

Abstract
HIV-1 integrase (IN) is a validated therapeutic target for the treatment of AIDS. However, the emergence of resistance to Raltegravir, the sole marketed FDA-approved IN inhibitor, emphasizes the need to develop second-generation inhibitors that retain efficacy against clinically-relevant IN mutants. We report herein bicyclic hydroxy-1H-pyrrolopyridine-triones as a new family of HIV-1 integrase inhibitors that were efficiently prepared using a key “Pummerer cyclization deprotonation cycloaddition” cascade of imidosulfoxides. In in vitro HIV-1 integrase assays the analogues showed low micromolar inhibitory potencies with selectively for strand transfer reactions as compared with 3’-processing inhibition. A representative inhibitor (5e) retained most of its inhibitory potency against the three major raltegravir-resistance mutant IN enzymes, G140S/Q148H, Y143R and N155H. In antiviral assays employing viral vectors coding these IN mutants, compound 5e was approximately 200-fold and 20-fold less affected than raltegravir against the G140S/Q148H and Y143R mutations, respectively. Against the N155H mutation 5e was approximately 10-fold less affected than raltegravir. Thus, our new compounds represent a novel structural class that may be further developed to overcome resistance to raltegravir, particularly in the case of the G140S/Q148H mutations.
Integrase (IN) catalyzes the incorporation of HIV-1 cDNA into host DNA in a process involving two sequential steps, 3’-processing (3’-P) and strand transfer (ST), (1, 2). In 2007 Merck’s Isentress™ (MK-0518 or raltegravir) (1) (3–6) became the first marketed drug targeting HIV-1 IN (Figure 1). Raltegravir shares with many other potent IN inhibitors the ability to target ST reactions, at pharmacological concentrations where it does not affect the IN 3’-P step (6). Inhibitors of this class typically contain an array of heteroatoms that efficiently chelate the two divalent metal ions associated with three conserved acidic residues in the IN protein (Asp64, Asp116 and Glu152; the ‘DDE motif’) (7). The mechanism of ST inhibition has recently been clarified by X-ray co-crystal structures of inhibitors bound to the IN of the prototype foamy virus (PFV) complexed with substrate DNA, which show that the inhibitors displace the processed dA of the 3’ end of the DNA, preventing ST and blocking integration of viral DNA (8, 9). This involves tight binding at the viral DNA-IN-Mg2+ interface (1, 2).
Figure 1.
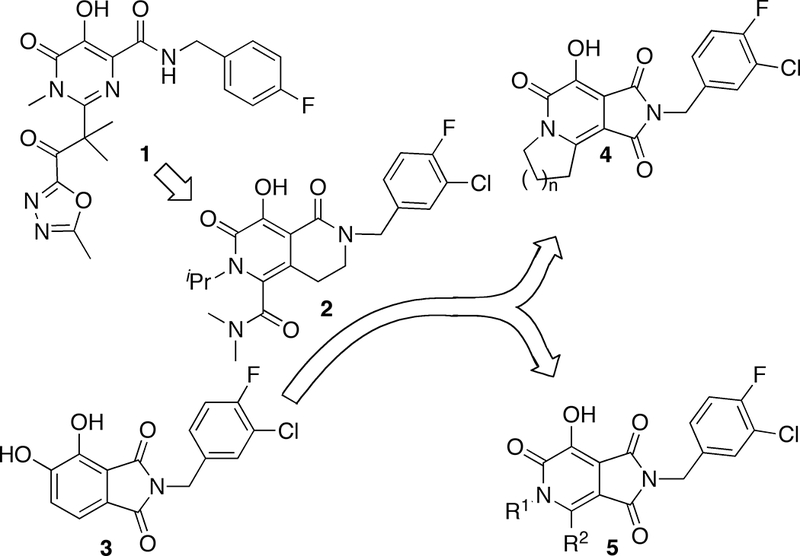
Conceptual approach to targets 5.
In response to the appearance of IN mutants that reduce IN sensitivity to raltegravir (10, 11), there is a strong need to develop “second-generation” ST inhibitors that are effective against raltegravir resistant mutants (7). For example, the bicyclic 2-pyridone-containing inhibitor MK-0536 (2) exhibits improved activity against raltegravir-resistant strains (12–14). Previously, we reported 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones (3) as structurally simple, yet potent HIV-1 IN inhibitors (15, 16). However, the therapeutic utility of these compounds is limited by their cytotoxicity, which could potentially arise from the catechol functionality. More recently we prepared and tested tricyclic hydroxy-1H-pyrrolopyridine-triones (4) as IN inhibitors. These compounds remove the catechol functionality by combining 3 with features of 2 (Figure 1) (17). However, although these compounds had the desired reduction in cytotoxicity, they suffered from a loss of IN inhibitory potency. It was unclear whether the observed potency reduction of inhibitor 4 is a feature inherent to their tricyclic scaffold and we wondered whether related bicylic structures might have improved potencies. Accordingly, the current study was undertaken with the objective of synthesizing and evaluating bicyclic hydroxy-1H-pyrrolopyridine-triones exemplified by 5 (Figure 1).
Methods and Materials
General synthetic
1H and 13C NMR data were obtained on a Varian 400 MHz spectrometer and are reported in ppm relative to TMS and referenced to the solvent in which the spectra were collected. Solvent was removed by rotary evaporation under reduced pressure and anhydrous solvents were obtained commercially and used without further drying. Purification by silica gel chromatography was performed using EtOAc–hexanes. Preparative high pressure liquid chromatography (HPLC) was conducted using a Waters Prep LC4000 system having photodiode array detection and Phenomenex C18 columns (250 mm × 21.2 mm, 10 μm particle size, 110 Å pore) at a flow rate of 10 mL/min. A binary solvent systems consisting of A = 0.1% aqueous TFA and B = 0.1% TFA in acetonitrile was employed with gradients as indicated. Products were obtained as amorphous solids following lyophilization. Electrospray ionization-mass spectra (ESI-MS) and atmospheric pressure chemical ionization-mass spectra (APCI-MS) were acquired using an Agilent LC/MSD system equipped with a multimode ion source. Matrix-assisted laser desorption/ionization (MALDI) mass spectra were acquired on a Shimadzu Biotech Axima-CFR time-of-flight instrument using α–cyano-4-hydroxycinnamic acid as matrix. High-resolution mass spectra (HRMS) were obtained from the UCR Mass Spectrometry Facility, University of California at Riverside.
2-(Ethylthio)-N-isopropylacetamide (6a):
To 2-ethylthioacetic acid (40 mmol) in anhydrous dichloromethane (20 mL) was added DMF (0.1 mL) followed by oxalyl chloride (120 mmol), dropwise at 0 °C and the mixture was stirred at room temperature (2 h). The mixture was concentrated under reduced pressure and then added dropwise to a solution of isopropylamine (40 mmol) in benzene (20 mL) with triethylamine (11 mL) at 0 °C and the resultant solution was allowed to come to ambient temperature with stirring (overnight). The mixture was partitioned between EtOAc and H2O and the organic phase was washed (brine) and dried (sodium sulfate) and take to dryness and the remaining residue was purified by silica gel column chromatography to provide 6a as a yellow solid (81% yield). 1H NMR (400 MHz, CDCl3) δ 6.69 (bs, 1H), 4.08 – 3.97 (m, 1H), 3.14 (s, 2H), 2.49 (q, J = 7.4 Hz, 2H), 1.20 (t, J = 7.4 Hz, 3H), 1.12 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.75, 41.54, 35.88, 26.96, 22.59 (2C), 14.40. APCI-MS m/z: 152.1 (M+H+), 323.1 (M2H+).
2-(Ethylthio)-N-(4-methoxybenzyl)acetamide (6b):
According to procedure reported above, 6b was obtained from 4-methoxybenzylamine as a solid in 14% yield. 1H NMR (400 MHz, CDCl3) δ 7.18 (d, J = 8.4 Hz, 2H), 7.08 (bs, 1H), 6.83 (d, J = 8.4 Hz, 2H), 4.37 (d, J = 5.7 Hz, 2H), 3.76 (s, 3H), 3.23 (s, 2H), 2.50 (q, J = 7.4 Hz, 2H), 1.20 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 168.59, 159.04, 130.00, 129.04 (2C), 114.07 (2C), 55.25, 43.24, 35.80, 27.04, 14.31. ESI-MS m/z: 240.10 (M+H+), 262.10 (M+Na+).
General Procedure A for the Synthesis of Ethylthioacetyl-containing Amides (7):
Acyl chloride (37 mmol) was added to amide 6 (20 mmol) in benzene (5 mL) and the resultant solution was stirred at reflux (6 h). In cases where significant starting material remained (TLC), triethylamine (74.4 mmol) was added carefully and the mixture was refluxed (overnight). The reaction mixture was cooled to room temperature and extracted (EtOAc, 300 mL) the organic phase was washed (brine), dried (Na2SO4), taken to dryness and the residue was purified by silica gel column chromatography.
2-(2-(Ethylthio)-N-isopropylacetamido)-2-oxoethyl acetate (7a):
According to General Procedure A, 7a was obtained from 6a and 2-chloro-2-oxoethyl acetate in 36% yield. 1H NMR (400 MHz, CDCl3) δ 4.87 (s, 2H), 4.12–4.05 (m, 1H), 3.44 (s, 2H), 2.62 (q, J = 7.4 Hz, 2H), 2.15 – 2.06 (m, 3H), 1.43 (d, J = 6.8 Hz, 6H), 1.26 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.72, 171.19, 170.53, 65.53, 51.14, 36.35, 26.42, 20.50, 20.16 (2C), 14.22. ESI MS m/z: 262.10 (M+H+).
N-(2-(Ethylthio)acetyl)-N-isopropyl-3-methylbutanamide (7b):
According to General Procedure A, 7b was obtained from 6a and 3-methylbutanoyl chloride in 38% yield. 1H NMR (400 MHz, CDCl3) δ 4.15 – 4.05 (m, 1H), 3.54 (s, 2H), 2.54 (q, J = 7.4 Hz, 2H), 2.38 (d, J = 6.9 Hz, 2H), 2.17–2.07 (m, 1H), 1.36 (d, J = 6.8 Hz, 6H), 1.19 (t, J = 7.4 Hz, 3H), 0.91 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 176.67, 173.73, 49.89, 46.55, 38.01, 26.35, 25.48, 22.50 (2C), 20.46 (2C), 14.28. APCI-MS m/z: 246.1 (M+H+).
Methyl 4-(2-(ethylthio)-N-isopropylacetamido)-4-oxobutanoate (7c):
According to General Procedure A, 7c was obtained from 6a and methyl 4-chloro-4-oxobutanoate in 30% yield. 1H NMR (400 MHz, CDCl3) δ 4.19–4.09 (m, 1H), 3.62 (s, 3H), 3.54 (s, 2H), 2.85 (t, J = 7.2 Hz, 2H), 2.61 (t, J = 6.5 Hz, 2H), 2.57–2.52 (m, 2H), 1.37 (d, J = 6.8 Hz, 6H), 1.20 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 175.58, 173.0, 172.93, 51.77, 50.08, 37.87, 32.82, 29.02, 26.33, 22.56, 20.37, 14.28. APCI-MS m/z: 276.1 (M+H+).
2-(2-(Ethylthio)-N-(4-methoxybenzyl)acetamido)-2-oxoethyl acetate (7d):
According to General Procedure A, 7d was obtained from 6b and 2-chloro-2-oxoethyl acetate in 70% yield. 1H NMR (400 MHz, CDCl3) δ 7.05 (d, J = 8.8, 1H), 6.83 (d, J = 8.7, 1H), 5.08 (s, 1H), 4.97 (s, 1H), 3.74 (s, 2H), 3.37 (s, 1H), 2.58 (q, J = 7.4 Hz, 1H), 2.14 (s, 2H), 1.21 (t, J = 7.4 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 172.08, 170.94, 170.57, 159.12, 127.94, 127.47 (2C), 114.42 (2C), 65.46, 55.25, 46.30, 35.85, 26.21, 20.50, 14.08. ESI-MS m/z: 362.1 (M+Na+).
General Procedure B for the Synthesis of Ethanesulfinylacetyl-containing Substrates (8):
Compound 7 (20 mmol) in MeOH (10 mL) was added dropwise to a solution of sodium periodate (26 mmol) in MeOH : H2O (20 mL, 1 : 1) and the mixture was stirred at room temperature (2 h), then extracted (CHCl3) and the organic extracts were dried (Na2SO4) and taken to dryness to afford crude 8 as a colorless oil. This was either used directly or purified by silica gel column chromatography.
N-(2-(Ethylsulfinyl)acetyl)-N-isopropyl-3-methylbutanamide (8a):
According to General Procedure B, 8a was obtained from 7a as a colorless oil in 63% yield. 1H NMR (400 MHz, CDCl3) δ 4.16–4.06 (m, 1H), 4.09 (d, J = 14.8 Hz, 1H), 3.90 (d, J = 14.7 Hz, 1H), 2.89–2.80 (m, 1H), 2.76–2.67 (m, 1H), 2.36 (d, J = 6.8 Hz, 2H), 2.15–2.05 (m, 1H), 1.36 (d, J = 6.8 Hz, 3H), 1.35 (d, J = 6.8 Hz, 3H), 1.28 (t, J = 7.5 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 176.67, 168.87, 61.20, 50.32, 46.23, 45.88, 25.32, 22.45 (2C), 20.52, 20.21, 6.54. ESI-MS m/z: 262.1 (M+H+), 284.1 (M+Na+).
2-(2-(Ethylsulfinyl)-N-isopropylacetamido)-2-oxoethyl acetate (8b):
According to General Procedure B, 8b was obtained from 7b as a colorless oil in 74% yield. 1H NMR (400 MHz, CDCl3) δ 4.85 (dd, J = 17.8, 16.2 Hz, 2H), 4.13 – 4.06 (m, 1H), 3.99 (d, J = 43.2, 14.6 Hz, 2H), 2.99 – 2.79 (m, 2H), 2.14 (s, 3H), 1.45 (dd, J = 6.8, 1.1 Hz, 6H), 1.36 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.81, 170.45, 168.49, 64.63, 58.86, 50.91, 46.30, 20.42, 20.40, 20.23, 6.64. ESI-MS m/z: 278.10 (MH+); 300.10 (M+Na+).
Methyl 4-(2-(ethylsulfinyl)-N-isopropylacetamido)-4-oxobutanoate (8c):
According to General Procedure B, 8c was obtained from 7c as a colorless oil in 61% yield. 1H NMR (400 MHz, CDCl3) δ 4.23–4.13 (m, 1H), 4.09 (d, J = 14.7 Hz, 1H), 3.95 (d, J = 14.7 Hz, 1H), 3.63 (s, 3H), 2.90 – 2.70 (m, 4H), 2.64–2.61 (m, 2H), 1.40 (d, J = 6.8 Hz, 3H), 1.39 (d, J = 6.8 Hz, 3H), 1.29 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 175.78, 172.72, 168.63, 60.62, 51.90, 50.41, 46.12, 32.09, 28.82, 20.39, 20.20, 6.57. APCI-MS m/z: 292.1 (M+H+).
2-(2-(Ethylsulfinyl)-N-(4-methoxybenzyl)acetamido)-2-oxoethyl acetate (8d):
According to General Procedure B, 8d was obtained from 7d as a colorless oil in 53% yield. 1H NMR (400 MHz, CDCl3) δ 7.06 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 4.98 (d, J = 2.3 Hz, 2H), 5.03 – 4.85 (m, 4H), 3.93 (dd, J = 31.8, 14.7 Hz, 2H), 3.73 (s, 3H), 2.86 (dq, J = 13.2, 7.5 Hz, 1H), 2.73 (dq, J = 13.2, 7.4 Hz, 1H), 2.12 (s, 3H), 1.27 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.53, 170.44, 168.56, 159.30, 127.61 (2C), 127.11, 114.58 (2C), 64.85, 58.03, 55.26, 46.34, 46.25, 20.40, 6.43. ESI-MS m/z: 356.1 (M+H+), 378.1 (M+Na+), 711.2 (M2H+), 733.2 (M2Na+).
General Procedure C for the [3+2] Cycloaddition-mediated Synthesis of Products 10:
To a refluxing solution of acetic anhydride (15 mmol), and N-(3-chloro-4-flourobenzyl)maleimide 9 (1.5 mmol) (17) and p-toluenesulfonic acid (2 mg) in toluene (20 mL) was added dropwise, a solution of ethanesulfinylacetyl-containing substrate 8 (1.5 mmol) in toluene (2 mL) and the mixture was stirred at reflux (1 h). The mixture was then concentrated and purified by silica gel column chromatography.
2-(3-Chloro-4-fluorobenzyl)-7-(ethylthio)-4,5-dimethyltetrahydro-1H-4,7-epoxypyrrolo[3,4-c]pyridine-1,3,6(2H,3aH)-trione (10a):
Reaction of N-acetyl-2-ethanesulfinyl-N-methylacetamide [prepared from N-methylacetamide (6a)] (18) and N-(3-chloro-4-flourobenzyl)maleimide (9) (17) according to General Procedure C, provided 10a in 37% yield. 1H NMR (400 MHz, CDCl3) δ 7.32 (dd, J = 6.9, 2.2 Hz, 1H), 7.16–7.12 (m, 1H), 7.00 (t, J = 8.7 Hz, 1H), 4.53 (q, J = 14.5 Hz, 2H), 3.17 (dd, J = 16.4, 6.7 Hz, 2H), 2.78 (s, 3H), 2.71 (q, J = 7.5 Hz, 2H), 1.71 (s, 3H), 1.20 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.37, 171.17, 169.50, 158.71 (d, J = 247.9 Hz), 131.98 (d, J = 4.6 Hz), 130.57, 128.24 (d, J = 6.9 Hz), 121.09 (d, J = 18.3 Hz), 116.74 (d, J = 21.4 Hz), 94.92, 94.61, 53.35, 49.97, 41.65, 25.76, 24.20, 15.29, 14.67. APCI-MS m/z: 413.0 (M+H+).
2-(3-Chloro-4-fluorobenzyl)-7-(ethylthio)-4-methyl-5-phenyltetrahydro-1H-4,7-epoxypyrrolo[3,4-c]pyridine-1,3,6(2H,3aH)-trione (10b):
Reaction of N-Icetyl-2-ethanesulfinyl-N-phenylacetamide [prepared from N-phenylacetamide (6b)] (18) and N-(3-chloro-4-flourobenzyl)maleimide (9) (17) according to General Procedure C, provided 10b in 41% yield. 1H NMR (400 MHz, CDCl3) δ 7.43 – 7.30 (m, 5H), 7.20 – 7.17 (m, 1H), 7.14 – 7.11 (m, 1H), 7.06 – 7.01 (m, 1H), 4.59 (q, J = 14.5 Hz, 2H), 3.56 (d, J = 6.7 Hz, 1H), 3.39 (d, J = 6.7 Hz, 1H), 2.85 (q, J = 7.5 Hz, 2H), 1.76 (s, 3H), 1.29 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.26, 171.04, 168.26, 157.25 (d, J = 248.0 Hz, 1C), 133.74, 131.96 (d, J = 4.6 Hz, 1C), 130.60, 129.70 (2C), 128.28 (d, J = 6.9 Hz, 1C), 128.11, 125.47 (2C), 121.16 (d, J = 17.6 Hz, 1C), 116.74 (d, J = 20.4 Hz, 1C), 96.54, 94.73, 54.89, 49.71, 41.72, 24.25, 16.57, 14.68. APCI-MS m/z: 475.0 (M+H+).
2-(3-Chloro-4-fluorobenzyl)-7-(ethylthio)-4-isobutyl-5-isopropyltetrahydro-1H-4,7-epoxypyrrolo[3,4-c]pyridine-1,3,6(2H,3aH)-trione (10c):
Reaction of 8a and N-(3-chloro-4-flourobenzyl)maleimide (9) (17) according to General Procedure C, provided 10c in 82% yield. 1H NMR (400 MHz, CDCl3) δ 7.28 (dd, J = 6.9, 2.2 Hz, 1H), 7.14–7.10 (m, 1H), 6.98 (t, J = 8.7 Hz, 1H), 4.53 (q, J = 14.6 Hz, 2H), 3.49–3.39 (m, 1H), 3.23 (d, J = 6.6 Hz, 1H), 3.11 (d, J = 6.6 Hz, 1H), 2.75 – 2.65 (m, 2H), 2.10–2.06 (m, 1H), 1.89–1.83 (m, 1H), 1.79–1.69 (m, 1H), 1.43 (d, J = 6.9 Hz, 3H), 1.22 (d, J = 6.6 Hz, 3H), 1.20 (t, J = 7.5 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H), 0.91 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.41, 171.19, 169.21, 157.63 (d, J = 248.7 Hz), 132.03 (d, J = 3.8 Hz), 130.25, 128.06 (d, J = 7.6 Hz), 121.08 (d, J = 18.3 Hz), 116.67 (d, J = 21.3 Hz), 98.48, 94.27, 56.28, 49.19, 46.25, 41.47, 36.47, 24.43, 24.08, 23.97, 22.95, 21.14, 18.05, 14.78. ESI-MS m/z: 483.10 (M+H+).
(2-(3-Chloro-4-fluorobenzyl)-7-(ethylthio)-5-isopropyl-1,3,6-trioxooctahydro-1H-4,7-epoxypyrrolo[3,4-c]pyridin-4-yl)methyl acetate (10d):
Reaction of 8b and N-(3-chloro-4-flourobenzyl)maleimide (9) (17) according to General Procedure C, provided 10d in 64% yield. 1H NMR (400 MHz, CDCl3) δ 7.33 (dd, J = 6.9, 2.2 Hz, 1H), 7.17–7.14 (m, 1H), 7.02 (t, J = 8.7 Hz, 1H), 4.67 (d, J = 13.8 Hz, 1H), 4.59 – 4.50 (m, 3H), 3.73–3.62 (m, 1H), 3.45 (d, J = 6.7 Hz, 1H), 3.22 (d, J = 6.7 Hz, 1H), 2.82 – 2.69 (m, 2H), 2.08 (s, 3H), 1.40 (d, J = 6.9 Hz, 3H), 1.29 (d, J = 6.7 Hz, 3H), 1.22 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.49, 170.82, 169.41, 168.15, 157.80 (d, J = 248.7 Hz), 131.78 (d, J = 3.9 Hz), 130.72, 128.39 (d, J = 7.6 Hz), 121.18 (d, J = 17.6 Hz), 116.78 (d, J = 21.4 Hz), 95.16, 94.96, 59.39, 54.75, 48.98, 46.51, 41.6, 24.0, 20.92, 20.49, 18.77, 14.63. ESI-MS m/z: 499.20 (MH+), 521.20 (M+Na+).
Methyl 3-(2-(3-chloro-4-fluorobenzyl)-7-(ethylthio)-5-isopropyl-1,3,6-trioxooctahydro-1H-4,7-epoxypyrrolo[3,4-c]pyridin-4-yl)propanoate (10e):
Reaction of 8c and N-(3-chloro-4-flourobenzyl)maleimide (9) (17) according to General Procedure C, provided 10e in 69% yield. 1H NMR (400 MHz, CDCl3) δ 7.28 (dd, J = 6.9, 2.2 Hz, 1H), 7.14–7.10 (m, 1H), 6.98 (t, J = 8.7 Hz, 1H), 4.51 (q, J = 14.5 Hz, 2H), 3.59 (s, 3H), 3.55 – 3.48 (m, 1H), 3.32 (d, J = 6.7 Hz, 1H), 3.16 (d, J = 6.7 Hz, 1H), 2.74 – 2.60 (m, 2H), 2.56 – 2.37 (m, 3H), 2.33–2.26 (m, 1H), 1.40 (d, J = 6.9 Hz, 3H), 1.22 (d, J = 6.6 Hz, 3H), 1.16 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.13 (2C), 171.03, 169.04, 157.67 (d, J = 248.7 Hz), 131.93 (d, J = 3.9 Hz), 130.52, 128.30 (d, J = 7.6 Hz), 121.02 (d, J = 18.3 Hz), 116.75 (d, J = 21.4 Hz), 97.21, 94.20, 55.37, 51.92, 49.66, 46.00, 41.60, 27.91, 24.00, 23.38, 21.30, 18.29, 14.84. ESI-MS m/z: 513.10 (M+H+), 535.10 (M+Na+).
(2-(3-Chloro-4-fluorobenzyl)-7-(ethylthio)-5-(4-methoxybenzyl)-1,3,6-trioxooctahydro-1H-4,7-epoxypyrrolo[3,4-c]pyridin-4-yl)methyl acetate (10f):
Reaction of 8d and N-(3-chloro-4-flourobenzyl)maleimide (9) (17) according to General Procedure C, provided 10f in 39% yield. 1H NMR (400 MHz, CDCl3) δ 7.31 (dd, J = 6.9, 2.2 Hz, 1H), 7.18 (d, J = 8.6 Hz, 2H), 7.14–7.11 (m, 1H), 7.00 (t, J = 8.7 Hz, 1H), 6.81 (d, J = 8.7 Hz, 2H), 4.80(d, J = 15.1Hz, 1H), 4.67 (d, J = 13.7 Hz, 1H), 4.52 (d, J = 13.7 Hz, 1H), 4.49 (s, 2H), 4.15 (d, J = 15.1 Hz, 2H), 3.77–3.71 (m, 4H), 3.06 (dd, J = 6.7, 0.7Hz, 1H), 2.81 (q, J = 7.5 Hz, 2H), 2.76 (d, J = 6.7 Hz, 1H), 2.05 (s, 3H), 1.25 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, cdcl3) δ 171.40, 170.60, 169.48, 168.91, 159.66, 157.79 (d, J = 249.8 Hz), 131.67 (d, J = 4.1 Hz), 130.79, 129.45 (2C), 128.43 (d, J = 7.4 Hz), 126.95, 121.16 (d, J = 17.8 Hz), 116.74 (d, J = 21.4 Hz), 114.57, 114.11, 95.31, 94.54, 59.00, 55.23, 53.98, 49.07, 44.03, 41.74, 24.29, 20.51, 14.70. ESI-MS m/z: 577.1 (M+H+), 599.1 (M+Na+).
General Procedure D for Oxide Bridge Cleavage to Form Products (5):
To a solution of the cycloadducts 10 (0.1 mmol) in anhydrous dichloromethane (4 mL) was added borontrifluoride etherate (50 μL, 0.4 mmol) at room temperature (19). The mixture was heated at reflux until the disappearance of starting material as indicated by TLC. The reaction was then quenched by the addition of brine (15 mL), extracted with CHCl3 (3 × 40 mL) and the combined organic phase was dried (Na2SO4), concentrated and purified by preparative HPLC.
2-(3-Chloro-4-fluorobenzyl)-7-hydroxy-4,5-dimethyl-1H-pyrrolo[3,4-c]pyridine-1,3,6(2H,5H)-trione (5a):
Treatment of cycloadduct 10a according to General Procedure D and purification by preparative HPLC (as indicated in the General Synthetic Procedures using a 00G-4435-E0 column with a linear gradient of 60% B to 90% B over 30 minutes; retention time = 18.2 minutes) afforded product 5a as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.06 (s, 1H), 7.49 (dd, J = 7.2, 2.1 Hz, 1H), 7.37 (t, J = 9.0 Hz, 1H), 7.30 – 7.26 (m, 1H), 4.67 (s, 2H), 3.51 (s, 3H), 2.68 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.01, 164.19, 160.25, 156.38 (d, J = 246.2 Hz), 141.91, 140.16, 134.54 (d, J = 3.8 Hz), 129.51, 128.12 (d, J = 7.6 Hz), 119.27 (d, J = 17.7 Hz), 116.90 (d, J = 21.0 Hz), 111.54, 104.20, 39.21, 31.21, 14.40. MALDI-MS m/z: 349.64 (M-H). HRMS calcd for C16H13N2O4FCl [M+H+]: 351.0542. Found: 351.0542.
2-(3-Chloro-4-fluorobenzyl)-7-hydroxy-4-methyl-5-phenyl-1H-pyrrolo[3,4-c]pyridine-1,3,6(2H,5H)-trione (5b):
Treatment of cycloadduct 10b according to General Procedure D and purification by preparative HPLC (as indicated in the General Synthetic Procedures using a 00G-4435-E0 column with a linear gradient of 50% B to 60% B over 30 minutes; retention time = 23.6 minutes) afforded product 5b as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.28 (bs, 1H), 7.61–7.55 (m, 3H), 7.53 – 7.50 (m, 1H), 7.42 – 7.35 (m, 3H), 7.32 – 7.29 (m, 1H), 4.71 (s, 2H), 2.20 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.42, 164.67, 161.07, 156.95 (d, J = 244.9 Hz), 143.65, 139.98, 137.57, 135.07 (d, J = 3.8 Hz), 130.15, 130.12 (d, J = 5.3 Hz), 130.09, 129.64, 128.72 (d, J = 7.5 Hz), 128.51 (2C), 119.83 (d, J = 18.3 Hz), 117.48 (d, J = 21.4 Hz), 112.37, 105.13, 40.06, 16.12. MALDI-MS m/z: 411.39 (M-H); HRMS calcd for C21H15N2O4FCl [MH+]: 413.0699. Found: 413.0705.
2-(3-Chloro-4-fluorobenzyl)-7-hydroxy-4-isobutyl-5-isopropyl-1H-pyrrolo[3,4-c]pyridine-1,3,6(2H,5H)-trione (5c):
Treatment of cycloadduct 10c according to General Procedure D and purification by preparative HPLC (as indicated in the General Synthetic Procedures using a 00G-4435-E0 column with a linear gradient of 50% B to 60% B over 30 minutes; retention time = 29.0 minutes) afforded product 5c as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.91 (bs, 1H), 7.43 (dd, J = 7.1, 2.2 Hz, 1H), 7.32 (t, J = 8.6 Hz, 1H), 7.24 – 7.20 (m, 1H), 4.62 (s, 2H), 4.56–4.50 (m, 1H), 3.12 (bs, 2H), 1.85–1.78 (m, 1H), 1.48 (d, J = 6.6 Hz, 6H), 0.88 (d, J = 5.7 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 166.69, 164.49, 161.12, 156.88 (d, J = 246.2 Hz), 143.50, 142.57, 135.08 (d, J = 3.8 Hz), 129.97, 128.56 (d, J = 7.5 Hz), 119.79 (d, J = 17.9 Hz), 117.41 (d, J = 21.1 Hz), 111.31, 106.29, 51.35, 40.03, 35.04, 28.46(2C), 19.40(3C). MALDI-MS m/z: 419.50 (M-H). HRMS calcd for C21H23N2O4FCl [M+H+]: 421.1325. Found: 421.1335.
(2-(3-Chloro-4-fluorobenzyl)-7-hydroxy-5-isopropyl-1,3,6-trioxo-2,3,5,6-tetrahydro-1H-pyrrolo[3,4-c]pyridin-4-yl)methyl acetate (5d):
Treatment of cycloadduct 10d according to General Procedure D and purification by preparative HPLC (as indicated in the General Synthetic Procedures using a 00G-4435-E0 column with a linear gradient of 30% B to 80% B over 30 minutes; retention time = 19.5 minutes) afforded product 5d as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.44 (dd, J = 2.2, 7.0 Hz, 1H), 7.30 – 7.26 (m, 1H), 7.03 (t, J = 8.7 Hz, 1H), 5.64 (s, 2H), 4.69 (s, 2H), 4.39–4.36 (m, 1H), 2.09 (s, 3H), 1.58 (d, J = 6.8 Hz, 6H). ESI-MS m/z: 437.1 (MH+). HRMS calcd for C20H19N2O6FCl [M+H+]: 437.0910. Found: 437.0898.
Methyl 3-(2-(3-chloro-4-fluorobenzyl)-7-hydroxy-5-isopropyl-1,3,6-trioxo-2,3,5,6-tetrahydro-1H-pyrrolo[3,4-c]pyridin-4-yl)propanoate (5e):
Treatment of cycloadduct 10e according to General Procedure D and purification by preparative HPLC (as indicated in the General Synthetic Procedures using a 00G-4435-E0 column with a linear gradient of 30% B to 70% B over 30 minutes; retention time = 28.3 minutes) afforded product 5e as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.43 (dd, J = 7.0, 2.1 Hz, 1H), 7.29 – 7.25 (m, 1H), 7.03 (t, J = 8.7 Hz, 1H), 4.68 (s, 2H), 4.50 (bs, 1H), 3.70 (s, 3H), 3.53 (t, J = 8.0 Hz, 2H), 2.57 (t, J = 8.0 Hz, 2H),, 1.60 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 171.40, 165.70, 164.08, 161.00, 157.73 (d, J = 249.4 Hz), 141.80, 140.92, 133.01 (d, J = 4.0 Hz), 131.09, 128.82 (d, J = 7.4 Hz), 121.09 (d, J = 17.9 Hz), 116.65 (d, J = 21.2 Hz), 110.84, 106.50, 77.16, 52.20, 51.98, 40.62, 32.76, 23.04, 19.12. MALDI-MS m/z: 449.35 (M-H). HRMS calcd for C21H21N2O6FCl [M+H+]: 451.1067. Found: 451.1071.
(2-(3-Chloro-4-fluorobenzyl)-7-hydroxy-1,3,6-trioxo-2,3,5,6-tetrahydro-1H-pyrrolo[3,4-c]pyridin-4-yl)methyl acetate (5f) and 2-(3-Chloro-4-fluorobenzyl)-7-hydroxy-4-(hydroxymethyl)-1H-pyrrolo[3,4-c]pyridine-1,3,6(2H,5H)-trione (5g):
Treatment of cycloadduct 10f according to General Procedure D and purification by preparative HPLC (as indicated in the General Synthetic Procedures using a 00G-4436-P0-AX column with a linear gradient of 40% B to 60% B over 30 minutes; retention time = 19.3 minutes for 5f and 13.8 minutes for 5g) afforded products 5f and 5g as a white solids: (5f): 1H NMR (400 MHz, DMSO-d6) δ 12.55 (s, 1H), 11.54 (s, 1H), 7.46 (dd, J = 7.1, 2.1 Hz, 1H), 7.36 – 7.29 (m, 1H), 7.26–7.22 (m, 1H), 5.14 (s, 5H), 4.61 (s, 5H), 2.02 (s, 7H). 13C NMR (100 MHz, DMSO-d6) δ 170.28, 165.38, 164.75, 160.83, 156.93 (d, J = 246.2 Hz), 145.79, 134.87 (d, J = 3.9 Hz), 133.30, 130.13, 128.76 (d, J = 7.6 Hz), 119.80 (d, J = 17.8 Hz), 117.41 (d, J = 21.0 Hz), 112.62, 106.97, 57.79, 39.94, 20.90. ESI-MS (+Ve) m/z: 395.0 (M+H+); ESI-MS (-Ve) m/z: 393.0 (M-H). (5g): 1H NMR (400 MHz, DMSO-d6) δ 12.10 (s, 1H), 11.26 (bs, 1H), 7.45 (dd, J = 7.1, 2.2 Hz, 1H), 7.33 (t, J = 8.6 Hz, 1H), 7.25 – 7.21 (m, 1H), 5.38 (t, J = 5.8 Hz, 1H), 4.62 (d, J = 5.6 Hz, 2H), 4.61 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.62, 164.94, 160.88, 156.91 (d, J = 246.3 Hz), 135.02 (d, J = 3.8 Hz), 130.10, 128.71 (d, J = 7.5 Hz), 119.79 (d, J = 17.8 Hz), 117.41 (d, J = 21.0 Hz), 55.62, 39.94. ESI-MS (+Ve) m/z: 353.0 (M+H+); ESI-MS (-Ve) m/z: 351.0 (M-H).
In Vitro Integrase Catalytic Assays
Expression and purification of the recombinant IN in Escherichia coli were performed as previously reported (10, 13, 20). Preparation of oligonucleotide substrates has been described (10). Integrase reactions were performed in 10 μL with 400 nM of recombinant IN, 20 nM of 5’-end [32P]-labeled oligonucleotide substrate and inhibitors at various concentrations. Reactions containing 10% DMSO were used as controls. Reactions were incubated at 37 °C (60 minutes) in buffer containing 50 mM MOPS, pH 7.2, 7.5 mM MgCl2, and 14.3 mM 2-mercaptoethanol. Reactions were terminated by addition of 10 μL of loading dye (10 mM EDTA, 98% deionized formamide, 0.025% xylene cyanol and 0.025% bromophenol blue). Reactions were then subjected to electrophoresis in 20% polyacrylamide–7 M urea gels. Gels were dried and reaction products were visualized and quantified with a Typhoon 8600 (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Densitometric analyses were performed using ImageQuant from Molecular Dynamics Inc. The concentrations at which enzyme activity was reduced by 50% (IC50) were determined using “Prism” software (GraphPad Software, San Diego, CA) for nonlinear regression to fit dose-response data to logistic curve models.
Cellular Cytotoxicity
The human osteosarcoma cell line, HOS, was obtained from Dr. Richard Schwartz (Michigan State University, East Lansing, MI) and grown in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 5% (v/v) fetal bovine serum, 5% newborn calf serum, and penicillin (50 units/mL) plus streptomycin (50 μg/mL; Quality Biological, Gaithersburg, MD). On the day prior to the screen, HOS cells were seeded in a 96-well luminescence cell culture plate at a density of 4000 cells in 100 μL per well. On the day of the screen, cells were treated with compounds at the appropriate concentration range chosen and incubated at 37 °C for 48 hrs. Cytotoxicity was measured by monitoring ATP levels using a luciferase reporter assay. Cells were lysed in 50 μL cell lysis buffer (PerkinElmer, Waltham, MA) and shaken at 700 rpm at room temperature for 5 minutes. After the addition of 50 μL of ATPlite buffer (PerkinElmer) directly onto the lysed cells and shaking at 700 rpm at room temperature for 5 minutes, ATP levels were monitored by measuring luciferase activity using a microplate reader. Activity was normalized to cytotoxicity in the absence of target compounds. KaleidaGraph (Synergy Software, Reading, PA) was used to perform regression analysis on the data. CC50 values were determined from the fit model.
Vector Constructs
pNLNgoMIVR−ΔEnv.LUC has been described previously (15). The integrase codon reading frame was removed from pNLNgoMIVR−ΔEnv.LUC (between KpnI and SalI sites) and placed between the KpnI and SalI sites of pBluescript II KS+. Using this construct as the wild-type template, the following HIV-1 integrase-resistant mutants were prepared via the QuikChange II XL (Stratagene, La Jolla, CA) site-directed mutagenesis protocol: Y143R, N155H, and G140S/Q148H. The following sense with cognate antisense (not shown) oligonucleotides (Integrated DNA Technologies, Coralville, IA) were used in the mutagenesis: Y143R, 5’-GCAGGAATTTGGCATTCCCCGCAATCCCCAAAGTCAAGGA-3’; N155H, 5’-CCAAAGTCAAGGAGTAATAGAATCTATGCATAAAGAATTAAAGAAAATTATAGGACA-3’; G140S, 5’-GGGGATCAAGCAGGAATTTAGCATTCCCTACAATC-3’; Q148H, 5’-CATTCCCTACAATCCCCAAAGTCATGGAGTAATAGAATCTA-3’. The double mutation G140S/Q148H was constructed by using the previously generated Q148H mutant and the appropriate oligonucleotides for the second mutation, G140S. The DNA sequence of each construct was verified independently by DNA sequence determination. The mutant integrase coding sequences from pBluescript II KS+ were then subcloned into pNLNgoMIVR−ΔEnv.LUC (between the KpnI and SalI sites) to produce the full-length mutant HIV-1 integrase constructs. These DNA sequences were additionally checked independently by DNA sequence determination.
Results and Discussion
Chemistry
Synthesis of the target analogues 5 (Scheme 1) relied on the “Pummerer cyclization deprotonation cycloaddition” cascade of imidosulfoxides (18, 21), similar to what we had previously employed for the preparation of tricyclic hydroxy-1H-pyrrolopyridine-triones (4, Figure 1) (17). A distinguishing feature in the current approach was the use of open-chain amide starting materials (6) rather than cyclic lactams. This allowed the synthesis of bicyclic rather than tricyclic products. Acylation of the open-chain amides gave the ethylthio-containing imides (7), which were oxidized to provide the corresponding imidosulfoxides (8, Scheme 1). Treatment of the latter compounds with acetic anhydride in the presence of N-(3-chloro-4-fluorobenzyl)maleimide (9) and a catalytic quantity of p-toluenesulfonic acid, gave the intermediate oxide-bridged cycloadducts (10). Refluxing 10 with boron trifluoride diethyl etherate in dichloromethane, yielded the corresponding bicyclic products, thereby completing a facile four-step synthesis of 5 (Scheme 1).
Scheme 1.
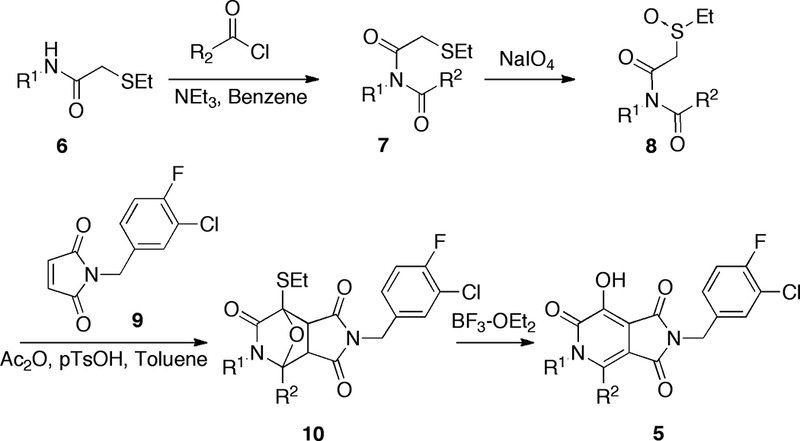
Synthesis of target bicycles 5.
Evaluation of Integrase Inhibitory Potencies
Analogues 5 were evaluated in biochemical IN assays that measure inhibitory potencies (IC50 values) against both the 3’-P and ST reactions (Table 1) (15). The potencies of the new compounds were similar to what we found for the original tricyclic series 4 (for example, 4a IC50 = 9 μM, Table 1) (15): All new compounds had IC50 values for the ST reactions in the low micromolar range (6 μM – 22 μM) with good selectively for ST reactions relative to the 3’-P reactions (IC50 > 111 μM). In general, we observed few differences in ST inhibitory potencies, in spite of the fact that the R1 and R2 groups varied considerably in size and composition.
Table 1.
Integrase inhibitory potencies of synthetic analogues.
 |
Evaluation against Raltegravir Resistant IN Mutants in in vitro Assays
Mutant forms of IN that show reduced sensitivity to raltegravir in in vitro assays have been identified in patient viruses that have develop resistance to raltegravir (22). In order to test the susceptibility of the bicyclic hydroxy-1H-pyrrolopyridine-triones to some of the known resistance mutants, in vitro assays were performed against a panel of enzymes that included the wild-type (WT) IN and the three key mutant forms G140S/Q148H, Y143R and N155H (Table 2) (13, 22). Assays were run using pre-cleaved (3’-processed) DNA with Mg2+ as the metal cofactor. Although none of the new compounds was as potent against WT IN as raltegravir, the inhibitory potency of 5e was less susceptible than raltegravir to the effects of these mutations. For example, while raltegravir was approximately 26-fold less potent against the Y143R mutant than WT, inhibitor 5e showed a smaller loss of potency (2-fold) (Table 2). Although interpreting this data should be done based on the understanding that the binding of 5e is suboptimal, the reduced sensitivity to the effects of the Y143R mutation are consistent with the X-ray crystal structure of the homologous prototype foamy virus (PFV) integrase in complex with raltegravir (Figure 2A) (8, 23, 24). Most structurally important features of the IN active site are shared by the PFV IN (25). These data show that Y212 (equivalent to Y143 in HIV-1 IN) interacts with an oxadiazole ring in raltegravir, which is in a position equivalent to the R2-group in 5e. In contrast to the oxadiazole ring in raltegravir, the 3-(methyl propanoate) R2-group in 5e with the Y143 residue in a way that contributes significantly to overall binding affinity of 5e, and for this reason, mutations at Y143 have a minimal impact on the potency of this compound.
Table 2.
Inhibitory potencies in vitro using wild-type (WT) and mutant integrase enzymes.a
| No. | ST IC50 (μM, WT) | Mutantsb | ||
|---|---|---|---|---|
| G140S/Q148H | Y143R | N155H | ||
| 1 | 0.067 | 105x | 26x | 7x |
| 5e | 7.1 ± 0.6 | 12x | 2x | 4x |
Data was obtained as described in Experimental Section.
Fold-loss of potency relative to virus containing WT enzyme.
Figure 2.
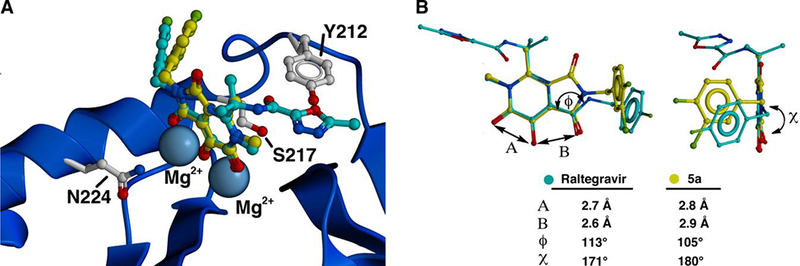
Structural comparison of raltegravir with inhibitor 5a. (A) Crystal structure of raltegravir (carbons in cyan) bound to PFV integrase showing the catalytic Mg2+ ions and residues S217, Y212 and N224 corresponding to HIV-1 IN residues Q148, Y143 and N155 [taken from PDB 3OYA (23)]. Inhibitor 5a (carbons in yellow) is overlaid onto raltegravir. (B) Superposition of raltegravir from Panel A onto inhibitor 5a comparing key angles and distances. Graphics were generated using ICM Chemist Pro software (24).
The G140S/Q148H double mutant caused approximately a 100–fold reduction in the potency of raltegravir. In contrast 5e was only 12–fold less potent (Table 3). The Q148 residue in HIV-1 IN corresponds to S217 in PFV IN, where it is situated on a 310 helix with its side chain directed between the catalytically essential residues D185 and E221 (Figure 2A) (23, 25). Binding of inhibitors to PFV IN bearing the S217H mutation, requires energetically unfavorable movement of the protein backbone to accommodate the His side chain. Similar steric considerations may contribute to a loss of potency by ST inhibitors against the Q148H mutation in HIV-1 IN. Overlaying 5a, a bicyclic hydroxy-1H-pyrrolopyridine-trione inhibitor, onto PFV-bound raltegravir shows that there would be differences in the interactions of the critical halobenzyl rings with the region proximal to S217 of PFV IN and Q148 of HIV IN. These differences in the interactions of the two compounds arise from variations in torsion angles associated with the respective benzylamide carbonyl groups, and which can be attributed to conformational rigidy in the bicyclic hydroxy-1H-pyrrolopyridine-trione nucleus not found in raltegravir (Figure 2B). In addition, the metal-dependency data (Table 2) suggets that there are subtle differences in the way raltegravir and 5e interact with the catalytic divalent ions. It is possible that the G140S/Q148H mutant alters inhibitor – metal binding, and that such alteration is less deleterious for 5e.
Table 3.
Antiviral potencies in cells infected with HIV-1 containing wild-type (WT) or mutant integrase enzymes.a
| No. | CC50 (μM) | EC50 (μM, WT) | Mutantsb | ||
|---|---|---|---|---|---|
| G140S/Q148H | Y143R | N155H | |||
| 1 | >100 | 0.004 ± 0.002 | 475x | 41x | 38x |
| 2 | >100 | 0.017 ± 0.004 | 5x | 0.4x | 1x |
| 3 | 5.4 ± 1.3 | 1.7 ± 0.5 | 5x | 1x | 3x |
| 5e | 392 ± 66 | 9.2 ± 1.9 | 2x | 2x | 3x |
Data was obtained as described in the Experimental Section.
Fold-loss of potency relative to virus containing WT enzyme.
Modeling studies on IN have also suggested that introduction of conformational rigidity into an inhibitor may lessen deleterious effects of the G140S/Q148H mutations (26). As indicated above, this is supported by our current work, where conformational restriction was a key design feature in our original 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones (3) (15, 16) that was carried over into the hydroxy-1H-pyrrolopyridine-triones (4 and 5) (17). Inhibitor 5e was also approximately 2–fold less sensitive to the effects of the N155H mutation than raltegravir (7–fold loss versus 4–fold loss, respectively) (Table 3). As with the G140S/Q148H mutation, this difference in sensitivity may be related to interactions of raltegravir and 5e with the metal ions at the active site. This idea is supported by observations that the resistance to raltegravir imparted by the N155H mutation arises from perturbations related to the divalent metal ions and to direct raltegravir – enzyme contacts (27). N224 in PFV integrase (which corresponds to N155 in HIV-1 IN) lies in an α-helix proximal to the divalent metal ions (Figure 2A). The histidine imidazole ring of the N224H mutant shifts the catalytic D128 and E221 carboxylates. It also causes movement of the DNA backbone by interacting with the phosphate group of the 3’-adenosine. Disrupting of these His-phosphate interactions incurred by inhibitor binding may be energetically unfavorable (23).
Evaluation against HIV-1 Vectors for Raltegravir Resistant IN Mutants in Cellular Assays
To complement the biochemical data, HIV-1 vectors replicating the resistant IN mutants G140S/Q148H, Y143R and N155H were expressed in cultured cells and the cytoprotecive effects of raltegravir (1), MK-0536 (2) and 5e were evaluated (Table 4). A key rationale in the design of both 4 and 5 was to reduce cytotoxicity by removing the catechol functionality. This was accomplished by introducing a nitrogen into the parent 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones (3). As shown in Table 4, the cytotoxicity of 5e (CC50 = 392 μM) is approximately 70–fold lower than the related catechol-containing analogue 3 (CC50 = 5.4 μM). Consistent with the in vitro data for the raltegravir-resistant IN mutants (Table 2), Raltegravir showed a greater than 400 – fold loss of antiviral efficacy against the vector carrying the G140S/Q148H double mutant as compared to the WT (Table 3). The second-generation IN inhibitor MK-0536 (2) showed a 5–fold loss of antiviral efficacy for this double mutant. In contrast, inhibitor 5e displayed only a 2–fold loss of potency against the G140S/Q148H mutant vector, thereby making it approximately 200–fold and 3–fold less susceptible, respectively, to the effects of this double mutant. Similarly, consistent with the in vitro data shown in Table 3, analogue 5e had loss of potency approximately 20–fold lower than raltegravir against vectors that individually carry the Y143R (approximately 40–fold loss for raltegravir and 2–fold loss for 5e) and 13–fold lower for N155H mutations (38–fold loss for Raltegravir and 3–fold loss for 5e).
Conclusions
We previously incorporated a 2(1H)-pyridone functionality into the catechol-based 1H-isoindole-1,3(2H)-dione-based analogues (3), which resulted in tricyclic hydroxy-pyrrolopyridine trione inhibitors (4) that exhibited significantly lower cytotoxicity than the parent compounds. In the current work we have applied “Pummerer cyclization deprotonation cycloaddition” cascades of imidosulfoxides to prepare bicyclic variants (5). These newly developed analogues maintain good selectivity for ST reactions as compared to the 3’-P reactions and exhibit reduced cytotoxicities relative to the parent catechol-containing compounds (3). In in vivo antiviral assays a representative member of the new series had a much smaller reduction in potency than raltegravir when challenged with vectors carrying the G140S/Q148H, Y143R and N155H mutations. Although the second-generation inhibitor, MK-0536 is more effective against these mutations, the new compound showed a smaller loss of potency when challenged with vectors carrying the G140S/Q148H mutations. Although the potency of the new compounds needs to be improved, we have made progress on the problem of toxicity and the fact that the hydroxy-pyrrolopyridine-trione nucleus can be used to create compounds that are relatively insensitive to important IN mutations may facilitate the development of second-generation inhibitors.
Acknowledgements
This work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick and the National Cancer Institute, National Institutes of Health and the Joint Science and Technology Office of the Department of Defense. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- 1.Pommier Y, Johnson AA, Marchand C (2005) Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discovery; 4: 236–248. [DOI] [PubMed] [Google Scholar]
- 2.Pommier Y, Cherfils J (2005) Interfacial inhibition of macromolecular interactions: Nature’s paradigm for drug discovery. Trends in Pharmacological Sciences; 26: 138–145. [DOI] [PubMed] [Google Scholar]
- 3.Anker M, Corales RB (2008) Raltegravir (mk-0518): A novel integrase inhibitor for the treatment of HIV infection. Expert Opin Invest Drugs; 17: 97–103. [DOI] [PubMed] [Google Scholar]
- 4.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M (2008) Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem; 51: 5843–5855. [DOI] [PubMed] [Google Scholar]
- 5.De Clercq E (2009) Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents; 33: 307–320. [DOI] [PubMed] [Google Scholar]
- 6.Marchand C, Maddali K, Metifiot M, Pommier Y (2009) HIV-1 in inhibitors: 2010 update and perspectives. Curr Top Med Chem; 9: 1016–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johns BA, Svolto AC (2008) Advances in two-metal chelation inhibitors of HIV integrase. Expert Opin Ther Pat; 18: 1225–1237. [Google Scholar]
- 8.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P (2010) Retroviral intasome assembly and inhibition of DNA strand transfer. Nature; 464: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cherepanov P, Maertens GN, Hare S (2011) Structural insights into the retroviral DNA integration apparatus. Curr Opin Struct Biol; 21: 249–256. [DOI] [PubMed] [Google Scholar]
- 10.Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y (2008) Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry; 47: 9345–9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fransen S, Gupta S, Danovich R, Hazuda D, Miller M, Witmer M, Petropoulos CJ, Huang W (2009) Loss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple nonoverlapping genetic pathways. J Virol; 83: 11440–11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muraglia E, Kinzel O, Gardelli C, Crescenzi B, Donghi M, Ferrara M, Nizi E, Orvieto F, Pescatore G, Laufer R, Gonzalez-Paz O, Di Marco A, Fiore F, Monteagudo E, Fonsi M, Felock PJ, Rowley M, Summa V (2008) Design and synthesis of bicyclic pyrimidinones as potent and orally bioavailable HIV-1 integrase inhibitors. J Med Chem; 51: 861–874. [DOI] [PubMed] [Google Scholar]
- 13.Metifiot M, Marchand C, Maddali K, Pommier Y (2010) Resistance to integrase inhibitors. Viruses; 2: 1347–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metifiot M, Vandegraaff N, Maddali K, Naumova A, Zhang X, Rhodes D, Marchand C, Pommier Y (2011) Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. AIDS; 25: 1175–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao XZ, Semenova EA, Vu BC, Maddali K, Marchand C, Hughes SH, Pommier Y, Burke TR Jr. (2008) 2,3-dihydro-6,7-dihydroxy-1I-isoindol-1-one-based HIV-1 integrase inhibitors. J Med Chem; 51: 251–259. [DOI] [PubMed] [Google Scholar]
- 16.Zhao XZ, Maddali K, Vu BC, Marchand C, Hughes Stephen H, Pommier Y, Burke Terrence R Jr. (2009) Examination of halogen substituent effects on HIV-1 integrase inhibitors derived from 2,3-dihydro-6,7-dihydroxy-1H-isoindol-1-ones and 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones. Bioorg Med Chem Lett; 19: 2714–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao XZ, Maddali K, Metifiot M, Smith SJ, Christie Vu B, Marchand C, Hughes SH, Pommier Y, Burke TR (2011) Development of tricyclic hydroxy-1H-pyrrolopyridine-trione containing HIV-1 integrase inhibitors. Bioorg Med Chem Lett; 21: 2986–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Padwa A, Heidelbaugh TM, Kuethe JT (1999) Synthesis of substituted 2-pyridones via the Pummerer cyclization‚ deprotonation‚ cycloaddition cascade of imidosulfoxides. J Org Chem; 64: 2038–2049. [DOI] [PubMed] [Google Scholar]
- 19.Miah S, Slawin AMZ, Moody CJ, Sheehan SM, Marino JP Jr., Semones MA, Padwa A, Richards IC (1996) Ligand effects in the rhodium(II) catalyzed reactions of diazoamides and diazoimides. Tetrahedron; 52: 2489–2514. [Google Scholar]
- 20.Zouhiri F, Mouscadet J-F, Mekouar K, Desmaele D, Savourr D, Leh H, Subra F, Le Bret M, Auclair C, d’Angelo J (2000) Structure-activity relationships and binding mode of styrylquinolines as potent inhibitors of HIV-1 integrase and replication of HIV-1 in cell culture. J Med Chem; 43: 1533–1540. [DOI] [PubMed] [Google Scholar]
- 21.Padwa A, Heidelbaugh TM, Kuethe JT (2000) Using the Pummerer cyclization‚ deprotonation‚ cycloaddition cascade of imidosulfoxides for alkaloid synthesis. J Org Chem; 65: 2368–2378. [DOI] [PubMed] [Google Scholar]
- 22.Croxtall JD, Scott LJ (2010) Raltegravir: In treatment-naive patients with HIV-1 infection. Drugs; 70: 631–642. [DOI] [PubMed] [Google Scholar]
- 23.Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P (2010) Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc Nat Acad Sci USA; 107: 20057–20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.ICM Chemist Pro Software v37–1f/MacOSX [https://molsoftcom/icm-chemistr-pro-html] MolSoft LLC, La Jolla, CA; [Google Scholar]
- 25.Cherepanov P (2010) Integrase illuminated. EMBO reports; 11: 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perryman AL, Forli S, Morris GM, Burt C, Cheng Y, Palmer MJ, Whitby K, McCammon JA, Phillips C, Olson AJ (2010) A dynamic model of HIV integrase inhibition and drug resistance. J Mol Biol; 397: 600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grobler JA, Stillmock KA, Miller MD, Hazuda DJ (2008) Mechanism by which the HIV integrase active-site mutation N155H confers resistance to raltegravir. Antiviral Ther; 13: A41–A41. [Google Scholar]