

Abstract
The fragment 2 domain (F2) of prothrombin and its interaction with factor (F) Va is known to contribute significantly to prothrombinase-catalyzed activation of prothrombin. The extent to which the F2-FVa interaction affects the overall thrombin generation, however, is uncertain. To study this interaction, nuclear magnetic resonance spectroscopy of recombinant F2 was used to identify seven residues within F2 that are significantly responsive to FVa binding. The functional role of this region in interacting with FVa during prothrombin activation was verified by the FVa-dependent inhibition of thrombin generation using peptides that mimic the same region of F2. Because six of the seven residues were within a 9-residue span, these were mutated to generate a prothrombin derivative (PT6). These mutations led to a decreased affinity for FVa as determined by surface plasmon resonance. When thrombin generation by an array of FXa containing prothrombinase components was monitored, a 54% decrease in thrombin generation was observed with PT6 compared with the wild-type, only when FVa was present. The functional significance of the specific low-affinity binding between F2 and FVa is discussed within the context of a dynamic model of molecular interactions between prothrombin and FVa engaging multiple contact sites.
Introduction
The immediate need to minimize blood loss upon vascular injury is met with the formation of a stable fibrin clot that is triggered by the rapid activation of the blood coagulation cascade. A common theme in such an emergency response is the involvement of multi-component complexes composed of an enzyme, a protein cofactor, the lipid surface and a divalent cation such as the prothrombinase and both the extrinsic and intrinsic tenase complexes1. The prothrombinase complex, formed in the penultimate step of the coagulation cascade, is composed of the serine protease factor (F) Xa and the protein cofactor FVa, assembled on negatively charged phospholipid membranes (e.g. activated platelets)2,3 and is mediated by calcium ions1. This complex accelerates prothrombin activation by several orders of magnitude4, leading to a rapid accumulation of thrombin and conversion of circulating fibrinogen into a fibrin clot.
It has been established that the full assembly of coagulation complexes can increase their catalytic efficiencies by several orders of magnitude, as in the cases of the prothrombinase5 and the full tenase complexes6. Underlying this, enzyme/protein components of each macromolecular complex appear to have many points of contact with one another with varying binding affinities7,8. For example, FVa has discrete binding contacts with each of the four structural domains of prothrombin9–12, and in turn prothrombin interacts with the multi-domain enzyme FXa13,14. This example is in concert with the concept of multivalent molecular assembly15 which has begun to emerge as a model for intracellular machineries16. One essential feature of multivalent assembly is the requirement for transient and weak component interactions to both confer highly dynamic molecular interactions15, and respond to “on-demand” regulation16–18. This emerging picture of macromolecular machinery is reminiscent of the dynamics of coagulation enzyme assembly and subsequent thrombin generation, as shown in a number of studies from our laboratory19,20 and others21. This dynamic regulation may also be required for physiological functions of thrombin generation other than haemostasis22–24.
While high-affinity interactions have been studied and characterized extensively, other, weaker but equally important interactions have largely been ignored for coagulation complexes due to the challenges in studying their binding properties. We sought to investigate weaker interactions in the context of the prothrombinase complex, specifically the binding interactions between FVa and the F2 domain of prothrombin (Fig. 1). These low-affinity protein-protein interactions in prothrombinase9,14,25 have been suggested to play an important functional role in the turnover of prothrombin26. We further characterize the binary interactions between FVa and prothrombin, in particular that between the heavy chain of FVa (FVa-HC) and the F2 domain of prothrombin. Multi-dimensional nuclear magnetic resonance (NMR) was used to identify the residues within the F2 domain that are perturbed upon FVa-HC binding, which were then subsequently mutated. The functional consequences of the loss of these F2 residues were then investigated. Significance of the specific binding between F2 and FVa-HC is discussed in the context of a dynamic network of domain interactions between prothrombin and FVa as part of the catalytic machinery of prothrombinase.
Figure 1.
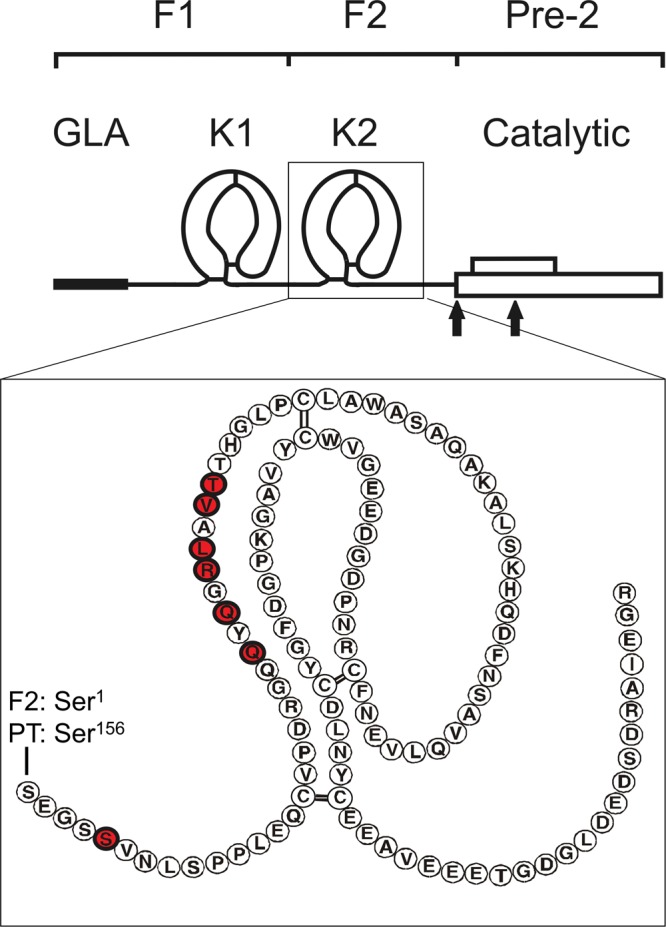
The structural organization of prothrombin. Prothrombin consists of fragment 1 (F1), which contains the γ-carboxyglutamic (GLA) and kringle 1 (K1) domains, fragment 2 (F2), which contains the kringle 2 (K2) domain, and the catalytic domain, which is also referred to as prethrombin-2 (Pre-2). The cleavage sites for activation of prothrombin by FXa (Arg271 and Arg320) are indicated with arrows. The close-up image of the K2 domain is shown with its amino acid sequence. Ser1 of K2 corresponds to Ser156 of full-length prothrombin sequence numbering. The seven residues identified using heteronuclear NMR in our study are highlighted in red.
Results
Identifying F2 residues involved in binding FVa-HC
Recombinant F2 domain of prothrombin and purified F2 from human plasma prothrombin (Fig. 1) exhibited essentially the same functional activities for enhancing the activation of prethrombin-2 by prothrombinase. Proton and 15N/1H HSQC NMR spectra of recombinant F2 exhibited progressive changes with increasing concentrations of FVa-HC from 10 to 30 μM (Fig. 2), indicating a binding affinity (Kd) in the low to middle μM range. 15N/1H HSQC spectra at the highest (30 μM) FVa-HC concentration collected for the 15N-labeled F2 showed that a majority of the 15N/1H cross-peaks for F2 were not affected by the presence of the FVa-HC (Fig. 2). However, there were 7 HSQC peaks with significant chemical shift perturbations for the backbone and/or side-chain 15N and/or 1H resonances of F2, with six of the seven located in the N-terminal Gln22-Thr30 segment of the kringle. For example, Gln24 and Leu27 (residues 179 and 182 in prothrombin numbering) displayed changes of ~0.10 and ~0.08 ppm for the proton chemical shift (ΔδH) and ~0.9 and ~0.7 ppm for the amide nitrogen resonance (ΔδN), respectively. Residues Arg26 and Thr30 (prothrombin residues 181 and 185) also exhibited perturbed resonances with ΔδH~0.08 and ΔδH~0.03; ΔδN~0.8 and ΔδN~0.9 ppm, respectively, while residues Ser5 and Val29 (prothrombin residues 160 and 184) were somewhat perturbed upon titration of the FVa-HC. Importantly, F2 residues Gln22-Thr30 (prothrombin residues 177–185) perturbed by FVa all reside on a highly exposed surface loop of the kringle structure of F2 in the non-covalent complexes of F2 with Phe-Pro-Arg-chloromethylketone-inhibited thrombin (FPRck-thrombin)27,28, prethrombin-229, as well as prethrombin-1 (prothrombin des F1) (Fig. 1)30. Therefore, all the highly perturbed resonances likely correspond to residues located at or near the binding interface31 within the binary molecular complex of F2 with FVa-HC.
Figure 2.
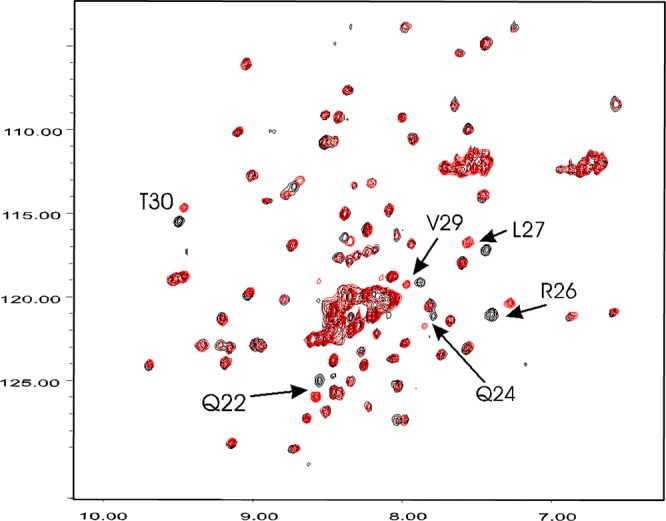
Overlay of the 15N HSQC spectra (at 500 MHz) of free F2 and FVa heavy chain-titrated F2. The HSQC spectra were recorded in 90% H2O and 10% D2O with 50 mM sodium phosphate at pH 7.0 and at 35 °C. The protein concentrations were 0.3 mM for F2 and 0.03 mM for the FVa heavy chain (FVa-HC). HSQC peaks of F2 most shifted by FVa are labelled by the assigned F2 residues. The arrows point to the center of resonance displacement between the free F2 (black) and F2 complexed with FVa (red). Peak displacements of the affected F2 residues decreased gradually (not shown) as the FVa-HC concentration varied between 0.03 mM and 0.01 mM.
Functional interactions of F2-derived peptides
To test whether the clustered residues on F2 of prothrombin identified by NMR represent the FVa-HC binding site, a 20-residue peptide that encompasses this region (F2P-(16-36) (prothrombin residues 171–191) was synthesized, with Cys36 replaced with a serine32. Compared with full-length F2, this truncated peptide did not exhibit pronounced proton resonance perturbations with up to 6-fold molar excess of FVa-HC. However, it did show a significant enhancement of nuclear Overhauser effects (NOEs) when comparing their two-dimensional NOE spectra (NOESY) in the absence and presence of the FVa-HC. Specifically, residues Val29 and Thr30 (Val184 and Thr185 in prothrombin numbering) in F2P-(16-36) exhibited the strongest FVa-HC-induced NOEs, i.e. transferred NOEs33 within the region of intact F2 that is the most responsive to FVa-HC binding in H-15N HSQC experiments (Fig. 2). Taken together, these data suggest that the 20-mer region contains, at least in part, the residues identifiable by NMR as a potential FVa-HC binding site.
To explore this region in greater detail, six peptides that encompass most of the F2 sequence were generated (Table S1). Four of these peptides (F2P1, F2P2, F2P3, and F2P8), when included at increasing concentrations, inhibited prothrombin activation in a dose-dependent manner, while two of the peptides (F2P6 and F2P7) did not (Fig. S1). The four peptides that showed dose-dependent effects were then further investigated for their ability to inhibit prothrombin activation in a FVa-dependent manner. In the absence of FVa, F2P1 and F2P8 inhibited prothrombin activation while no effects were observed for F2P2 and F2P3 (Fig. 3A). Therefore, F2P1 and F2P8 were considered not to be involved in binding FVa and thus omitted in subsequent studies. In the presence of FVa (i.e. prothrombinase), F2P2 and F2P3, which also contain sequence overlaps with the longer peptide F2P-(16-36), inhibited prothrombin activation with estimated Ki values of 520 ± 92 μM and 31 ± 6 μM, respectively (Fig. 3B). Linear transformation using the Lineweaver-Burke plots showed a non-competitive type inhibition with comparable Ki values of 427 ± 4 μM and 34 ± 4 μM for F2P2 and F2P3, respectively (Fig. S2). Similar Ki values were also observed for these interactions in equilibrium binding using light scattering (Fig. S3). F2P2 and F2P3, respectively, showed 5- and 2-fold inhibition of prothrombin-FVa binding at saturation, which is noteworthy considering the multiple contacts engaged by prothrombin for binding FVa in prothrombinase5.
Figure 3.
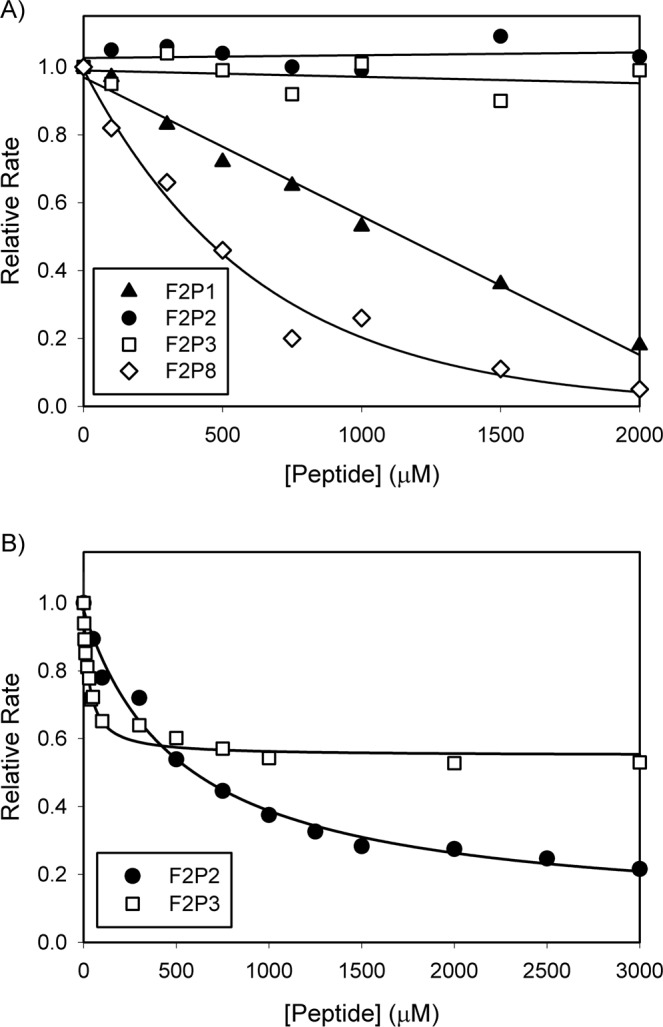
Inhibition of FXa-mediated prothrombin activation by synthetic peptides derived from F2. (A) Relative rates of prothrombin activation in the presence of 300 nM prothrombin, 5 mM CaCl2, 5 μM DAPA, 50 μM PCPS, and F2P1 (triangles), F2P2 (circles), F2P3 (squares) or F2P8 (diamonds) at varying concentration are plotted with respect to inhibitor concentrations. Reactions were started by addition of 20 nM FXa. (B) Prothrombin activation was carried out as above, but in the presence of 5 nM FVa and F2P2 (circles), or F2P3 (squares) at varying concentrations. Reactions were initiated by addition of 0.1 nM FXa. Non-linear regression analysis was carried out to determine the IC50 and the apparent Ki was calculated using the Cheng-Prusoff equation.
Prothrombin activation monitored using DAPA
The six residues that comprise the potential FVa binding site on F2 were then mutated (Gln177Ala, Gln179Ala, Arg181Ala, Leu182Thr, Val184Thr and Thr185Ala, in prothrombin numbering) creating a prothrombin mutant we refer to as PT6 (see experimental procedures for further detail). The functional consequences of these mutations were quantified and compared with the wild-type prothrombin using DAPA as the thrombin-specific probe34. The activation time course of prothrombin was used to measure the initial rates of thrombin generation by various combinations of prothrombinase components (Table 1). Significant differences in thrombin generation between the wild-type prothrombin and PT6 were observed only when FVa was present in the activation complex. When activated by FXa/FVa (Fig. 4A), the initial rates of thrombin generation measured from wild-type or PT6 prothrombin were (1.8 ± 0.5) × 10−2 s−1 and (5.3 ± 3.7) × 10−3 s−1, respectively; PT6 activation rate being 29% of that observed with the wild-type prothrombin. The reactions, however, were not complete even after 4 h, indicating that the overall thrombin generation is slow without the membrane surface. Total thrombin generated from PT6 was 35% of that observed with wild-type prothrombin, suggesting that the ability to generate mature thrombin is affected by the loss of these key residues within the F2 domain of prothrombin. With FXa/PCPS as the activator (Fig. 4B), the initial rates of activation were not significantly different between wild-type prothrombin (1.8 ± 0.5 × 10−2 s−1) and PT6 (1.4 ± 0.1 × 10−2 s−1). Similarly, with FXa alone (Fig. 4C), there were no significant differences between wild-type prothrombin (3.9 ± 4.3 × 10−4 s−1) and PT6 (5.1 ± 5.8 × 10−4 s−1). The differences in total thrombin levels generated between the two prothrombin variants were also minimal with either FXa/PCPS or FXa alone as the activator. With full prothrombinase (i.e. FXa/FVa/PCPS/Ca2+), a statistically significant difference in the initial rates of wild-type prothrombin (66.6 ± 9.6 s−1) or PT6 (55.1 ± 8.7 s−1) activation was observed (p < 0.05). Total thrombin generated was 42% lower for PT6 than wild-type prothrombin (Fig. 5). Taken together, these data suggest that the differences in activation between wild-type and PT6 prothrombin are apparent only in the presence of FVa.
Table 1.
Initial rates of wild-type or PT6 prothrombin by FXa with varying components of prothrombinase.
| Wild-type | PT6 | ||||
|---|---|---|---|---|---|
| Rate | Fold | Rate | Fold | p | |
| s − 1 | s − 1 | ||||
| FXa | 3.9 ± 4.3 × 10−4 | 1 | 5.1 ± 5.8 × 10−4 | 1 | 0.08 |
| FXa, PCPS | 1.8 ± 0.5 × 10−2 | 44.9 | 1.4 ± 0.1 × 10−2 | 27.3 | 0.29 |
| FXa, FVa | 1.8 ± 0.5 × 10−2 | 46.9 | 5.3 ± 3.7 × 10−3 | 10.4 | <0.005 |
| FXa, PCPS, FVa | 66.6 ± 9.6 | 1.7 × 105 | 55.1 ± 8.7 | 1.1 × 105 | <0.05 |
Wild-type or PT6 was activated by prothrombinase, FXa/FVa, FXa/PCPS, or FXa alone (see methods for concentrations). All reactions contain 5 mM CaCl2. Fold differences in initial rates are calculated separately for wild-type and PT6, relative to activation by FXa alone. Statistical significance was measured between wild-type and PT6 for each set of experiments using Student’s t-test. Values represent mean ± SD of at least two separate experiments in duplicates.
Figure 4.
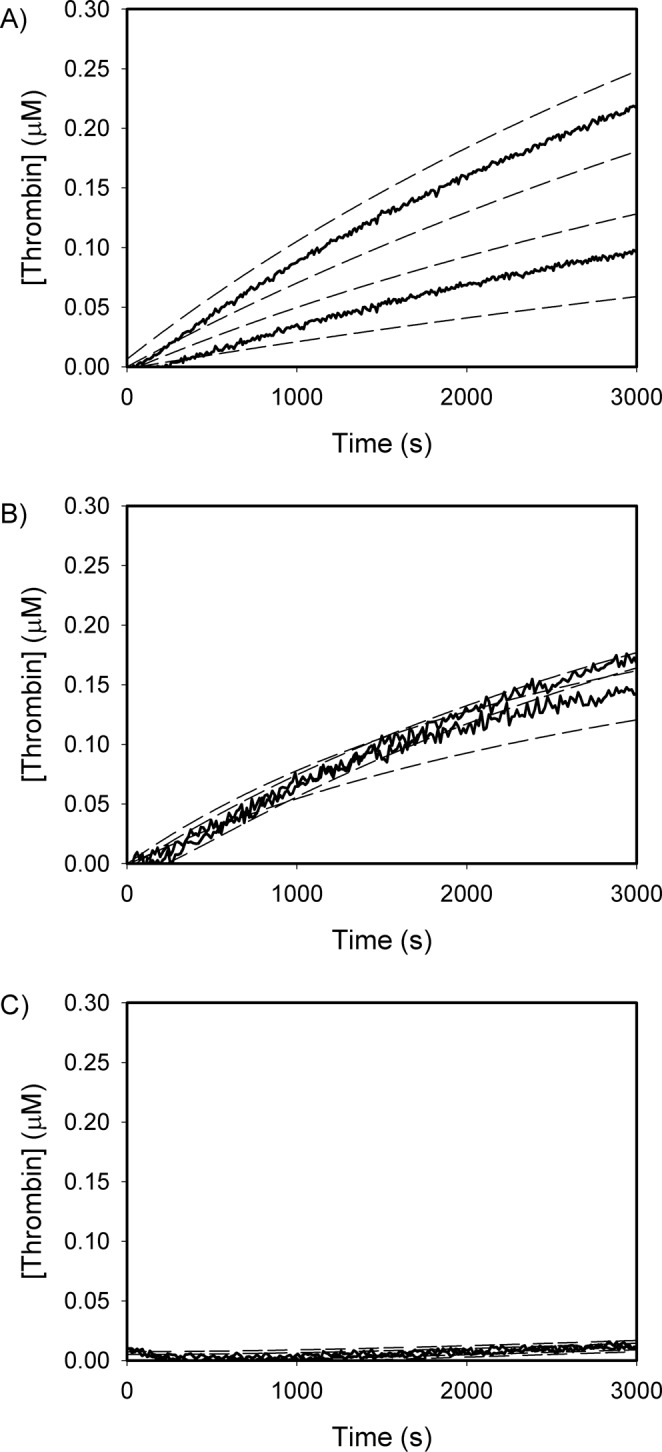
Thrombin generation profile of wild-type prothrombin and PT6 (1 μM) activation by (A) FXa/FVa, (B) FXa/PCPS, or (C) FXa alone, measured by FRET analysis using DAPA. Reaction mixtures for each set of conditions were as follows: (A) FXa (5 nM) and FVa (20 nM) (FXa/FVa), (B) FXa (5 nM) and PCPS (50 μM) (FXa/PCPS), and (C) FXa alone (5 nM). Under all conditions, experiments were initiated with the addition of FXa in HBST buffer with CaCl2 (5 mM) and DAPA (10 μM). Fluorescence was measured by a SpectraMax M2 fluorescent plate reader, with an excitation wavelength of 280 nm, emission wavelength of 545 nm and an emission cutoff at 530 nm at 20 s intervals.
Figure 5.
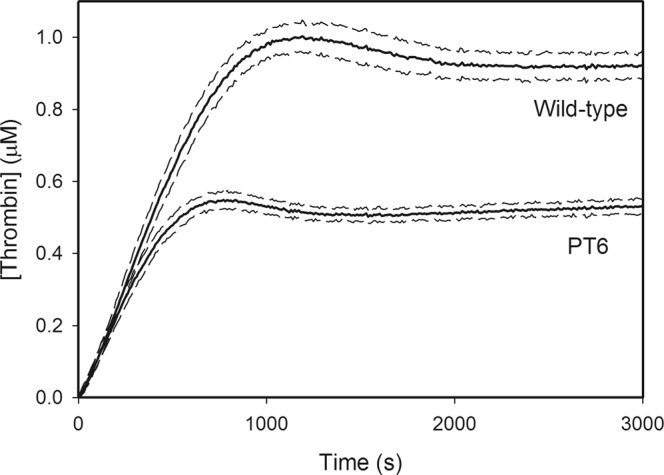
Thrombin generation profile of wild-type prothrombin and PT6 (1 μM) activation by the prothrombinase complex, measured by FRET analysis using DAPA. This reaction mixture contained FXa (20 pM), FVa (20 nM) and PCPS (50 μM). Under all conditions, experiments were initiated with the addition of FXa in HBST buffer with CaCl2 (5 mM) and DAPA (10 μM). Fluorescence was measured by a SpectraMax M2 fluorescent plate reader, with an excitation wavelength of 280 nm, emission wavelength of 545 nm and an emission cutoff at 530 nm at 20 s intervals.
Effects of selected residue mutations on the binding of prothrombin to FVa
The binding of wild-type prothrombin or PT6 to FVa was monitored in real-time and quantified using surface plasmon resonance (SPR). With FVa coupled onto a CM5 sensor chip, wild-type prothrombin showed a rapid association that was saturable, followed by a rapid and complete dissociation (Fig. 6A). PT6 demonstrated slower association, followed by slow dissociation (Fig. 6B). The maximum response unit (RU) change observed at 60 s was, on average, 33% less in PT6 compared with wild-type prothrombin of equal concentration. Kinetic rate analyses were used to estimate the affinity between prothrombin and FVa, as PT6 did not reach saturation and was not suitable for steady state analyses. The dissociation constant (Kd) for PT6 (Kd = 2.6 μM) was 2.6-fold higher than wild-type prothrombin (Kd = 1 μM). Taken together, these data demonstrate that binding of prothrombin to FVa clearly involves the six residues identified within the F2 domain of prothrombin.
Figure 6.
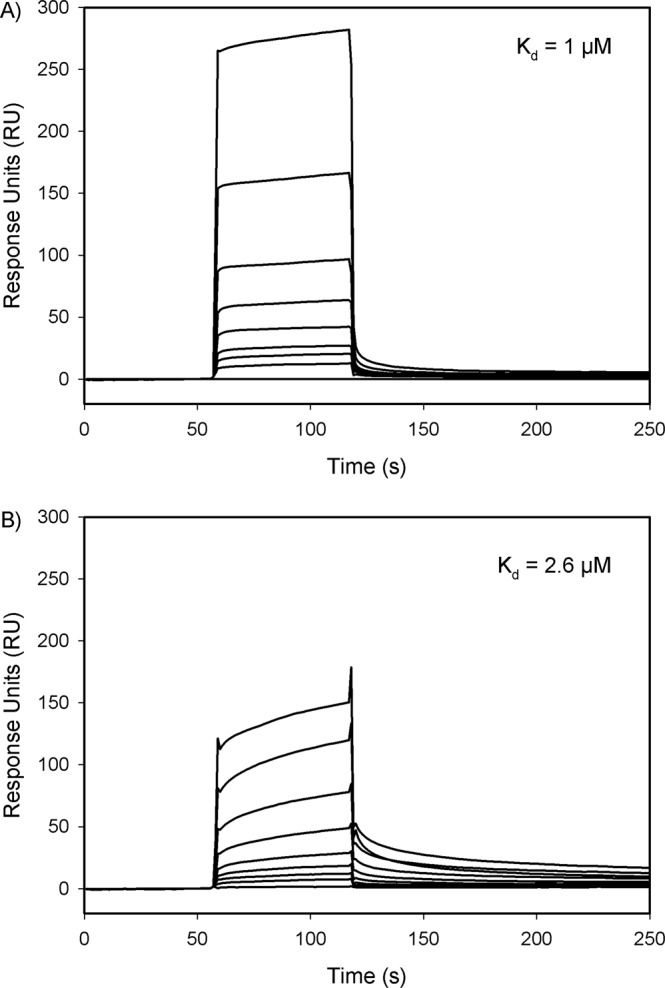
Binding of (A) wild-type or (B) PT6 prothrombin to FVa using surface plasmon resonance. Wild-type (A) or PT6 (B) prothrombin at varying concentrations (0, 78, 156, 313, 625, 1250, 2500, 5000 and 10,000 nM) were injected into a flow cell containing immobilized FVa for 60 s prior to washing with HBSC Tween-80. Kd values were determined using a kinetic 1:1 model.
Discussion
To date, the functional role of F2 in the prothrombin-FVa interaction has mostly been studied by the use of prothrombin variants with or without the F2 domain, and quantifying its activation by FXa/FVa. In particular, inclusion of isolated F2 was found to accelerate the FVa-dependent activation of prethrombin-2 both in the absence35,36 and presence37,38 of phospholipid membranes. One other study reported that F2 inhibits the cleavage of the full-length prothrombin by the FXa/FVa complex and that isolated F2 appears to have a comparable affinity of binding to FVa as that of prothrombin39. Binding quantified by NMR reveals specific interactions between the FVa-HC and residues located in the kringle portion of F2 (Fig. 2). Additional NMR studies also show the involvement of residues Asp223–Lys236 and the C-terminal peptide region of F2 in binding to thrombin32, which is in full agreement with the structural characteristics of the F2-thrombin complex formed non-covalently27,28 or covalently40. The FVa- and thrombin-binding sites on F2 are also spatially distinct from a sequence locus of F2 identified for binding FXa13,14. In all, residues of F2 perturbed by binding of the FVa-HC are different from those responsible for interactions with thrombin, which suggests that the same residues of the F2 domain of prothrombin and F2-containing zymogen intermediates, such as prethrombin-19,35 or the functionally equivalent F2-prethrombin-2 complex27,28,38,41 should also be available for binding FVa.
While the difference in initial rates of prothrombin activation by prothrombinase between wild-type and PT6 were minimal (Table 1), there was a large difference in total thrombin generation at completion (Figs 4 and 5), comparable with the effect observed in the peptide inhibition studies. A similar observation has been reported previously by our group42, whereby decreases in total thrombin generation are not accompanied by proportional decreases in the initial rates of prothrombin activation. Effects on the initial rates of thrombin generation, as well as total thrombin generation were accentuated only when prothrombin was activated in the presence of FVa, indicating FVa dependence of inhibition. This is consistent with the results obtained using SPR whereby the loss of key residues within the F2 region significantly weakened the binding of prothrombin to FVa.
The main function of FVa in the prothrombinase complex is to promote the efficient conversion of prothrombin into thrombin25, in part by mediating the interaction of a mature exosite I in meizothrombin43 with a negatively-charged C-terminal region of the FVa-HC44–46. Binding between prothrombin and FVa is also thought to involve the F2 domain26,37,47 and the F1 domain10,39. Such binding interactions of prothrombin with FVa involving multiple contact sites are normally interpreted10 as having complete tertiary structural assembly between multi-domain proteins48,49. However, the initial interaction between FVa-HC and prothrombin appears to be partially dispensable. Thrombin generation remains largely unaffected when prothrombinase is composed of truncated forms of FVa lacking portions of the C-terminal tails of the heavy chain44,46. As well, potently inhibiting peptides derived from the C-terminus of the FVa-HC only partially reduced the enhanced thrombin generation by the FXa/FVa complex, compared with membrane-bound FXa alone44,50. Taken together, this suggests the functional interaction between FVa and prothrombin can also engage a subset of all possible contact sites within prothrombin, including proexosite I, Gla- and kringle 1 domain of F1, and F2.
It has been widely accepted that FVa alone increases the rate of thrombin generation by 3 orders of magnitude while the membrane surface alone increases the catalytic efficiency of thrombin generation by 2 orders of magnitude, which together increases its catalytic efficiency by 5 orders of magnitude5. These individual contributions of the prothrombinase components toward overall thrombin generation, however, were not observed in our experiments (Table 1). The difference in rate enhancement of thrombin generation afforded by FVa or PCPS alone was similar (~45-fold). The 5-orders of magnitude enhancement as reported in literature was only observed upon the full assembly of prothrombinase. A similar trend has previously been reported during FX activation by the intrinsic tenase complex (FIXa/FVIIIa/membrane surface), whereby addition of either the cofactor FVIIIa or the membrane surface each results in the enhancement of tenase by 2 orders of magnitude, but assembly of the full tenase complex enhances its activity by 6 orders of magnitude6. These findings demonstrate that the effect of individual components of multivalent enzymatic complexes do not always show simple multiplicative effects of independent binding events. The overall reaction rate is reflective of all binding modes between the enzyme, cofactor, lipid surface, and the substrate that must all work in concert for maximal efficiency of a fully-assembled catalytic complex. This idea has also been shown in the context of the prothrombinase complex, where prothrombin variants lacking membrane binding potential (desGla) showed very modest decreases in the rate of thrombin generation (5-fold)51, compared with the 2-orders of magnitude decrease upon loss of membrane binding historically described in literature. Interestingly, desGla prothrombin was shown to be more efficiently cleaved at Arg271 than wild-type, demonstrating that alternate binding modes were being utilized in processing prothrombin by prothrombinase. This information is in agreement with our new results and rationalizes the overall enhancement that is greater than the multiplicative of the individual contributions provided by each prothrombinase component during thrombin generation.
Multivalent molecular complexes can engage in partial/dynamic binding modes, and has been observed with polyvalent biopolymers15, prothrombinase19,20, and other coagulation complexes21. These partial binding modes would allow for thrombin generation to occur, even without a complete association of the enzyme-substrate complex, a phenomenon quite prevalent in the enzymatic processing of polymeric or multi-domain substrates20,21,52. Such intrinsic heterogeneity of multivalent prothrombin-FVa interactions may explain the reported weak inhibitory capacities of the FVa-binding Gla- and kringle 1 domains of prothrombin, despite their high affinities for FVa10,39. This may also explain the partial inhibitory effect observed for a monoclonal antibody specific for the F2 domain of prothrombin26,47. As well, a peptide derived from residues 55–65 of hirudin was found to have much higher inhibitory effect on prothrombin activation by the FXa/FVa complex in the absence of a membrane surface53–55. Partial assembly of multivalent molecular complexes provides opportunity for accelerated rate of complex dissociation15. This may be advantageous for modulating its activity rather than being dependent on inhibition by a specific inhibitor. Multivalent complexes also offer the substrate (i.e. prothrombin) to have a greater contribution in its activation mechanism20,22,23 and opportunities for dynamic control of thrombin generation under physiological requirements other than blood coagulation22–24. Together with our data that identify residues of prothrombin F2 that interact with FVa, we therefore demonstrate that the overall enhancement of thrombin generation afforded by full prothrombinase complex formation is consistent with other reported multivalent molecular complexes whereby all interactions, including the substrate, can work in concert to provide the overall mechanism and ultimate efficiency of substrate processing.
Experimental Procedures
Materials
Various materials and their sources are listed in the Supplementary Materials.
Synthetic Peptides
Peptides were synthesized by the solid-phase method using standard FMOC chemistry on an Applied Biosystems 431A peptide synthesizer (Sheldon Biotechnology Center, McGill University). The details are outlined in Supplementary Materials.
Preparation of FVa heavy chain and bovine thrombin
The FVa heavy chain for NMR studies was prepared from purified bovine FVa, following a procedure as described previously45. Briefly, FV was activated to FVa by the addition of thrombin (1.96 units/mL) at 37 °C for 25 min and stopped with Phe-Pro-Arg-chloromethylketone (FPRck, 1 μM). The heavy and light chains of FVa were isolated using immunoaffinity column chromatography as described previously56. The chains were then pooled separately, precipitated against 80% ammonium sulfate, and stored at 4 °C.
Molecular cloning and protein expression with Pichia pastoris
Recombinant expression of human prothrombin F2 encompassing residues 156–271 of prothrombin was performed in the yeast Pichia pastoris as described previously32. Briefly, the cDNA encoding the sequence of F2 was amplified by PCR using a subcloned segment of the prothrombin cDNA as a template. cDNA encoding F2 was inserted into the pPIC9 expression vector, whereby the final F2 expressed contains six-histidine tags in its C-terminus separated by the natural FXa cleavage site. The expressed F2 protein was first purified by nickel-agarose chromatography and then treated by FXa to remove the His-tag. After removing FXa using benzamidine Sepharose, purity was assessed by SDS-PAGE and its molecular mass determined (12,605 Da) using both the ESI (Perkin-Elmer Sciex) and MALDI-TOF (Kratos Analytical) methods.
NMR sample preparation
Uniform 15N and 13C/15N isotopic labeling of F2 was achieved as described previously32. Once labeled, F2 preparations prepared and pH adjusted, 25 μL of D2O was added to the protein solution to provide the NMR deuterium lock signal. For the titration experiments, small aliquots of the FVa-HC or FPRck-thrombin, both in 2 mM HEPES, 50 mM sodium phosphate buffer, pH 7, were added to samples of 15N-labeled F2 up to a molar ratio of ~10:1. A thrombin-derived peptide (see Synthetic Peptides in Supplementary Materials) (in 2 mM HEPES, 50 mM sodium phosphate buffer at pH 7) was also added to the 15N-labeled F2 to a molar ratio of 1:3 with the thrombin peptide in molar excess. 1H-15N HSQC experiments were utilized to follow binding interactions of F2 with the addition of FVa-HC, thrombin and the thrombin-derived peptide.
NMR experiments
All NMR experiments were carried out at 35° C on a Bruker Avance500 or an Avance800 spectrometer equipped with triple-resonance (1H, 13C, and 15N) and three-axis gradient probes. Two-dimensional and three-dimensional NMR data collected for 15N- and 13C/15N-labeled F2 included 1H-15N HSQC, HNCO, HN(CA)CO, HNCA, HN(CO)CA, CBCA(CO)NH and HNCACB57–59. Water suppression was achieved using the WATERGATE method with a 3:9:19 selective pulse incorporated in all the three-dimensional pulse sequences60. To improve water suppression, water magnetization was re-aligned to the +Z-axis before acquisition by setting the phase of the last 90° 1H pulse to −X. For the HNCA experiment, SEDUCE-1 decoupling with a field of 2.5 kHz61 was used to decouple 13CO-15N interactions in the t1 and t2 evolution periods. All data sets were processed using NMRPipe62 with 90°-shifted sine-square weighting functions in all three dimensions. Spectral display and initial assignments were carried out using the XEASY software package63. Sequence-specific assignments of the backbone (HN, 15N, 13Cα and some 13Cβ) resonances of human F2 perturbed by FVa were achieved by use of a combined analysis of the two-dimensional TOCSY/NOESY and three-dimensional HNCO, HN(CA)CO, HNCA, HN(CO)CA, CBCA(CO)NH and HNCACB experiments.
Effects of F2 and peptides on prethrombin-2 and prothrombin activation
Recombinant and plasma-derived F2 were characterized through the activation of prethrombin-2 (100 nM) by prothrombinase (FXa at 50 pM, FVa at 100 pM, PCPS vesicles at 10 μM and calcium ion at 2.5 mM in 20 mM Tris-HCl, 0.15 M NaCl, pH 7.4 (TBS) at 37 °C in the absence or presence of F2 (200 nM). Activation of prothrombin (300 nM) was carried out in the presence of PCPS (50 μM), FVa (5 nM), and DAPA (5 μM) and various concentrations of each peptide in TBS with 5 mM CaCl2 and 0.01% Tween-80. Reactions were carried out in 96-well plates which had been pre-treated with TBS with 1% Tween-80 and rinsed thoroughly with water. Reactions were started with addition of FXa (0.1 nM), and monitored in SpectraMax Gemini fluorescence plate reader (Molecular Devices, Sunnyvale, California) at excitation and emission wavelengths of 280 nm and 545 nm, respectively, with a 530 nm emission cut off filter in the emission beam at 25 °C. In the absence of FVa, reactions were carried out as described above; however, the reactions were started with the addition of 20 nM FXa. The Ki values were estimated using the Cheng-Prusoff equation (IC50 = [1 + ([S]/KM)] Ki) based on the IC50 values obtained by nonlinear regression of a rectangular hyperbola, whereby [S] is the prothrombin concentration and KM is the Michaelis constant of prothrombinase for prothrombin determined in a separate experiment as described previously19.
Affinities of prothrombin variants for FVa determined using SPR
Binding interactions were studied by SPR as described previously64,65 using a Biacore T200 (GE Healthcare). Briefly, FVa was immobilized on a CM5 sensor chip (GE Healthcare) using an amine coupling kit (GE Healthcare). After injecting the 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride/N-hydroxysuccinimide mixture into the flow cell at a rate of 10 μL/min for 420 s, 28 μg/mL FVa in 10 mM acetate buffer, pH 5.5 was injected at a rate of 5 μL/min until 8000–9000 response units (RU) were immobilized. Flow cells were then washed with 1 M ethanolamine for 420 s, followed by 0.02 M HEPES, 0.15 M NaCl, pH 7.4 (HBS) with 5 mM CaCl2 and 0.01% Tween-80.
To measure the binding of prothrombin to FVa, increasing concentrations (0, 78, 156, 313, 625, 1250, 2500, 5000 and 10,000 nM) of wild-type or PT6 prothrombin were injected into the flow cells at a flow rate of 30 μL/min for 60 s, followed by buffer for 300 s. Flow cells were regenerated with 500 mM CaCl2 between sample injections. Kd values were calculated by estimating the individual on- and off-rate constants based on fitting of the sensorgrams to 1:1 Langmuir binding model using BIAevaluation software (GE Healthcare). Samples were analyzed twice in duplicate.
Generation and isolation of a prothrombin derivative, PT6
Six out of the seven residues of F2 within a 9-residue cluster identified by heteronuclear NMR were altered by site-directed mutagenesis (Gln177Ala, Gln179Ala, Arg181Ala, Leu182Thr, Val184Thr and Thr185Ala; prothrombin numbering). using the QuikChange Lightening Site-Directed Mutagenesis kit as described previously66. Mutations of the six residues were introduced in two-steps, with three mutations per reaction, using primers 5′-GATCGGGGGCAGGCGTACGCGGGGGCCCTGGCGGTGACCACAC-3′ and 5′-GCGTACGCGGGGGCCACGGCGACGGCCACACATGGGCTCCCC-3′. DNA was then isolated using the QIAprep Spin Miniprep Kit and was sequenced and amplified using Plasmid Maxi Kit. All DNA fragments were sequenced at Robarts Research Institute (London, Canada). PT6 cDNA was then transfected into baby hamster kidney cells, and the expressed PT6 was isolated as described previously19. The purified PT6 was then subjected to precipitation by 80% ammonium sulfate and stored at −20 °C in 50% glycerol.
Thrombin generation measured by DAPA fluorescence
Prothrombin activation was quantified using DAPA as described previously34. Wild-type or PT6 prothrombin (1 μM) was diluted into HBS with 0.01% Tween-80 and 0.1% Prionex (Pentapharm, Switzerland), and subsequently mixed with CaCl2 (5 mM), DAPA (10 μM) and varying mixtures of FVa and PCPS. To initiate the reaction, these mixtures were then transferred to FXa-containing wells in a 96-well microtitre plate. Experiments using full prothrombinase included PCPS (50 μM), FVa (20 nM) and FXa (20 pM). The incomplete prothrombinase compositions were: (1) FXa (5 nM) and FVa (20 nM) (FXa/FVa), (2) FXa (5 nM) and PCPS (50 μM) (FXa/PCPS), and (3) FXa alone (5 nM). Fluorescence was measured by a SpectraMax M2 fluorescent plate reader, with an excitation wavelength of 280 nm, emission wavelength of 545 nm and an emission cutoff at 530 nm at 20 s intervals. The fluorescence quantum yield of DAPA-thrombin complex was determined experimentally to be 2065.9 RFUs/μM thrombin.
Supplementary information
Acknowledgements
We thank Tom Abbot for assistance in the preparation of the FVa heavy chain, Willem Stevens for preparing the pPIC9-F2 plasmid and initial characterizations of the recombinant F2 protein, and Ping Wang for NMR spectra of peptide interactions with the FVa heavy chain. This paper is dedicated to the memories of Professor Michael E. Nesheim and his doctoral student, Dr. Nicole Brufatto. This work would not have been possible without Michael’s tremendous insights on the dynamics and complexity of macromolecular interactions and without Nicole’s astute characterizations of weak biomolecular interactions. This work was supported in part by the Natural Sciences and Engineering Research Council of Canada (grant #CGP064726 to F.N.), and by a Genomics and Health Initiative (GHI) program of the National Research Council of Canada (NRCC Publication No. NRC-HHT_53372 for the work of F.N. and P.X.), sponsored by the Government of Canada. This work was also supported by Natural Sciences and Engineering Research Council of Canada (RGPIN-2017-05347 to PYK). PYK holds a Hamilton Health Sciences Early Career Award.
Author Contributions
A.P.F. carried out experiments relating to functional characterization and wrote the manuscript. P.L.G. and P.Y.K. were responsible for experiment design, data analyses and wrote the manuscript. C.W. was involved in mutagenesis and experimental design relating to PT6 prothrombin. A.K., P.X. and F.N. were responsible for experimental design, execution, analyses, and write-up for NMR and associated protein productions. J.C.F. and J.I.W. were responsible for experimental design, execution, analyses, and write-up for S.P.R. All authors wrote and edited the manuscript. All data are available upon request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-019-38857-4.
References
- 1.Mann KG, Jenny RJ, Krishnaswamy S. Cofactor Proteins in the Assembly and Expression of Blood Clotting Enzyme Complexes. Annu. Rev. Biochem. 1988;57:915–956. doi: 10.1146/annurev.bi.57.070188.004411. [DOI] [PubMed] [Google Scholar]
- 2.Mann KG, Brummel-Ziedins K, Orfeo T, Butenas S. Models of blood coagulation. Blood Cells, Mol. Dis. 2006;36:108–117. doi: 10.1016/j.bcmd.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 3.Libby P, Theroux P. Pathophysiology of Coronary Artery Disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 4.Mann KG. The assembly of blood clotting complexes on membranes. Trends Biochem. Sci. 1987;12:229–233. doi: 10.1016/0968-0004(87)90115-0. [DOI] [Google Scholar]
- 5.Rosing J, Zwaal RF, Tans G. Formation of meizothrombin as intermediate in factor Xa-catalyzed prothrombin activation. J. Biol. Chem. 1986;261:4224–8. [PubMed] [Google Scholar]
- 6.Rawala-Sheikh R, Ahmad SS, Ashby B, Walsh PN. Kinetics of coagulation factor X activation by platelet-bound factor IXa. Biochemistry. 1990;29:2606–11. doi: 10.1021/bi00462a025. [DOI] [PubMed] [Google Scholar]
- 7.Qureshi SH, Yang L, Rezaie AR. Contribution of the NH2-terminal EGF-domain of factor IXa to the specificity of intrinsic tenase. Thromb. Haemost. 2012;108:1154–64. doi: 10.1160/TH12-06-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L, Manithody C, Yang L, Rezaie AR. Zymogenic and enzymatic properties of the 70–80 loop mutants of factor X/Xa. Protein Sci. 2004;13:431–42. doi: 10.1110/ps.03406904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luckow EA, Lyons DA, Ridgeway TM, Esmon CT, Laue TM. Interaction of clotting factor V heavy chain with prothrombin and prethrombin 1 and role of activated protein C in regulating this interaction: analysis by analytical ultracentrifugation. Biochemistry. 1989;28:2348–54. doi: 10.1021/bi00431a055. [DOI] [PubMed] [Google Scholar]
- 10.Blostein MD, Rigby AC, Jacobs M, Furie B, Furie BC. The Gla domain of human prothrombin has a binding site for factor Va. J. Biol. Chem. 2000;275:38120–6. doi: 10.1074/jbc.M007174200. [DOI] [PubMed] [Google Scholar]
- 11.Kalafatis M, Beck DO, Mann KG. Structural Requirements for Expression of Factor Va Activity. J. Biol. Chem. 2003;278:33550–33561. doi: 10.1074/jbc.M303153200. [DOI] [PubMed] [Google Scholar]
- 12.Bock PE, Panizzi P, Verhamme IMA. Exosites in the substrate specificity of blood coagulation reactions. J. Thromb. Haemost. 2007;5:81–94. doi: 10.1111/j.1538-7836.2007.02496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim BJ, Koo SY, Kim SS. A peptide derived from human prothrombin fragment 2 inhibits prothrombinase and angiogenesis. Thromb. Res. 2002;106:81–7. doi: 10.1016/S0049-3848(02)00086-5. [DOI] [PubMed] [Google Scholar]
- 14.Taneda H, Andoh K, Nishioka J, Takeya H, Suzuki K. Blood coagulation factor Xa interacts with a linear sequence of the kringle 2 domain of prothrombin. J. Biochem. 1994;116:589–97. doi: 10.1093/oxfordjournals.jbchem.a124565. [DOI] [PubMed] [Google Scholar]
- 15.Mammen M, Choi SK, Whitesides GM. Angew. Chemie Int. Ed. 1998. Polyvalent Interactions Occur Throughout Biology Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors; pp. 2754–2794. [DOI] [PubMed] [Google Scholar]
- 16.Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alberti S. The wisdom of crowds: regulating cell function through condensed states of living matter. J. Cell Sci. 2017;130:2789–2796. doi: 10.1242/jcs.200295. [DOI] [PubMed] [Google Scholar]
- 18.Zhou H-X, Nguemaha V, Mazarakos K, Qin S. Why Do Disordered and Structured Proteins Behave Differently in Phase Separation? Trends Biochem. Sci. 2018;43:499–516. doi: 10.1016/j.tibs.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim PY, Nesheim ME. Further evidence for two functional forms of prothrombinase each specific for either of the two prothrombin activation cleavages. J. Biol. Chem. 2007;282:32568–81. doi: 10.1074/jbc.M701781200. [DOI] [PubMed] [Google Scholar]
- 20.Boskovic DS, Bajzar LS, Nesheim ME. Channeling during prothrombin activation. J. Biol. Chem. 2001;276:28686–93. doi: 10.1074/jbc.M101813200. [DOI] [PubMed] [Google Scholar]
- 21.Wolberg AS, Morris DP, Stafford DW. Factor IX activation by factor XIa proceeds without release of a free intermediate. Biochemistry. 1997;36:4074–9. doi: 10.1021/bi962274y. [DOI] [PubMed] [Google Scholar]
- 22.Billy D, Willems GM, Hemker HC, Lindhout T. Prothrombin contributes to the assembly of the factor Va-factor Xa complex at phosphatidylserine-containing phospholipid membranes. J. Biol. Chem. 1995;270:26883–9. doi: 10.1074/jbc.270.45.26883. [DOI] [PubMed] [Google Scholar]
- 23.Billy D, Speijer H, Willems G, Hemker HC, Lindhout T. Prothrombin activation by prothrombinase in a tubular flow reactor. J. Biol. Chem. 1995;270:1029–34. doi: 10.1074/jbc.270.3.1029. [DOI] [PubMed] [Google Scholar]
- 24.Posma JJN, Posthuma JJ, Spronk HMH. Coagulation and non-coagulation effects of thrombin. J. Thromb. Haemost. 2016;14:1908–1916. doi: 10.1111/jth.13441. [DOI] [PubMed] [Google Scholar]
- 25.Boskovic D, Giles A, Nesheim ME. Studies of the role of factor Va in the factor Xa-catalyzed activation of prothrombin, fragment 1.2-prethrombin-2, and dansyl-L-glutamyl- glycyl-L-arginine-meizothrombin in the absence of phospholipid. J. Biol. Chem. 1990;265:10497–10505. [PubMed] [Google Scholar]
- 26.Kotkow KJ, Deitcher SR, Furie B, Furie BC. The second kringle domain of prothrombin promotes factor Va-mediated prothrombin activation by prothrombinase. J. Biol. Chem. 1995;270:4551–4557. doi: 10.1074/jbc.270.9.4551. [DOI] [PubMed] [Google Scholar]
- 27.Arni RK, Padmanabhan K, Padmanabhan KP, Wu TP, Tulinsky A. Structure of the non-covalent complex of prothrombin kringle 2 with PPACK-thrombin. Chem. Phys. Lipids. 1994;67–68:59–66. doi: 10.1016/0009-3084(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 28.Arni RK, Padmanabhan K, Padmanabhan KP, Wu TP, Tulinsky A. Structures of the noncovalent complexes of human and bovine prothrombin fragment 2 with human PPACK-thrombin. Biochemistry. 1993;32:4727–4737. doi: 10.1021/bi00069a006. [DOI] [PubMed] [Google Scholar]
- 29.Adams TE, Huntington JA. Structural transitions during prothrombin activation: On the importance of fragment 2. Biochimie. 2016;122:235–242. doi: 10.1016/j.biochi.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Z, Pelc LA, Di Cera E. Crystal structure of prethrombin-1. Proc. Natl. Acad. Sci. USA. 2010;107:19278–83. doi: 10.1073/pnas.1010262107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foster MP, et al. Chemical shift as a probe of molecular interfaces: NMR studies of DNA binding by the three amino-terminal zinc finger domains from transcription factor IIIA. J. Biomol. NMR. 1998;12:51–71. doi: 10.1023/A:1008290631575. [DOI] [PubMed] [Google Scholar]
- 32.Koutychenko, A. Interactions of human prothrombin fragment 2 with thrombin and factor Va. Ph.D. thesis, McGill University (2002).
- 33.Ni F. Recent developments in transferred NOE methods. Prog. Nucl. Magn. Reson. Spectrosc. 1994;26:517–606. doi: 10.1016/0079-6565(94)90000-0. [DOI] [Google Scholar]
- 34.Nesheim ME, Prendergast FG, Mann KG. Interactions of a fluorescent active-site-directed inhibitor of thrombin: dansylarginine N-(3-ethyl-1,5-pentanediyl)amide. Biochemistry. 1979;18:996–1003. doi: 10.1021/bi00573a010. [DOI] [PubMed] [Google Scholar]
- 35.Esmon CT, Jackson CM. The conversion of prothrombin to thrombin. IV. The function of the fragment 2 region during activation in the presence of factor V. J. Biol. Chem. 1974;249:7791–7. [PubMed] [Google Scholar]
- 36.Heldebrant CM, Butkowski RJ, Bajaj SP, Mann KG. The activation of prothrombin. II. Partial reactions, physical and chemical characterization of the intermediates of activation. J. Biol. Chem. 1973;248:7149–63. [PubMed] [Google Scholar]
- 37.Krishnaswamy S, Walker RK. Contribution of the Prothrombin Fragment 2 Domain to the Function of Factor Va in the Prothrombinase Complex. Biochemistry. 1997;36:3319–3330. doi: 10.1021/bi9623993. [DOI] [PubMed] [Google Scholar]
- 38.Bajaj SP, Butkowski RJ, Mann KG. Prothrombin fragments. Ca2+binding and activation kinetics. J. Biol. Chem. 1975;250:2150–6. [PubMed] [Google Scholar]
- 39.Deguchi H, Takeya H, Gabazza EC, Nishioka J, Suzuki K. Prothrombin kringle 1 domain interacts with factor Va during the assembly of prothrombinase complex. Biochem. J. 1997;321:729–35. doi: 10.1042/bj3210729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin PD, Malkowski MG, Box J, Esmon CT, Edwards BF. New insights into the regulation of the blood clotting cascade derived from the X-ray crystal structure of bovine meizothrombin des F1 in complex with PPACK. Structure. 1997;5:1681–93. doi: 10.1016/S0969-2126(97)00314-6. [DOI] [PubMed] [Google Scholar]
- 41.Myrmel KH, Lundblad RL, Mann KG. Characteristics of the association between prothrombin fragment 2 and alpha-thrombin. Biochemistry. 1976;15:1767–73. doi: 10.1021/bi00653a027. [DOI] [PubMed] [Google Scholar]
- 42.Kim PY, Nesheim ME. Down regulation of prothrombinase by activated protein C during prothrombin activation. Thromb. Haemost. 2010;104:61–70. doi: 10.1160/TH09-09-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bianchini EP, Orcutt SJ, Panizzi P, Bock PE, Krishnaswamy S. Ratcheting of the substrate from the zymogen to proteinase conformations directs the sequential cleavage of prothrombin by prothrombinase. Proc. Natl. Acad. Sci. USA. 2005;102:10099–104. doi: 10.1073/pnas.0504704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirbawi J, Bukys MA, Barhoover MA, Erdogan E, Kalafatis M. Role of the acidic hirudin-like COOH-terminal amino acid region of factor Va heavy chain in the enhanced function of prothrombinase. Biochemistry. 2008;47:7963–74. doi: 10.1021/bi800593k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim PY, Manuel R, Nesheim ME. Differences in prethrombin-1 activation with human or bovine factor va can be attributed to the heavy chain. Thromb. Haemost. 2009;102:623–33. doi: 10.1160/TH09-04-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toso R, Camire RM. Role of Hirudin-like factor Va heavy chain sequences in prothrombinase function. J. Biol. Chem. 2006;281:8773–9. doi: 10.1074/jbc.M511419200. [DOI] [PubMed] [Google Scholar]
- 47.Church WR, Ouellette LA, Messier TL. Modulation of human prothrombin activation on phospholipid vesicles and platelets using monoclonal antibodies to prothrombin fragment 2. J. Biol. Chem. 1991;266:8384–8391. [PubMed] [Google Scholar]
- 48.Banner DW, et al. The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature. 1996;380:41–46. doi: 10.1038/380041a0. [DOI] [PubMed] [Google Scholar]
- 49.Ruf W, et al. Importance of factor VIIa Gla-domain residue Arg-36 for recognition of the macromolecular substrate factor X Gla-domain. Biochemistry. 1999;38:1957–66. doi: 10.1021/bi982254r. [DOI] [PubMed] [Google Scholar]
- 50.Bukys MA, Kim PY, Nesheim ME, Kalafatis M. A control switch for prothrombinase: characterization of a hirudin-like pentapeptide from the COOH terminus of factor Va heavy chain that regulates the rate and pathway for prothrombin activation. J. Biol. Chem. 2006;281:39194–39204. doi: 10.1074/jbc.M604482200. [DOI] [PubMed] [Google Scholar]
- 51.Bradford HN, Orcutt SJ, Krishnaswamy S. Membrane binding by prothrombin mediates its constrained presentation to prothrombinase for cleavage. J. Biol. Chem. 2013;288:27789–800. doi: 10.1074/jbc.M113.502005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morris DP, Stevens RD, Wright DJ, Stafford DW. Processive post-translational modification. Vitamin K-dependent carboxylation of a peptide substrate. J. Biol. Chem. 1995;270:30491–8. doi: 10.1074/jbc.270.51.30491. [DOI] [PubMed] [Google Scholar]
- 53.Anderson PJ, Nesset A, Dharmawardana KR, Bock PE. Role of Proexosite I in Factor Va-dependent Substrate Interactions of Prothrombin Activation. J. Biol. Chem. 2000;275:16435–16442. doi: 10.1074/jbc.M001255200. [DOI] [PubMed] [Google Scholar]
- 54.Bukys MA, Orban T, Kim PY, Nesheim ME, Kalafatis M. The interaction of fragment 1 of prothrombin with the membrane surface is a prerequisite for optimum expression of factor Va cofactor activity within prothrombinase. Thromb. Haemost. 2008;99:511–22. doi: 10.1160/TH07-08-0532. [DOI] [PubMed] [Google Scholar]
- 55.Chen L, Yang L, Rezaie AR. Proexosite-1 on prothrombin is a factor Va-dependent recognition site for the prothrombinase complex. J. Biol. Chem. 2003;278:27564–9. doi: 10.1074/jbc.M302707200. [DOI] [PubMed] [Google Scholar]
- 56.Kalafatis M, Krishnaswamy S, Rand MD, Mann KG. Factor V. Methods Enzymol. 1993;222:224–36. doi: 10.1016/0076-6879(93)22016-9. [DOI] [PubMed] [Google Scholar]
- 57.Ikura M, Kay LE, Bax A. Improved three-dimensional 1H-13C-1H correlation spectroscopy of a 13C-labeled protein using constant-time evolution. J. Biomol. NMR. 1991;1:299–304. doi: 10.1007/BF01875522. [DOI] [PubMed] [Google Scholar]
- 58.Bax A, Ikura M. An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the alpha-carbon of the preceding residue in uniformly 15N/13C enriched proteins. J. Biomol. NMR. 1991;1:99–104. doi: 10.1007/BF01874573. [DOI] [PubMed] [Google Scholar]
- 59.Mori S, Abeygunawardana C, Johnson MO, van Zijl PC. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B. 1995;108:94–8. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 60.Piotto M, Saudek V, Sklenár V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–5. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 61.Matsuo H, Kupce E, Li H, Wagner G. Increased sensitivity in HNCA and HN(CO)CA experiments by selective C beta decoupling. J. Magn. Reson. B. 1996;113:91–6. doi: 10.1006/jmrb.1996.0161. [DOI] [PubMed] [Google Scholar]
- 62.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 63.Bartels C, Xia TH, Billeter M, Güntert P, Wüthrich K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 64.Vu TT, et al. Histidine-rich glycoprotein binds DNA and RNA and attenuates their capacity to activate the intrinsic coagulation pathway. Thromb. Haemost. 2016;115:89–98. doi: 10.1160/TH15-04-0336. [DOI] [PubMed] [Google Scholar]
- 65.Petrera NS, et al. Long range communication between exosites 1 and 2 modulates thrombin function. J. Biol. Chem. 2009;284:25620–9. doi: 10.1074/jbc.M109.000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu C, et al. The roles of selected arginine and lysine residues of TAFI (Pro-CPU) in its activation to TAFIa by the thrombin-thrombomodulin complex. J. Biol. Chem. 2009;284:7059–67. doi: 10.1074/jbc.M804745200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.