

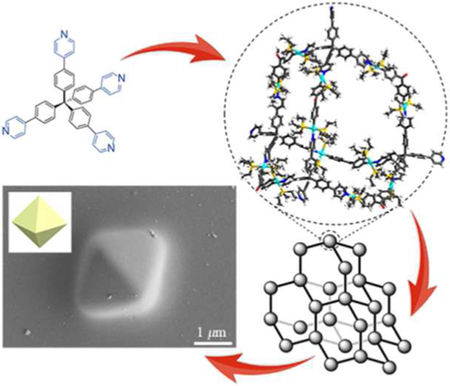
Abstract
Recently, porous framework materials with various network-type structures have been constructed via several different approaches, such as coordination interactions, reversible covalent bonds, and non-covalent interactions. Here, we have combined the concepts of supramolecular coordination complex (SCC) and metal-organic framework (MOF) to offer a new strategy to construct a diamondoid supramolecular coordination framework (SCF) from an adamantanoid supramolecular coordination cage as the tetrahedral node and a difunctional Pt(II) ligand as the linear linker via the stepwise orientation-induced supramolecular coordination. The adamantanoid supramolecular coordination cage has four uncoordinated pyridyl groups, which serve as the four vertexes of the tetrahedral geometry in the diamondoid framework. As a result, this diamondoid SCF exhibits an adamantanoid-to-adamantanoid substructure with two sets of pores including the interior cavity of the adamantanoid cage and the extended adamantanoid space in between the individual cages in the framework. In addition, the shape-controllable and highly-ordered self-assembly of nanometer-sized diamondoid SCF is observed as micrometer-sized regular octahedrons by evaporation under heating in DMSO. This study demonstrates the potential application of supramolecular coordination complexes in the precise construction of highly-regulated porous framework materials.
Graphical Abstract
INTRODUCTION
Although diamond has been known for a long time, scientists pay increasing attention to its atomic-level framework structure called a diamond lattice, where each carbon atom as a tetrahedral center is bonded covalently with other four carbon atoms and they show an arrangement of a variation of the face-centered cubic structure.1 Compared to its allotrope, graphite, diamond possesses several distinctive properties due to its framework structure, such as extreme hardness, high stability and thermal conductivity, broad optical transparency, and wide band gap.2 Therefore, controlling the structures in the synthesis process is crucial for the design of a variety of framework materials in order to obtain the desired properties. Inspired by diamond’s framework structure, chemists and material scientists have always been interested in this highly-ordered structure with Td symmetry, and attempt to construct similar diamondoid frameworks through molecular building blocks in a well-controlled manner while building up their own framework materials.3–17 In recent decades, porous framework materials have received considerable attention from the chemical, biological, and material science communities.18 Particularly, metal-organic frameworks (MOFs),19 covalent organic frameworks (COFs)20 and supra-molecular organic frameworks (SOFs),21 as prominent framework materials, have been widely studied and used for gas adsorption and separation,22–23 energy storage,24–25 heterogeneous catalysis,26 drug delivery,27 and luminescent materials.28 These frameworks have various network-type structures including two-dimensional (2D) layers and three-dimensional (3D) frameworks, the structures of which include triangle-, square-, rhombus-, hexagon-, cube-, and diamond-type shapes.29 Specifically, diamondoid frameworks have been constructed directly from tetrahedral precursors with metals or inorganic clusters (e.g. MOFs),3–5 complementary reactants (e.g. COFs),6–9 and complementary hosts or supramolecular building blocks (e.g. SOFs)10–17 via coordination interactions, reversible covalent bonds, and non-covalent interactions, respectively, and have exhibited excellent selective adsorption properties.7–9 However, to the best of our knowledge, the reports about stepwise construction of 3D diamondoid frameworks with multiple-level substructures are limited. Compared to the direct construction of 3D diamondoid frameworks, the geometry and structure of the framework materials could be controlled easily in the step-wise construction, but significant challenges remain in the designed and orientation-induced implementation of supra-molecular forces, which can express self-sorted assembly in the stepwise process.30
Supramolecular coordination is a powerful tool for constructing novel supramolecular coordination complexes (SCCs), which are typically obtained by simply mixing soluble metals and ligands that spontaneously form metal-ligand bonds to generate a discrete thermodynamically favored self-assembled structure.31–37 This methodology allows for the preparation of fascinating supramolecular architectures of predesigned and controlled shape, size, and functionality. For examples, Stang, Raymond, Fujita, Nitschke, and others have already developed a series of rationally designed supramo lecular coordination complexes with various shapes including triangle,38 square,39 rectangle,40 and higher polygonal 2D SCCs,41–43 as well as tetrahedron,44 cube,45 octahedron,46 cub-octahedron,47–49 dodecahedron,40 and other 3D SCCs.51–52 These SCCs have been used in host–guest chemistry, catalysis, supramolecular polymers, bio-applications, and so on. With the combined concepts of discrete SCCs and infinite MOFs,53–54 we offer a new strategy to stepwise construct a novel type of framework material from discrete supramolecular coordination complexes as the nodes via supramolecular interactions, called supramolecular coordination framework (SCF). In this strategy, an ideal SCC node, which should possess directional binding sites with highly-symmetrical geometry, can be connected predictably to neighboring SCCs with linkers via directional supramolecular interactions to generate a highly-ordered framework material with multiple-level substructures. Based on this idea, we report here the fabrication and construction of a diamondoid supramolecular coordination framework based on an adamantanoid supramolecular coordination cage as the node and a linear difunctional platinum(II) ligand as the linker. In fact, this adamantanoid cage, which possesses four uncoordinated pyridines as the four vertexes of the tetrahedral geometry, self-assembles with two equivalents of complementary linear difunctional Pt(II) ligands to form a diamondoid supramolecular coordination framework with an adamantanoid-to-adamantanoid substructure, which possesses two sets of porous structures from cages and frameworks, respectively. More interestingly, these nanometer-sized frameworks achieve a shape-controllable and highly-ordered self-assembly to form micrometer-sized regular octahedrons by evaporation under heating in DMSO.
RESULTS AND DISCUSSION
Design for diamondoid supramolecular coordination framework.
To exploit our strategy for constructing diamondoid supramolecular coordination framework, a suitable tetrahedral subunit, tetra(4-(4-pyridinyl)phenyl)methane (1)55 with the directing angle of nearly 108°, which is the optimal angle for the tetrahedral geometry, and a difunctional Pt(II) linker with a directing angle of 120°, bis[4,4’-(trans-Pt(PEt3)2OTf)]diphenylmethanone (2),56 were prepared according to literature reports (Scheme 1). Based on the specific angularity and geometry of the binding sites of 1 and 2, we envisioned that 1 undergoes supramolecular coordination with 2 to give a discrete adamantanoid platinum(II) cage (3) with four tetrahedrally-oriented and uncoordinated pyridine groups. Next, the assembly of adamantanoid platinum(II) cage 3 as the SCC node and bis[1,4-(trans-Pt(PEt3)2OTf)]ethynylbenzene (4),50 a difunctional Pt(II) ligand with a directing angle of 180°, as the linear linker, leads to the formation of a self-assembled intermediate, adamantanoidto-adamantanoid fragment (5), which further extend into a diamondoid supramolecular coordination frameworks (6). Furthermore, micrometer-sized regular octahedrons (SCF-1) with SCF features are self-assembled from diamondoid framework 6 by evaporation under heating (Scheme 1).
Scheme 1.

Schematic illustration for the stepwise construction process of adamantanoid supramolecular coordination cage 3, diamondoid supramolecular coordination framework 6, and micrometer-sized octahedron SCF-1.
Self-assembly of adamantanoid Pt(II) cage 3.
As the simplest diamondoid,1 adamantane consists of three connected cyclohexane-like rings in the “chair” configuration that comprise the highest symmetry group Td. Hence, an adamantanoid Pt(II) cage can be prepared via edge-directed assembly from four tetradentate angular subunits with an approximately 108° directing angle combined with six difunctional Pt(II) linkers with directing angels of 108~120°.57 When 2 was added into a solution of 1 in DMSO-d6 in a ratio of 6:4 while the stoichiometry was carefully monitored by 1H and 31P NMR, adamantanoid supramolecular coordination cage 3 was obtained in 68% isolated yield.
In the 1H and COSY NMR spectra of 3 (Figure 1a and Figure S5), the protons (H’a-d) of the coordinated pyridyl arms showed obvious downfield shifts (Δδ = 0.18–0.37 ppm), compared with those of ligand 1, consistent with the coordination of the N atoms to the platinum centers. More importantly, the protons (H*a-d) of the partial pyridyl arms in 3 only showed slight downfield shifts (Δδ = 0.03–0.04 ppm), indicating that these pyridyl groups at the four vertexes are uncoordinated. Moreover, the 31P{1H} spectra of 2 and 3 exhibited a sharp singlet with concomitant 195Pt satellites at 20.14 ppm for 2 and 14.45 ppm for 3, respectively. The significant upfield shift of 5.69 ppm is attributed to the coordination between Pt and N atoms, which indicates the formation of a highly-symmetric adamantanoid Pt(II) cage. Temperature-dependent 1H NMR spectra also showed that 3 is a stable SCC in DMSO-d6 at high temperature (up to 85 °C) (Figure S6). In the 2D diffusion-ordered spectroscopy (DOSY) experiment of 3, the observation of a single band confirmed that only a single product was formed, and the diffusion coefficient was calculated as (1.176 ± 0.025) × 10−10 m2 s−1 (Figure S7). The discrete nature of 3 was further evidenced by ESI-TOF-MS, which exhibits seven- and nine-positively charged peaks at m/z = 1,359.74 for [M-7OTf]7+ and 1,024.44 for [M-9OTf]9+, moreover the isotopic spacing of these peaks are consistent with their theoretical distributions (Figure 1c and Figure S8). Hence, the observed 1H, 31P, COSY, and DOSY NMR, and ESITOF-MS spectra are all consistent with the formation of a single and highly-symmetric adamantanoid Pt(II) cage.
Figure 1.

a) Partial 1H NMR spectra (500 MHz, DMSO-d6, 298 K) recorded for: i) 1, ii) 2, iii) 3, iv) 4 (CD2Cl2), v) 6; b) 31P{1H} NMR spectra (121.4 MHz, DMSO-d6, 298 K) recorded for: i) 2, ii) 3, iii) 4 (CD2Cl2), iv) 6; c) Calculated (i) and experimental (ii) ESI-TOF-MS spectra of 3 [M – 9OTf]9+; d) MMFF calculated structure of 3, Color: C, grey; N, blue; O, red; P, yellow; H, white; Pt, turquoise. Here, primes (‘) and stars (*) denote resonances for the coordinated and uncoordinated pyridyl arms of 3, respectively; double primes (“) denote resonances for 6.
Self-assembly of diamondoid supramolecular coordination framework 6.
The molecular mechanics force field (MMFF) calculated structure of 3 shows a cavity with an inside diameter of about 3.0 nm and an outside diameter of about 5.2 nm, and four uncoordinated pyridyl groups as the four vertexes (Figures 1d and S9). Therefore, 3, like carbon atoms in diamond, is an ideal SCC node for constructing diamondoid SCF materials. As expected, diamondoid supramolecular coordination framework 6 further assembled from 3 and 4 in DMSO-d6 with 62% isolated yield (Scheme 1).
The structure and photophysical properties of 6 were unambiguously elucidated by 1H, 31P, COSY, and DOSY NMR, infrared (IR), Raman, UV-vis, and fluorescence experiments. Firstly, 1H and COSY NMR experiments in DMSO-d6 provided important insight into the structure of 6 (Figures 1a and S10). Comparisons of the 1H NMR spectra of 3, 4, and 6, revealed that the protons (H*a-d) of the uncoordinated pyridyl arms in 3 shift downfield to the same chemical shift of the protons (H”a-d) of the coordinated pyridyl arms in 6, and the proton (Hz) of the phenyl ring in 4 became slightly downfield-shifted and broad in 6 (Figure 1a). Secondly, compared with those of 2 and 4, the 31P{1H} spectrum of 6 exhibited two sharp singlets with concomitant 195Pt satellites at 14.49 ppm and 16.81 ppm, corresponding to two distinctive sets of phosphorous environments in the adamantanoid cage (upfield shift: Δδ = 5.65 ppm) and on the coordinated linear linker (upfield shift: Δδ = 5.37 ppm), indicating the formation of a highly-symmetric framework 6 (Figure 1b). The phosphorous chemical shifts of subunit 2 in 3 (14.45 ppm) and 6 (14.49 ppm) are nearly the same, indicating that the adamantanoid cage remains intact in the framework structure of 6. Thirdly, IR and Raman spectroscopies confirmed the presence of the introduced carbon-carbon triple bond on the linker 4 in the framework structure (Figures S11-S12). Moreover, Uv-vis and fluorescence experiments showed that the absorbance maxima at 282 nm for 1 was red-shifted to 311 nm for 3 and 317 nm for 6, and the fluorescent intensities were decreasing during the stepwise formation of adamantanoid cage 3 and diamondoid framework 6 from tetrahedral subunit 1 (Figure S13).
Additionally, the diffusion coefficient of 6 in DMSO-d6 was calculated as (4.087 ± 0.250) × 10−11 m2 s−1, indicating the high uniformity of framework 6 (Figure S14). If we assume that 3 and 6 are roughly spherical, then the ratio of the measured diffusion coefficients can be converted into a ratio of molecular weights by the Stokes−Einstein equation,58 which in turn gives a degree of polymerization of about 24. Hence, the construction of framework 6 is about a 288-component assembly process, likely stepwise tuned by the angularity and directionality between the binding sites of the pyridyl donors with tetrahedral geometry and two different difunctional Pt(II) ligands with directing angles of 120° and 180°, respectively. The results of dynamic light scattering (DLS) experiments confirmed that the average hydrodynamic diameter (DH) of 3 and 6 are 4.9 nm and 91.3 nm, respectively, indicating the for mation of large supramolecular coordination frameworks (Figure 2). Despite the large size and molecular weight, framework 6 is still soluble (up to ~10 mg/mL) in DMSO, presumably due to the extensive solvation of its multiple charged surface. In summary, all of the above-mentioned data show that the assembly of adamantanoid platinum(II) cage 3 and linear linker 4 leads to the formation of a highly-symmetric supramolecular coordination framework 6. This stepwise assembly process was under the precise geometry-control of the binding sites of the subunits, making the overall frameworks comparable to a big protein in terms of size (~90 nm) and molecular weight (~253,300 Da).
Figure 2.

DLS profiles of the solution of 3 (2.5 mg/mL) and 6 (2.5 mg/mL) in DMSO.
As the smallest structural unit in 6, the MMFF calculated structure of adamantanoid fragment 5 showed that the adamantanoid-to-adamantanoid substructure of 6 has two sets of pores including the interior cavity (~3.0 nm) of adamantanoid cage and the extended adamantanoid space (~10.0 nm) in between the individual cages in the framework (Figure S15). As a result, the framework structure of 6 has a two-stage adamantanoid-to-adamantanoid substructure: The first-stage adamantanoid is cage 3 based on tetrahedral subunit 1 as primary nodes, while the second-stage adamantanoid is fragment 5 based on cage 3 as secondary nodes. Additionally, thermogravimetric analysis (TGA) of 6 showed a rapid weight loss of trapped solvents (3.1% observed) in the range of 30–70°C, followed by a stable platform until 300 °C (Figure S16). Based on its high stability, nitrogen sorption measurements of 6 were performed. However, only 2.86 cm3/g N2 was adsorbed at 77 K, and the pore size distribution (PSD) curve demonstrated that the average pore size of 6 was only about 2.0 nm (Figure S17). The low N2 adsorption and small pore size may be due to the fact that a number of OTf− counter ions reside in the framework structure.
Self-assembly of micrometer-sized regular octahedron SCF-1.
We also directly observed the morphology of the self-assembled framework 6 by scanning electron microscopy (SEM), atomic force microscopy (AFM) and transmission electron microscopy (TEM) experiments. The images obtained from the SEM experiments of 3 showed only discrete particles with irregular shapes (Figure S18). However, SEM images obtained from a solution of 6 in DMSO by evaporation at 50°C showed that nanometer-sized SCF 6 self-assembled into a micrometer-sized regular octahedron (SCF-1) with a side length of ~2.12 μm and a calculated volume of ~4.49 μm3 (Figure 3a–d). The size of micrometer-sized octahedron SCF-1 with particle size d defined as the distance between opposing vertexes ranges from ~2.65 μm to ~3.55 μm (Figure S19). Furthermore, the thickness of SCF-1 as determined by AFM was ~216 nm at the same concentration, indicating the 3D feature of SCF-1 (Figure S20). TEM images confirmed that these micrometer-sized regular octahedrons are solid (Figure S21). Therefore, we surmised that the nanometer-sized SCF 6 as the seeds grow can self-assemble or crystallize during the evaporation process under heating to SCF-1, which possess an extended diamondoid supramolecular coordination framework (Figure 3e). In the growth process, once SCF-1 has grown past a critical size, structural fluctuations become energetically costly so that the self-assembly becomes locked into a well-defined regular octahedron. In Nature, natural crystals, such as diamond, alum and fluorite, are octahedral, due to the framework of their atoms with diamond cubic geometry which also possess the tetrahedral-octahedral structure. In the shape-controlled synthesis of metal nanoparticles, only one geometry of the nanocrystal can be obtained when the population of seeds with specific internal structure is tightly controlled.59 Therefore, 6, bearing diamondoid framework units, tended to self-assemble or crystallize into an entropically favored and well-ordered arrangement that finally extended to a micrometer-sized regular octahedron controlled by its internal diamondoid framework structure. For the shape-controlled self-assembly of nanoparticles,30,60 it is important to provide an effective approach to construct highly-ordered self-assembled 3D framework structures with regular geometry for the design and synthesis of a variety of functional materials, including energy storage, gas absorption, and catalytic materials.
Figure 3.

SEM (a-d) images of SCF-1 prepared from the solution of 6 (2.5 mg/mL) in DMSO; e) Schematic illustration for the possible mechanism of SCF-1 formed from 6.
Conclusion
Supramolecular coordination is a powerful tool for constructing supramolecular coordination complexes (SCCs) with a variety of shapes and functionalities, due to its strong, highly directional, and dynamic nature. However, compared with targeting infinite arrays of MOFs, SCCs have only been utilized in the past in constructing discrete supramolecular assemblies. In this paper, we combined the concepts of discrete SCCs and infinite MOFs to offer a new strategy to stepwise construct a novel type of diamondoid supramolecular coordination framework (SCF). SCFs, with both the fundamental characteristics of MOFs and SCCs, can possess both a predictable and well-designed structure, when compared to MOFs, and open space and highly-ordered arrangements, when compared to SCCs. Therefore, this work will help draw the field of framework material science and supramolecular coordination chemistry closer together by showing the stepwise construction of a diamondoid supramolecular coordination framework based on an adamantanoid supramolecular coordination cage as the node and a linear difunctional platinum(II) ligand as the linker where both hold binding sites with specific angularity and geometry. Furthermore, these nanometer-sized SCFs undergo a shape-controllable and highly-regulated self-assembly to form micrometer-sized regular octahedrons defined by its diamondoid structure. We expect this approach to provide many other new frameworks with various geometries and valuable properties (e.g. significant porosity, well-defined structure, host-guest recognition, photopsychical and catalytic activity), which are attractive in designing and preparing new functional materials for potential applications such as energy storage, gas separation, drug delivery, sensing, heterogeneous catalysis, etc.
EXPERIMENTAL SECTION
Synthesis of adamantanoid cage 3.
To a solution of 1 (4.16 mg, 6.62 μmol) in DMSO-d6 (1.5 mL) was added 2 (13.31 mg, 9.93 μmol), and the reaction mixture was stirred at room temperature for 24 h. Then, the mixture was poured into ethyl ether (10 mL) to give a yellow precipitate, which was washed with CH2Cl2 (0.2 mL) and centrifuged, and washed again with CH2Cl2/Et2O (0.2 mL/5.0 mL) and centrifuged. The precipitate was dried under high vacuum to give yellow powder 3 (12.01 mg, 1.13 μmol, 68%). 1H NMR (500 MHz, DMSO-d6): 8.84 (s, 24H), 8.66 (s, 8H), 8.09 (s, 24H), 8.03 (s, 24H), 7.87 (s, 8H), 7.74 (s, 8H), 7.70–7.35 (m, 80H), 1.45–1.25 (m, 144H), 1.15–0.95 (m, 216H). 31P{1H} NMR (121.4 MHz, DMSO-d6): 14.45 (s, 195Pt satellites, 1JPt-P = 2647.7 Hz). ESI-TOF-MS: m/z 1359.74 for [3-7OTf]7+ and 1024.44 for [3-9OTf−]9+.
Synthesis of diamondoid framework 6.
To a solution of 3 (10.40 mg, 0.99 μmol) in DMSO-d6 (1.0 mL) was added 4 (6.03 mg, 4.69 μmol), and the reaction mixture was stirred at room temperature for 8 days. The mixture was poured into ethyl ether (5.0 mL) to give a yellow precipitate, which was washed with CH2Cl2 (0.2 mL) and centrifuged, and washed again with CH2Cl2/Et2O (0.2 mL/5.0 mL) and centrifuged. The precipitate was dried under high vacuum to give yellow powder 6 (9.20 mg, 62%). 1H NMR (500 MHz, DMSO-d6, n is the number of repeated unit): 8.81 (s, 32nH), 8.50–7.80 (m, 64nH), 7.80–6.80 (m, 88nH), 1.90–1.65 (m, 48nH), 1.50–1.20 (m, 144nH), 1.2–0.90 (m, 288nH). 31P{1H} NMR (121.4 MHz, DMSO-d6): 16.81 (s, 195Pt satellites, 1JPt-P = 2309.0 Hz), 14.49 (s, 195Pt satellites, 1 JPt-P = 2640.5 Hz).
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Natural Science Foundation of China (21771145 and 21472149). L.C. thanks Prof. Yi Liu (Lawrence Berkeley National Laboratory) for trying solution-phase synchrotron small-angle X-ray scattering. P.J.S. thanks the NIH (Grant R01-CA215157) for financial support.
Footnotes
Supporting Information.
The supporting information is available free of charge via the Internet at http://pubs.acs.org.
NMR, UV-vis, fluorescence, IR, Raman, SEM, AFM, and TEM, data (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Schwertfeger H; Fokin AA; Schreiner PR Angew. Chem. Int. Ed 2008, 47, 1022. [DOI] [PubMed] [Google Scholar]
- (2).Pierson HO, Handbook of carbon, graphite, diamonds and fuller-enes: Properties, processing and applications Noyes Publications: Park Ridge, New Jersey, 1993. [Google Scholar]
- (3).Carrington EJ; McAnally CA; Fletcher AJ; Thompson SP; Warren M; Brammer L Nat. Chem 2017, 9, 882. [DOI] [PubMed] [Google Scholar]
- (4).Evans OR; Xiong RG; Wang ZY; Wong GK; Lin WB Angew. Chem. Int. Ed 1999, 38, 536. [DOI] [PubMed] [Google Scholar]
- (5).Evans OR; Lin WB Chem. Mater 2001, 13, 2705. [Google Scholar]
- (6).Beaudoin D; Maris T; Wuest JD Nat. Chem 2013, 5, 830. [DOI] [PubMed] [Google Scholar]
- (7).Uribe-Romo FJ; Hunt JR; Furukawa H; Klöck C; O’Keeffe M; Yaghi OM J. Am. Chem. Soc 2009, 131, 4570. [DOI] [PubMed] [Google Scholar]
- (8).Li Z; Li H; Guan X; Tang J; Yusran Y; Li Z; Xue M; Fang Q; Yan Y; Valtchev V; Qiu S J. Am. Chem. Soc 2017, 139, 17771. [DOI] [PubMed] [Google Scholar]
- (9).Han X; Huang J; Yuan C; Liu Y; Cui Y J. Am. Chem. Soc 2018, 140, 892. [DOI] [PubMed] [Google Scholar]
- (10).Lindeman SV; Hecht J; Kochi JK J. Am. Chem. Soc 2003, 125, 11597. [DOI] [PubMed] [Google Scholar]
- (11).Ermer O J. Am. Chem. Soc 1988, 110, 3747. [Google Scholar]
- (12).Ermer O; Eling A Angew. Chem. Int. Ed 1988, 27, 829. [Google Scholar]
- (13).Simard M; Su D; Wuest JD J. Am. Chem. Soc 1991, 113, 4696. [Google Scholar]
- (14).Copp SB; Subramanian S; Zaworotko MJ J. Am. Chem. Soc 1992, 114, 8719. [Google Scholar]
- (15).Gunawardana CA; Dakovic M; Aakeroy CB Chem. Commun 2018, 54, 607. [DOI] [PubMed] [Google Scholar]
- (16).Reddy DS; Craig DC; Desiraju GR J. Am. Chem. Soc 1996, 118, 4090. [Google Scholar]
- (17).Zaworotko MJ Chem. Soc. Rev 1994, 23, 283. [Google Scholar]
- (18).(a) Yaghi OM; O’Keeffe M; Ockwig NW; Chae HK; Eddaoudi M; Kim J Nature 2003, 423, 705; [DOI] [PubMed] [Google Scholar]; (b) Chatterjee B; Noveron JC; Resendiz MJE; Liu J; Yamamoto T; Parker D; Cinke M; Nguyen CV; Arif AM; Stang PJ J. Am. Chem. Soc 2004, 126, 10645. [DOI] [PubMed] [Google Scholar]
- (19).Li H; Eddaoudi M; O’Keeffe M; Yaghi OM Nature 1999, 402, 276. [Google Scholar]
- (20).Côté AP; Benin AI; Ockwig NW; O’Keeffe M; Matzger AJ; Yaghi OM Science 2005, 310, 1166. [DOI] [PubMed] [Google Scholar]
- (21).Zhang K-D; Tian J; Hanifi D; Zhang Y; Sue AC-H; Zhou T-Y; Zhang L; Zhao X; Liu Y; Li Z-T J. Am. Chem. Soc 2013, 135, 17913. [DOI] [PubMed] [Google Scholar]
- (22).Li J-R; Sculley J; Zhou H-C Chem. Rev 2012, 112, 869. [DOI] [PubMed] [Google Scholar]
- (23).Wu H; Gong Q; Olson DH; Li J Chem. Rev 2012, 112, 836. [DOI] [PubMed] [Google Scholar]
- (24).Getman RB; Bae Y-S; Wilmer CE; Snurr RQ Chem. Rev 2012, 112, 703. [DOI] [PubMed] [Google Scholar]
- (25).Suh MP; Park HJ; Prasad TK; Lim D-W Chem. Rev 2012, 112, 782. [DOI] [PubMed] [Google Scholar]
- (26).Yoon M; Srirambalaji R; Kim K Chem. Rev 2012, 112, 1196. [DOI] [PubMed] [Google Scholar]
- (27).Lu K; He C; Lin W J. Am. Chem. Soc 2014, 136, 16712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cui Y; Yue Y; Qian G; Chen B Chem. Rev 2012, 112, 1126. [DOI] [PubMed] [Google Scholar]
- (29).Tranchemontagne DJ; Ni Z; O’Keeffe M; Yaghi OM Angew. Chem. Int. Ed 2008, 47, 5136. [DOI] [PubMed] [Google Scholar]
- (30).(a) Bosch M; Yuan S; Rutledge W; Zhou H-C Acc. Chem. Res 2017, 50, 857; [DOI] [PubMed] [Google Scholar]; (b) Sudik AC; Côté AP; Wong‐Foy AG; O’Keeffe M; Yaghi OM Angew. Chem. Int. Ed 2006, 45, 2528; [DOI] [PubMed] [Google Scholar]; (c) Ahmad N; Younus HA; Chughtai AH; Van Hecke K; Danish M; Gaoke Z; Verpoort F Sci. Rep 2017, 7, 832; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li Y; Dong Y; Miao X; Ren Y; Zhang B; Wang P; Yu Y; Li B; Isaacs L; Cao L Angew. Chem. Int. Ed 2018, 57, 729. [DOI] [PubMed] [Google Scholar]
- (31).Stang PJ; Olenyuk B Acc. Chem. Res 1997, 30, 502. [Google Scholar]
- (32).Seidel SR; Stang PJ Acc. Chem. Res 2002, 35, 972. [DOI] [PubMed] [Google Scholar]
- (33).Pluth MD; Bergman RG; Raymond KN Acc. Chem. Res 2009, 42, 1650. [DOI] [PubMed] [Google Scholar]
- (34).Harris K; Fujita D; Fujita M Chem. Commun 2013, 49, 6703. [DOI] [PubMed] [Google Scholar]
- (35).Fujita M; Tominaga M; Hori A; Therrien B Acc. Chem. Res 2005, 38, 369. [DOI] [PubMed] [Google Scholar]
- (36).Wiester MJ; Ulmann PA; Mirkin CA Angew. Chem. Int. Ed 2011, 50, 114. [DOI] [PubMed] [Google Scholar]
- (37).Smulders MMJ; Riddell IA; Browne C; Nitschke JR Chem. Soc. Rev 2013, 42, 1728. [DOI] [PubMed] [Google Scholar]
- (38).Li S; Huang J; Zhou F; Cook TR; Yan X; Ye Y; Zhu B; Zheng B; Stang PJ J. Am. Chem. Soc 2014, 136, 5908. [DOI] [PubMed] [Google Scholar]
- (39).Fujita M; Sasaki O; Mitsuhashi T; Fujita T; Yazaki J; Yamaguchi K; Ogura K Chem. Commun 1996, 1535. [Google Scholar]
- (40).Gong J-R; Wan L-J; Yuan Q-H; Bai C-L; Jude H; Stang PJ Proc. Natl. Acad. Sci. USA 2005, 102, 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Grishagin IV; Pollock JB; Kushal S; Cook TR; Stang PJ; Olenyuk BZ Proc. Natl. Acad. Sci. USA 2014, 111, 18448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhou Z; Yan X; Saha ML; Zhang M; Wang M; Li X; Stang PJ J. Am. Chem. Soc 2016, 138, 13131. [DOI] [PubMed] [Google Scholar]
- (43).Yang H-B; Ghosh K; Zhao Y; Northrop BH; Lyndon MM; Muddiman DC; White HS; Stang PJ J. Am. Chem. Soc 2008, 130, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Musser AJ; P. Neelakandan P; Richter JM; Mori H; Friend RH; Nitschke JR J. Am. Chem. Soc 2017, 139, 12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Pilgrim BS; Roberts DA; Lohr TG; Ronson TK; Nitschke JR Nat. Chem 2017, 9, 1276. [Google Scholar]
- (46).Müller IM; Spillmann S; Franck H; Pietschnig R Chem. – Eur. J 2004, 10, 2207. [DOI] [PubMed] [Google Scholar]
- (47).Olenyuk B; Whiteford JA; Fechtenkotter A; Stang PJ Nature 1999, 398, 796. [DOI] [PubMed] [Google Scholar]
- (48).Rizzuto FJ; Nitschke JR Nat. Chem 2017, 9, 903. [DOI] [PubMed] [Google Scholar]
- (49).Sun Q-F; Sato S; Fujita M Nat. Chem 2012, 4, 330. [DOI] [PubMed] [Google Scholar]
- (50).Olenyuk B; Levin MD; Whiteford JA; Shield JE; Stang PJ J. Am. Chem. Soc 1999, 121, 10434. [Google Scholar]
- (51).Wang Q-Q; Gonell S; Leenders SHAM; Dürr M; Ivanović-Burmazović I; Reek JNH Nat. Chem 2016, 8, 225. [DOI] [PubMed] [Google Scholar]
- (52).Fujita M; Yu S-Y; Kusukawa T; Funaki H; Ogura K; Yamaguchi K Angew. Chem. Int. Ed 1998, 37, 2082. [DOI] [PubMed] [Google Scholar]
- (53).Chakrabarty R; Mukherjee PS; Stang PJ Chem. Rev 2011, 111, 6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Cook TR; Stang PJ Chem. Rev 2015, 115, 7001. [DOI] [PubMed] [Google Scholar]
- (55).Caputo CB; Vukotic VN; Sirizzotti NM; Loeb SJ Chem. Commun 2011, 47, 8545. [DOI] [PubMed] [Google Scholar]
- (56).Wei P; Cook TR; Yan X; Huang F; Stang PJ J. Am. Chem. Soc 2014, 136, 15497. [DOI] [PubMed] [Google Scholar]
- (57).Schweiger M; Seidel SR; Schmitz M; Stang PJ Org. Lett 2000, 2, 1255. [DOI] [PubMed] [Google Scholar]
- (58).Cohen Y; Avram L; Frish L Angew. Chem. Int. Ed 2005, 44, 520. [DOI] [PubMed] [Google Scholar]
- (59).Xia Y; Xiong Y; Lim B; Skrabalak SE Angew. Chem. Int. Ed 2009, 48, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Avci C; Imaz I; Carné-Sánchez A; Pariente JA; Tasios N; Pérez-Carvajal J; Alonso MI; Blanco A; Dijkstra M; López C; Maspoch D Nat. Chem 2017, 10, 78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.