

Abstract
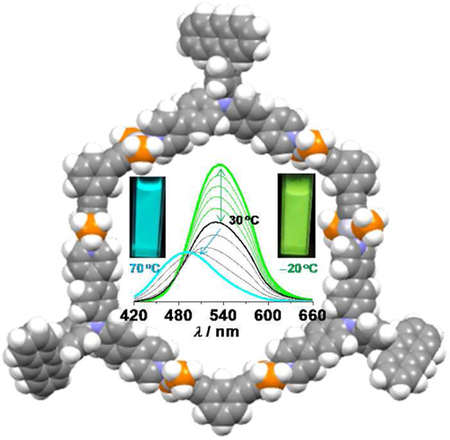
The synthesis, characterization, and temperature-responsive properties of two fluorescent organoplatinum(II) metal-lacycles are reported. Metallacycles M1 and M2 were prepared via the coordination-driven self-assembly of a 120° triarylamine ligand L1 and a 120° diplatinum(II) acceptor Pt-1 or 180° diplatinum(II) acceptor Pt-2, respectively. M1 and M2 are hexagonal metallacycles, comprising of three or six freely rotating anthracene pendants on their periphery, respectively. In response to the temperature variation between −20 and 60 °C, the ligand displays irregular emission changes, whereas both metallacycles show reversible absorption and emission spectral changes in THF. The changes in their green emission intensity also exhibit a linear correlation to the temperature variation, with an average sensitivity of −0.67% and −0.77% per °C for M1 and M2, respectively. Furthermore, in coordinating solvents, such as DMF and CH3CN, M1 and M2 show different behaviors: in the lower temperature range, i.e. below 30 °C, their spectral changes are similar to what was observed in THF, however, at a higher temperature the metal-lacycles were destroyed by the solvents and displayed ratiometric fluorescent responses including a cyan emission of the ligand L1.
Graphical Abstract
INTRODUCTION
Temperature-responsive fluorescence phenomenon has a wide array of applications. To date, organic dyes,1–4 polymers,5–7 biomaterials,8–9 nanomaterials,10,11 coordination metal complexes10–12 and polymers13 have been developed for temperature sensing14–17 and bioapplications.15–20 In general, two typesof fluorescent changes are observed in response to temperature. In most cases, the emission intensity of the material increases or decreases as the temperature varies. This response is simple and straightforward and the material functions as a thermometer by monitoring the emission intensity changes. In contrast, a ratiometric fluorescent response is based on the change of the emission intensity ratio at two different wavelengths, which improves the measuring accuracy and precision via a self-calibration.18–19 Current efforts have been focused on the design and synthesis of new materials with improved accuracy and reproducibility by diminishing the background interference or invoking a ratiometric response.13
Coordination-driven self-assembly is a well-established methodology for fabricating well-defined supramolecular coordination complexes (SCCs) by the spontaneous formation of multiple metal-ligand bonds among carefully selected Lewis-acid metallic acceptors and Lewis-base organic donors.20–29
Due to their diverse shape and size, SCCs have been used in a wide range of applications including host-guest chemistry,25,30–39 catalysis,40–44 biological and chemical sensing.45–52 Recent studies show that the incorporation of photo-active motifs into 2D or 3D SCCs leads to new fluorescent materials with intriguing photophysical properties.35,39,53–57 We envisioned that the relatively rigid geometry of SCCs would be beneficial for developing sensitive and reproducible temperature-responsive materials that can diminish the background interference. Although, some temperature-responsive metal complexes have been reported,12,15 thermal responsive fluorescent SCCs remain a largely unexplored area.
In this work, we present the synthesis and temperature-responsive emissions of two hexagonal metallacycles M1 and M2 assembled from a 120° organic donor L1 and 120° (Pt-1) or 180° (Pt-2) diplatinum(II) acceptor, respectively (Figure 1). Ligand L1 is a triarylamine derivative containing one anthracene and two pyridine moieties. The presence of the anthracene unit makes L1 highly emissive via an internal charge transfer (ICT) excitation. The attachment of two pyridine units further allows L1 to function as a 120° organic donor for the coordination assembly with Pt-1 or Pt-2. M1 and M2 were designed and synthesized to examine the effect of size and different number of rotating anthracene pendants on the emission properties and responsive behavior of these SCCs.
Figure 1.

Synthetic routes to metallacycles M1 and M2.
RESULTS AND DISCUSSION
Synthesis and Characterization.
The organic ligand L1 was synthesized by a Pd-catalyzed Suzuki-Miyaura cross-coupling reaction between 4-bromo-N,N-bis(p-(pyrid-4-yl)phenyl)aniline58 and 9-anthracenylboronic acid. The hexagonal metallacycles M1 and M2 were readily prepared by stirring an equimolar mixture of L1 and Pt-1 or Pt-2 in a mixed solvent of CH2Cl2/CH3OH (2/1, v/v) for 10 h.23 The formation of discrete metallacyclic assemblies were confirmed by 1H NMR, 31P{1H} NMR and electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS) data. As shown in Figure 2a, the metallacycles M1 and M2 show very similar 1H NMR spectral changes compared to the free ligand L1. The Hβ protons of the pyridyl groups exhibit a large downfield shift after the formation of the metal-ligand coordination bonds (Δδ = −0.45 ppm for both M1 and M2), while the Hα protons display minor changes. The assignments of these protons are further supported by 1H-1H COSY NMR analysis (see Figures S1–S3). The 31P{1H} NMR spectra of M1 and M2 both display a sharp singlet with concomitant 195Pt satellites (δ = 16.60 ppm for M1 and 16.84 ppm for M2, respectively, Figure 2b), indicative of a single phosphorous environment for forming discrete and highly symmetric metallacyclic assemblies. These peaks are around 6 ppm upfield shifted relative to those of the relevant starting material Pt-1 and Pt-2. This change and the decreased coupling of the flanking 195Pt satellites in M1 and M2, compared to those of Pt-1 and Pt-2, respectively, reflect the electron back-donation from the platinum centers (ΔJ = −45.2 Hz for M1 and −26.2 Hz for M2). The stoichiometry of the formation of discrete metallacycles was further confirmed by the ESI-TOF-MS analysis, which showed peaks at m/z at 967.31 and 865.68 Da for the [M1 - 5OTf]5+ and [M2 −11OTf]11+ species, respectively (Figure S4). This supports the stoichiometry of M1 and M2 as shown in Figure 1.
Figure 2.

(a) 1H NMR (500 MHz, CD2Cl2, 298 K) and (b) 31P{1H} NMR (121.4 MHz, CD2Cl2, 298 K) spectra of compounds studied in this work.
To gain insight into the structural characteristics of the obtained metallacycles, PM6 semi-empirical calculations were performed with the PEt3 ligands on platinum being modeled as PH3 ligands (Figure 3). These results suggest that M1 and M2 are roughly planar hexagonal structures with three or six appended anthracene moieties. The simulated cavity size of M1 and M2 is about 3.2 and 6.6 nm, respectively.
Figure 3.

PM6-simulated geometrical structures of (a) PH3-M1 and (b) PH3-M2.
Photophysical Properties and Computational Results.
The photophysical properties of L1, M1, and M2 were investigated in different solvents (Figure 4 and Table 1). Ligand L1 shows an absorption band at 370 nm in THF. In contrast, the absorptions of M1 and M2 are bathochromically shifted about 50 nm and showed a significantly enhanced molar absorption coefficients due to the inclusion of multiple ligands in one metallacyclic structure.59 The absorption band at 335 nm for M2 is likely derived from the Pt-2 moieties (Figure S5). The effects of solvent polarity on the absorption spectra of these three compounds are insignificant (Figures S6–S8). However, the emission band of L1 is distinctly shifted to the lower-energy region in solvents with increasing polarity. For instance, L1 displays an emission maximum (λ,em,ma) at 436 nm in toluene and it shifts to around 500 nm in CH3CN and DMF. The emissions of M1 and M2 are also bathochromically shifted in polar solvents (Figure 4c and 4d). However, the degree of shift is much smaller with respect to that of L1, indicating that the formation of metal-ligand coordination bonds likely mitigates the effects to a large extent. These results also suggest that the emissions of these compounds are associated with the ICT excited states.60–61 The emission quantum yield (Φ) of L1 is over 25% in all solvents examined (Table 1), while those of M1 and M2 are in the range of 2.9% –12.9%. The decreased Φ of the metallacycles is attributed to a heavy atom effect which was also observed in similar SCC systems.54 The emission lifetime of L1, M1, and M2 are all around a few ns with two exponential components in THF (τ1 = 2.4 ns, τ2 = 7.5 ns for L1; τ1 = 2.9 ns, τ2 = 1.0 ns for M1; τ1 = 2.3 ns, τ2 = 7.5 ns for M2; Figure S9). The presence of two exponential components may be caused by the coexistence of different CT excited states from different conformations.4,62
Figure 4.

(a) Absorption spectra of L1, M1 and M2 in THF. (b,c,d) Normalized emission spectra of (b) L1, (c) M1, and (d) M2 in different solvents. (λex = 370 nm, c = 2.0 × 10−5 M for L1; λex = 400 nm, c = 2.0 × 10−6 M for M1 and M2).
Table 1.
Photophysical data in different solvents.a
| Toluene | THF | Ethyl acetate | CH3CN | DMF | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | λem/nm | Φ/% | λem/nm | Φ/% | λem/nm | Φ/% | λem/nm | Φ/% | λem/nm | Φ/% |
| L1 | 436 | 39.0 | 458 | 34.5 | 454 | 36.5 | 501 | 26.4 | 498 | 33.1 |
| M1 | 503 | 12.9 | 526 | 10.1 | 512 | 9.1 | 529 | 7.5 | 531 | 7.4 |
| M2 | 505 | 2.9 | 520 | 8.4 | 511 | 4.9 | 527 | 6.9 | 529 | 6.8 |
Emission quantum yield was estimated by referencing to that of quinine sulfate in 0.1 M H2SO4 (Φ = 56%).
DFT calculations (B3LYP/LANL2DZ/6–31G*) suggest that the LUMO of L1 is localized on the anthracene unit, while the HOMO is delocalized over the anthracene-triphenylamine segment (Figure 5). To gain insight into the electronic properties of M1 and M2, DFT calculations were performed on a model compound Pt-3, which holds L1 and two [(PMe3)2Pt(C≡CPh)] moie-ties via coordination bonds (Figure 5). The metallacycles M1 and M2 are expected to have similar electronic properties as Pt-3. The results suggest that the electronic properties of L1 changed after coordination to the platinum centers. In Pt-3, the LUMO is associated with the phenylpyridine segments, while the HOMO is localized on the anthracene unit. These changes are likely because the coordination bonds that comprise the phenylpyridine and triphenylamine segments are electron-deficient compared to L1.63 TDDFT results on the ground state of L1 and Pt-3 indicate that the HOMO → LUMO transition is mainly responsible for the lowest excitation (Table S1 and Figures S10 and S11). In addition, TDDFT results of the singlet excited state of L1 and Pt-3 suggest that their emissions are associated with the triphenylamine to anthracene charge transfer and the anthracene to diphenylpyridine charge transfer, respectively (Table S2 and Figures S12 and S13).
Figure 5.

Isodensity plots of the LUMO and HOMO of L1 and the model diplatinum complex Pt-3.
Temperature-Responsive Properties.
A number of organic dyes with ICT character have successfully been reported as fluorescent thermometers.4,16,64 However, the use of fluorescent SCCs in temperature sensing has not been exploited to date. The unique metallacyclic structures of M1 and M2 prompted us to examine their thermo-responsiveness, because temperature may have a strong effect on the degree of concerted rotation of these appended anthracene groups and thus on the emission properties of the complexes. As shown in Figure 6, upon increasing temperature from −20 to 60 °C, the absorptions at 400 nm of M1 and M2 in THF significantly decreased, while those between 300 and 350 nm remain essentially unchanged. Accordingly, the emission intensity of M1 and M2 at 520 nm significantly decreased. In particular, the emission intensity of M2 dropped by 60% over this temperature range. The heating-cooling cycling experiments indicated that these spectral change processes good reversibility (Figure 6c and S14). The temperature-dependent emission response of M1 and M2 can be fitted with a linear decay (Figure 6f), with a average sensitivity SA of −0.66% and −0.76% per °C for M1 and M2 respectively2 (where SA = ΔI/(Iref × ΔT)). Upon increasing temperature, the rotations of the appended anthracene groups are expected to speed up, which will disrupt the anthracene-associated charge transfer absorptions and emissions and thereby decrease the absorbance and emissions of M1 and M2. This is further supported by the fact that M2 with six anthracene groups is slightly more temperature sensitive relative to M1, having only three anthracene groups.
Figure 6.

Temperature-responsive absorption and emission properties of M1 and M2 in THF recorded between −20 and 60 °C (λex = 400 nm; c = 2.0 μM; 10 °C temperature intervals). (a,d) Absorption spectral response of (a) M1 and (d) M2. (b,e) Emission spectral response of (b) M1 and (e) M2. The inset shows the image of the solution under UV illumination (365 nm) at −20 and 60 °C, respectively. (c) The cyclic switching of fluorescence intensity upon heating and cooling for M1. The cycling switching of M2 is shown in Figure S12. (f) The temperature dependence of the normalized emission intensity at 520 nm. R2 = 0.98 for the linear fitting of both series of data.
In contrast, the emission spectrum and intensity of the ligand L1 shows rather irregular changes in response to temperature variations from −20 to 60 °C in THF (Figure S15). Unlike the metallacycles M1 and M2, all structural units of L1 can rotate freely. This may account for the irregular emission changes of L1 in response to temperature, thus suggesting the advantages of metallacycles in thermo-responsiveness.
When the temperature response of M1 was investigated in DMF, a two-step spectral change was observed. In the temperature range between −20 and 30 °C, M1 behaves in a similar manner to what was described for THF (Figures 7 and S16). When temperature was further increased up to 70 °C, the absorption between 400 and 450 nm significantly decreased and new absorption bands between 300 and 370 nm appeared. At the same time, the emission maximum gradually shifted from 530 to 490 nm. This change can be seen by the emission color change from green to cyan. The emission change cannot be reversed by decreasing the temperature of the DMF solution. However, upon removal of the solvent, the emission of M1 can be essentially recovered (Figure S17). As is shown in Figure 7c, the ratio between the emission intensity at 460 and 530 nm (I460/I530) remains almost constant below 30°C. However, when the temperature was further elevated (particularly above 40 °C), the I460/I530 ratio increased sharply, and indicated that the temperature has surpassed a specific limit (in this case 40 °C), suggesting the potential application of the two-step emission response as ratiometric sensing of temperature. Similar temperature-responsive two-step emission spectral changes were observed for M2 in DMF and M1 in CH3CN, indicating the generality of this method (Figures S18 and S19). The 1H NMR spectrum of M1 at 60 °C in CD3CN exhibits distinct changes relative to it at 25 °C, where the Hβ signal of M1 decreases and shows the 1H NMR characteristics of L1 (Figures S20), suggesting a small mount of decomposing of M1. These results indicate that the metallacycle may decompose at elevated temperature by the coordinating solvents. A similar self-destructive mechanism has previously been reported for amino acid sensing using fluorescent platinum(II) cages.45
Figure 7.

Temperature-responsive absorption and emission properties of M1 in DMF recorded between −20 and 70 °C (λex = 400 nm; c = 2.0 μM; 10 °C temperature intervals). (a) absorption spectral changes. (b) Emission spectral changes. The inset shows the image of the solution under UV illumination (365 nm) at −20 and 70 °C, respectively. (c) Temperature dependence of the emission intensity at 530 nm (I530) and 460 nm (I460) and the ratio between I460 and I530.
CONCLUSION
Two hexagonal platinum(II) metallacycles M1 and M2 with three and six appended anthracene groups respectively, were designed and synthesized by the supramolecular coordination of a 120° organic donor L1 with a 120° or 180° diplatinum(II) acceptor, respectively. Complexes M1 and M2 are emissive in common organic solvents. In THF, M1 and M2 exhibited reversible absorption and emission spectral changes in response to temperature variation between −20 and 60 °C. The larger metallacycle M2 displayed a slightly higher sensitivity than M1, while the precursor ligand L1 only showed irregular fluorescent changes in the same temperature range. On the other hand, two-step spectral changes were observed for M1 and M2 in solvents such as DMF and CH3CN. In the lower temperature range, emission spectral changes followed the trend as observed in THF. At elevated temperature, these complexes displayed ratiometric emission spectral changes via a self-destruction mechanism. This work demonstrates that fluorescent metallacycles possess interesting and promising temperature-responsive properties and it opens up a new arena for the development of stimuli-responsive materials.
EXPERIMENTAL SECTION
Materials and Methods.
All reagents were commercially available and used as supplied without further purification. 4-bromo-N,N-bis(p-(pyrid-4-yl)phenyl)aniline58 and diplatinum acceptor Pt-1 and Pt-2 were prepared according to the reported procedures.29 1H NMR and 13C NMR spectra were recorded in the designated solvents on a Varian Inova 500 or 400 MHz spectrometer. 31P{1H} NMR spectra were recorded on a Varian Unity 300 MHz spectrometer, using an external unlocked sample of 85% H3PO4 (δ = 0) as reference. Mass spectra were recorded on a Micromass Quattro II triple-quadrupole mass spectrometer using electrospray ionization with a MassLynx operating system. Absorption and fluorescence spectra were recorded on a Hitachi U-4100 and Hitachi F-7000 Spectrophotometer, equipped with 1 cm quartz cuvette from Starna Cells, Inc. The temperature-dependent fluorescent experiments were conducted on a Hitachi F-3000 fluorescence spectrophotometer. The luminescence lifetime was obtained on Quantaurus-Tau Fluorescence lifetime spectrometer C11367 from Hamamatsu Photonics.
Synthesis of 4-bromo-N,N,-bis(p-(pyrid-4-yl)phenyl)aniline.
A suspension of tri(p-bromophenyl)amine (2.41 g, 5.0 mmol), pyrid-4-yl boronic acid (0.93 g, 7.5 mmol), Pd(PPh3)4 (115.6 mg, 0.10 mmol) and K2CO3 (1.66 g, 12.0 mmol) in a mixture of 50 mL of 1,4-dioxane and 25 mL of H2O was refluxed at 100 °C for 24 h under N2 atmosphere. After cooling to room temperature, the reaction mixture was extracted with copious CH2Cl2 (3 × 50 mL). The combined organic layers were concentrated and subjected to flash column chromatography on silica gel (eluent: ethyl acetate/methanol, 100/4, v/v) to afford 900 mg of 4-bromo-N,N-bis(p-(pyrid-4-yl)phenyl)aniline as a pale yellow solid in 38% yield. 1H NMR (400 MHz, CDCl3): δ 8.64 (d, J = 5.5 Hz, 4H), 7.56 (d, J = 8.7 Hz, 4H), 7.47 (d, J = 8.0 Hz, 4H), 7.42 (d, J = 8.9 Hz, 2H), 7.19 (d, J = 8.6 Hz, 4H), 7.05 (d, J = 8.8 Hz, 2H). ESI-TOF-HRMS calcd for [M + H]+ C28H20BrN3 (m/z): 478.0919. Found: 478.0919.
Synthesis of ligand L1.
A suspension of 4-bromo-N,N-bis(p-(pyrid-4-yl)phenyl)aniline (239.2 mg, 0.50 mmol), 9-anthracenylboronic acid (222.1 mg, 1.0 mmol), Pd(PPh3)4 (57.8 mg, 0.05 mmol) and K2CO3 (207.4 mg, 1.5 mmol) in a mixture of 20 mL 1,4-dioxane and 10 mL of H2O was refluxed at 100 °C for 24 h under N2 atmosphere. After cooling to room temperature, the reaction mixture was extracted with copious CH2Cl2 (3 × 50 mL). The combined organic layers were concentrated and subjected to flash column chromatography on silica gel (eluent: ethyl acetate/methanol, 100/4, v/v) to afford 120 mg of L1 as a yellow solid in 42% yield. 1H NMR (500 MHz, CD2Cl2): δ 8.62 (J = 6.6 Hz, 4H), 8.53 (s, 1H), 8.07 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 9.8, 2H), 7.68 (d, J = 8.6 Hz, 4H), 7.54 (d, J = 6.2 Hz, 4H), 7.48 (t, J = 8.2 Hz, 2H), 7.45 – 7.39 (m, 10H). 13C NMR (125 MHz, CD2Cl2): δ 150.55, 148.57, 147.52, 146.48, 136.75, 134.44, 132.70, 132.59, 131.71, 130.56, 128.64, 128.20, 126.92, 126.82, 125.69, 125.44, 125.15, 124.54, 121.14. ESI-TOF-HRMS calcd for [M + H]+ C42H29N3 (m/z): 576.2440. Found: 576.2437.
Synthesis of M1.
To a mixture of 4 mL of CH2Cl2 and 2 mL of CH3OH were added ligand L1 (5.76 mg, 10.0 μmol) and Pt-l (12.8 mg, 10.0 μmol). After stirring at room temperature for 10 h, the system was concentrated by flushing with N2 gas, followed by the addition of 7 mL of diethyl ether. The resulting precipitate was collected by centrifugation to give 14 mg of M1 in 90% yield. 1H NMR (500 MHz, CD2Cl2): δ 8.62 (J = 5.6 Hz, 12H), 8.55 (s, 3H), 8.09 (d, J = 8.5 Hz, 6H), 7.97 (d, J = 5.8, 12H), 7.89 (d, J = 8.9 Hz, 12H), 7.80 (d, J = 8.8 Hz, 6H), 7.18 – 7.53 (m, overlapped, 48H), 1.83–1.86 (m, overlapped, 72H), 1.17–1.24 (m, overlapped, 108H). 31P{1H} NMR (121.4 MHz, CD2Cl2): δ 16.60 (s, 195Pt satellites, 1JPt-P = 2333.6 Hz). ESI-TOF-MS calcd for [M – 5OTf]5+ (m/z): 967.33. Found: 967.31.
Synthesis of M2.
According to the same procedure for the synthesis of M1, M2 was prepared from ligand L1 (5.76 mg, 10 μmol) and Pt-2 (12.85 mg, 10 μmol) in 91% yield as a yellow solid (16 mg of M2 was isolated). 1H NMR (500 MHz, CD2Cl2): δ 8.60 (J = 5.6 Hz, 24H), 8.57 (s, 6H), 8.11 (d, J = 8.5 Hz, 12H), 7.98 (d, J = 6.1 Hz, 24H), 7.91 (d, J = 8.5 Hz, 24H), 7.81 (d, J = 9.7 Hz, 12H), 7.45 – 7.55 (m, overlapped, 72H), 7.26 (s, 24H), 1.85–1.88 (m, overlapped, 144H), 1.15–1.25 (m, overlapped, 216H). 31P{1H} NMR (121.4 MHz, CD2Cl2, 298 K) δ: 16.84 (s, 195Pt satellites, 1JPt-P = 2323.8 Hz). ESI-TOF-MS calcd for [M – 11OTf]11+ (m/z): 865.66. Found: 865.68.
Calculation Methods.
DFT calculations were carried out using the B3LYP exchange correlation function and implemented in the Gaussian 09 package. The electronic structures were optimized using a general basis set with the Los Alamos effective core potential LANL2DZ basis set for Pt and 6–31G* for other atoms.65 No symmetry constraints were used in the optimization (nosymm keyword was used). All orbitals have been computed at an isovalue of 0.02 e/bohr3. TDDFT calculations were performed on the DFT-optimized structures on the same level of theory.
Supplementary Material
ACKNOWLEDGMENT
Y.-W.Z. thank the National Natural Science Foundation of China (grants 91622120, 21601194, 21472196, and 21521062), the Strategic Priority Research Program of the Chinese Academy of Sciences (grant XDB12010400), and the program of China Scholarships Council (grant 201704910576) for funding support. X.L. is thankful for financial support from the National Science Foundation (CHE-1506722). P.J.S. thanks the NIH (Grant R01 CA215157) for financial support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Additional experimental data and characterization spectra, including Figures S1 – S28.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Homma M; Takei Y; Murata A; Inoue T; Takeoka S Chem. Commun 2015, 51, 6194–6197. [DOI] [PubMed] [Google Scholar]
- (2).Pais VF; Lassaletta JM; Fernández R; El - Sheshtawy HS; Ros A; Pischel U Chem. Eur. J 2014, 20, 7638–7645. [DOI] [PubMed] [Google Scholar]
- (3).Liu X; Li S; Feng J; Li Y; Yang G Chem. Commun 2014, 50, 2778–2780. [DOI] [PubMed] [Google Scholar]
- (4).Li K; Liu Y; Li Y; Feng Q; Hou H; Tang BZ Chem. Sci 2017, 8, 7258–7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Okabe K; Inada N; Gota C; Harada Y; Funatsu T; Uchiyama S Nat. Commun 2012, 3, 705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chen Z; Zhang KY; Tong X; Liu Y; Hu C; Liu S; Yu Q; Zhao Q; Huang W Adv. Funct. Mater 2016, 26, 4386–4396. [Google Scholar]
- (7).Gota C; Okabe K; Funatsu T; Harada Y; Uchiyama SJ Am. Chem. Soc 2009, 131, 2766–2767. [DOI] [PubMed] [Google Scholar]
- (8).Ebrahimi S; Akhlaghi Y; Kompany-Zareh M; Rinnan Å ACS Nano. 2014, 8, 10372–10382. [DOI] [PubMed] [Google Scholar]
- (9).Wong FH; Banks DS; Abu-Arish A; Fradin CJ Am. Chem. Soc 2007, 129, 10302–10303. [DOI] [PubMed] [Google Scholar]
- (10).Yam VW-W; Au VK-M; Leung SY-L Chem. Rev 2015, 115, 7589–7728. [DOI] [PubMed] [Google Scholar]
- (11).Chakraborty S; Hong W; Endres KJ; Xie T-Z; Wojtas L; Moorefield CN; Wesdemiotis C; Newkome GR J. Am. Chem. Soc 2017, 139, 3012–3020. [DOI] [PubMed] [Google Scholar]
- (12).Chan AK-W; Ng M; Wong Y-C; Chan M-Y; Wong W-T; Yam VW-W J. Am. Chem. Soc 2017, 139, 10750–10761. [DOI] [PubMed] [Google Scholar]
- (13).Rao X; Song T; Gao J; Cui Y; Yang Y; Wu C; Chen B; Qian GJ Am. Chem. Soc 2013, 135, 15559–15564. [DOI] [PubMed] [Google Scholar]
- (14).Liebsch G; Klimant I; Wofbeis OS Adv. Mater 1999, 11, 1296–1299. [Google Scholar]
- (15).Wang X.-d; Wofbeis OS; Meier RJ Chem. Soc. Rev 2013, 42, 7834–7869. [DOI] [PubMed] [Google Scholar]
- (16).Feng J; Tian K; Hu D; Wang S; Li S; Zeng Y; Li Y; Yang G Angew. Chem. Int. Ed 2011, 50, 8072–8076. [DOI] [PubMed] [Google Scholar]
- (17).Plakhotnik T; Doherty MW; Cole JH; Chapman R; Manson NB Nano Lett. 2014, 14, 4989–4996. [DOI] [PubMed] [Google Scholar]
- (18).Takei Y; Arai S; Murata A; Takabayashi M; Oyama K; Ishiwata S. i.; Takeoka S; Suzuki M ACS Nano. 2013, 8, 198–206. [DOI] [PubMed] [Google Scholar]
- (19).Brites CDS; Lima PP; Silva NJO; Millan A; Amaral VS; Palacio F; Carlos LD Nanoscale 2012, 4, 4799–4829. [DOI] [PubMed] [Google Scholar]
- (20).Avestro AJ; Belowich ME; Stoddart JF Chem. Soc. Rev 2012, 41, 5881–5895. [DOI] [PubMed] [Google Scholar]
- (21).Fujita M; Tominaga M; Hori A; Therrien B Acc. Chem. Res 2005, 38, 369–378. [DOI] [PubMed] [Google Scholar]
- (22).Sun Q-F; Sato S; Fujita M Nat. Chem 2012, 4, 330. [DOI] [PubMed] [Google Scholar]
- (23).Lescop C Acc. Chem. Res 2017, 50, 885–894. [DOI] [PubMed] [Google Scholar]
- (24).Huang SL; Hor TSA; Jin GX Coord. Chem. Rev 2017, 333, 1–26. [Google Scholar]
- (25).Xu L; Wang YX; Chen LJ; Yang HB Chem. Soc. Rev 2015, 44, 2148–2167. [DOI] [PubMed] [Google Scholar]
- (26).Li H; Yao ZJ; Liu D; Jin GX Coord. Chem. Rev 2015, 293, 139–157. [Google Scholar]
- (27).Chen L; Chen QH; Wu MY; Jiang FL; Hong MC Acc. Chem. Res 2015, 48, 201–210. [DOI] [PubMed] [Google Scholar]
- (28).Saha ML; Yan X; Stang PJ Acc. Chem. Res 2016, 49, 2527–2539. [DOI] [PubMed] [Google Scholar]
- (29).Cook TR; Stang PJ Chem. Rev 2015, 115, 7001–7045. [DOI] [PubMed] [Google Scholar]
- (30).Winter A; Hoeppener S; Newkome GR; Schubert US Adv. Mater 2011, 23, 3484–3498. [DOI] [PubMed] [Google Scholar]
- (31).Wang QQ; Gonell S; Leenders S; Durr M; Ivanovic-Burmazovic I; Reek JNH Nat. Chem 2016, 8, 225–230. [DOI] [PubMed] [Google Scholar]
- (32).Therrien B; Suss-Fink G; Govindaswamy P; Renfrew AK; Dyson PJ Angew. Chem. Int. Ed 2008, 47, 3773–3776. [DOI] [PubMed] [Google Scholar]
- (33).Zhou Z; Yan X; Saha ML; Zhang M; Wang M; Li X; Stang PJ J. Am. Chem. Soc 2016, 138, 13131–13134. [DOI] [PubMed] [Google Scholar]
- (34).Zhukhovitskiy AV; Zhong MZ; Keeler EG; Michaelis VK; Sun JEP; Hore MJA; Pochan DJ; Griffin RG; Willard AP; Johnson JA Nat. Chem 2016, 8, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Howlader P; Das P; Zangrando E; Mukherjee PS J. Am. Chem. Soc 2016, 138, 1668–1676. [DOI] [PubMed] [Google Scholar]
- (36).Browne C; Ramsay WJ; Ronson TK; Medley-Hallam J; Nitschke JR Angew. Chem. Int. Ed 2015, 54, 11122–11127. [DOI] [PubMed] [Google Scholar]
- (37).Zhang MM; Yin SC; Zhang J; Zhou ZX; Saha ML; Lu CJ; Stang PJ Proc. Natl. Acad. Sci. USA 2017, 114, 3044–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chakraborty S; Newkome GR Chem. Soc. Rev 2018, [DOI] [PubMed] [Google Scholar]
- (39).Zhou Z; Yan X; Cook TR; Saha ML; Stang PJ J. Am. Chem. Soc 2016, 138, 806–809. [DOI] [PubMed] [Google Scholar]
- (40).Toste FD; Sigman MS; Miller SJ Acc. Chem. Res 2017, 50, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Qiao YP; Zhang L; Li J; Lin W; Wang ZQ Angew. Chem. Int. Ed 2016, 55, 12778–12782. [DOI] [PubMed] [Google Scholar]
- (42).Mondal P; Sarkar S; Rath SP Chem. Eur. J 2017, 23, 7093–7103. [DOI] [PubMed] [Google Scholar]
- (43).Kaphan DM; Levin MD; Bergman RG; Raymond KN; Toste FD Science 2015, 350, 1235–1238. [DOI] [PubMed] [Google Scholar]
- (44).Neelakandan PP; Jimenez A; Thoburn JD; Nitschke JR Angew. Chem. Int. Ed 2015, 54, 14378–14382. [DOI] [PubMed] [Google Scholar]
- (45).Yeung MC-L; Yam VW-W Chem. Soc. Rev 2015, 44, 4192–4202. [DOI] [PubMed] [Google Scholar]
- (46).Eryazici I; Moorefield CN; Newkome GR Chem. Rev 2008, 108, 1834–1895. [DOI] [PubMed] [Google Scholar]
- (47).Zhang MM; Saha ML; Wang M; Zhou ZX; Song B; Lu CJ; Yan XZ; Li XP; Huang FH; Yin SC; Stang PJ J. Am. Chem. Soc 2017, 139, 5067–5074. [DOI] [PubMed] [Google Scholar]
- (48).Li XZ; Wu JG; Chen LY; Zhong XM; He C; Zhang R; Duan CY Chem. Commun 2016, 52, 9628–9631. [DOI] [PubMed] [Google Scholar]
- (49).Liu CL; Zhang RL; Lin CS; Zhou LP; Cai LX; Kong JT; Yang SQ; Han KL; Sun QF J. Am. Chem. Soc 2017, 139, 12474–12479. [DOI] [PubMed] [Google Scholar]
- (50).Vardhan H; Yusubov M; Verpoort F Coord. Chem. Rev 2016, 306, 171–194. [Google Scholar]
- (51).Ahmad N; Younus HA; Chughtai AH; Verpoort F Chem. Soc. Rev 2015, 44, 9–25. [DOI] [PubMed] [Google Scholar]
- (52).Wang J; He C; Wu PY; Wang J; Duan CY J. Am. Chem. Soc 2011, 133, 12402–12405. [DOI] [PubMed] [Google Scholar]
- (53).Ni XL; Chen SY; Yang YP; Tao ZJ Am. Chem. Soc 2016, 138, 6177–6183. [DOI] [PubMed] [Google Scholar]
- (54).Zhang QW; Li DF; Li X; White PB; Mecinovic J; Ma X; Agren H; Nolte RJM; Tian HJ Am. Chem. Soc 2016, 138, 13541–13550. [DOI] [PubMed] [Google Scholar]
- (55).Zhu MR; Yang CL Chem. Soc. Rev 2013, 42, 4963–4976. [DOI] [PubMed] [Google Scholar]
- (56).Yan XZ; Cook TR; Wang P; Huang FH; Stang PJ Nat. Chem 2015, 7, 342–348. [DOI] [PubMed] [Google Scholar]
- (57).Tian Y; Yan X; Saha ML; Niu Z; Stang PJ J. Am. Chem. Soc 2016, 138, 12033–12036. [DOI] [PubMed] [Google Scholar]
- (58).Zhang MD; Shi ZQ; Chen MD; Zheng HG Dalton Trans. 2015, 44, 5818–5825. [DOI] [PubMed] [Google Scholar]
- (59).Yan X; Wang M; Cook TR; Zhang M; Saha ML; Zhou Z; Li X; Huang F; Stang PJ J. Am. Chem. Soc 2016, 138, 4580–4588. [DOI] [PubMed] [Google Scholar]
- (60).Liu X; Li SY; Feng J; Li Y; Yang GQ Chem. Commun 2014, 50, 2778–2780. [DOI] [PubMed] [Google Scholar]
- (61).Rao M,R; Liao C-W; Su W-L; Sun S-SJ Mater. Chem. C 2013, 1, 5491–5501. [Google Scholar]
- (62).Fang Q; Li J; Li S; Duan R; Wang S; Yi Y; Guo X; Qian Y; Huang W; Yang G Chem. Commun 2017, 53, 5702–5705. [DOI] [PubMed] [Google Scholar]
- (63).Chen J-S; Zhao G-J; Cook TR; Han K-L; Stang PJ J. Am. Chem. Soc 2013, 135, 6694–6702. [DOI] [PubMed] [Google Scholar]
- (64).Wang X Chem. Soc. Rev 2013, 42, 7834. [DOI] [PubMed] [Google Scholar]
- (65).Hay PJ; Wadt WR J. Chem. Phys 1985, 82, 299–310. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.