

Abstract
Benzynes formed by heating a suitable triyne (or tetrayne) substrate are shown to react with in situ-generated alkynyl copper species. The latter are compatible with the polyyne substrates and two types of chemistries have been achieved: (i) 1-bromo-1-alkynes efficiently undergo net bromoalkynylation of the (unsymmetrical) benzynes and (ii) in situ-generated alkynylcopper species give rise to hydroalkynylation products. The regiochemical preferences of these two modes of reaction are complementary to one another with respect to the position of alkynyl substituent in the final products.
Keywords: metal-catalyzed aryne trapping, copper catalysis, alkynylations
Graphical Abstract

Cu-catalyzed bromo- and hydro-alkynylations of unsymmetrical arynes was described. These two mechanistically distinct modes of reaction are efficient, broad in scope, and giving complementary alkynylation products. Mechanisms are proposed and potential for synthetic elaboration is demonstrated.
1,2-Haloalkynylation of arynes is a potentially valuable way to install a considerable degree of complexity into the aryne family of reactive intermediates. We are aware of only one report describing such a transformation (Figure 1a). In 2010 Yoshida[1] and coworkers showed that (symmetrical) benzynes generated by the Kobayashi protocol[2] from 2-trimethylsilylaryl triflates (1) could engage bromoalkynes in the presence substoichiometric amounts of CuBr2. The major products 2 arose from the (presumably sequential) introduction of two molecules[3] of benzyne; these were sometimes accompanied by lesser amounts of the 1:1 adducts 3, depending upon reaction conditions. We report here that benzynes 7, generated by the hexadehydro-Diels-Alder (HDDA) cycloisomerization reaction,[4] efficiently undergo bromo- and hydro-alkynylation in the presence of copper (I) catalysts (Figure 1b). A hallmark of the HDDA reaction is that the reactive benzyne intermediates are generated in the absence of any external reagent, rendering a pristine environment for the trapping event. The hydroalkynylation of benzynes has been studied more extensively and, again, the (mostly symmetric) benzynes have been generated by the Kobayashi protocol of fluoride treatment of o-trimethylsilylaryl triflates.[5] Here, we not only show the generality of this reaction under essentially neutral conditions, but also provide mechanistic insights that can be inferred from the products arising from the, necessarily, unsymmetrical HDDA-benzynes. In addition, the regiochemical preferences for the hydroalkynylation and the bromoalkynylation are complementary to one another (cf. 8/8’ vs. 9/9’)—C–C bond formation occurs preferentially at different carbon atoms of the benzynes 7.
Figure 1.
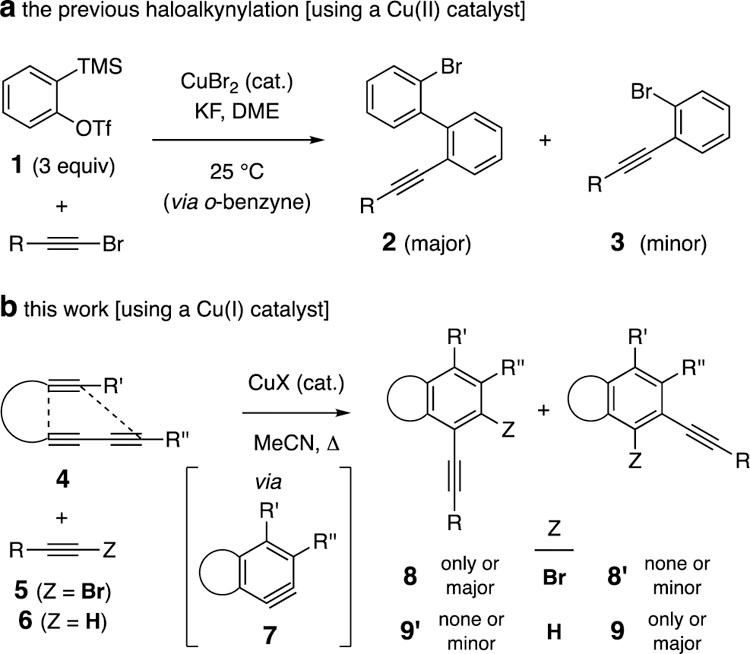
[a] Previous reports by Yoshida et al. [b] This work.
More generally,[6] metal-catalyzed functionalization of an aryne is inherently challenging because the steady state concentration of both the aryne and the active organometallic must be sufficiently high to allow for their mutual capture. We have found that bromoalkynylation occurs smoothly in the presence of CuBr in MeCN solvent. For example (Figure 2a), the tetrayne benzyne precursor 10, is trapped by 1-bromo-2-phenylethyne (5a) in excellent yield when heated (80 °C) in the presence of 10 mol% of CuBr (structural assignment discussed below). Benzyne 11 is known to preferentially undergo nucleophilic attack at C6, which is consistent with both the easier steric access of the nucleophilic atom to C6 vs. C7 as well as with the larger internal bond angle[7] at C6 computed for (the N-methanesulfonyl analog of)[8] the N-toluenesulfonylbenzyne 11.
Figure 2.
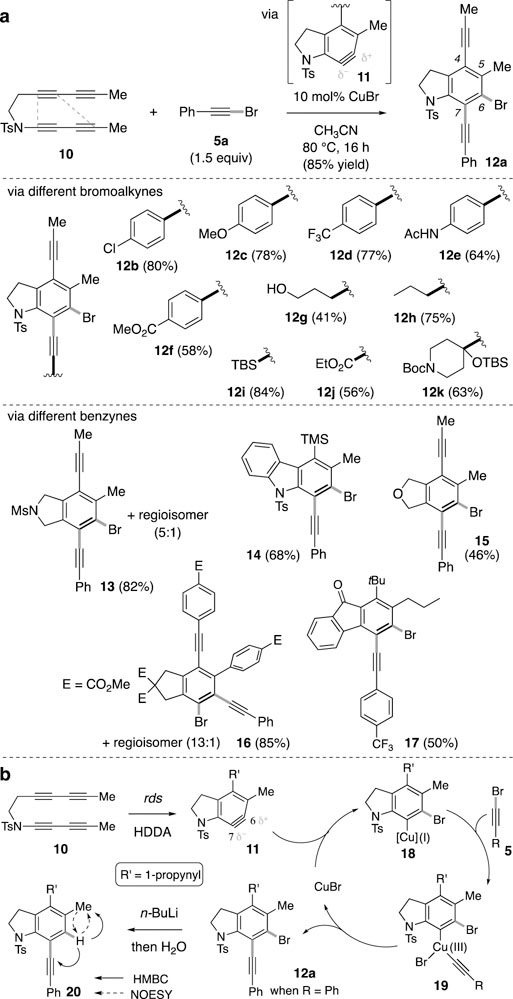
[a] Scope of the bromoalkynylation reaction. [b] Proposed catalytic cycle and basis for assignment of the constitution of 12a (cf. 20).
The scope of the bromoalkynylation reaction is quite broad (Figure 2a). Aliphatic and aromatic bromoalkynes with a variety of functionalities are tolerated (cf. 12a–12k). We have also examined several polyyne substrates that incorporate different tethers; these led to the bromoalkynylation products 13–17. It is noteworthy that we have never observed a product indicating premature reaction of the HDDA polyyne precursor with any of the active organometallic species involved in this chemistry.[9] Moreover, we observed no desired product formation in the absence of the copper catalyst.[10]
We envision the reaction proceeding through a mechanism such as that depicted in Figure 2b. The initial thermal cycloisomerization is the rate-determining step in the overall cycle, because the reaction rate is similar for triyne 10 regardless of the trapping agent used.[11] We propose that benzyne 11 initially undergoes nucleophilic addition with CuBr to produce the aryl copper(I) species 18, which then reacts with a molecule of the bromoalkyne to generate the Cu(III) adduct 19.
This view is consistent with the fact that trace amounts of chloro- or iodo-alkynylation products were observed (GC-MS) when CuCl or CuI was used as the catalyst instead of CuBr. Reductive elimination of the alkyne and arene ligands produces product 12. Two minor byproducts often detected were those corresponding to net hydrobromination and/or dibromination of the benzyne. These can be accounted for by hydrolytic capture of the ArCu species 18 by adventitious water or competitive (and, fortunately, less facile) reductive elimination of bromo and arene ligands from 19. It is noteworthy that when iodoalkynes was used, efficient diiodination rather than iodoalkynylation of the benzyne was observed. Presumably, the reductive elimination of iodo and arene ligands from the ArCuI2 species (analogous to 19) became the dominate pathway (for more details, see Supporting Information, page S19).[12] Purposely adding water into the reaction mixture at the outset led to decreased overall yield of 12 with concomitant formation of the net hydrobromination adduct. The structure of 12 was confirmed by its conversion to the reductively debrominated derivative 20. The site of the aromatic proton was definitively identified based on the indicated nOe and HMBC analyses. This mode of regioselectivity supported the above mechanistic view—namely, that the initial bromination by CuBr had occurred at the more electrophilic site in benzyne 11.
We briefly explored several potential synthetic applications of the bromoalkynylation products (Figure 3). One-pot halogen exchange and global deprotections of, for example, bromocarbazole 14 gave rise to alkynylcarbazole 21, whereas heating compound 14 with sodium sulfide efficiently provided the deprotected thienocarbazole 22.[13] Palladium-catalyzed cross-couplings of the bromobenzene moiety were also tested.[14] For example, Sonogashira and Suzuki reactions of the bromoalkyne 15 efficiently afforded the trialkynylbenzene 23 and the biaryl 24, respectively.
Figure 3.
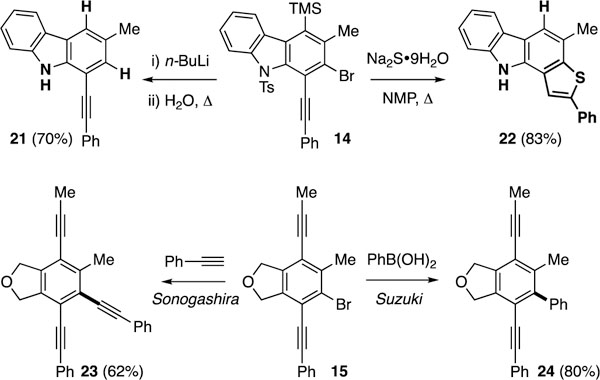
Potential synthetic applications [see Supporting Information (SI) for more experimental details].
Many alkyne cross-coupling reactions rely on the robustness of copper acetylides.[15] We envisioned that when formed in situ, these could serve as nucleophiles to arynes and that a net hydroalkynylation process could ensue. This had the potential to be complementary to the bromoalkynylation reaction with respect to the point of attachment of the alkyne in the product arene. However, an organic amine is often required to promote Cu-acetylide formation, but (even tertiary) amines are excellent nucleophilic trapping agents for benzynes.[16] Cu-acetylides can be formed and trapped in situ by the action of a fluoride base under conditions where arynes are being co-produced.[5] We were pleased to see that in the absence of any added base, copper chloride alone (5 mol%), in acetonitrile solution, induces the addition of terminal alkynes (e.g., 6a) to HDDA benzyne intermediates (e.g., 11 from 10) to give net hydroalkynylation products (e.g., 25a; see SI for the reaction optimization studies leading to the choice of conditions shown in the top of Figure 4a).
Figure 4.

[a] Scope of the hydroalkynylation reaction revealed by outcomes with various alkynes and benzyne precursors. [b] Cu-catalyzed hydroalkynylation occurs faster than other potential aryne trapping reactions; in the absence of catalyst, aryl ether 31 is the major product.
Notably, only a slight excess (1.1 equiv) of alkyne needs to be used because its oxidative dimerization can be mitigated either by careful deoxygenation of the reaction solution or, more conveniently, using sodium ascorbate, even under an ambient atmosphere, to prevent formation of Cu(II) species. Presumably these conditions allow for the Cu-acetylide to be relatively long-lived; the use of just 5 mol% of copper loading gave high enough steady-state concentration to effectively trap the reactive benzyne. The structure of 25a is the complement to that of 20 (Figure 2b) and is consistent with the proposal that Cu-acetylide addition is involved in this transformation. Namely, the distorted nature of the HDDA-benzyne 11 (Figure 2a) is consistent with an initial alkynyl-cupration of the benzyne.
Given the quite good isolated yield, mild reaction conditions, and the easy experimental setup, we were encouraged to explore the generality of this hydroalkynylation reaction. Products of both aromatic (25a–25g) and aliphatic (25h–25k) alkyne addition are readily formed. For aromatic alkynes, the impact of electronic effects from substituents was minimal and ortho-substitution (25g) was tolerated.[17] As with the bromoalkynylation, we also tested the behavior of several HDDA-benzyne precursors containing different tethers in the hydroalkynylation. These gave products 26–30 in high overall yield. The level of regioselectivity was uniformly lower for the hydroalkynylation. The reaction leading to 29 was performed on a relatively large scale and showed no erosion in efficiency (88% yield). The flanking aryl substituent in the intermediate benzyne substantially inhibits approach of the nucleophile to the proximal sp-carbon, leading to a reversal in the location of the alkyne in 29 vs. 30 (cf., also, 16).
Orthogonal chemoselectivity can be achieved simply by performing the reaction in the presence or absence of the Cu catalyst. For example (Figure 4b), the hydroxy group in alkynol 6n traps benzyne 11 (from 10) to give the ether 31 when no CuCl is used.[18] In contrast, under the copper catalysis conditions described here, the complementary alkyne 25l was formed highly selectively, indicating that the copper acetylide trapping event of benzynes is much faster than that by OH. In neither reaction was any of the product of the alternative constitution observed by NMR analysis of the crude product mixture. Additional selectivity was seen when natural product derivatives were used as trapping agents.[19] Namely, the hydroalkynylation products 25m and 25n were efficiently produced in the presence of CuCl when terminal alkynes derived from estradiol and cinchonidine (see SI for details) were used as the trapping agents. This is especially notable because these molecules contain additional potentially competing reactive moieties that include phenol,[8] alcohol,[18] tertiary amine,[19] ether,[20] alkene,[21] cyclic alkane,[22] and quinoline sites[23].[24]
A mechanistic proposal for the hydroalkynylation reaction is shown in Figure 5. At the top are possible ways by which an initial copy of the requisite Cu-acetylide (34) could be formed. Sacrificial reaction of one benzyne with CuCl would give the arylcopper species 32, which could then engage in proton exchange with the terminal alkyne 6.; indeed, we have detected low levels of the benzyne + HCl adduct 33 by GCMS analysis of a crude reaction mixture. Alternatively, acetonitrile could function as a base to deprotonate a CuCl•6 complex, leading to the HCl adduct 35[25] or its iminium hydrochloride salt 35•HCl.[26] Indeed, we have observed that the choice of acetonitrile as solvent is critical for success of the hydroalkynylation reaction. Once produced, 34 could then enter the cycle highlighted by the gray box. Alkynyl-cupration of benzyne 11 would give 36 and proton exchange with 6 would lead to 25 and close the cycle.
Figure 5.

Proposed initiation event(s) and catalytic cycle for the hydroalkynylation reaction.
In conclusion, we have developed an efficient copper-catalyzed protocol for installation of an alkynyl substituent onto a HDDA-generated benzyne. 1-Bromoalkynes lead to products of bromoalkynylation in which the bromide has preferentially engaged the more electrophilic carbon of the (necessarily) unsymmetrical benzyne (Figure 2a). The hydroalkynylation reaction of terminal alkynes occurs in complementary fashion; namely, the alkyne carbon is now attached to the more electrophilic carbon (Figure 4). Catalytic cycles for each of these two reaction processes are proposed (Figures 2b and 5). The potential utility of the ortho-alkynylbromobenzene products as substrates in further transformations of interest is also demonstrated (Figure 3).
Supplementary Material
Acknowledgements
This study was supported by the U.S. Dept. of Health and Human Services [National Institute of General Medical Sciences (R01 GM65597 then R35 GM127097)] and the National Science Foundation (CHE-1665389). A portion of the NMR data were obtained with an instrument funded by the NIH Shared Instrumentation Grant program (S10OD011952).
References
- [1].Morishita T, Yoshida H, Ohshita J, Chem. Commun 2010, 46, 640. [DOI] [PubMed] [Google Scholar]
- [2].Himeshima Y, Sonoda T, Kobayashi H, Chem. Lett 1983, 12, 1211. [Google Scholar]
- [3].Various Cu(I) salts have been reported to oligomerize Kobayashi benzynes: Mizukoshi Y, Mikami K, Uchiyama M, J. Am. Chem. Soc 2015, 137, 74. [DOI] [PubMed] [Google Scholar]
- [4].a)Miyawaki K, Suzuki R, Kawano T, Ueda I, Tetrahedron Lett 1997, 38, 3943; [Google Scholar]; b)Bradley AZ, Johnson RP, J. Am. Chem. Soc 1997, 119, 9917; [Google Scholar]; c)Tsui JA, Sterenberg BT, Organometallics 2009, 28, 4906; [Google Scholar]; d)Hoye TR, Baire B, Niu D, Willoughby PH, Woods BP, Nature 2012, 490, 208; [DOI] [PMC free article] [PubMed] [Google Scholar]; e)Karmakar R, Mamidipalli P, Yun SY, Lee D, Org. Lett 2013, 15, 1938; [DOI] [PubMed] [Google Scholar]; f) Diamond OJ, Marder TB, Org. Chem. Front 2017, 4, 891. [Google Scholar]
- [5].a)Xie CS, Liu LF, Zhang YH, Xu PX, Org. Lett 2008, 10, 2393; [DOI] [PubMed] [Google Scholar]; b)Jeganmohan M, Bhuvaneswari S, Cheng CH, Angew. Chem. Int. Ed 2009, 48, 391; [DOI] [PubMed] [Google Scholar]; c)Yoshida H, Morishita T, Nakata H, Ohshita J, Org. Lett 2009, 11, 373; [DOI] [PubMed] [Google Scholar]; d)Akubathini SK, Biehl E, Tetrahedron Lett 2009, 50, 1809; [Google Scholar]; e)Kerisit N, Toupet L, Larini P, Perrin L, Guillemin J-C, Trolez Y, Chem. Eur. J 2015, 21, 6042. [DOI] [PubMed] [Google Scholar]
- [6].Dhokale RA, Mhaske SB, Synthesis 2018, 50, 1. [Google Scholar]
- [7].a)Cheong PHY, Paton RS, Bronner SM, Im GYJ, Garg NK, Houk KN, J. Am. Chem. Soc 2010, 132, 1267; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Garr AN, Luo DH, Brown N, Cramer CJ, Buszek KR, VanderVelde D, Org. Lett 2010, 12, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang JT, Niu DW, Brinker VA, Hoye TR, Org. Lett 2016, 18, 5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Perez D, Pena D, Guitian E, Eur. J. Org. Chem 2013, 2013, 5981. [Google Scholar]
- [10].Instead, we observed benzocyclobutadiene-derived products that arise from trapping of the benzyne by an aryl alkyne: Xiao X, Woods BP, Xiu W, Hoye TR, Angew. Chem. Int. Ed 2018, 57, 9901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen JH, Palani V, Hoye TR, J. Am. Chem. Soc 2016, 138, 4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xiao X, Hoye TR, Nat. Chem 2018, 10, 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nakano M, Takimiya K, Chem. Mater 2017, 29, 256. [DOI] [PubMed] [Google Scholar]
- [14].Seechurn CCCJ, Kitching MO, Colacot TJ, Snieckus V, Angew. Chem. Int. Ed 2012, 51, 5062, and refs therein. [DOI] [PubMed] [Google Scholar]
- [15].a)Adeleke AF, Brown APN, Cheng LJ, Mosleh KAM, Cordier CJ, Synthesis 2017, 49, 790; [Google Scholar]; b)Copper-Mediated Cross-Coupling Reactions, Eds.: Evano G, Blanchard N, Wiley-VCH: Hoboken, 2013. [Google Scholar]
- [16].Karmakar R, Yun SY, Wang KP, Lee D, Org. Lett 2014, 16, 6. [DOI] [PubMed] [Google Scholar]
-
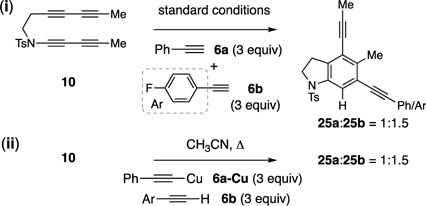
[17]. Competition
experiments showed that: (i) phenylacetylene (6a) and
4-fluorophenylacetylene (6b) reacted at very similar rates and (ii)
exchange (direct or indirect, the latter through the catalytic
cycle) between a preformed Cu-acetylide (from 6a) and a second,
terminal alkyne (6b) was occurring.
- [18].Willoughby PH, Niu DW, Wang T, Haj MK, Cramer CJ, Hoye TR, J. Am. Chem. Soc 2014, 136, 13657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ross SP, Hoye TR, Nat. Chem 2017, 9, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brewer JPN, Heaney H, Jablonski JM, Tetrahedron Lett 1968, 42, 4455. [Google Scholar]
- [21].Karmakar R, Mamidipalli P, Yun SY, Lee D, Org. Lett 2013, 15, 1938. [DOI] [PubMed] [Google Scholar]
- [22].Niu DW, Willoughby PH, Woods BP, Baire B, Hoye TR, Nature 2013, 501, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bhunia A, Roy T, Pachfule P, Rajamohanan PR, Biju AT, Angew. Chem. Int. Ed 2013, 52, 10040. [DOI] [PubMed] [Google Scholar]
- [24].García-López JA, Greaney MF, Chem. Soc. Rev 2016, 45, 6766, and refs. 3–16 therein to previous reviews. [DOI] [PubMed] [Google Scholar]
- [25].a)Murray FE, Schneider WG, Can. J. Chem 1955, 33, 797; [Google Scholar]; b)Bajwa JS, Jiang XL, Slade J, Prasad K, Repic O, Blacklock TJ, Tetrahedron Lett 2002, 43, 6709. [Google Scholar]
- [26].a)Janz GJ, Danyluk SS. J. Am. Chem. Soc 1959, 81, 3850;b)Williams JM, Peterson SW, Brown GM, Inorg. Chem 1968, 7, 2577.A neutron diffraction study in ref 26b established the formulation of 35•HCl as MeC(Cl)=NH2+Cl– rather than MeC(Cl)2NH2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.