confirmed mouse spermatogonia and human spermatogonia, respectively, allowed us to delineate corresponding human sper- matogonial subtypes.
A recent report of single-cell transcriptomes of individual human spermatogonia analyzed SSEA4+ cells that those authors suggested were enriched for SSCs (Guo et al., 2017). Our results confirm that subsets of human SSEA4+ spermatogonia are phenotypically equivalent to mouse SSCs, and others bear more similarity to mouse progenitors and early differentiating spermatogonia. Thus, our data indicate that SSEA4+ human testis cells include a mixture of SSCs, progenitors, and early differentiating spermatogonia. Indeed, our results suggest that precise resolution of subpopulations of SSCs, progenitors, and differentiating spermatogonia in either mice or men requires simultaneous identification of two or more marker genes known to be expressed at elevated levels in SSCs (e.g., ID4 and NANOS2), in conjunction with the use of multiple marker genes known to be expressed at elevated levels in progenitors and differentiating spermatogonia (e.g., NANOS3 and PLPPR5).
Our refined assignments of spermatogonial subtypes facilitated investigation of the elusive mechanisms regulating the balance between the alternate SSC fates of self-renewal and initiation of differentiation. We confirmed that mouse and human SSCs exhibit conserved upregulation of glial cell line-derived neurotrophic factor (GDNF) signaling, as expected, but also enhanced translational control (EIF2, mTOR, and p70S6K signaling), suggesting that one mechanism driving self-renewal may involve selective translation of transcripts. We also found a distinct spermatogonial state that features elevated expression of genes associated with the hepatic stellate cell activation pathway, including the key transcription factor, KLF6, which may be related to initial specification and/or ongoing maintenance of SSC fate.
To provide an effective diagnostic asset for either basic studies of spermatogenesis or clinical detection of specific spermatogenic cell types, we identified convenient, 3-gene expression signatures unique to cell types or subtypes in the mouse and human spermatogenic lineages, respectively. These signatures can be used to simultaneously confirm the presence or absence of each of 11 spermatogenic cell types or subtypes in any sample of mouse or human testis tissue or isolated cells. This can be useful for assessing spermatogenic phenotypes associated with naturally occurring mutations or gene knockouts in the mouse orfor rapid detection of spermatogenic cell types in testes of subfertile men. The latter could improve resolution of diagnoses of closely related male subfertility pathologies, such as hypospermatogenesis, Sertoli cell only, or maturation arrest (McLachlan et al., 2007), and corresponding prognoses for the potential to use testicular sperm extraction (TESE) to facilitate the ability of these men to father their own children. An ultimate solution to male infertility may be the advent of successful spermatogenesis in vitro, and these cell-type-specific gene expression signatures should prove useful to validate progression through either the murine or human spermatogenic lineage.
In summary, the single-cell transcriptome data provided by this study represent an extensive, publicly accessible resource that will support future experimental studies of spermatogenesis in general and of male infertility or male contraception strategies in particular. We have made this resource particularly user friendly by providing these data in the form of fully analyzed and annotated Loupe Cell Browser files, allowing their easy download to query for individual genes to determine in which spermatogenic cell type(s) or subtype(s) any particular gene is expressed (see Key Resources Table). Importantly, this resource also provides a comprehensive gene expression roadmap to be emulated as efforts progress to optimize protocols for in vitro spermatogenesis (Saitou and Miyauchi, 2016; Irie and Surani, 2017).
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Biotin anti-mouse CD9 Antibody (Clone MZ3; 1 μg/106 cells) | Biolegend | Cat# 124804 |
| Biotin Rat IgG2a, κ Isotype Ctrl Antibody (Clone RTK2758; 1μg/106 cells) | Biolegend | Cat# 400504 |
| Streptavidin-APC (0.5μg/106 cells) | Biolegend | Cat# 405207 |
| APC Anti-Human HLA-ABC (Clone G46–2.6; 20μl/106 cells) | BD Biosciences | Cat# 562006 |
| APC Mouse IgG1 κ Isotype Control (Clone MOPC-21; 20μl/106 cells) | BD Biosciences | Cat# 555751 |
| APC anti-human CD49e Antibody (Clone NKI-SAM-1; 20μl/106 cells) | Biolegend | Cat# 328011 |
| APC Mouse IgG2b, κ Isotype Ctrl Antibody (Clone MPC-11; 10μl/106 cells) | Biolegend | Cat# 400321 |
| Biotin Mouse Anti-Human CD90 (Clone 5E10, 1μg/106 cells) | BD PharMingen | Cat# 555594 |
| Biotin Mouse IgG1 κ Isotype Control (Clone MOPC-21; 2μl/106 cells) | BD PharMingen | Cat# 555747 |
| PE/Cy7 Streptavidin (2.5μl/106 cells) | Biolegend | Cat# 405206 |
| Pacific Blue anti-human/mouse CD49f Antibody (Clone GoH3; 20μl/106 cells) | Biolegend | Cat# 313619 |
| Pacific Blue Rat IgG2a, κ Isotype Ctrl Antibody (Clone RTK2758; 8μl/106 cells) | Biolegend | Cat# 400527 |
| PE anti-human CD326 (EpCAM) Antibody (Clone 9C4; 20μl/106 cells) | Biolegend | Cat# 324205 |
| PE Mouse IgG2b, κ Isotype Ctrl Antibody (Clone MPC-11; 2.5μl/106 cells) | Biolegend | Cat# 400313 |
| Alexa Fluor 488 anti-human SSEA-4 Antibody (Clone MC813–70; 5μl/106 cells) | Biolegend | Cat# 330412 |
| Alexa Fluor 488 Mouse IgG3, κ Isotype Ctrl Antibody (Clone MG3–35; 4μl/106 cells) | Biolegend | Cat# 401324 |
| Rabbit anti-KCTD9 antibody [EPR13909], 0.142 μg/ml | Abcam | Cat# ab180937 |
| Donkey Anti-Rabbit IgG H&L (Alexa Fluor 555) preadsorbed, 1:500; Figure S1I & NDRG4 (Figure 6L) | Abcam | Cat# ab150062 |
| Rat anti-Germ cell-specific antigen antibody [TRA98], 1 Rabbit anti-KCTD9 antibody μg/ml; Figure S1I | Abcam | Cat# ab82527 |
| Donkey Anti-Rat Alexa Fluor 647 (IgG H&L) preadsorbed; 1:500; Figure S1I | Abcam | Cat# ab150155 |
| Goat anti-GATA4 antibody (C-20), 2 μg/ml; Figures 6K and S1H | Santa Cruz | Cat# sc-1237 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed, Alexa Fluor 555; 1:500; Figures 6K (GFRA1), S1H (GATA4), and S6C (GATA4) | ThermoFisher Scientific | Cat# A21432 |
| Rabbit anti-DDX4/MVH antibody (1 μg/ml); Figures S1J and S2Q | Abcam | Cat# ab13840 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed, Alexa Fluor 647; 1:500; Figures S1J and S2Q | ThermoFisher Scientific | Cat# A31573 |
| Goat anti-GFRA1 antibody, 0.25 μg/ml; Figure 6K | R&D Systems | Cat# AF560 |
| Rabbit anti-NDRG4 antibody; Figure 6L | Proteintech | Cat# 12184–1-AP |
| Goat anti-Human VASA antibody (diluted to 2μg/ml); Figure S2N | R&D Systems | Cat# AF2030 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed, Alexa Fluor 568 (diluted 1:200); Figure S2N | ThermoFisher Scientific | Cat# A11057 |
| Rabbit anti-SOX9 antibody (diluted to 4μg/ml); Figure S2N | EMD Millipore | Cat# AB5535 |
| Donkey anti-Rabbit IgG (H+L) Cross-Adsorbed, Alexa Fluor 488 (diluted 1:200); Figure S2N | ThermoFisher Scientific | Cat# A21206 |
| Rabbit Anti-DUSP6 antibody [EPR129Y] (diluted to 2.5μg/ml); Figure S3I | Abcam | Cat# 76310 |
| chicken anti-rabbit IgG-Biotin; Figure S3I | Santa Cruz Biotechnology | Cat# sc-2986 |
| Streptavidin HRP; Figure S3I | Sigma-Aldrich | Cat# S5512 |
| Rabbit Anti-RHCG antibody (diluted to 4μg/ml); Figure S4J | GeneTex | Cat# GTX60193 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed, Alexa Fluor 488 (diluted 1:200); Figure S4J | ThermoFisher Scientific | Cat# A11108 |
| Rabbit anti-ACTL7B antibody (diluted 1:750); Figures S5I and S5J. | Proteintech Group | Cat#13537–1-AP |
| Biological Samples | ||
| Human testicular tissue biopsies | Infertility Center of St. Louis | Table S5 |
| Human testicular tissue from organ donors | University of Texas Transplant Center & Texas Organ Sharing Alliance | Table S5 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3’ v2 Reagent Kits & Chip Kits | 10X Genomics | Cat#s 120237,120267, 1000009, 120236 |
| SMARTer Ultra Low RNA Kit for the Fluidigm C1 System (v2) | Takara/Clontech | Cat#s 634832, 634833 |
| Deposited Data | ||
| Raw and analyzed P6 ID4-EGFP spermatogonia C1 scRNA-seq data | This paper | GEO: GSE108970 |
| Raw and analyzed Adult ID4-EGFP spermatogonia C1 scRNA-seq data | This paper | GEO: GSE108974 |
| Raw and analyzed Adult Human Spermatogonia C1 scRNA-seq data | This paper | GEO: GSE108977 |
| Raw and analyzed P6 ID4-EGFP testis and spermatogonia 10X Genomics Drop-Seq data | This paper | GEO: GSE109049 |
| Raw and analyzed Adult Mouse spermatogenic cell 10X Genomics Drop-Seq data | This paper | GEO: GSE109033 |
| Raw and analyzed Adult Human spermatogenic cell 10X Genomics Drop-Seq data | This paper | GEO: GSE109037 |
| Mendeley Data - Queryable single-cell RNA-seq (Drop-Seq) datasets of Human and Mouse spermatogenic cells; Loupe Cell Browser files (from 9 datasets) for easy retrieval and interrogation of the resource data in this manuscript. | This paper | Mendeley Data: https://doi.org/10.17632/kxd5f8vpt4.1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX:000664 |
| Mouse: C57BL/6NTac | Taconic Farms | TAC:b6 |
| Mouse: B6-Cg-Id4-Egfp(LT-11) | Chan et al., 2014 | N/A |
| Mouse: B6;129S-Gt(ROSA)26Sor/J | Friedrich and Soriano, 1991 | JAX:002073 |
| Mouse: 129S1/SvImJ | The Jackson Laboratory | JAX:002448 |
| Oligonucleotides | ||
| Primers for qRT-PCR (Figure 7) | This paper | Table S7 |
| Software and Algorithms | ||
| FlowJo v9 | FlowJo, LLC | N/A |
| AxioVision 4.8.2 | Zeiss | N/A |
| Galaxy | Giardine et al., 2005; https://usegalaxy.org | SCR_006281 |
| TopHat (Galaxy v0.9) | Trapnell et al., 2012 | SCR_013035 |
| Cufflinks/Cuffnorm (Galaxy v.2.2.1.0) | Trapnell et al., 2012 | SCR_014597 |
| Picard tools (Collectrnaseqmetrics) | Broad Institute, http://broadinstitute.github.io/picard | SCR_007073 |
| SINGuLar analysis toolset 3.1 | Fluidigm | SCR_015685 |
| Seurat2.3.0 | Butler, 2018. https://github.com/satijalab/seurat | N/A |
| Monocle2 | Trapnell et al., 2014 and Qiu et al., 2017. http://cole-trapnelllab.github.io/monocle-release/ | N/A |
| Ingenuity Pathway Analysis, Build 477929M, Content versions 43605602 (3/2018) and 44691306 (6/2018) | QIAGEN | SCR_008653 |
| Cell Ranger v1.3.0 and v2.0.1 | 10X Genomics, https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | N/A |
| Loupe Cell Browser v2.0.0 | 10X Genomics, https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | N/A |
| Cell Ranger R kit v2.0.0 | 10X Genomics, https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/rkit | N/A |
| Real-time PCR Analysis Software | Fluidigm | SCR_015686 |
| Other | ||
| Human reference genome NCBI build 37, GRCh37 (hg19) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13/ |
| Mouse reference genome NCBI build 38, GRCm38 (mm10) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ |
| Human reference GRCh38 dataset required for Cell Ranger, v1.2.0 | 10X Genomics, https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | N/A |
| Mouse reference GRCm38 (mm10) dataset required for Cell Ranger, v1.2.0 | 10X Genomics, https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | N/A |
| Human GRCh37 (hg19) and Mouse GRCm38 (mm10) reference dataset required for Cell Ranger, v1.2.0 | 10X Genomics, https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Inquiries regarding reagents and resource sharing should be directed by email (Brian.Hermann@utsa.edu) to the Lead Contact for this study. All datasets (raw and analyzed) produced in the conduct of these studies are publically available at NIH GEO and Mendeley Data (Key Resources Table). There are no restrictions for the use of these data in future studies.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human testicular tissue
De-identified, surgical excess normal adult human testicular tissue was obtained through the Infertility Center of St. Louis from patients undergoing a diagnostic testicular biopsy (n = 28) in preparation for microscopic vasectomy reversal (MVR) or testicular sperm extraction (TESE) for obstructive azoospermia with the informed consent of patients (see Table S5). De-identified testicular tissue was also recovered from male organ donors (n = 2) through the Texas Organ Sharing Alliance and the University of Texas Transplant Center with informed consent of the next of kin. The University of Texas at San Antonio IRB determined that research on these tissues did not constitute human subjects research (IRB #12–098N and #17–074N). The age of the individuals was recorded (median = 42yr, mean = 41.6 ± 1.3yr, Table S5), and while organ donors tended to be younger (34.3 ± 7.2yr) than biopsy patients (42.4 ± 1.2yr), the difference was not significant (p = 0.32). Procured tissue was transported to the laboratory on ice in either Lactated Ringer’s solution or minimal essential medium alpha (MEMa) containing 10% fetal bovine serum. The amount of time tissues were exposed to cold ischemia priorto processing was recorded (Table S5, median = 18.7hr; mean = 24.7 ± 3.8 hr). In all cases, a portion of the testicular tissue was snap frozen for RNA extraction and a portion was fixed in 4% paraformaldehyde and/or Bouin’s solution. For many specimen, though, the bulk of the tissue was used for cell isolation as noted below.
Mice
All experiments utilizing animals were approved by the Institutional Animal Care and Use Committees of the University of Texas at San Antonio (Assurance A3592–01), East Carolina University (Assurance A3469–01) or Washington State University (Assurance A3485–01) and were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. All animals were maintained under conditions of ad libitum water and food with constant light-dark cycles. For single-cell and transplant studies, testes from 6-day postpartum (P6) or adult F1 male offspring (at least two per experiment) from a cross between Id4-eGfp [LT-11B6; (Chan et al., 2014)] and either C57BL/6J or B6;129S-Gt(ROSA)26Sor/J [(Friedrich and Soriano, 1991); both from The Jackson Laboratory] were used to generate suspensions of cells following enzymatic digestion, as described (Oatley and Brinster, 2006; Hermann et al., 2015). Recipient mice for spermatogonial stem cell transplant were 129 x C57BL/6J F1 hybrid male offspring (originally from The Jackson Laboratory). Pregnant female C57BL/6NTac mice or lactating female mice with litters (both from Taconic Farms) were used as a source of male offspring for gene expression studies in testes from postnatal day (P) 6 through P30. Adult male C57BL/6NTac mice were also used for this experiment (Taconic Farms) and in all cases, testicular tissue was snap frozen for RNA extraction.
METHOD DETAILS
Generation of cell suspensions
Testes from P6 mice were used to generate suspensions of cells following enzymatic digestion as described previously (Ogawa et al., 1997; Oatley and Brinster, 2006). Briefly, testicular parenchyma from at least two pups were pooled and digested with 0.25% trypsin/EDTA (ThermoFisher Scientific) for ~6–8 minutes at 37°C, quenched with 10% FBS (v/v, Sigma). Single-cell suspensions of seminiferous tubules were generated from adult mouse testes using a two-step enzymatic digestion approach. Briefly, testicular parenchyma from 2 or more adult mice were digested with 1μg/ml Collagenase Type IV (Worthington Biochemicals) for 2–3 minutes at 37°C, washed with Hank’s Buffered Salt Solution (HBSS) to remove interstitial cells, digested with 0.25% trypsin/EDTA containing 1.4μg/ml DNase I (Sigma) for 7–9 minutes at 37°C, and quenched with 10% FBS. Suspensions of human seminiferous tubule cells were prepared from adult testicular tissue parenchyma by a two-step enzymatic digestion as described previously (Hermann et al., 2007; Hermann et al., 2009; Dovey et al., 2013). Briefly, testis tissue was digested with 2μg/ml collagenase type IV at 37°C with vigorous agitation, washed with HBSS to remove interstitial cells, followed by digestion with 0.25% trypsin/EDTA containing 1.4μg/ml DNase I for 15 minutes at 37°C with trituration every 5 minutes, and quenched with 10% FBS. In all cases, cell suspensions were strained (40μm for mouse, 70μm for humans) and suspended in MEMα containing 10% FBS.
Spermatogonia isolation by FACS
Testis cell suspensions were used for FACS to enrich spermatogonia essentially as described (Hermann et al., 2015). Briefly, cells were suspended (5–20 × 106 cells/ml) in ice-cold Dulbecco’s phosphate-buffered saline (DPBS) containing 10% FBS (DPBS+S), labeled with antibodies (see Key Resources Table), and subjected to flow cytometry using an LSRII cytometer (BD) or FACS using either a FACS Aria (BD) or SY3200 (Sony). Positive antibody labeling was determined by comparison to staining with isotype control antibodies (see Key Resources Table). Positive ID4-EGFP epifluorescence was determined by comparison to testis cells from Id4- eGfp-negative littermates (for experiments at P6) or C57BL/6 males (for experiments in adults). Dead cells were discriminated with propidium iodide (Biolegend). Spermatogonia from P6 mice were isolated by FACSs based on ID4-EGFP+ gating (and in some cases, subgating into EGFPbright and EGFPdim sub-populations). To isolate adult mouse spermatogonia, seminiferous tubule cells were preenriched for spermatogonia by density centrifugation in DPBS+S over a 30% Percoll cushion (Sigma) for 8 minutes at 600xg without braking. Pelleted cells were subjected to FACS and spermatogonia isolated based on gating CD9-bright and ID4-EGFP double-positive cells (and in some cases, subgating into EGFPbright and EGFPdim sub-populations). Human spermatogonia were isolated by sorting cells with the phenotype HLA-ABCnegative/CD49enegative/THY1dim/ITGA6+/EpCAMdim dim (see Figure 2F). These markers were chosen because previous results indicate they specifically label undifferentiated spermatogonia, including some with colonization potential based on xenotransplantation (Dovey et al., 2013; Valli et al., 2014). In some cases, human cells were also stained with an SSEA4 antibody in addition to the 5-marker panel noted above. All antibodies and their dilution for staining is noted in the Key Resources Table. Seminiferous tubule suspensions used for 10X Genomics analysis were sorted for viable cells (PI-negative selection) prior to use.
Spermatocyte/spermatid enrichment by StaPut
Cells from adult Id4-eGfp+ mouse and adult human seminiferous tubules were enriched for spermatocytes and spermatids based on sedimentation velocity at unit gravity (Romrell et al., 1976; Bellvé et al., 1977b). Briefly, testis cells (106-107) suspended in 2 mL of buffer plus 0.5% BSA were loaded onto a 50 mL gradient of 2%−4% BSA (McCarrey et al., 1992) and allowed to sediment for 2.5 hr at 4°C. Approximately one hundred 0.5 mL fractions were then collected in microcentrifuge tubes and analyzed for content of spermatocytes or spermatids on the basis of morphology under phase contrast optics (which typically yields ≥ 85% purity). Fractions containing spermatocytes or spermatids were pooled separately, concentrated (to ~2 ×106 cells/ml) and stored in buffer containing FBS on ice until use.
Fluidigm C1 Single-cell RNA-seq
ID4-EGFP+ spermatogonia from P6 mice, CD9bright/ID4-EGFP+ spermatogonia adult mice or HLA-ABCnegative / CD49enegative / THY1dim / ITGA6+ / EpCAMdim human spermatogonia were used for single-cell RNA-seq facilitated by the Fluidigm C1 instrument essentially as described (Hermann et al., 2015; Wu et al., 2014). Briefly, single cells were captured on 10–17μm integrated fluidic circuit chips using the C1 Single-Cell Autoprep System (Fluidigm), stained with ethidium homodimer, imaged on an AxioImager M1 (Zeiss), and used to prepare cDNA with SMARTer Ultra Low RNA Kit for the Fluidigm C1 System (v2 chemistry; Takara). Dead cells (ethidium+), multiplets and cells contaminated with debris were excluded from further analysis. Mouse ID4-EGFP+ spermatogonia were stratified based on the EGFP epifluorescence intensity using the interactive measurement module of AxioVision 4.8.2 (Zeiss) and images taken of each cell. The densitometric mean value of the EGFP channel for each cell was normalized on a scale from 0 to 1 (1 representing the brightest EGFP+ cell) and cells were grouped into quartiles for retrospective evaluations. Routine 250 cell and 0-cell off-chip controls were performed in parallel with each experiment. Amplified cDNA was quantified by PicoGreen flu-orometry (ThermoFisher Scientific) on a Synergy II (Biotek) based on manufacturer recommendations and normalized cDNA mass from each cell was used for Nextera XT dual-index library preparation (Illumina) with modifications from manufacturer recommendations essentially as described (Mutoji et al., 2016). Single-cell libraries were pooled, qualified for fragment size and distribution on a 2100 Bioanalyzer (Agilent), quantified and sequenced on an Illumina Hiseq2500 generating 100bp paired-end reads at the UT Southwestern Microarray and Genomics Core. Primary analysis of C1 single-cell RNA-Seq data was performed using a public-use server (https://usegalaxy.org) running the Galaxy NGS analysis environment [see https://galaxyproject.org/; (Giardine et al., 2005)]. Paired FASTQ files from each sample were trimmed and the quality confirmed using FASTQC, aligned to the mouse genome GRCm38 (mm10) or the human genome GRCh37 (hg19) with TopHat (Galaxy v0.9) and transcript abundance was determined with Cufflinks (Galaxy v.2.2.1.0) (Trapnell et al., 2012). Cuffnorm (Galaxy v.2.2.1.0) was used to merge transcript abundance values [Fragments per Kilobase per Million mapped (FPKM)] for each cell into a single matrix. Quality control with the Picard tool Collectrnaseqmetrics was used to eliminate poorly quality cells (http://broadinstitute.github.io/picard) and quality control metrics from retained cells are found in Table S6.
Single-cell transcriptomes derived using the 10X Genomics Chromium
Cell suspensions were loaded into Chromium microfluidic chips with 3´ v2 chemistry and used to generate single-cell gelbead emulsions (GEMs) using the Chromium controller (10X Genomics) per manufacturer recommendations (Zheng et al., 2017). In all cases, suspensions containing ~8700 cells were loaded on the instrument with the expectation of collecting up to 5,000 GEMs containing single cells. For the multiplet test (Figures S1E-S1G), equal numbers of mouse and human cells were loaded. GEM-RT was performed in aT100 Thermal cycler (Bio-Rad) and all subsequent steps to generate single-cell libraries were performed according to manufacturer recommendations. Libraries were sequenced at the Genome Sequencing Facility (GSF) at Greehey Children’s Cancer Research Institute in the UT Health San Antonio (UTHSA) on either a HiSeq3000 or NextSeq 500 instrument (Illumina). Trimmed FASTQ files (26bp Cell barcode and UMI Read1, 8bp i7 index, and 100bp Read2), were generated using the CellRanger mkfastq command (a 10X Genomics wrapper around BCL2Fastq). Primary data analysis (alignment, filtering, and UMI counting) to determine gene transcript counts per cell (producing a gene-barcode matrix), quality control, clustering and statistical analysis were performed using CellRanger count (10X Genomics) and either GRCh38 (human hg38), GRCm38 (mouse mm10) ora combined human GRCh37 (hg19) + mouse GRCm38 (mm10) (for multiplet tests) genome assembly/annotation references (see Key Resources Table). Outputs from multiple independent samples of single-cells were combined using CellRanger aggr (10X Genomics) based on mapped read counts to normalize sequencing depth and produce aggregated gene x cell barcode matrices and clustering models. The Loupe Cell Browser v2.0.0 (10X Genomics) was used to visualize results. Quality control metrics from these data are found in Table S6.
Single-cell RNA-seq secondary analyses
Raw count matrices (10X Genomics) or FPKM matrices (Fluidigm C1) were imported to Seurat2.3.0 (Butler et al., 2018), filtered (cells expressing ≥ 200 detected genes, genes expressed in ≥ 3 cells) and gene expression values were log normalized and scaled. Unsupervised cell clustering and tSNE analysis were performed in Seurat based on the statistically significant principal components. The top 10 differentially-expressed genes (marker genes) of each cell cluster were determined by log fold change ≥ 0.25 using a default Wilcoxon rank-sum test. After identification of cell clusters, raw count matrices without cluster subsets (e.g., without testicular somatic cells) were imported to Monocle2 (Trapnell et al., 2014; Qiu et al., 2017) and used for additional combined t-SNE and unsupervised density peaks clustering. Differentially expressed genes or significantly variable genes among cells were identified and used for dynamic trajectory analysis which ordered cells in pseudotime. Lists of differentially- expressed genes were analyzed by Ingenuity Pathway Analysis [QIAGEN, Build 477929M, content versions 43605602 (3/2018) and 44691306 (6/2018)] to identify biological pathways that are significantly over-represented among the genes in each list. For some GO analyses, the Bioconductor OrgDb or KEGG databases were used. Comparison between human and mouse cells was performed by first limiting the respective gene-cell matrices to include only the 16,859 orthologous genes annotated in both species (Ensembl) and further limited in a cell-type-specific manner to include only the orthologous genes expressed in either species in that cell type. Seurat was then used to perform differential expression analysis of merged matrices based on the non-parametric Wilcoxon rank-sum test.
Testis tissue immunostaining
For KCTD9/TRA98, GATA4/DDX4, GFRA1, and NDRG4 immunofluorescent staining experiments (Figures S1H, S1I, S4J, 6K, and 6L), staining was performed as described (Hermann et al., 2015). Briefly, P6 and Adult Id4-eGfp+ testes were fixed with 4% PFA at 4°C for 2 hr or O/N, respectively, washed extensively with DPBS, soaked in 30% sucrose, embedded in OCT medium and frozen. Frozen sections (5μm) were cut and placed on positively-charged slides and stored at −80°C prior to use. Sections were blocked for 1 hr at room temperature in 1X PBS containing 3% BSA + 0.1% Triton X-100, stained for 1 hr in antibody diluted in blocking buffer (see list of antibodies and dilutions in Key Resources Table), and washed with 1X PBS + 0.1% Triton X-100. Indirect immunofluorescence labeling was then performed with secondary antibodies (see Key Resources Table) plus phalloidin-635 (1:500, Life Technologies) for 1 hr at room temperature. Primary antibody was omitted as a negative control. After additional stringency washes, sections were mounted with Vectastain either with or without DAPI (Vector Laboratories), coverslipped, and images obtained using a Fluoview FV1000 confocal laser-scanning microscope (Olympus America). EGFP signal was from epifluorescence, not antibody labeling. Each staining was performed in triplicate on testes from at least 2 different animals. Primary antibodies were omitted as a negative control. Immunofluorescent staining for the proteins DDX4/SOX9 in adult human testis sections and RHCG in adult mouse sections (Figure S2N) was as described (Hermann et al., 2009). Briefly, adult mouse or human testis fragments were fixed with 4% PFA at 4°C O/N, washed extensively with DPBS, paraffin embedded and sectioned (5μm). Sections were de- paraffinized, rehydrated, subjected to antigen retrieval in sodium citrate buffer (10mM Sodium citrate pH 6.0, 0.05% Tween-20) or EDTA buffer (1mM EDTA pH 8.0, 0.05% Tween-20), rinsed, and blocked in antibody diluent (DPBS + 0.1% Triton X-100, 5% normal serum from host species of secondary antibody, 3% BSA). Blocked sections were labeled concurrently with the noted antibodies for 90 minutes at ambient temperature, washed (DPBS + 0.1% Tween-20), stained with secondary antibodies for 45 minutes at ambient temperature and washed again. Sections were counterstained with 1ug/ml Hoechst 33342 (Sigma-Aldrich) to identify nuclei during the secondary antibody incubation. Positive immunoreactivity was validated by omission of primary antibody. Fluorescently stained sections were mounted with FluoromountG (Southern BioTech) and imaged at 20X magnification using an AxioImager M1 (Zeiss) and an AxioCam MRm (Zeiss). For mouse ACTL7B immunohistochemical (IHC) staining, testes were fixed overnight with Bouin’s solution (Sigma) and washed extensively with 70% EtOH prior to embedding in paraffin. For DUSP6 and ACTL7B IHC staining in human and mouse testes, respectively (Figures S3I, S5I, and S5J), tissues were PFA-fixed and paraffin-embedded. Sections for IHC staining were deparaffinized, rehydrated, subjected to citrate antigen retrieval, and non-specific peroxidases were blocked with 6% (v/v) H2O2 for 15 minutes at ambient temperature, and then blocked with 5% (w/v) bovine serum albumin in DPBS for 30 minutes at ambient temperature. Primary antibody incubation was at 4°C overnight (non-immune IgG antibody served as negative control) followed by washes, and incubation with biotinylated secondary antibody, and lastly a streptavidin-conjugated HRP (S5512). Staining was visualized using 3,3’-diaminobenzidine (DAB) as chromogen, and hematoxylin as nuclear counterstain.
Spermatogonial stem cell transplantation
Cells from adult Id4-eGfp+ / Rosa-LacZ F1 hybrid male mice were sorted and transplanted into the seminiferous tubules of busulfan- treated recipient mice, essentially as described (Oatley and Brinster, 2006). Briefly, sorted cell suspensions were diluted in medium to 0.5×106 cells/ml and ~10 μL was microinjected into the seminiferous tubules of each adult 129XC57 F1 hybrid busulfan-treated (60 μg/kg) recipient mouse testis. One testis of each recipient received CD9Bright/ID4-EGFPBright cells and the contralateral testis received CD9Bright/ID4-EGFPDim cells. Presence of donor-derived colonies of spermatogenesis was detected 2–3 mo post-transplantation by staining with X-Gal and spermatogenic colonies were counted. Results shown are from 20 recipient testes and 3 replicate cell sorting and transplant experiments and statistically-significant results were identified using Student’s t tests.
qRT-PCR
Cells or testis tissue were homogenized in Trizol (ThermoFisher Scientific) and RNA was extracted according to the manufacturer recommendations. Genomic DNA was removed with the Turbo DNA-free kit (ThermoFisher Scientific). Complementary DNA was synthesized from DNase-treated RNA as described (Lovelace et al., 2016) using SuperScript III reverse transcriptase (ThermoFisher Scientific) and oligo-dT18 priming. Primers were designed using consensus coding sequences (CCDS) from NCBI according to gene ID using PrimerQuest (Integrated DNA Technologies) and Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/), selecting the most specific primer sets with low self-complementarity, favoring intron-spanning pairs where possible (Hermann and Heckert, 2005). Primer validation was performed by qRT-PCR on the BioMark HD System (Fluidigm, see details below) using 6 ×10-fold serial dilutions of cDNA from either mouse or human testis samples, selecting only primer sets which exhibited 85%−100% efficiency (Table S7), single products on melt-curve analysis, and lack of amplification in -RT and water negative control samples. An aliquot of each cDNA (250 ng) was subjected to 18-cycles of pre-amplification using Preamp Master Mix (Fluidigm) in 5 μl reaction volumes with pools of all forward and reverse primers for the amplicons of interest (500 μM) to produce sufficiently-concentrated samples for high-throughput microfluidic qPCR. A 2.25 μL aliquot of each preamplified, diluted (1:10) cDNA was mixed with 2.5 μL of 2X SsoFast EvaGreen Supermix with Low ROX (Bio-Rad) and 0.25 μL of 20X DNA Binding Dye Sample Loading Reagent (Fluidigm), which was then pipetted into an individual sample inlet in a 96.96 Dynamic Array integrated fluidic circuit (IFC) chip (Fluidigm). Individual qPCR primer pairs (pool forward and reverse, 100 μM each, Table S7) were diluted 1:10 with TE (10μM Tris pH 8.0, 0.1μM EDTA; 2.5 mL total volume), mixed with 2.5 mL Assay Loading Reagent (Fluidigm), and then individually pipetted into individual assay inlets in the same 96.96 Dynamic Array IFC chip. Samples and assays were loaded into the IFC chambers with an IFC Controller HX (Fluidigm) and qPCR was performed with the BioMark HD real-time PCR reader (Fluidigm) following the manufacturer’s instructions using standard fast cycling conditions and melt-curve analysis, generating an amplification curve for each gene of interest in each sample (9,216 reactions per IFC). Quantitative PCR results were analyzed using Fluidigm’s Real-time PCR Analysis software with the following parameters: 0.65 curve quality threshold, linear derivative baseline correction, automatic thresholding by assay (gene), and manual melt curve exclusion. Cycle threshold (Ct) values for each reaction were exported for further analysis. For the mouse gene expression signature experiment (Figures 7A-7C), data were from two biological replicate samples for each mouse age and two technical replicates per sample (from two independent IFCs). For the human gene expression signature experiment (Figures 7D and 7E), data were from four technical replicates per sample (two replicates on each of two independent IFCs). The relative mRNA abundance for each gene of interest was calculated using the ΔΔCt method where Rps2 cDNA (mouse) or RPL7 cDNA (human) amplification was used for normalization to determine the fold-change value (2-ΔΔCt) relative to the adult mouse sample or the average of all intact human testis samples, respectively. Significant differences between samples were identified using t tests.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analyses of single-cell transcriptomes
Raw count matrices generated by Cell Ranger (10X Genomics) or FPKM matrices (C1) were imported to Seurat2.3.0 and filtered for only high-quality cells. Briefly, we removed cells with less than 200 detected genes and genes detected in 3 or fewer cells. Gene expression values were log normalized and scaled before further downstream analyses. Cell clustering and tSNE analysis were performed based on the statistically significant principal components. Marker genes of each cell cluster were determined by Log Fold Change threshold above 0.25 using the default Wilcoxon rank-sum test (top-ten DEGs and full DEG lists are shown in Tables S1 and S2). After identification of cell clusters, raw count matrices of data subsets (i.e., without somatic cells or including only spermatogonia) were imported to Monocle2 and only expressed genes above threshold (0.1) were used for analyses (of note, this generally led to very little additional filtering above those genes already excluded by Seurat). Differentially expressed genes or significantly variable genes among cells were identified by Monocle2 and used for ordering cells in pseudotime. Only genes with a dispersion ratio above 0.1 were used for training the pseudotime trajectories (of note, using a FDR q-value cutoff ≤ 0.1 produced nearly identical results). To generate the pseudotime heatmaps, DEGs among cell clusters in pseudotime with qval <0.1 were included and clustered hierarchically based on their expression trends.
Identifying the number of persistently-expressed genes in spermatogenesis
Using the human unselected spermatogenic cell dataset, we found that the total number of expressed (detected) genes was 28,625. Based on scaled expression levels, 9,400 of these genes exhibited lower variance than average (mean variance = 0.76550), and thus, were considered constantly expressed, and 19,225 genes with higher variance are differentially expressed during human spermatogenesis.
Using the mouse unselected spermatogenic cell dataset, 20,939 genes were detected (expressed), and based on scaled expression, 7,031 genes exhibited lower variance than average (mean variance = 0.76787) were considered to be constantly expressed, and 13,908 genes with higher variance were differentially expressed during mouse spermatogenesis.
Single-cell transcriptome comparisons between mouse and human cell types
Using default parameters, statistically-significant differential gene expression was calculated using Seurat based on the non-para- metric Wilcoxon rank-sum test. Only genes that exhibited log fold-change values of ≥ 1 and p values < 0.01 were considered significant.
qRT-PCR analyses
Raw Fluidigm qRT-PCR data were analyzed with the following parameters: 0.65 curve quality threshold, linear derivative baseline correction, automatic thresholding by assay (gene), and manual melt curve exclusion. Cycle threshold (Ct) values for each reaction were exported for further analysis and relative mRNA abundance for each gene of interest was calculated using the ΔΔCt method where Rps2 cDNA (mouse) or RPL7 cDNA (human) amplification was used for normalization to determine the fold-change value (2-ΔΔCt) relative to the adult mouse sample or the average of all intact human testis samples, respectively. Significant differences between samples were identified using Student’s t tests.
Supplementary Material
Highlights.
Single-cell transcriptomes of >62,000 spermatogenic cells from mice and humans
Human SSC fate regulation pathways match those of functionally defined mouse SSCs
The hepatic stellate cell activation pathway is associated with SSC fate
Unique 3-gene identifiers distinguish 11 spermatogenic cell types in mice and humans
ACKNOWLEDGMENTS
The authors are grateful for computational support from Armando Rodiguez and Jeremy Mann in the UTSA Research Computing Support Group, as well as the Shamu HPC cluster operated by the UTSA Office of Information Technology. Sequencing data were generated by Chaoying Liang and Kasthuribai Viswanathan in the UT Southwestern Genomics and Microarray Core, as well as Dawn Garcia and Zhou Lai from the Genome Sequencing Facility (GSF) at Greehey Children’s Cancer Research Institute in the UT Health San Antonio (UTHSA). The graphical abstract was produced by Victoria Foor. This work was supported by (in no particular order) NIH grants P30-CA054174 (UTHSA), P30-GM092334 (J.R.M.), R01-HD061665 (J.M.O.), R00-HD062687 (B.P.H.), R01-HD078679 (J.R.M.), R01-HD90007 (B.P.H.), R01-HD90083 (C.B.G.), G12-MD007591 (UTHSA), S10-OD021805 (UTHSA), and UL1-RR025767 (UTHSA); NSF grant DBI-1337513 (B.P.H.); CPRIT Core Facility Award RP160732 (UTHSA); a fellowship from the Deutsches Primatenzentrum (B.W.); a UTHSA IIMS/CTSA Translational Technology Supplement (B.P.H.); the Max and Minnie Tomerlin Voelcker Fund (B.P.H.); the Helen Freeborn Kerr Charitable Foundation (B.P.H.); and the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation (J.R.M.).
Footnotes
DATA AND SOFTWARE AVAILABILITY
All software used for this study were publically available as noted in the Key Resources Table, and no new software was produced for the conduct of these studies. All newly-generated genomics data reported in this study are available through the NIH Gene Expression Omnibus (GEO) and Sequence Read Archive (SRA) databases under accession numbers GSE108970, GSE108974, GSE108977, GSE109049, GSE109033, and GSE109037 (see Key Resources Table). To facilitate ease of interrogation of these data by the scientific community, analyzed and annotated datasets in Loupe Cell Browser format were deposited in the Mendeley Data repository (https://data.mendeley.com) under https://doi.org/10.17632/kxd5f8vpt4.1. Users should download the appropriate Loupe Cell Browser verison (see https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest) to facilitate queries of these data. Please note that due to software limitations, these Loupe files present the results of this study in a different format than as they appear in the manuscript figures.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and seven tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.10.026.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ames K, Da Cunha DS, Gonzalez B, Konta M, Lin F, Shechter G, Starikov L, Wong S, Bulow HE, and Melendez A (2017). A non-cell-autonomous role of BEC-1/BECN1/Beclin1 in coordinating cell-cycle progression and stem cell proliferation during germline development. Curr. Biol. 27, 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellvé AR, Cavicchia JC, Millette CF, O’Brien DA, Bhatnagar YM, and Dym M (1977a). Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. J. Cell Biol. 74, 68–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellvé AR, Millette CF, Bhatnagar YM, and O’Brien DA (1977b). Dissociation of the mouse testis and characterization of isolated spermatogenic cells. J. Histochem. Cytochem. 25, 480–494. [DOI] [PubMed] [Google Scholar]
- Bolcun-Filas E, Bannister LA, Barash A, Schimenti KJ, Hartford SA, Eppig JJ, Handel MA, Shen L, and Schimenti JC (2011). A-MYB (MYBL1) transcription factor is a master regulator of male meiosis. Development 138, 3319–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busada JT, Chappell VA, Niedenberger BA, Kaye EP, Keiper BD, Hogarth CA, and Geyer CB (2015). Retinoic acid regulates Kit translation during spermatogonial differentiation in the mouse. Dev. Biol. 397, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caunt CJ, and Keyse SM (2013). Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 280, 489–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan F, Oatley MJ, Kaucher AV, Yang QE, Bieberich CJ, Shashikant BS, and Oatley JM (2014). Functional and molecular features of the Id4+ germline stem cell population in mouse testes. Genes Dev. 28, 1351–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zheng Y, Gao Y, Lin Z, Yang S, Wang T, Wang Q, Xie N, Hua R, Liu M, et al. (2018). Single-cell RNA-seq uncovers dynamic processes and critical regulators in mouse spermatogenesis. Cell Res. 28, 879–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij DG (2017). Organization of the seminiferous epithelium and the cycle, and morphometric description of spermatogonial subtypes (rodents and primates) In The Biology of Mammalian Spermatogonia, Oatley JM and Griswold MD, eds. (New York, NY: Springer; ), pp. 3–20. [Google Scholar]
- Dovey SL, Valli H, Hermann BP, Sukhwani M, Donohue J, Castro CA, Chu T, Sanfilippo JS, and Orwig KE (2013). Eliminating malignant contamination from therapeutic human spermatogonial stem cells. J. Clin. Invest. 723, 1833–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannigan R, and Schlegel PN (2017). Genetic diagnostics of male infertility in clinical practice. Best Pract. Res. Clin. Obstet. Gynaecol. 44, 26–37. [DOI] [PubMed] [Google Scholar]
- Friedrich G, and Soriano P (1991). Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 5, 1513–1523. [DOI] [PubMed] [Google Scholar]
- Gawriluk TR, Hale AN, Flaws JA, Dillon CP, Green DR, and Rucker EB 3rd. (2011). Autophagy is a cell survival program for female germ cells in the murine ovary. Reproduction 747, 759–765. [DOI] [PubMed] [Google Scholar]
- Geyer CB (2017). Setting the stage: the first round of spermatogenesis In The Biology of Mammalian Spermatogonia, Oatley JM and Griswold MD, eds. (New York, NY: Springer; ), pp. 39–63. [Google Scholar]
- Ghiassi-Nejad Z, Hernandez-Gea V, Woodrell C, Lang UE, Dumic K, Kwong A, and Friedman SL (2013). Reduced hepatic stellate cell expression of Kruppel-like factor 6 tumor suppressor isoforms amplifies fibrosis during acute and chronic rodent liver injury. Hepatology 57, 786–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. (2005). Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 75, 1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Grow EJ, Yi C, Mlcochova H, Maher GJ, Lindskog C, Murphy PJ, Wike CL, Carrell DT, Goriely A, et al. (2017). Chromatin and singlecell RNA-seq profiling reveal dynamic signaling and metabolic transitions during human spermatogonial stem cell development. Cell Stem Cell 27, 533–546.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurzov EN, Stanley WJ, Brodnicki TC, and Thomas HE (2015). Protein tyrosine phosphatases: molecular switches in metabolism and diabetes. Trends Endocrinol. Metab. 26, 30–39. [DOI] [PubMed] [Google Scholar]
- Handel MA, and Schimenti JC (2010). Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat. Rev. Genet. 77, 124–136. [DOI] [PubMed] [Google Scholar]
- Helsel AR, and Oatley JM (2017). Transplantation as a quantitative assay to study mammalian male germline stem cells. Methods Mol. Biol. 7463, 155–172. [DOI] [PubMed] [Google Scholar]
- Helsel AR, Yang QE, Oatley MJ, Lord T, Sablitzky F, and Oatley JM (2017). ID4 levels dictate the stem cell state in mouse spermatogonia. Development 744, 624–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann BP, and Heckert LL (2005). Silencing of Fshr occurs through a conserved, hypersensitive site in the first intron. Mol. Endocrinol. 79, 2112–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann BP, Sukhwani M, Lin CC, Sheng Y, Tomko J, Rodriguez M, Shuttleworth JJ, McFarland D, Hobbs RM, Pandolfi PP, et al. (2007). Characterization, cryopreservation, and ablation of spermatogonial stem cells in adult rhesus macaques. Stem Cells 25, 2330–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann BP, Sukhwani M, Simorangkir DR, Chu T, Plant TM, and Or- wig KE (2009). Molecular dissection of the male germ cell lineage identifies putative spermatogonial stem cells in rhesus macaques. Hum. Reprod. 24, 1704–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann BP, Mutoji KN, Velte EK, Ko D, Oatley JM, Geyer CB, and McCarrey JR (2015). Transcriptional and translational heterogeneity among neonatal mouse spermatogonia. Biol. Reprod. 92, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano M, Yamada S, Tanaka H, Nishimune Y, and Nozaki M (2003). Genomic structure and promoter activity of the testis haploid germ cell-specific intronless genes, Tact1 and Tact2. Mol. Reprod. Dev. 65, 148–156. [DOI] [PubMed] [Google Scholar]
- Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, and Passegue E (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie N, and Surani MA (2017). Efficient induction and isolation of human primordial germ cell-like cells from competent human pluripotent stem cells. Methods Mol. Biol. 1463, 217–226. [DOI] [PubMed] [Google Scholar]
- Johnson L, Petty CS, and Neaves WB (1980). A comparative study of daily sperm production and testicular composition in humans and rats. Biol. Reprod. 22, 1233–1243. [DOI] [PubMed] [Google Scholar]
- Koehler KE, Cherry JP, Lynn A, Hunt PA, and Hassold TJ (2002). Genetic control of mammalian meiotic recombination. I. Variation in exchange frequencies among males from inbred mouse strains. Genetics 162, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiho A, Kotaja N, Gyenesei A, and Sironen A (2013). Transcriptome profiling of the murine testis during the first wave of spermatogenesis. PLoS ONE 8, e61558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen Y, Mo R, Hui C, Cheng JF, Mohandas N, and Huang CH (2000). Characterization of human RhCG and mouse Rhcg as novel noneryth- roid Rh glycoprotein homologues predominantly expressed in kidney and testis. J. Biol. Chem. 275, 25641–25651. [DOI] [PubMed] [Google Scholar]
- Lovelace DL, Gao Z, Mutoji K, Song YC, Ruan J, and Hermann BP (2016). The regulatory repertoire of PLZF and SALL4 in undifferentiated spermatogonia. Development 143, 1893–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukassen S, Bosch E, Ekici AB, and Winterpacht A (2018). Characterization of germ cell differentiation in the male mouse through single-cell RNA sequencing. Sci. Rep. 8, 6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarrey JR, Berg WM, Paragioudakis SJ, Zhang PL, Dilworth DD, Arnold BL, and Rossi JJ (1992). Differential transcription of Pgk genes during spermatogenesis in the mouse. Dev. Biol. 154, 160–168. [DOI] [PubMed] [Google Scholar]
- McCarrey JR (2017). Transition of prenatal prospermatogonia to postnatal spermatogonia In The Biology of Mammalian Spermatogonia, Oatley J and Griswold M, eds. (Springer Publishing; ), pp. 23–38. [Google Scholar]
- McLachlan RI, Rajpert-De Meyts E, Hoei-Hansen CE, de Kretser DM, and Skakkebaek NE (2007). Histological evaluation of the human testis- approaches to optimizing the clinical value of the assessment: mini review. Hum. Reprod. 22, 2–16. [DOI] [PubMed] [Google Scholar]
- Mutoji K, Singh A, Nguyen T, Gildersleeve H, Kaucher AV, Oatley MJ, Oatley JM, Velte EK, Geyer CB, Cheng K, et al. (2016). TSPAN8 expression distinguishes spermatogonial stem cells in the prepubertal mouse testis. Biol. Reprod. 95, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa SH, Park PJ, Zhang LF, Shima JE, McCarrey JR, Griswold MD, and Lee JT (2006). Postmeiotic sex chromatin in the male germ- line of mice. Curr. Biol. 16, 660–667. [DOI] [PubMed] [Google Scholar]
- Neuhaus N, Yoon J, Terwort N, Kliesch S, Seggewiss J, Huge A, Voss R, Schlatt S, Grindberg RV, and Scholer HR (2017). Single-cell gene expression analysis reveals diversity among human spermatogonia. Mol. Hum. Reprod. 23, 79–90. [DOI] [PubMed] [Google Scholar]
- Oatley JM, and Brinster RL (2006). Spermatogonial stem cells. Methods Enzymol. 419, 259–282. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Aréchaga JM, Avarbock MR, and Brinster RL (1997). Transplantation of testis germinal cells into mouse seminiferous tubules. Int. J. Dev. Biol. 41, 111–122. [PubMed] [Google Scholar]
- Pekarsky Y, Koval A, Hallas C, Bichi R, Tresini M, Malstrom S, Russo G , Tsichlis P, and Croce CM (2000). Tcl1 enhances Akt kinase activity and mediates its nuclear translocation. Proc. Natl. Acad. Sci. USA 97, 3028–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma YA, and Trapnell C (2017). Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 14, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romrell LJ, Bellvé AR, and Fawcett DW (1976). Separation of mouse spermatogenic cells by sedimentation velocity. A morphological characterization. Dev. Biol. 49, 119–131. [DOI] [PubMed] [Google Scholar]
- Royo H, Prosser H, Ruzankina Y, Mahadevaiah SK, Cloutier JM, Baumann M, Fukuda T, Hoog C, Toth A, de Rooij DG, et al. (2013). ATR acts stage specifically to regulate multiple aspects of mammalian meiotic silencing. Genes Dev. 27, 1484–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M, and Miyauchi H (2016). Gametogenesis from pluripotent stem cells. Cell Stem Cell 18, 721–735. [DOI] [PubMed] [Google Scholar]
- Shima JE, McLean DJ, McCarrey JR, and Griswold MD (2004). The murine testicular transcriptome: characterizing gene expression in the testis during the progression of spermatogenesis. Biol. Reprod. 71, 319–330. [DOI] [PubMed] [Google Scholar]
- Song HW, Bettegowda A, Lake BB, Zhao AH, Skarbrevik D, Babaja- nian E, Sukhwani M, Shum EY, Phan MH, Plank TM, et al. (2016). The homeobox transcription factor RH0X10 drives mouse spermatogonia! stem cell establishment. Cell Rep. 17, 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel G, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida T, and Friedman SL (2017). Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411. [DOI] [PubMed] [Google Scholar]
- Turner JM, Mahadevaiah SK, Ellis PJ, Mitchell MJ, and Burgoyne PS (2006). Pachytene asynapsis drives meiotic sex chromosome inactivation and leads to substantial postmeiotic repression in spermatids. Dev. Cell 10, 521–529. [DOI] [PubMed] [Google Scholar]
- Valli H, Sukhwani M, Dovey SL, Peters KA, Donohue J, Castro CA, Chu T, Marshall GR, and Orwig KE (2014). Fluorescence- and magnetic-activated cell sorting strategies to isolate and enrich human spermato- gonial stem cells. Fertil. Steril. 102, 566–580.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kopylow K, Schulze W, Salzbrunn A, and Spiess AN (2016). Isolation and gene expression analysis of single potential human spermatogonial stem cells. Mol. Hum. Reprod. 22, 229–239. [DOI] [PubMed] [Google Scholar]
- Wandernoth PM, Mannowetz N, Szczyrba J, Grannemann L, Wolf A, Becker HM, Sly WS, and Wennemuth G (2015). Normal fertility requires the expression of carbonic anhydrases II and IV in sperm. J. Biol. Chem. 290,29202–29216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, and Navin NE (2015). Advances and applications of single-cell sequencing technologies. Mol. Cell 58, 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PJ, Page DC, and McCarrey JR (2005). Differential expression of sex-linked and autosomal germ-cell-specific genes during spermatogenesis in the mouse. Hum. Mol. Genet. 14, 2911–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward WS (2010). Function of sperm chromatin structural elements in fertilization and development. Mol. Hum. Reprod. 16, 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtasz L, Cloutier JM, Baumann M, Daniel K, Varga J, Fu J, Anastas- siadis K, Stewart AF, Remenyi A, Turner JM, and Toth A (2012). Meiotic DNA double-strand breaks and chromosome asynapsis in mice are monitored by distinct HORMAD2-independent and -dependent mechanisms. Genes Dev. 26, 958–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, and Quake SR (2014). Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 11,41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsenko AN, Georgiadis AP, Ropke A, Berman AJ, Jaffe T, Olszew- ska M, Westernstroer B, Sanfilippo J, Kurpisz M, Rajkovic A, et al. (2015). X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N. Engl. J. Med. 372, 2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Zir- aldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8,14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Nie R, Li Y, Friel P, Mitchell D, Hess RA, Small C, and Griswold MD (2008). Expression of stimulated by retinoic acid gene 8 (Stra8) in spermatogenic cells induced by retinoicacid: an in vivo study in vitamin A-suf- ficient postnatal murine testes. Biol. Reprod. 79, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.