

Summary
The ability to induce pluripotency in somatic cells is one of the most important scientific achievements in the fields of stem cell research and regenerative medicine. This technique allows researchers to obtain pluripotent stem cells without the controversial use of embryos, providing a novel and powerful tool for disease modeling and drug screening approaches. However, using viruses for the delivery of reprogramming genes and transcription factors results in integration into the host genome causing random mutations within the target cell, thus limiting the use of these cells for downstream applications. To overcome this limitation, various non-integrating techniques, including Sendai virus, mRNA, minicircle, and plasmid-based methods, have recently been developed. Utilizing a newly developed codon optimized 4-in-1 minicircle (CoMiC), we were able to reprogram human adult fibroblasts using chemical defined media and without the need for feeder cells.
Keywords: human induced pluripotent stem cells (hiPSC), minicircle, reprogramming, disease modeling, chemically defined, differentiation
1. Introduction
Somatic cells, such as human adult fibroblasts, can be reverted to an embryonic stem cell (ESC)-like state by inducing the expression of specific genes and transcription factors that regulate the maintenance of pluripotency in embryonic stem cells (1). These so-called “core factors” of pluripotency, namely OCT4, SOX2, NANOG, and two oncogenes (c-MYC and LIN28), initiate the resetting of the epigenetic profile of a terminal differentiated somatic cell and activate the molecular circuitry of pluripotency (2–4). The resultant induced pluripotent stem cells (iPSCs) have similar morphology, growth characteristics, and gene expression profiles as embryonic stem cells (ESCs) (5). Furthermore, the capacity of iPSCs to differentiate into all other cell types of the human body has opened up new clinical opportunities for developing successful personalized stem cell-based therapies and drug discovery approaches. Initially iPSCs were produced using retroviral and lentiviral vectors that are known to integrate into the chromosomes of the target cells, potentially causing disruption of gene transcription and subsequent tumor formation (6). Therefore, various laboratories have developed additional non-integrating reprogramming techniques such as Sendai virus, mRNA, and protein- or DNA-based methods (7–11).
In order to induce the transformation of a nullipotent somatic cell into a pluripotent state, it is necessary to express the right stoichiometry of the reprogramming factors within the same transfected cell (12). One elegant approach to achieve this requirement uses polycistronic constructs in which the four reprogramming factors are controlled by one promoter separated by a different self-cleaving 2a peptide sequence (13). The disadvantage of such polycistronic plasmids is their large size, which might decrease the transfection efficiency in the adult human fibroblast. To overcome this problem we cloned the polycistronic expression cassette into a minicircle plasmid backbone. Minicircles are special episomal DNA vectors devoid of any bacterial plasmid backbone (Figure 1A). Therefore minicircles are significantly smaller than standard plasmids, which in turn enhances their transfection efficiency and the survival rate of the target cells (14). Furthermore, it has been shown that minicircle constructs sustain a higher expression rate of their harboring transgenes over a longer period of time. Although the minicircle theoretically should not integrate into the genome of the target cell, there is still a relatively small chance of integration. Therefore, it is important to screen the derived iPSCs for random integration using specific PCR primers or southern blot experiments (15).
Figure 1.

(A) Map of parental 4-in-1 reprogramming minicircle before and after arabinose induction. (B) Control digest to verify quality of the minicircle production. (C) Gel showing bands representing the minicircle after induction, both before and after purification steps.
The following protocol details the entire process of reprogramming human adult fibroblasts into human iPSCs: from dissociating fibroblasts from patient skin biopsy samples, to the reprogramming and characterization of the newly derived induced pluripotent stem cell line. The time from induction of pluripotency to expansion of the subsequent iPSC line to passage 3 is approximately one month (Figure 2B). The workflow of the reprogramming process is summarized in Figure 4. The final part of the protocol explains the pluripotency of the iPSCs is confirmed using immunostaining (Figure 3A). However, it will also be necessary to perform further experiments, including directed differentiation into specific germ layers and teratoma formation experiments, to verify pluripotency (Figures 3B-C) (16). It should also be mentioned that the minicircle reprogramming technique still has a very low efficiency and therefore should be only used by researchers with iPSC experience as an alternative method for obtaining integration-free induced pluripotent stem cells, or if it is necessary to compare different reprogramming techniques.
Figure 2.
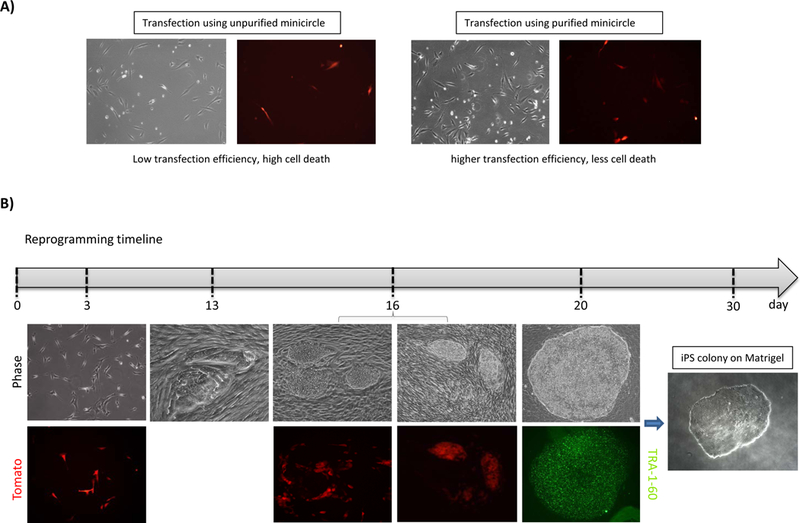
(A) Representative bright field and fluorescent (Tomato) images demonstrating the transfection efficiency and cell survival of unpurified and purified minicircle. (B) Timeline showing the expected phase contrast and fluorescent images at various stages of the reprogramming process. At day 20, pluripotency was confirmed by TRA-1–60 immunofluorescent staining.
Figure 4.

Timeline for reprogramming adult human fibroblasts into induced pluripotent stem cells using CoMiC.
Figure 3.
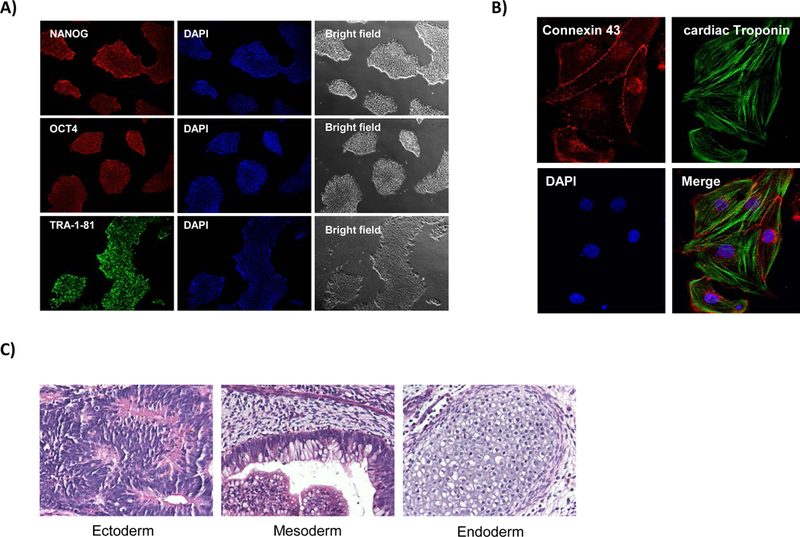
Immunofluorescence staining demonstrating (A) pluripotency markers NANOG, OCT4 and TRA-1–81, DAPI, and bright field images. (B) Immunofluorescent staining of iPSC-derived cardiomyocytes demonstrating positive staining of cardiac markers connexin 43 and cardiac troponin as well as DAPI. (C) Representative H&E staining from teratomas to confirm the pluripotency status of the generated iPSCs.
2. Materials
2.1. Media
Fibroblast media: Low glucose Dulbecco’s Modified Eagle Media (DMEM low glucose) with 20% fetal bovine serum (FBS).
iPSC media: Essential 8 (combine supplement with base media) and Essential 7(Essential 6 combined with base media and supplemented with 100 ng/mL FGF2)
2.2. Reagents
Phosphate buffered saline (PBS).
Collagenase IV.
Penicillin/streptomycin.
Lennox L Broth (LB) base.
Sodium hydroxide (NaOH) solution, 1M.
L-(+)-Arabinose ≥99%, distilled water (dH2O).
Kanamycin sulfate, 100X.
Matrigel—Growth factor reduced, hESC-qualified.
DMEM/F12.
TrypLE Express, store at room temperature (RT), there is no need to warm to 37°C before use.
0.25% Trypsin/ Ethylenediaminetetraacetic acid (EDTA).
Accutase.
Y27632 (ROCK inhibitor): 10mM stock in Ultrapure water, store at −20°C.
Ultrapure water.
Sodium butyrate (NaB).
Antibodies (see Table 1).
4% paraformaldehyde (PFA) solution.
Triton-X 100.
Bovine serum albumin (BSA).
Goat Serum.
Tween-20.
Vectashield mounting medium with DAPI.
Qiagen Plasmid Plus Maxi Kit.
DNAase.
Gelatin.
Neon Buffer R.
Qiagen Plasmid Plus Maxi kit.
4% Paraformaldehyde (PFA).
Table 1.
List of antibodies and dilutions used for characterization of human induced pluripotent stem cells and differentiated cardiomyocytes. Staining protocols were optimized with the antibodies from the listed suppliers. Optimization may be required if different antibodies are used.
| Antigen | Supplier | Product no. |
Dilution | Raised in | Secondary (1:400) | Supplier | Product no. |
|---|---|---|---|---|---|---|---|
| Connexin-43 | SIGMA | C6219 | 1:400 | rabbit | Alexa Fluor® 488 Goat Anti-Rabbit IgG (H+L) |
Life Technologies |
A-11037 |
| c Troponin-T | Thermo Scientific |
MS-295-P | 1:400 | mouse | Alexa Fluor® 488 Goat Anti-Mouse IgG (H+L) |
Life Technologies |
A-11001 |
| NANOG | Santa Cruz |
SC-33759 | 1:100 | rabbit | Alexa Fluor® 488 Goat Anti-Rabbit IgG (H+L) |
Life Technologies |
A-11037 |
| OCT4 | Santa Cruz |
SC-9081 | 1:100 | rabbit | Alexa Fluor® 488 Goat Anti-Rabbit IgG (H+L) |
Life Technologies |
A-11037 |
| Tra-1–81 | Millipore | mab4381 | 1:250 | mouse | Alexa Fluor® 488 Goat Anti-Mouse IgG (H+L) |
Life Technologies |
A-11001 |
2.3. Equipment
6-well cell culture plates (surface area = 9.8 cm2) coated with 2 mL Matrigel.
12-well cell culture plates (surface area = 3.8 cm2) coated with 1 mL Matrigel.
10 cm tissue culture plates (surface area = 58.95 cm2) coated with 12 mL Matrigel.
90 mm petri dishes.
15 and 50 mL polypropylene conical tubes.
2 mL plastic aspiration pipettes.
5, 10, 25, and 50 mL plastic pipettes.
250 and 500 mL polyethersulfone (PES) media filters.
8-chamber slides.
T225 flasks.
Coverslips.
Parafilm.
Countess Cell Counter, slides, and trypan blue.
Tissue culture incubator capable of 37°C, 5% CO2, and 85% relative humidity (such as New Brunswick Galaxy 170R).
Dual gas tissue culture incubator capable of 37°C, 5% CO2, 5% O2, and 85% relative humidity with split inner door.
Centrifuge.
Inverted tissue culture microscope (such as Nikon Ti) with heated stage.
Autoclave.
Orbital shaker.
Spectrophotometer.
Waterbath.
Electroporation.
3. Methods
3.1. Minicircle production
Prepare 1200 mL of LB Broth media by adding 24 grams of LB Broth Base to 1200 mL of distilled water (hH2O) and divide this over the three culture flasks.
Autoclave the flasks using the “liquid cycle.”
After cooling down of the flasks to room temperature, add 4 mL of Kanamycin 100X to each one of the culture flasks.
In the morning, pick one clone from a parental plasmid transformed bacterial plate in 5mL LB media containing kanamycin (50 μg/mL) and grow this pre-culture until the evening at 37°C with shaking at 250 rpm.
In the evening, grow an overnight culture by combining 100 μL of the pre-culture with each of the flasks containing media with kanamycin and culture overnight (16–18 hours) at 37°C with shaking at 250 rpm (see Note 1).
The optical density (OD) of the overnight culture should be between 3.75 and 4.25.
Prepare a minicircle induction mix by comprised of 1 volume of fresh LB Broth (1200 mL), 4% 1N NaOH (48 mL) and 1% L-Arabinose (12 grams) and mix well.
Add 400 mL of the induction mix to each of the three flasks with the overnight culture and mix well.
Incubate the combined mix at 32°C with shaking at 250 rpm for 6–8 hours.
Harvest all the culture flasks, transfer them to spinning tubes and pellet the bacteria.
Isolate minicircles according to the Qiagen Plasmid Plus Maxi kit instructions with certain modifications (see Notes 2 and 3).
Confirm the successful Minicircle induction by digestion using HindIII and Nde1. Nde1 linearizes only the parental backbone but not the minicircle. Therefore you should expect one band around 7.7 kb when digesting the Minicircle with both enzymes (Figure 1B).
3.2. Fibroblast isolation from a patient skin punch biopsy
All of these steps should be performed in a sterile tissue culture hood. Prepare a fibroblast digestion solution by dissolving 1 mg/mL Collagenase IV in DMEM/F12 and sterile filter it through a 0.2 μm pore size filter.
Transfer the skin punch biopsy into a 15 mL conical tube containing 3mL PBS + penicillin/streptomycin (1:100), repeat this step 3 times.
Transfer the tissue into a sterile 10 cm plate and add 1 mL Collagenase IV. Use two scalpels to cross cut the tissue into small pieces while keeping the tissue immersed in Collagenase IV.
Additionally, add 2 mL Collagenase IV solution to the minced tissue and pipette it up and down. Transfer the Collagenase IV/tissue suspension into a 15 mL conical tube and add 25μL of 0.5 M EDTA and 20μL of DNase. Afterwards, incubate the mixture for 3 hours in a tissue culture incubator at 37°C.
Following the 3-hour incubation time, pipette the tissue pieces up and down several times to obtain single-cell fibroblasts.
Add 12mL fibroblast media containing 1X penicillin/streptomycin to stop the collagenase IV digestion.
Centrifuge for 4 minutes at 1000 rpm.
Re-suspend the pellet in 2 mL fibroblast media containing 1X penicillin/streptomycin solution, and plate it in one well of a 0.1% gelatin-coated 6-well plate. Depending on the size of your tissue sample, you may need to plate the cells in more than one well of a 6-well plate. To prepare gelatin coated plates, gelatin stock is dissolved in XY, Z ml of this solution is pipetted into a well of a 6-well plate and incubated for Q hr at R temperature.
Incubate the cells for at least 4 days without changing the media, until some of the fibroblasts start to attach and propagate. After this, change the media every other day.
Once the cells become confluent, split using 0.25% Trypsin-EDTA for 10 minutes. Plate the cells onto a 0.1% gelatin coated T225 flask to further expand and freeze multiple vials of fibroblast after the flask become confluent.
3.3. Prepare Matrigel Plates
Thaw stock bottle of Matrigel at 4°C overnight.
Keep Matrigel on ice, and add 300 μL Matrigel to 50mL of cold DMEM/F12 media. Add 2mL to each well of a 6-well plate. (Alternatively use one Matrigel aliquot per 20 mL cold DMEM/F12 media and cover four 10 cm plates using 5 mL).
Place the plates in the fridge and allow the Matrigel to polymerize overnight.
3.4. Reprogramming of human fibroblasts using Neon transfection and CoMiC
The target fibroblast cell line should be thawed and cultured for at least 3 days before starting the reprogramming experiment. Additionally it is beneficial to further purify the minicircle DNA using the Zymoclean Gel DNA Recovery Kit to eliminate residual chromosomal bacterial DNA of the minicircle preparation (Figure 1C). This step will increase the cell survival after the electroporation (Figure 2A).
Plate 1.1×106 human fibroblast in a 10 cm tissue culture plate one day before starting the reprogramming experiment.
On the following day (day 0), trypsinize the cells using Trypsin 0.25% EDTA for 10 minutes to obtain a single cell suspension.
Wash off the trypsin using 10 mL of DMEM plus 20% FBS and centrifuge for 3 minutes at 1000 rpm.
Aspirate the media, and dissolve the cell pellet in 100–105 μL Neon Buffer R (the volume depends on the amount of plasmid you have to use to get 10 – 12 μg DNA). The final volume of buffer, DNA and cells should be around 110 μL.
Perform the transfection using a 100 μL Neon tip and the following settings: 1600 volt, 10 ms and 3 pulses (transfection efficiency should be more than 50%).
Plate the cells equally distributed onto two Matrigel coated plates (5.5 × 105 cells) in fibroblast media.
On the following morning (day 1), change the media to fibroblast media and add Sodium butyrate (0.2 mM) plus 50μg/mL ascorbic acid.
One day 3, change the media to fibroblast media and add sodium butyrate (0.2 mM) plus 50 μg/mL ascorbic acid.
On day 5, change the media to 50:50 fibroblast media:Essential 7 media and add sodium butyrate (0.2 mM) plus 50μg/mL ascorbic acid.
On days 7–13, change the media every other day until the first pre-iPSC colonies appear. Change the media to Essential 7 and add sodium butyrate (0.2 mM) plus 50 μg/mL ascorbic acid.
Between days 14 and 20, after you recognize the first colonies, switch to Essential 8 media and hypoxic culture conditions. Around day 20 the iPS colonies should be big enough for manual picking under the microscope. Pick 6 individual iPS clones using the Vitrolife stem cell cutting tool with a 10 μL tip and transfer them into 6 different wells of a Matrigel-coated plate with Essential 8 media in presence of 10 μM ROCK Inhibitor (Y-27632). If it is difficult to detach the iPS colonies from the surrounding fibroblasts, you also can scrape those fibroblasts away to make room for the outgrowing iPS colony. After two days, the iPS clone should be large enough to pick.
3.5. Colony Purification and Expansion
After 5–7 days, the individual colonies become big enough to dissociate into single cells using 0.5 mL TrypLE for 8 minutes. Detach the remaining colonies by pipetting them up and down with a 1 mL pipette.
Wash the cells once with 5 mL Essential 8 media.
Centrifuge for 3 minutes at 1000 rpm.
Passage the cells into one well of a Matrigel coated 6-well plate containing 2 mL Essential 8 media.
After 5–7 days, colonies will have grown out and become dense enough to split onto a Matrigel-coated 10 cm plate. Therefore incubate the cells with 1mL Accutase for 8 min, wash using 9 mL Essential 8 media, and centrifuge for 3 minutes at 1000 rpm.
Seed the cells onto a Matrigel-coated 10 cm plate in Essential 8 media in the presence of 10 μM Y27632.
Once the 10 cm plate become confluent, freeze down 5 vials (passage 3 iPS cells) and continue to grow at a 1:6 split ratio, freezing vials every 10 passages.
Grow cells to at least passage 5 to test for the expression of the pluripotency marker genes and to passage15 before testing for efficient differentiation.
3.6. Immunofluorescence Staining Protocol
Coat 8-chamber slides with Matrigel (200 μL per chamber) as described above in 3.3.
Seed iPSCs (1×104 cells/chamber) or differentiated cardiomyocytes (1.75×104 cells/chamber) as a negative control, on 8-chamber slides in a final volume of 200μL of the appropriate medium.
Place the chamber slides in 90 mm Petri dishes and store in a 37oC incubator.
Allow cells to attach and grow for ~24–72 hours and fix as follows (see Note 4).
Aspirate medium and wash cells 3X with PBS.
Add to each chamber 100 μL 4% Paraformaldehyde (PFA) in PBS for 20 minutes at RT.
Aspirate PFA and wash 3X with 200 μL PBS.
Aspirate PBS and replace with 500 μL of fresh PBS.
Seal slides with parafilm and place at 4ºC until needed for staining (keep for up to 7 days)
Incubate with 100μL of permeabilization solution (0.2% Triton X-100 in PBS), for 15 minutes at room temperature (see Note 5).
Wash twice with PBS, for 5 minutes at room temperature.
Incubate with blocking solution (2% BSA or 4% Goat Serum or 2% FBS in PBS), for 1 hour at room temperature or overnight at 4ºC.
Wash once in PBS, for 5 minutes at room temperature.
Incubate overnight at 4°C with primary antibody (Table 1) diluted appropriately in blocking solution (100 μL per chamber).
Wash in 1% goat serum, 0.1% Tween-20 in PBS four times, for 10 minutes
Incubate with the appropriate secondary antibody diluted in blocking solution, for 1 hour at RT (100 μL per chamber). Wrap in aluminum foil as soon as secondary antibody is added.
Wash in 1% Goat Serum, 0.1% Tween-20 in PBS four times, for 10 minutes
Remove the chambers from the slide.
Place a small amount of Vectashield mounting medium with DAPI over the area where each chamber used to be.
Place a clean coverslip over the slide and gently blot off any liquid remaining after the staining protocol.
Seal with nail varnish.
Store slides at 4°C in the dark.
Image at least 3 hours later.
4. Notes
Do not start off with too much parental plasmid, as an overgrown culture will limit the induction efficiency.
To ensure a high yield, make sure that the pellet is completely re-suspended in Qiagen Maxi Kit P1 buffer and use the same volume of P2 and S3. For example, when using 400 mL spinning tubes, re-suspend the pellet in 10 mL P1, P2 and S3 buffer instead of 8mL.
To avoid bacterial genomic DNA contamination, avoid excessive shaking of the re-suspended pellet when adding buffer P2.
Solutions should be made fresh for each experiment. Keep at 4ºC until day 2.
The permeabilization step is only required for the detection of nuclear protein (e.g. OCT4, SOX2 or NANOG), when surface markers are detected, skip the permeabilization step and go straight to the primary antibody incubation.
References
- 1.Takahashi K, et al. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131(5):861–872. [DOI] [PubMed] [Google Scholar]
- 2.Mitalipov S & Wolf D (2009) Totipotency, pluripotency and nuclear reprogramming. Adv Biochem Eng Biotechnol 114:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng B, Ng JH, Heng JC, & Ng HH (2009) Molecules that promote or enhance reprogramming of somatic cells to induced pluripotent stem cells. Cell stem cell 4(4):301–312. [DOI] [PubMed] [Google Scholar]
- 4.Maherali N, et al. (2007) Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell stem cell 1(1):55–70. [DOI] [PubMed] [Google Scholar]
- 5.Wernig M, et al. (2007) In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448(7151):318–324. [DOI] [PubMed] [Google Scholar]
- 6.Sommer CA, et al. (2010) Excision of reprogramming transgenes improves the differentiation potential of iPS cells generated with a single excisable vector. in Stem Cells, pp 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stadtfeld M & Hochedlinger K (2010) Induced pluripotency: history, mechanisms, and applications. Genes Dev 24(20):2239–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stadtfeld M, Nagaya M, Utikal J, Weir G, & Hochedlinger K (2008) Induced pluripotent stem cells generated without viral integration. Science 322(5903):945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warren L, et al. (2010) Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell stem cell 7(5):618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou H, et al. (2009) Generation of induced pluripotent stem cells using recombinant proteins. Cell stem cell 4(5):381–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okita K, et al. (2011) A more efficient method to generate integration-free human iPS cells. Nature methods 8(5):409–412. [DOI] [PubMed] [Google Scholar]
- 12.Tiemann U, et al. (2011) Optimal reprogramming factor stoichiometry increases colony numbers and affects molecular characteristics of murine induced pluripotent stem cells. Cytometry A 79(6):426–435. [DOI] [PubMed] [Google Scholar]
- 13.Warlich E, et al. (2011) Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol Ther 19(4):782–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia F, et al. (2010) A nonviral minicircle vector for deriving human iPS cells. Nature methods 7(3):197–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez F, Boue S, & Izpisua Belmonte JC (2011) Methods for making induced pluripotent stem cells: reprogramming a la carte. Nat Rev Genet 12(4):231–242. [DOI] [PubMed] [Google Scholar]
- 16.Lan F, et al. (2013) Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell stem cell 12(1):101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]