

Abstract
Ligands that coordinate via dianionic phosphonate groups have not been widely utilized in radiopharmaceuticals. N-(phosphonomethyl)iminodiacetic acid (1, PMIDA) and N-(phosphonomethyl)glycine (2, PMG) were investigated as new chelators for the 99mTc/Re-tricarbonyl “core” (fac-M(CO)3, M = 99mTc, Re) present in a major class of radiopharmaceuticals. fac-M(CO)3(PMIDA) and fac-M(CO)3(PMG) complexes were studied by HPLC and 1H/13C/31P NMR methods for M = Re (Re-1 and Re-2) and by HPLC for M = 99mTc (99mTc-1 and 99mTc-2). Re-1 and 99mTc-1 complexes exhibit a similar pH-dependent equilibrium between geometric linkage isomers (M-1a and M-1b). However, only one isomer exists for M-2 under all conditions. Structural characterization by X-ray crystallography reveals the presence of a bond between a phosphonate oxygen and the Re(I) center in fac-Re(CO)3(PMG) (Re-2). Detailed comparisons of NMR data for Re-2 conclusively demonstrate that the phosphonate group is coordinated in Re-1b (isomer favored at high pH) but not in Re-1a, which has a dangling N-(phosphonomethyl) group. To our knowledge, Re-1b and Re-2 and their 99mTc analogs are the first well-documented examples of complexes with these metal-tricarbonyl cores having a dianionic phosphonate group directly coordinated in a fac-M(CO)3-O-P grouping. Pharmacokinetic studies using Sprague-Dawley rats reveal that 99mTc-2 is a robust tracer. Hence, phosphonate groups should be considered in designing 99mTc and 186/188Re radiopharmaceuticals, including agents with bioactive moieties attached to dangling carboxylate or phosphonate groups.
Keywords: Rhenium(I), Technetium(I), Tricarbonyl complexes, Phosphonate ligands, X-ray structure, Geometric isomers
Graphical Abstract
Formation of linkage isomers; these equilibrate in a pH-dependent fashion with phosphonate group binding favored at pH > 6 and carboxylate group binding highly favored at pH4.
1. Introduction
Metal complexes play an important role in molecular imaging, and radiometals are fully integrated into single photon emission computed tomography (SPECT: 99mTc, 111In, 186Re, etc.) and positron emission tomography (PET: 64Cu, 68Ga, etc.) scans [1, 2]. The 99mTc radionuclide is the most widely used isotope in diagnostic nuclear medicine, owing to its optimal physical properties (t1/2 = 6 h, γ = 142 keV); in the U.S. alone, nearly 80% of all radiopharmaceuticals used in nuclear imaging procedures are 99mTc-labeled complexes [3]. The organometallic fac-[99mTc(CO)3]+ core has particular merit, not only because of facile accessibility from an available kit but also because of its versatile coordination chemistry [4–6]. The development of new metal-based radiotracers has required ligands that can provide sufficient stability under physiological conditions. Flexible multidentate ligands containing a variety of neutral (e.g., amine, thioether, and phosphine) and anionic (carboxylate) donor groups and suitable for facial tridentate coordination to the fac-[99mTc(CO)3]+ core are generally used [4, 5, 7–18]. Because Tc and Re have essentially identical coordination parameters, the development of 99mTc radiopharmaceuticals benefits from an understanding of the features of their Re analogs.
In our ongoing studies to elucidate the fundamental coordination chemistry for potential renal radiopharmacutical applications, we have reported many examples in which multidentate aminopolycarboxylate ligands formed stable fac-[M(CO)3] (M = 99mTc and Re) complexes; the iminodiacetic acid (IDA) unit has been especially useful in producing well-defined monomeric fac-[M(CO)3] products [11, 13, 19, 20]. For example, the nitrilotriacetic acid (NTAH3) ligand employs its IDA chelating moiety for tridentate ONO coordination to the fac-[99mTc(CO)3]+ core, thereby forming a single, highly hydrophilic complex, 99mTc(CO)3-nitrilotriacetic acid (99mTc(CO)3(NTA), Scheme 1, Eq. 1) [21]. 99mTc(CO)3(NTA) has proved to be a very promising renal tracer with excellent pharmacokinetic properties in rats and humans [21–23]. Given the success and versatility of studies using carboxylate ligands, a worthwhile goal became to explore the utility of other anionic donor groups such as phosphonates. Even though phosphonates are commonly used as labels in 99mTc bone-imaging agents in nuclear medicine [24–27], the 99mTc/Re-tricarbonyl chemistry of IDA analogs with a phosphonate in place of carboxyl donors (i.e., the aminopolyphosphonate ligands) has not been well studied. In fact, no direct phosphonate coordination to the /ac-[M(CO)3]+ core has yet been confirmed; only one example of a 99mTc/Re(CO)3-O-P bond has been reported, but that complex was formed by a bound phosphonic acid ester [28]. In that study, aminophosphonate ligands formed 99mTc/Re- tricarbonyl products that appeared to be difficult to purify and thus to characterize. Hence, improving our understanding of phosphonate group coordination in 99mTc/Re-tricarbonyl complexes has merit. It is worth noting that there are very few reports demonstrating binding to the Re-tricarbonyl core by the phosphate group [29], a group closely related to the phosphonate group.
Scheme 1.

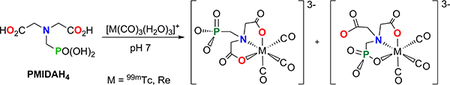
Summary of the coordination reaction of NTAH3 (Eq. 1), PMIDAH4 (1, Eq. 2) and PMGH3 (2, Eq. 3) with a metal-tricarbonyl precursor, [M(CO)3(H2O)3]+ (M = 99mTc, Re).
The nitrogen-containing carboxyphosphonates, close analogs to aminopolycarboxylates, are known to be efficient chelating ligands possessing versatile coordination properties toward various metals [30–32]. One of the most promising aminocarboxyphosphonate ligands is N-(phosphonomethyl)iminodiacetic acid (PMIDAH4, 1, Scheme 1, Eq. 2) because it is a tetradentate ligand analogous to the NTAH3 ligand, with one of the three acetate acid groups in NTAH3 replaced by the methylphosphonic acid group. Because PMIDAH4 contains the IDA moiety like NTAH3, PMIDAH4 could thus form monomeric stable 99mTc/Re-tricarbonyl complexes. In addition, the study of the PMIDAH4 ligand allows direct comparison with the NTAH3 ligand, because, unlike the highly symmetrical NTAH3 ligand, the less symmetrical PMIDAH4 ligand could possibly form 99mTc/Re-tricarbonyl complexes having linkage isomers. This capacity of the PMIDAH4 ligand affords the opportunity to assess phosphonate group binding and also the overall relative stability of the complexes. Understanding the chemistry of these isomers, as well as that of the 99mTc/Re-tricarbonyl complexes of the smaller and less complicated tridentate N-(phosphonomethyl)glycine (PMGH3, 2) ligand, could provide insight into the chemistry of phosphonate complexes in general and lead to novel radiopharmaceutical development.
2. Experimental
2.1. Materials
Both N-(phosphonomethyl)iminodiacetic acid (PMIDAH4, 1, called PMIDA ) and N-(phosphonomethyl)glycine (PMGH3, 2, called PMG) were used as received from Aldrich. An aqueous stock solution (0.1 M) of [Re(CO)3(H2O)3]OTf was prepared as previously reported [9]. All other reagent-grade chemicals and solvents were obtained from commercial suppliers and used without further purification. H/ P NMR spectra were recorded on Inova 400 MHz or Mercury 300 MHz spectrometers, and C NMR spectra were recorded on an Inova 600 MHz spectrometer; chemical shifts are reported in δ units, with the residual solvent peak used as reference. Electrospray mass spectrometry (ESI-MS, negative mode) was performed on a Thermo Finnigan LTQ-FT instrument. HPLC analyses of Re-tricarbonyl complexes (monitored at 254 nm) were performed on a Waters Breeze system equipped with a Waters 2487 detector, Waters 1525 binary pump, and XTerra MS C18 column (5 pm; 4.6 × 250 mm). The mobile phase was comprised of 0.05 M triethylammonium phosphate at pH 2.5 aqueous buffer (solvent A) and methanol (solvent B), the flow rate was 1 mL/min, and the gradient methods used were the same as previously reported [13]. 99mTc-pertechnetate (99mTcO4−) in 0.9% saline was received from Triad Isotopes. The “CRS Isolink kit” (Center for Radiopharmaceutical Science, Paul Scherrer Institute, Villigen, Switzerland) was used to prepare the [99mTc(CO)3(H2O)3]+ precursor according to the manufacturer’s insert. HPLC separation and quality control of the 99mTc tracers were performed using a Beckman Gold Nouveau system equipped with a Model 166 ultraviolet light-visible light detector (monitored at 254 nm), a Model 170 radioisotope detector, and a Beckman C18 RP Ultrasphere octyldecyl silane column (5 pm, 4.6 × 250 mm); data were acquired by using the 32 Karat software (Beckman Coulter). The mobile phase and the flow rate were identical as for Re complexes, but the gradient method was slightly modified as reported earlier [33].131I-OIH, which served as an internal control in our biodistribution studies, was prepared by the isotope exchange reaction between non-radioactive hippuran (OIH) and radioactive sodium iodide (Na131I) according to the method reported by Anghileri [34] and modified as previously described [21]. All animal experiments followed the principles of laboratory animal care and were approved by the Institutional Animal Care and Use Committee of Emory University. Tissue/organ radioactivity was measured with an automated 2480 Wizard 2 gamma counter (Perkin Elmer) that corrects for spillover from 131I into the 99mTc window based on a prior normalization process.
Terminology: because all specific 99mTc/Re(CO)3 complexes mentioned in this work have a facial geometry, the fac- designation is usually omitted. The uncomplexed and coordinated ligands are designated in CAPS, with the total number of dissociable protons indicated by a subscript (e.g., PMIDAH4). Also, the M(CO)3(L) or M-L nomenclature (M = 99mTc, Re; L = ligand) is used as a shorthand notation for the 99mTc and Re complexes, whereas a specific form with its protonation state defined by our work is designated as [M(CO)3(L)]2− or [M-L]2− (dianion).
2.2. Synthesis
2.2.1. fac-Re(CO)3(PMIDA) (Re-1): (Re(CO)3(N-(phosphonomethyl)iminodiacetate)
An aqueous solution of 1 (55 mg, 0.25 mmol) was neutralized with 1 M NaOH and combined with a stirred solution of 0.1 M [Re(CO)3(H2O)3]OTf (2.5 mL, 0.25 mmol). A small aliquot of the reaction mixture was heated at 70 °C for 5 min, which pushed the reaction to completion (according to HPLC). At room temperature, the reaction proceeded to completion within 4 h. During the course of the reaction the pH was not adjusted further, and the final pH of the reaction mixture was 4. HPLC analysis revealed one major product peak with a retention time of 10.2 min. The reaction mixture was concentrated to 1 mL and purified over Sephadex G-15 gel. UV-active fractions were analyzed by HPLC, and those that contained the major product were combined and concentrated to yield Re-1a as a white crystalline solid (88 mg, 0.17 mmol, 68 %). HRMS (M−, ESI) Calcd for CgH7O10NNaP187Re: 517.92683, found: 517.92735 (Δ = 0.52 mmu, 1.01 ppm).
When the pH of the reaction mixture was maintained at ≥7 by addition of 1 M NaOH, HPLC analysis revealed two peaks (~2:3 ratio) having retention times of 10.2 min (Re-1a) and 12.2 min (Re-1b). The reaction mixture was purified as described above for Re-1a. The 1H NMR spectrum of the isolated product showed two sets of peaks in a 2:3 ratio similar to that found by HPLC. Differences between spectra for the mixture of products (pH 7) and that for pure Re-1a (pH 4) were used to determine which signals were from Re-1b.
Re-1a: 1H NMR (D2O, pH 4) δ: 4.08 (d, 2H, J = 16 Hz), 3.91 (d, 2H, J = 16 Hz), 3.76 (d, 2H, J = 12 Hz). 13C NMR (D2O, pH 4) δ: 197.6 (2 C≡O), 196.6 (C≡O), 183.9 (2 CO2), 67.8 (d, CH2-P), 64.4 (2 CH2-CO2). 31P NMR (D2O, pH 4) δ: 12.99 ppm.
Re-1b: 1H NMR (D2O, pH 7) δ: 4.02 (d, 1H, J = 18 Hz), 3.98 (s, 2H), 3.79 (d, 1H, J = 18 Hz), 3.37 (dd, 1H, J = 15/12 9/6 Hz), 2.98 (dd, 1H, J = 15/12 Hz). 13C NMR (D2O, pH 7) δ: 198.6 (C≡O), 197.2 (C≡O), 196.9 (C≡O), 186.1 (CO2), 175.8 (CO2), 71.6 (CH9-CO9). 62.5 (CH9-CO9). 57.6 (d, CH2-P). 31P NMR (D2O, pH 7) δ: 30.35 ppm.
2.2.2. fac-Re(CO)3(PMG) (Re-2): (Re(CO)3(N-phosphonomethyl)glycine)
An aqueous solution of N-phosphonomethyl glycine (PMGH3, 2) (34 mg, 0.2 mmol) was neutralized with 1 M NaOH before the addition of an 0.1 M stock solution of [Re(CO)3(H2O)3]OTf (2.0 mL, 0.2 mmol) gave a slightly acidic solution (3 mL). After 30 min, the pH of the reaction had dropped to 4. The pH was again neutralized and the tridentate coordination was complete within 1 h at room temperature. The reaction mixture was concentrated and filtered over Sephadex G-15 gel. The UV-active fractions were analyzed by HPLC, combined, and concentrated to yield the Re-2 product as the disodium salt (55 mg, 0.12 mmol, 60%). 1H NMR (D2O, pH 7): δ: 6.05 (tt, 1H, J = 8/5 Hz), 3.7 (dd, 1H, J = 17/8 Hz), 3.39 (d, 1H, J = 17 Hz), 2.81 (m, 2H). 13C NMR (D2O, pH 7) δ: 197.45 (C≡O), 196.98 (C≡O), 195.86 (C≡O), 185.46 (CO2), 175.8 (CO2), 54.27 (CH2-CO2), 48.75 and 47.81 (CH2-P). 31P NMR (D2O, pH 7): δ: 31.3. HRMS ESI-MS [M+Na]−, m/z: Calcd for C6H5O8NNa187ReP: 459.92135, found: 459.92140; [M+H]2−, m/z: Calcd for C6H6O8N187ReP: 437.93940, found: 437.93961.
2.3. X-ray Structural Determination of fac-Re(CO)3(PMG) (Re-2)
Colorless needle-shaped crystals of Re-2 were recrystallized from a mixture of water and ethanol by slow evaporation. A suitable crystal (0.22×0.21×0.10 mm3 ) was selected and mounted on a loop with Paratone oil on a Rigaku SYNERGY diffractometer equipped with an Oxford Cryosystems low-temperature device. The crystal was cooled to 100(2) K during data collection. The structure was solved with the XT [35] structure solution program by using the Intrinsic Phasing solution method and by using Olex2 [36] as the graphical interface. The model was refined with version 2014/7 of XL [35] (Sheldrick, 2008) by using Least Squares minimization.
2.3.1. Crystal Data for Re-2:
C6H15NNa2O13PRe, Mr = 572.34, monoclinic, C2/c (No. 15), a = 28.956(11) Å, b = 10.087(9) Å, c = 11.306(2) Å, β = 90.50(2)°, α = γ = 90°, V = 3302(3) Å3, T = 100(2) K, Z = 8, Z’ = 1, μ(MoKa) = 7.574, 15196 reflections measured, 4570 unique (Rint = 0.0488) which were used in all calculations. The final wR2 was 0.0590 (all data), and R1 was 0.0244 (I > 2σ(Γ)).
2.4. 99mTc Radiolabeling
The PMIDAH4 (1) and PMGH3 (2) ligands were both labeled in a similar manner to form 99mTc(CO)3 tracers as previously described [20, 21]. Briefly, the freshly prepared [99mTc(CO)3(H2O)3]+ precursor was added to a sealed vial containing ~0.2 mg of ligand in 0.2 mL of water. The pH of the solution was adjusted to ~7 with 1 M NaOH, and the labeling mixture was heated at 70 °C for 30 min, cooled to room temperature, and analyzed by HPLC. Retention times of the 99mTc tracers were nearly identical to those of their Re analogs when the 99mTc and Re complexes were co-injected. Both 99mTc(CO)3 tracers, designated as 99mTc(CO)3(PMIDA) (99mTc-1) and 99mTc(CO)3(PMG) (99mTc-2), were isolated by HPLC as described above. The 99mTc-1a and 99mTc-1b isomers had very similar retention times and were collected together. The aqueous fraction of 99mTc-2 collected was diluted in a physiological buffer (pH 7.4) to obtain a final concentration of 3.7 MBq/mL for in vivo experiments. The buffered solutions of the 99mTc tracers were evaluated by HPLC for up to 6 h to assess tracer in vitro integrity.
2.5. Biodistribution Studies in Rats
The biodistribution, in vivo stability, plasma protein binding (PPB), and erythrocyte uptake (RBC) studies of 99mTc(CO)3(PMG) (99mTc- 2) were conducted as previously reported [21]. Briefly, the pharmacokinetics of 99mTc- 2 were evaluated in Sprague-Dawley rats injected via tail vein at 10 and 60 min with 0.2 mL of a solution containing 99mTc- 2 (100 μCi/mL) and 131I-OIH (25 pCi/mL) in phosphate-buffered saline (PBS) pH 7.4. The bladder was catheterized for urine collection. Animals were sacrificed and tissues of interest, along with blood and urine, were collected and placed in counting vials. Each sample and the standards were counted for radioactivity by using an automated gamma-counter using the 99mTc/131I dual-label program. The percentage of the dose in each tissue or organ was calculated by dividing the counts in each tissue or organ by the total injected counts. The percentage of injected dose in whole blood was estimated by assuming a blood volume of 6.5% of total body weight. For metabolite studies, urine of two rats injected with ~18 MBq of 99mTc- 2 was collected, filtered with a 0.2 μm Millex-LG filter to remove foreign particles and analyzed by reversed-phase HPLC to determine whether the complex was metabolized or excreted unchanged in the urine.
3. Results and discussion
The N-(phosphonomethyl)iminodiacetic acid (PMIDAH4, 1) ligand is an analog of NTAH3 with a phosphonate group in place of one of the three carboxylates of NTAH3 (Scheme 1). The PMIDAH4 ligand possesses four acidic protons capable of deprotonation at pH values indicated in Figure 1 [37, 38]. Like NTAH3, 1 is a potential tetradentate ligand and is expected to bind as a tridentate ligand to form a robust mononuclear complex (Scheme 1, Eq. 2). However, replacing a carboxylate group with a phosphonate group leads to important differences. For example, the carboxylate group is planar, whereas the phosphonate group is tetrahedral. PMIDAH4, like other phosphonic acids containing amino group(s), exists in the zwitterionic forms with the N atom protonated by the one proton of the phosphonic acid group at the positive center and the monodeprotonated phosphonic acid group at one negative end. Because of its lower acidity compared to the carboxylic acid group, the monoanionic phosphonate group (-PO3H”) remains monodeprotonated over a large pH range, in contrast to the carboxyl groups (-CO2H) (Figure 1). Near neutral pH, the dianionic phosphonate group (- PO3 2−) is more basic than the monoanionic carboxylate group.
Figure 1.

Most prevalent protonation states of PMIDAH4 at various pH ranges, according to the previously reported pKa data [37, 38].
Because the normal chemical characterization of the radioactive 99mTc(CO)3(PMIDA) (99mTc-1) product cannot be performed owing to the very low concentration and short half-life of 99mTc, we relied on the nonradioactive rhenium analog, Re(CO)3(PMIDA) (Re-1). The formation of Re-1 at room temperature was initiated by the addition of a [Re(CO)3(H2O)3]OTf solution to an aqueous solution of 1 adjusted to pH ~ 7 with 1 M NaOH. As the reaction proceeded to completion within 4 h (at 70 °C the reaction was complete within 5 min), the mixture gradually became more acidic, reaching a final pH of ~4; only one major HPLC peak (~ 95%, retention time of 10.2 min) was observed. However, when the reaction mixture pH was maintained at ≥ 7, it gave two HPLC peaks, Re-1a and Re-1b (designated by retention times 10.2 min and 12.2 min, respectively), in a ratio of 2:3. No changes in this ratio were observed for 3 days at pH 7–12.
To definitively characterize the Re-1a and Re-1b product mixture, we relied on NMR data from samples at pH 4 and pH 7 (Figure 2). At pH 4, the 1H NMR spectrum (top of Figure 2A) clearly shows that the product exists primarily as the symmetrical isomer, Re-1a, with ligand 1 coordinated to Re via the two deprotonated carboxylates and the amine group (Scheme 1, Eq. 2). The Re-1a spectrum has an AB pattern (two “doublets” with J = 16 Hz integrating for two protons each) for the two equivalent coordinated -CH2CO2− groups (a and b, Scheme 1).
Figure 2.

(A) 1H NMR spectra in D2O of [Re-1a]2− at pH 4 (top) and a mixture of [Re-1a]3− and [Re-1b]3− at pH 7 (bottom). (B) 13C NMR (B) spectra in D2O of [Re-1a]2− at pH 4 (top) and a mixture of [Re-1a]3− and [Re-1b]3− at pH 7 (bottom). Selected signals from the symmetrical isomer [Re-1a]2− (blue) and the unsymmetrical isomer [Re-1b]3− (red) are labeled to highlight significant differences for both 1H NMR and 13C NMR spectra.
The diastereotopic protons in these groups point toward (endo) or away (exo) from the carbonyl ligands. From the characteristic shift relationship of these signals [13], we assign the downfield doublet (4.08 ppm) to the exo-H protons and the upfield doublet (3.91 ppm) to the endo-H protons. The methylene signal from the dangling methylphosphonate protons (c, Scheme 1) is a doublet at 3.76 ppm ( 2JPH = 12 Hz). These results and additional NMR and HPLC data lead us to conclude that, above pH 3 and below pH 6, the dominant complex is the symmetrical and dianionic isomer, [Re-1a]2−, with a trianionic chelate ligand (PMIDAH3−) having two deprotonated carboxylates and a (primarily monoprotonated) phosphonate (Scheme 1, Eq. 2). In this pH range, the deprotonated monoanionic carboxylate groups are able to coordinate much more favorably than the monoprotonated monoanionic phosphonate group.
The 1H NMR spectrum at pH 7 (bottom of Figure 2A) is more complicated than at pH 4 and indicates the presence of a second species. Of particular note, the CH2(c) doublet for the dangling CH2PO3H” group of [Re-1a] − is at 3.76 ppm at pH 4, but this doublet is farther upfield at 3.68 ppm at pH 7 (Figure 2A); the upfield shift is consistent with the fully deprotonated CH2PO3 − dangling group of the symmetrical [Re-1a]3− trianion. Thus, at pH 7 and above, Re-1 exists as a mixture of the fully deprotonated, trianionic symmetrical ([Re-1a]3−) and (as shown next) unsymmetrical ([Re-1b]3−) isomers, both present in significant amounts (Scheme 1, Eq. 2). Note that both linkage isomers at pH 7 and above have a fully deprotonated, bound tetra-anionic chelate ligand (PMIDA4−).
The identification and assignments for signals of [Re-1a]3− at pH 7, which are easily made from the assignments for [Re-1a]2− at pH 4, aided the signal assignment for [Re-1b]3− (Figure 2). These 1H NMR signals of [Re-1b]3− reveal that this isomer is unsymmetrical because one acetate (a) and one methylphosphonate (c) are coordinated to the fac-[Re(CO)3]+ core (Scheme 1). The [Re-1b]3− methylene groups (a and c, Scheme 1) give rise to 1H NMR AB patterns (CH2(a), 4.0 and 3.8 ppm, J = 18 Hz; and CH2(c), 3.4 and 2.9 ppm, J = 15.0 Hz). The methylphosphonate (c) “doublets” are further split by two-bond coupling to the adjacent 31P atom. The “doublet” at 2.9 ppm appears as a pseudo triplet, as the JPH and JHH coupling constants are almost identical. The CH2(b) singlet of the [Re-1b]3− dangling -CH2CO2− group is at 3.98 ppm.
Solution 1H NMR studies of Re-1 isomers at pH 4 and 7 were supplemented with 13C and 31P NMR experiments. The 13C NMR chemical shifts for the two Re-1 isomers differ significantly (Figure 2B), confirming the very different coordination mode of 1 in each isomer. At pH 4 the [Re-1a]2− spectrum displays two 13C NMR peaks (197.6 and 196.6 ppm) with a 2:1 peak ratio (top of Figure 2B), consistent with the two types of CO ligands in this symmetrical isomer. At pH 7 there are three additional carbonyl ligand C NMR peaks in a 1:1:1 ratio (198.6, 197.2 and 196.9 ppm) from the unsymmetrical isomer [Re-1b]3− (bottom of Figure 2B). Coordination of the two equivalent carboxylate groups in the Re-1a isomer at both pH 4 and pH 7 ([Re-1a] 2− and [Re-1a]2−, respectively) is also confirmed by a single peak for carboxyl (at 183.9 ppm) and CH2 (at 64.4 ppm ) carbons of the coordinated acetate chelate rings. At pH 7, [Re-1b]3− has -CO2− signals at 186.1 and 175.8 ppm (Figure 2B), as expected for a five- membered chelate ring and a dangling chain, respectively. An upfield signal (62.5 ppm) for [Re-1b]3− is assigned to the CH2 group of the coordinated acetate (a) because it has a similar shift to the CH2 signal of the coordinated acetate groups in [Re-1a] −; thus, the downfield [Re-1b]3–13 C NMR signal (71.6 ppm) is assigned to the dangling acetate CH2 group (b). At pH 7, the shift of the -CH2PO3 13C NMR resonance for the coordinated -CH2PO3 group in [Re-1b]3− (57.6 ppm) is upfield to that for the dangling -CH2PO3 group in [Re-1a]3− (68.2 ppm). The dangling - CH2PO3 13C NMR signal has a similar shift (67.8 ppm) at pH 4 (Figure 2B). The -CH2PO3 13C 31 NMR signal is a doublet in all cases owing to the one-bond 31P coupling.
The 31P NMR chemical shift of the phosphonate group depends on whether or not the phosphonate group is bound to a metal atom or remains uncoordinated [28, 39]. At pH 4, the 31P NMR spectrum (Supporting Information, Fig. S1A) showed only a single peak at 12.99 ppm attributable to the major product, [Re-1a]2−, confirming that the monoprotonated phosphonate group is not coordinated in this symmetrical isomer [39]. At pH 7 (Supporting Information, Fig. S1B), the signal of the now dianionic phosphonate group of [Re-1a]3− is shifted only slightly upfield (12.28 ppm). This pH 7 31P NMR spectrum has an additional peak from [Re-1b]3− at lower field (30.35 ppm) characteristic of a fully deprotonated phosphonate group bound to the metal atom in a chelate ring [28, 39].
The NMR spectroscopic and synthetic results described above indicate clearly that the Re-1 product contains a pH-dependent equilibrium mixture of geometric linkage isomers. To gain insight into the pH-dependence of this equilibrium, we examined solutions of Re-1 over a very wide pH range (1 to 12) by using HPLC. We divided the aqueous solution of Re-1a, the product purified by Sephadex and isolated from the reaction mixture at pH 4, into several fractions and adjusted the pH of each separate fraction to values ranging from 1–12. The ratio of linkage isomers over the course of 1 day at room temperature was monitored by HPLC. For each fraction, changes in the ratio of peaks ceased within 30 min and remained unchanged thereafter throughout the experiment, indicating that equilibrium had been established (Table 1). Then, after 1 d, the pH of the fraction equilibrated at pH 7 was adjusted back to pH 4, and the ratio of peaks eventually returned to the initial 24:1 ratio as shown in Figure 3 (see also Table 1). Note that the ratio of HPLC peaks did not change significantly for the six traces from pH 7 to 12 or for the three traces between pH 3 and 5. However, the HPLC retention times vary slightly in Figure 3 because the column equilibration time differed slightly between runs. Also, HPLC chromatograms together with 1H NMR spectra of the pH 4 sample before and after isomerization at pH 7 showed no difference in product ratios, indicating that ratio of isomers was not significantly affected by the HPLC method at these pH’s. Although an acidic component of the HPLC eluent in a gradient method could influence the isomer distribution (as observed in at least one report [40]), no evidence for such an effect was found here for M-1 complexes.
Table 1.
Percentages of isomers Re-1a and Re-1b present in the equilibrated fractions at different pH values according to HPLC (see Figure 3). The highlighted range shows the pH values at which the symmetrical isomer Re-1a, with both carboxylates coordinated to the metaltricarbonyl core [structurally similar to 99mTc/Re(CO)3(NTA)], is the major species present at ≥ 90%.
| pH | Peak 1 (Re-1a) | Peak 2 (Re-1b) |
|---|---|---|
| 12 | 41% | 59% |
| 11 | 41% | 59% |
| 10 | 37% | 63% |
| 9 | 39% | 61% |
| 8 | 38% | 62% |
| 7 | 38% | 62% |
| 6 | 52% | 48% |
| 5 | 94% | 6% |
| 4 | 96% | 4% |
| 3 | 90% | 10% |
| 2 | 81% | 19% |
| 1 | 74% | 26% |
Figure 3.

HPLC chromatograms of equilibrated fractions of Re(CO)3(PMIDA) (Re-1) showing the ratio of peaks of Re-1a and Re-1b, designated by order of elution, at various pH values. (Note that the HPLC retention times of Re-1a and Re-1b vary slightly owing to differences in the column equilibration time between runs.)
Analyses of the HPLC traces of each sample showed that from pH 3 to 5, Re-1 contains ~90% or more of Re-1a, e.g. 96% at pH 4 (Re-1a:Re-1b = 24:1). However, in samples more basic than pH 5 and more acidic than 3, the abundance of Re-1b increased. The Re-1a:Re-1b ratio was ~ 1:1 at pH 6 and ~ 2:3 at pH ≥ 7 (Figure 3, Table 1). At pH ≤ 3 the Re-1b peak also began to grow and continued to become more significant as the pH was lowered. At pH 1, the most acidic solution studied, the Re-1a:Re-1b ratio was 3:1 (Figure 3, Table 1).
These HPLC data (Figure 3, Table 1) are consistent with NMR results for linkage isomerism at pH 4 and 7 (Scheme 1, Eq. 2). The effect of changes in pH on chelate ligand coordination mode from pH 1 to 12 (Table 1) can be understood from the various protonation states of the PMIDAH4 ligand (Figure 1). As the extent of full phosphonate group deprotonation increases, the phosphonate group competes more effectively for the metal binding site than the deprotonated carboxylate groups. From the data at pH ≥ 7 in Table 1, the dangling dianionic phosphonate group is 50% better at coordinating than the one dangling deprotonated monoanionic carboxylate group. At pH ≤ ~ 7 but ≥ ~ 3, the one dangling monoanionic carboxylate group competes very favorably versus the monoprotonated monoanionic phosphonate group. At pH ≤ 3, protonated carboxylate and monoprotonated phosphonate groups both compete to form a bond with the metal center to give a mixture of isomers with a mononegative charge. At these pH ≤ 3 conditions, the phosphonate group again begins to compete well, but not so favorably as at pH ≥ 6.
Although direct coordination of the phosphonate group to the /ac-[Re(CO)3]+ core in the Re-1b isomer is consistent with the HPLC results and is undoubtedly established by the above NMR solution studies, we sought additional evidence because such direct coordination has not also been demonstrated in an X-ray structure. Re analogs of isomeric mixtures of 99mTc radiopharmaceuticals, especially those with dangling chains, are difficult to crystallize. Thus, we decided to utilize a smaller ligand in order to obtain X-ray quality crystals of a relevant Re complex. N-(phosphonomethyl)glycine (PMGH3, 2), the product of a metal-catalyzed O2 oxidation of 1 [41], is widely used as a commercially available herbicide known as Glyphosate or Roundup [42]. Several PMG complexes have been structurally characterized [43–47]. Thus, 2 could also be expected to coordinate in a tridentate fashion to the /ac-[M(CO)3]+ core and to form a single mononuclear product, M(CO)3(PMG) (M-2) (Scheme 1, Eq. 3).
The reaction of equimolar amounts of 2 and [Re(CO)3(H2O)3]OTf afforded Re-2 at pH 7 (Scheme 1, Eq. 3) as well as pH 4 and 10 proceeded over time to form a single product, as shown by HPLC analysis. The product crystallized as a disodium salt with 5 molecules of water, Na2[Re-2]•5H2O, in the C2/c space group. Single-crystal X-ray analysis of the molecular structure of the distorted octahedral [Re-2] − dianion confirmed that 2 binds as a trianion in a facial ONO coordination mode (Figure 4A; the water molecules and sodium atoms omitted for clarity in Figure 4A are shown in Figure 4B). Crystallographic details appear in the Supporting Information (Table S1). All P-O bonds have virtually the same length (~ 1.52 Å), consistent with a dianionic phosphonate donor. All bond lengths and angles involving the Re atom fall within expected values and are comparable to those reported for other octahedral complexes containing the _/ac-[Re(CO)3]+ core and similar donor atoms [13, 28].
Figure 4.

ORTEP view of (A) the [Re(CO)3(PMG)]2− dianion ([Re-2]2−) and (B) Na2[Re(CO)3(PMG)]•5H2O (Na2[Re-2]•5H2O) with 50% probability for thermal ellipsoids. Selected bond lengths [Å] and angles [°]: Re(1)-N(1) 2.231(2), Re(1)-O(4) 2.154(2), Re(1)-O(6) 2.545(2), Re(1)-C(1) 1.887(3), Re(1)-C(2) 1.904(3), Re(1)-C(3) 1.917(3), C(1)-Re(1)-N(1) 97.59(12), C(2)-Re(1)-N(1) 93.86(12), C(3)-Re(1)-N(1) 174.94(11), C(1)-Re(1)-O(6) 90.48(12), C(2)-Re(1)-O(4) 96.22(12), C(1)-Re(1)-O(4) 171.63(12), C(2)-Re(1)-O(6) 175.51(11), O(4)- Re(1)-N(1) 76.54(9), O(6)-Re(1)-N(1) 81.65(8), O(6)-Re(1)-O(4) 82.81(9), P(1)-C(6)-N(1)-C(4) 85.17, H(4A)-C(4)-N(1)-H(1) 22.09, H(4B)-C(4)-N(1)-H(1) 95.21, H(6A)-C(6)-N(1)-H(1) 77.80, H(6B)-C(6)-N(1)-H(1) 40.43.
The 1H, 13C and 31P NMR data for [Re-2]2− at pH 7 are fully consistent with the solid- state molecular structure and establish that the tri-anionic PMG ligand in [Re-2] − has the same unsymmetrical ONO coordination as indicated by the NMR results for the tetra-anionic PMIDA ligand in [Re-1b]3−. Specifically, [Re-2]2− exhibits a single 31P NMR peak (31.3 ppm) from the directly coordinated phosphonate group (Supporting Information Fig. S2); this shift is very similar to the P NMR signal (30.35 ppm) of [Re-1b] − at pH 7, confirming that both complexes have a fully deprotonated phosphonate group coordinated directly to Re. Also, as observed for [Re-1b]3−, the 1H NMR spectrum of [Re-2]2− has two AB-spin systems both with J = ~17 Hz arising from geminal coupling of methylene protons of the acetate and methylphosphonate groups. The methylphosphonate group’s AB components have almost the same shift (~ 2.8 ppm) and are split further by P coupling ( JPH) and additional NH coupling (Supporting Information Fig. S3). The two [Re-2]2− P-CH2 1H NMR AB signals have shifts (~2.8 ppm) similar to the ~2.9 ppm shift of the upfield P-CH2 AB signal of [Re-1b]3−; in contrast, the shift of the other P- CH2 AB signal of [Re-1b] − is relatively downfield (3.4 ppm), a finding that most likely results from the proximity of that exo-H proton to the dangling carboxylate group [13]. It is worth pointing out that one carboxymethyl group doublet (3.7 ppm) in [Re-2] − was also split further by coupling with the NH proton (J = 8 Hz). The same coupling constant can be found for the NH signal at 6.05 ppm, which appears as a pair of triplets since that signal not only shows coupling with the adjacent methylene protons but is also further split by long-range three-bond 31P coupling (3JPH = 20 Hz). According to the torsion angles in the molecular structure of Re-2 (Figure 4), NH signal coupling occur will involve one carboxymethylene proton, the endo-CH(4A) proton (H(4A)-C(4)-N(1)-H(1) torsion angle = 22.1°; J ~ 8 Hz), and one phosphonomethylene proton, the endo-CH(6B) proton ( H(6B)-C(6)-N(1)-H(1) torsion angle = 40.3°; J ~ 5 Hz). The exo-CH(4B) and exo-CH(6A) protons have torsion angles near 90° with, a value consistent with a coupling constant of essentially zero (for more details, see Supporting Information Fig. S4).
Studies of the formation of radiotracers with the fac-[99mTc(CO)3]+ core by b oth aminocarboxyphosphonate ligands (1 and 2) provided a valuable comparison of the species formed for the group 7 congeners. 1 and 2 were efficiently radiolabeled with 99mTc under mild conditions to produce high yields of well-defined tracers with the fac-[99mTc(CO)3]+ core, as found by reversed phase HPLC utilizing a radiometric detector. Because mild aqueous pH ~ 6–7 conditions were employed in the radiolabeling reaction of 1, two isomers of 99mTc(CO)3(PMIDA) (99mTc-1) were formed during the labeling process, a result similar to the formation of two Re-1 isomers under comparable aqueous conditions. The presence of two 99mTc-1 isomers was evident from the radiochromatograms of the HPLC-purified 99mTc-1; however, these isomers could not be separated because of their very similar retention times. In contrast to the 99mTc-1 tracer, the 99mTc(CO)3(PMG) (99mTc-2) radiotracer was obtained as a single product with radiochemical purity > 99%. The identities of the 99mTc-1 and 99mTc-2 radiotracers were confirmed by comparing the HPLC profile of the technetium complexes with the traces of their respective rhenium analogs; 99mTc/Re analogs have almost identical retention times, and the slight difference is attributed to the in-line configuration of the UV-vis and radiometric detectors. No measurable decomposition was observed for either of the 99mTc products when incubated at physiological pH for up to 6 h, confirming the in vitro stability of the radiotracers.
The nature of the chelating ligands and minor configurational changes can significantly alter the physicochemical and pharmacokinetic properties of radiotracers [48]; consequently, we did not conduct an in vivo evaluation of 99mTc-1 owing to its existence as a mixture of isomers, each with unique pharmacokinetic properties. Instead, we confined our investigation to the biological behavior of 99mTc-2 only, which exists as a single species.
The biodistribution studies of 99mTc-2 in normal rats were performed by simultaneous intravenous administration of the 99mTc radiotracer along with 131I-orthoiodohippurate (131I- OIH), followed by analysis of radioactivity in various organs at 10 and 60 min post-injection. Because 99mTc-2 is a small, polar complex with the dianionic charge at physiological pH, we expected that it would be eliminated mainly via the urinary pathway; thus, I-OIH, the radioactive standard for measurement of effective renal plasma flow (ERPF), was used as the internal control. The results of the biodistribution studies (Table 2) revealed a high specificity for the renal elimination. The activity of 99mTc-2 in the urine as a percentage of 131I-OIH was 62 ± 3% and 84 ± 2% at 10 and 60 min, respectively. 99mTc-2 showed good clearance from all organs and tissues, with less than 1% of the total activity present in the spleen, heart and lungs at either 10 or 60 min post-injection. The percent of the injected dose present in the liver at 10 min and 60 min post-injection was similar (~ 4%), which could be attributed more to the slight retention of the tracer in the blood rather than the hepatobiliary elimination because there was minimal gastrointestinal activity at those time points (1.3 ± 0.1% and 0.9 ± 0.2%, respectively).
Table 2.
Biodistribution of 99mTc(CO)3(PMG) (99mTc-2) and co-injected 131I-OIH in normal rats at 10 and 60 minutes post-injection. * Results are expressed as %ID ± SD in blood, urine and selected organs.
| 10 min |
60 min |
|||
|---|---|---|---|---|
| 99mTc-2 | 131I-OIH | 99mTc-2 | 131I-OIH | |
| Blood | 8.6 ± 0.9 | 4.7 ± 0.5 | 2.8 ± 0.6 | 0.8 ± 0.4 |
| Liver | 4.2 ± 0.8 | 2.7 ± 0.5 | 3.9 ± 0.4 | 0.6 ± 0.2 |
| Bowela | 1.3 ± 0.1 | 1.5 ± 0.1 | 0.9 ± 0.2 | 1.1 ± 0.2 |
| Spleen | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 |
| Heart | 0.2 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.1 | 0.1 ± 0.1 |
| Lung | 0.6 ± 0.2 | 0.4 ± 0.1 | 0.3 ± 0.0 | 0.1 ± 0.0 |
| Kidney | 8.9 ± 1.2 | 6.1 ± 1.9 | 2.5 ± 1.0 | 1.2 ± 0.6 |
| Urine | 33.1 ± 6.9 | 53.0 ± 9.2 | 73.9 ± 4.3 | 88.1 ± 2.6 |
| %Urineb | 62 ± 3 | 84 ± 2 | ||
Data are presented as mean ± SD; 10 min n = 5, 60 min n = 4.
Bowel includes intestines and stomach.
%Urine is expressed as a 99mTc-2/131I-OIH ratio.
In a related rat study designed to assess the in vivo stability of the 99mTc-2 radiotracer, the urine of two rats injected with 99mTc-2 was collected and analyzed by HPLC. More than 99% of the activity in the urine HPLC trace could be assigned to the parent 99mTc-2 tracer, establishing the high metabolic stability and high in vivo robustness of the 99mTc(CO)3(PMG) agent.
4. Conclusions
We found that M(CO)3(PMIDA) complexes exist as two distinct geometric isomers (M-1a and M-1b) in a pH-dependent equilibrium governed by the protonation state of the phosphonate and carboxylate groups. This finding establishes that at pH 7 the phophonate group binds to the fac-99mTc/Re(CO)3 cores with an affinity twice that observed for the carboxylate group. We conclude that chelate ligand analogs with phosphonate in place of carboxylate can be used to design complexes that will be sufficiently robust in vivo for use in radiopharmaceuticals. In support of our conclusions reached from the results with M(CO)3(PMIDA) complexes, the M(CO)3(PMG) (M-2) complexes exist in solution as only a single robust isomer under all conditions. The biodistribution results with the 99mTc(CO)3(PMG) tracer establish that the PMG ligand ONO donors are sufficient to withstand competition from plasma constituents in vivo.
We have crystallized and structurally characterized a rhenium complex of the PMG ligand for the first time. To our knowledge, both Re-1b and Re-2 and their 99mTc analogs are the first examples of stable complexes with a phosphonic acid group (deprotonated) directly coordinated via oxygen to the metal-tricarbonyl core (M(CO)3-O-P). Re-2 is the first well- defined, isolated monomeric complex with a phosphonate group directly coordinated to the fac- [Re(CO)3]+ core. Mundwiler et al. previously reported a similar P-O-Re(CO)3 coordination, but the bond was formed by a phosphonic acid ester, not a phosphonate group; the authors could not isolate any Re-tricarbonyl complexes with the phosphonate group acting directly as a donor group [28].
Various combinations of acetate and phosphonate donor groups anchored by an amine donor suggest a novel approach for the development of new bifunctional chelating agents for radiolabeling specific biomolecules with the fac-[99mTc(CO)3]+ core. Although 99mTc(CO)3(PMIDA) is not well suited to serve as a renal tracer because it exists as two geometric isomers at physiologically relevant pH, the phosphonate or carboxylate groups in the dangling chain of M-1a and M-1b isomers, respectively, could be employed to attach a bioactive moiety through the formation of ester or amide bonds. Introduction of phosphodiester or amide groups would bypass the linkage isomer problem because our results indicate that neither the phosphodiester nor the amide groups would compete with phosphonate and carboxyl donor groups for the fac-99mTc/Re(CO)3 cores. Such bioconjugation chemistry designed by employing chelate ligands anchored by an amine donor could be employed in the development of robust target-specific 99mTc and 186/188Re radiopharmaceuticals for imaging and therapeutic applications.
Supplementary Material
Highlights.
Aminocarboxyphosphonates chelate tridentately to fac-[99mTc/Re(CO)3]+ cores.
X-ray/NMR/HPLC data unambiguously establish phosphonate binding to the cores.
Some chelates form linkage isomers that interconvert in a pH-dependent ratio.
Isomers with phosphonate binding are favored over carboxylate binding at pH > 6.
Phosphonate-to-fac-[99mTc(CO)3]+ binding is totally robust in in vivo rat studies.
Acknowledgments
This research was supported by the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases grant R37 DK038842. The authors thank Eugene Malveaux for his excellent technical assistance with all animal studies. The authors also thank Dr. John Bacsa, Emory X-ray Crystallography, for the X-ray structural analysis and Dr. Patricia A. Marzilli for her invaluable comments during the preparation of the manuscript. We also acknowledge the use of the Rigaku SYNERGY diffractometer, supported by the National Science Foundation under grant CHE-1626172.
Footnotes
Supplementary data
CCDC 1846394 contains the supplementary crystallographic data for Re-2. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Jurisson S, Berning D, Jia W, and Ma D. Coordination compounds in nuclear medicine. Chem. Rev 1993;93:1137–56. [Google Scholar]
- [2].Cutler CS, Hennkens HM, Sisay N, Huclier-Markai S, and Jurisson SS. Radiometals for combined imaging and theraphy. Chem. Rev 2013;113:858–83. [DOI] [PubMed] [Google Scholar]
- [3].National Academies of Sciences, Engineering, and Medicine. Molybdenum-99 for Medical Imaging, Washington, DC: The National Academies Press; DOI: 10.17226/23563.,2016. [DOI] [PubMed] [Google Scholar]
- [4].Alberto R, Schibli R, Waibel R, Abram U, and Schubiger PA. Basic aqueous chemistry of [M(OH2)3(CO)3]+ (M =-Re, Tc) directed towards radiopharmaceutical application. Coord. Chem. Rev. 1999;190–192:901–19. [Google Scholar]
- [5].Schibli R and Schubiger PA. Current use and future potential of organometallic radiopharmaceuticals. Eur. J. Nucl. Med 2002;29:1529–42. [DOI] [PubMed] [Google Scholar]
- [6].Alberto R The chemistry of technetium-water complexes within the manganese triad: challenges and perspectives. Eur. J. Inorg. Chem 2009:21–31. [Google Scholar]
- [7].Schibli R, La Bella R, Alberto R, Garcia-Garayoa E, Ortner K, Abram U, et al. Influence of the denticity of ligand systems on the in vitro and in vivo behavior of 99mTc(I)-tricarbonyl complexes: A hint for the future functionalization of biomolecules. Bioconjugate Chem. 2000;11:345–51. [DOI] [PubMed] [Google Scholar]
- [8].Pietzsch H-J, Gupta A, Reisgys M, Drews A, Seifert S, Syhre R, et al. Chemical and biological characterization of technetium(I) and rhenium(I) tricarbonyl complexes with dithioether ligands serving as linkers for coupling the Tc(CO)3 and Re(CO)3 moieties to biologically active molecules. Bioconjugate Chem. 2000;11:414–24. [DOI] [PubMed] [Google Scholar]
- [9].He H, Lipowska M, Xu X, Taylor AT, Carlone M, and Marzilli LG. Re(CO)3 complexes synthesized via an improved preparation of aqueous _fac-[Re(CO)3(H2O)3]+ as an aid in assessing 99mTc imaging agents. Structural characterization and solution behavior of complexes with thioether-bearing amino acids as tridentate ligands. Inorg. Chem 2005;44:5437–46. [DOI] [PubMed] [Google Scholar]
- [10].Banerjee SR, Maresca KP, Francesconi L, Valliant J, Babich JW, and Zubieta J. New directions in the coordination chemistry of 99mTc: a reflection on technetium core structures and a strategy for new chelate design. Nucl. Med. Biol 2005;32:1–20. [DOI] [PubMed] [Google Scholar]
- [11].Lipowska M, He H, Xu X, Taylor AT, Marzilli PA, and Marzilli LG. Coordination modes of multidentate ligands in fac-[Re(CO)3(polyaminocarboxylate)] analogues of 99mTc radiopharmaceuticals. Dependence on aqueous solution reaction conditions. Inorg. Chem 2010;49:3141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shen Y, Schottelius M, Zelenka K, De Simone M, Pohle K, Kessler H, et al. Orthogonally protected artificial amino acid as tripod ligand for automated peptide synthesis and lableing wit [99mTc(OH2)3(CO)3]+. Bioconjugate Chem. 2013;24:26–35. [DOI] [PubMed] [Google Scholar]
- [13].Klenc J, Lipowska M, Abhayawardhana PL, Taylor AT, and Marzilli LG. Structure and properties of fac-[ReI (CO)3(NTA)]2− (NTA3− = trianion of nitrilotriacetic acid) and fac-[ReI(CO)3(L)]n− analogues useful for assessing the excellent renal clearance of the fac-[99mTci(CO)3(NTA)]2− diagnostic renal agent. Inorg. Chem 2015;54:6281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Santos I, Paulo A, and Correia. Rhenium and technetium complexes anchored by phosphines and scorpionates fr radiopharmaceutical applications. Top Curr. Chem 2005;252:45–84. [Google Scholar]
- [15].He H, Morley JE, Twamley B, Groeneman RH, Bucar D-K, MacGillivray LR, et al. Investigation of the coordination interactions of S-(pyridin-2-ylmethyl)-L-cysteine ligands with M(CO)3+ (M= Re, 99mTc). Inorg. Chem 2009;48:10625–34. [DOI] [PubMed] [Google Scholar]
- [16].Mylonas I, Triantis C, Panagiotopoulou A, Patsis G, Raptopoulou CP, Terzis A, et al. A new bifunctional tridentate NSN ligand leading to cationic tricarbonyl fac-[M(NSN)(CO)3]+ (M =Re, 99mTc) complexes. Inorg. Chim. Acta 2013;400:2–6. [Google Scholar]
- [17].Abhayawardhana PL, Marzilli PA, Fronczek FR, and Marzilli LG. Complexes possessing rare “tertiary” sulfonamide nitrogen-to-metal bonds of normal length: fac-[Re(CO)3(N(SO2R)dien)]PF6 complexes with hydrophilic sulfonamide ligands. Inorg. Chem. 2014;53:1144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mokolokolo PP, Frei A, Tsosane MS, Kama DV, Schutte-Smith M, Brink A, et al. Nuclearity manipulation in Schiff-base fac-tricarbonyl complexes of Mn(I) and Re(I). Inorg. Chim. Acta 2018;471:249–59. [Google Scholar]
- [19].Klenc J, Lipowska M, Taylor AT, and Marzilli LG. Synthesis and characterization of fac-Re(CO)3-aspartic-N-monoacetic acid: structural analogue of a potential renal tracer, fac- 99mTc(CO)3(ASMA). Eur. J. Inorg. Chem 2012:4334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lipowska M, Klenc J, Jarkas N, Marzilli LG, and Taylor AT. Monoanionic 99mTc-tricarbonyl-aminopolycarboxylate complexes with uncharged pendant groups: Radiosynthesis and evaluation as potential renal tubular trecer. Nucl. Med. Biol 2017;47:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lipowska M, Marzilli LG, and Taylor AT. 99mTc(CO)3-nitrilotriacetic acid: a new renal radiopharmaceutical showing pharmacokinetic properties in rats comparable to those of 131I-OIH. J. Nucl. Med 2009;50:454–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Taylor AT, Lipowska M, and Marzilli LG. 99mTc(CO)3(NTA): a 99mTc renal tracer with pharmacokinetic properties comparable to those of 131I-OIH in healthy volunteers. J. Nucl. Med 2010;51:391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Taylor AT, Lipowska M, and Cai H. 99mTc(CO)3(NTA) and 131I-OIH: comparable plasma clearances in patients with chronic kidney disease. J. Nucl. Med 2013;54:578–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Subramanian G, McAfee JG, Blair RJ, Kallfelz FA, and Thomas FD. Technetium-99m- methylene diphosphonate - a superior agent for skeletal imaging: comparison with other technetium complexes. J. Nucl. Med 1975;16:744–55. [PubMed] [Google Scholar]
- [25].Domstad PA, Coupal JJ, Kim EE, Blake JS, and DeLand FH. 99mTc-hydroxymethane diphosphonate: a new bone imaging agent with a low tin content. Radiology 1980;136:209–11. [DOI] [PubMed] [Google Scholar]
- [26].Ogawa K and Ishizaki A. Well-designed bone-seeking radiolabeled compounds for diagnosis and therapy of bone metastases. Biomed. Res. Int 2015;2015:676053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Love C, Din AS, Tomas MB, Kalapparambath TP, and Palestro CJ. Radionuclide Bone Imaging: An illustrative review. RadioGraphics 2003;23:341–58. [DOI] [PubMed] [Google Scholar]
- [28].Mundwiler S, Waibel R, Spingler B, Kunze S, and Alberto R. Picolylamine-methylphosphonic acid esters as tridentate ligands for the labeling of alcohols with the fac-[M(CO)3]+ core (M = 99mTc, Re): synthesis and biodistribution of model compounds and of a 99mTc-labeled cobinamide. Nucl. Med. Biol 2005;32:473–784. [DOI] [PubMed] [Google Scholar]
- [29].Adams KM, Marzilli PA, and Marzilli LG. Reaction offac-[Re(CO)3(H2O)3]+ with nucleoside diphosphates and thiamine diphosphates in aqueous solution investigated ny multinuclear NMR spectroscopy. Inorg. Chem 2007;46:9172–81. [DOI] [PubMed] [Google Scholar]
- [30].Appleton TG. Donor atom preferences in complexes of platinum and palladium with amino acids and related molecules. Coord. Chem. Rev 1997;166:313–59. [Google Scholar]
- [31].Stone AT, Knight MA, and Nowack B. Speciation and chemical reactions of phosphonate chelating agents in aqueous media In: Lipnick RL, Mason RP, Phillips ML, and Pittman CU Jr. editors. ACS Symposium Series 806. Washington, DC: American Chemical Society; 2002, p. 59–94. [Google Scholar]
- [32].Mateescu A, Gabriel C, Raptis RG, Baran P, and Salifoglou A. pH - Specific synthesis, spectroscopic, and structural characterization of an assembly of species between Co(II) and A,A-bis(phosphonomethyl)glycine. Gaining insight into metal-ion phosphonate interactions in aqueous Co(II)-organophosphonate systems. Inorg. Chim. Acta 2007;360:638–48. [Google Scholar]
- [33].He H, Lipowska M, Christoforou AM, Marzilli LG, and Taylor AT. Initial evaluation of new 99mTc(CO)3 renal imaging agents having carboxyl-rich thioether ligands and chemical characterization of Re(CO)3 analogues. Nucl. Med. Biol 2007;34:709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Anghileri LJ. A simplified method for preparing high specific activity I-labeled hippuran. Int. J. Appl. Radiat. Isot 1964:15–95. [Google Scholar]
- [35].Sheldrick GM. A short history of SHELX. Acta Crystallogr. 2008;A64:112–22. [DOI] [PubMed] [Google Scholar]
- [36].Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, and Puschmann H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst 2009;42:339–41. [Google Scholar]
- [37].Schwarzenbach G, Ackermann A, and Ruckstuhl P. Komplexone XV. Neue Derivate der Imino-diessigsäure und ihre Erdalkalikomplexe. Beziehungen zwischen Acidität und Komplexbildung. Helv. Chim. Acta 1949;32:1175–86. [Google Scholar]
- [38].Shkolnikowa LM, Porai-Koshits MA, Dyatlova NM, Yaroshenko GF, Rudomino MV, and Kolova EK. X-ray structural study of organic ligands of the complexone type. III. Crystal and molecular structure of phosphonomethylglycine and iminodiacetic- monoethylphosphonic acid. J. Struct. Chem 1982;23:737–46. [Google Scholar]
- [39].Appleton TG, Hall JR, and McMahon IJ. NMR spectra of iminobis(methylenephosphonic acid), HN(CH2PO3H2)2, and related ligands and of their complexes with platinum(II). Inorg. Chem 1986;25:726–34. [Google Scholar]
- [40].Bottorff SC, Moore AL, Wemple AR, Bucar D-K, MacGillivray LR, and Benny PD. pH- Controlled coordination mode rearrangements of “clickable” Huisgen-based multidentate ligands with[MI(CO)3]+ ( M = Re, 99mTc). Inorg. Chem 2013;52:2939–50. [DOI] [PubMed] [Google Scholar]
- [41].Riley DP, Fields DL, and Rivers W. Vanadium(IV, V) salts as homogeneous catalysts for the oxygen oxidation of N-(phosphonomethyl)iminodiacetic acid to N-(phosphonomethyl)glycine. Inorg. Chem 1991;30:4191–7. [Google Scholar]
- [42].Duke SO and Powles SB. Glyphosate: a once-in-a-century herbicide. Pest Manag. Sci. 2008;64:319–25. [DOI] [PubMed] [Google Scholar]
- [43].Motekaitis RJ and Martell AE. Metal chelate formation by N-phosphonomethyleneglycine and related ligands. J. Coord. Chem 1985;14:139–49. [Google Scholar]
- [44].Smith PH and Raymond KN. Solid-state and solution chemistry of calcium N- (phosphonomethyl)glycinate. Inorg. Chem 1988;27:1056–61. [Google Scholar]
- [45].Clark ET, Rudolf PR, Martell AE, and Clearfield A. Structural investigation of the Cu(II) chelate of N-phosphonomethylglycine, X-ray crystal structure of Cu(II)[O2CCH2NHCH2PO3].Na(H2O)3.5. Inorg. Chim. Acta 1989;164:59–63. [Google Scholar]
- [46].Heineke D, Franklin SJ, and Raymond KN. Coordination chemistry of glyphosate: structural and spectroscopic characterization of bis(glyphosphate)metal(III) complexes. Inorg. Chem 1994;33:2413–21. [Google Scholar]
- [47].Structure Szabo Z., equilibrium and ligand exchange dynamics in the binary and ternary dioxouranium(VI)-glyphosate-fluoride system. A multinuclear NMR study. J. Chem. Soc., Dalton Trans 2002:4242–7. [DOI] [PubMed] [Google Scholar]
- [48].Lipowska M, Klenc J, Folks RD, and Taylor AT. Initial evaluation of 99mTc(CO)3(ASMA) as a renal tracer in healthy human volunteers. Nucl. Med. Mol. Imag 2014;48:216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.