

Abstract
Boron trifluoride is observed to promote a variety of C–H insertion reactions of benzynes bearing pendant alkyl groups. Computations and various mechanistic studies indicate that BF3 engages the strained π-bond to confer carbene-like character on the adjacent, non-coordinated benzyne carbon. This represents an unprecedented catalytic role for a non-transition metal like BF3.
Graphical Abstract
Alkynes can, formally, be viewed as vicinal dicarbenes. The importance of this resonance contributor increases for strained (e.g., cyclic) alkynes.1 For example, dicarbene-like reactivity has been observed in the formation of spirocyclic, cyclopropane-containing carbene intermediates during the reaction of, e.g., ethylene with the alkyne in, e.g., norbornyne.1a,bVarious transition metal catalysts or promoters can induce alkynes to show reactivities that are an expression of 1,2-dicarbene character, including C–H insertion behavior.1c,d In a few instances, 1,2-dicarbenoids involving two metals at each of the original alkyne carbons have even been observed.2
ortho-Benzynes, useful and versatile building blocks in organic chemistry,3 owe their high reactivity to their highly strained formal triple bond. Lee and coworkers have described C–H insertion reactions of benzyne derivatives promoted by Ag(I) that were rationalized by the intermediacy of silver carbenoid species (Figure 1a).4 We describe here an unprecedented boron trifluoride-catalyzed process in which carbene-like reactivity is exhibited by one of the two sp-hybridized benzyne carbon atoms. We are unaware of any example of a non-transition metal eliciting this kind of behavior. In Figure 1b we show the results of DFT calculations that foreshadow the experimental results we present below. The computed potential energy surface (PES) for the prototypical reaction between o-benzyne (1) and methane, catalyzed by BF3, suggests the energetic accessibility of requisite intermediates and activation barriers. Specifically, the conversion of 1 + BF3 to 2 is computed to be a nearly equienergetic event. Adduct 2 has a highly distorted geometry—the internal bond angles at Cα and Cβ are 103° and 148°, respectively. The electronic configuration for this adduct was closed shell (stable to spin-symmetry breaking). The HOMO of 2 showed most of the pi-electron density above/below the plane of the benzyne; correspondingly, the LUMO has a large in-plane component at Cβ, suggesting significant phenyl cation character (cf. 2 vs. 2’). When methane is permitted to engage 2, the system collapses to a new adduct 3, the result of exergonic (29 kcal•mol−1) insertion of Cβ into the strong C–H bond of methane in a process computed to proceed through the transition structure TS2➝3, which is only 5.4 kcal•mol−1 higher in energy than 2. The 1,2-migration of the hydrogen atom in 3 proceeds as a very low-barrier 1.1 kcal•mol−1) process via transition structure TS3➝4 in which i) both bonds to the migrating light hydrogen atom were similar in length but ii) the C–B was only 0.01 Å longer than that in 3 (see SI for details). This TS collapsed directly to toluene (4) with simultaneous ejection of BF3. The overall exergonicity of o-benzyne (1) plus methane to toluene (4) was computed to be >75 kcal•mol-1.
Figure 1.
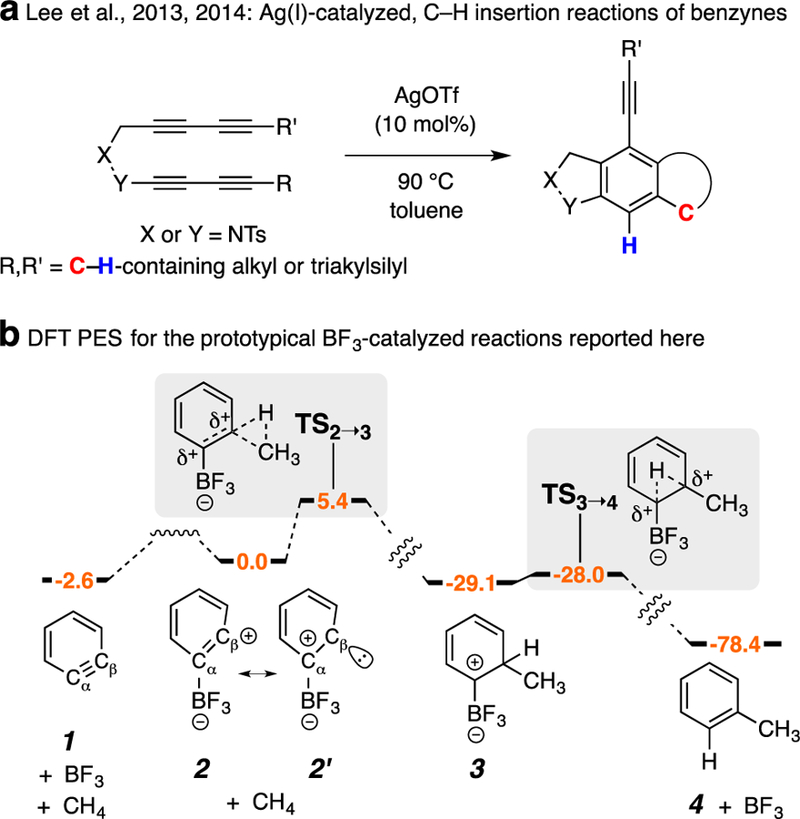
(a) Previous work demonstrating transition metal-mediated C–H insertion of a benzyne carbon. (b) Calculated {DFT [SMD(toluene)//M06–2X/6–311+G**]} energy profile for the BF3-catalyzed insertion of o-benzyne (1) into H–CH3 to give toluene (4). Ts = p-toluenesulfonyl; OTf = O3SCF3.; DFT, density functional theory; PES, potential energy surface.
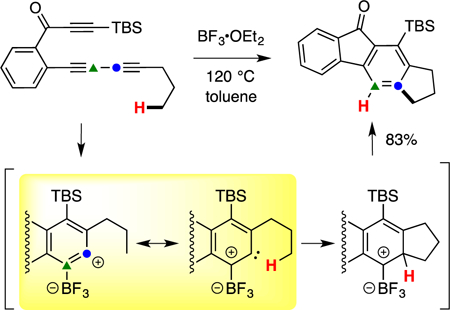
One of our earliest observations suggesting the occurrence of this novel BF3-promoted, C–H insertion process arose while studying the reaction of triyne 5 (Figure 2). When exposed to BF3•OEt2 (1.0 equiv) in toluene at 120 °C, the indane derivative 7a was isolated in 82% yield. We view BF3•OEt2 as a pre-catalyst, providing a low equilibrium concentration of dissociated BF3 to the reaction environment (some of which would be volatilized into the headspace of the sealed reaction vial). Further investigation showed that 7a, the product of net C–H insertion into the methyl group of the pendant n-propyl substituent, occurred in competition with trapping of the intermediate benzyne 6 by the toluene solvent, giving 8a and 8b. This competing pathway serves as a clock reaction that allowed us to evaluate the dependency of the reaction on the initial concentration of BF3•OEt2 (summarized in the Figure 2 insert). With an initial load of 150 mol% of the Lewis acid, formation of the toluene adducts 8 was completely suppressed, and at 50 mol% a nearly 1:1 mixture of 7a:8 was observed. This dependency on catalyst loading suggests that there is a significant amount of product inhibition—that is, that the fluorenones 7a and 8 out-compete the benzyne for engaging free BF3.
Figure 2.
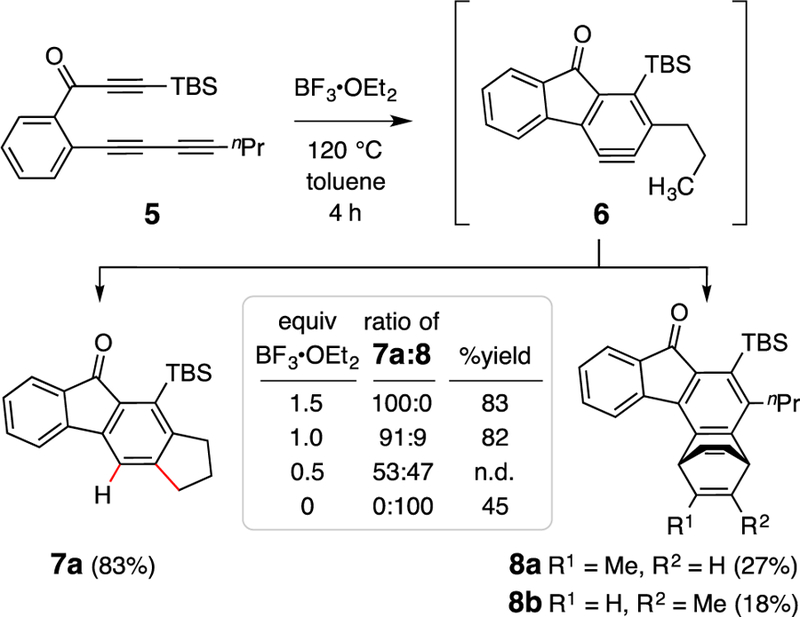
Results showing that the ratio of C–H insertion and Diels-Alder trapping (by PhMe solvent) products (7a:8) is [BF3]-dependent. TBS = tert-butyldimethylsilyl; nPr = 1-propyl; Et = ethyl; Me = methyl.
Because of the unprecedented nature of this Lewis-acid-promoted, carbene-like C–H insertion chemistry, further studies to probe aspects of the mechanism were warranted. First (Figure 3a), we prepared the trideuterated isotopomer 5-d3 to study whether the CD3 group is the source of the new aromatic hydrogen atom in the product 7a. Indeed, ≥95% of 7a-d3 was measured to have three deuterium atoms (following desilylation to 9; see SI for details). Second (Figure 3b), the deuterated triyne 5-d3 and the non-deuterated tetrayne 10, hexadehydro-Diels-Alder (HDDA) substrates that react at a similar rate, were simultaneously subjected to the reaction conditions and there was no evidence for crossover isotopomers in either of the products 11 or 7a-d3. This indicates that the reaction is intramolecular in nature. Third (Figure 3c), substrate 12, bearing a 1,1,1-trideutero-3-pentyl substituent, was converted to the ethyl-substituted indane derivative 13. The reaction proceeded with a small (and inverse) kH/kD kinetic isotope effect (KIE, see SI for details). Carbene insertions into C–H bonds frequently proceed with KIEs of low magnitude, encompassing both normal and inverse natures.5 Such insertions are often highly exergonic, presumably proceeding, accordingly, through relatively early transition state structures (cf. 2 to 3, Figure 1b) with only a small degree of C–H bond rupture. Moreover, the edge-on nature of those TS geometries minimizes the contribution from the C–H stretching vibration and accentuates the importance of bending vibrations. Fourth, phenyl carbenium ions are known to promote C–H insertion reactions,6 and one recently reported example6d of such a reaction shows a similarly small isotope effect for insertion into cyclohexane. We designed a control experiment to test whether BF3 itself was responsible for these reactions, because a BF3-activated Brønsted acid derived from, say, adventitious water in the reaction medium could be invoked as an alternative promoter. Tetrayne 10 was reacted under the conditions given in Figure 3b but now also containing an additional 0.5 equiv of diisopropylethylamine (DIPEA), which we presumed would buffer trace levels of Brønsted acid species such as H2O•BF3. As with the experiment done in the absence of amine, we again observed that 11 was formed as the major product. Finally, we did not detect any Friedel-Crafts arylation products arising from trapping by toluene, a behavior for phenyl cations recently reported by Nelson and coworkers.6d In aggregate, these experiments are consistent with the mechanistic framework laid out in Figure 1b.
Figure 3.
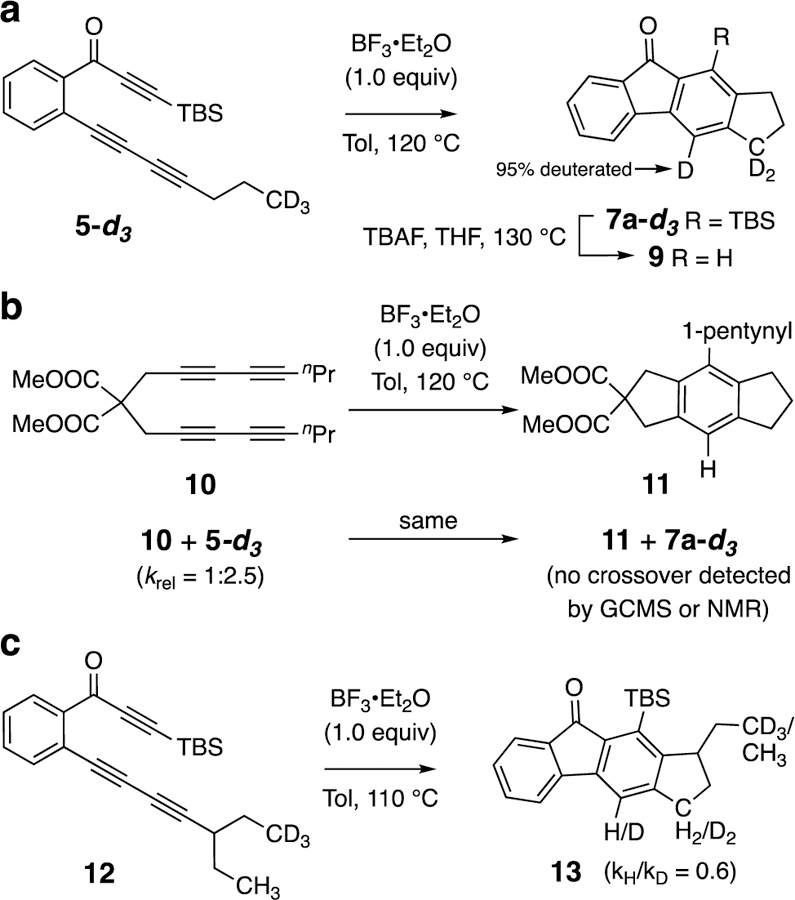
Experiments to probe mechanistic aspects of the reaction. (a) Use of substrate 5-d3 establishes the origin of the new aromatic hydrogen atom. (b) Crossover experiment using a mixture of substrates 10 and 5-d3. (c) Kinetic isotope effect measured with substrate 12. TBAF = tetra-n-butylammonium fluoride; Tol = toluene.
We returned to DFT computations for some final validation of the mechanistic framework we put forward at the outset. Specifically, we computed the BF3-catalyzed transformation of benzyne 6 to product 7 (but with structures truncated by removal of the TBS substituent for easier computation). The results (energies and geometries) directly paralleled those given in Figure 1b for the parent reaction (see Figure S1 in SI for details).
We proceeded to study this benzyne C–H insertion reaction with, first, a series of ynone HDDA substrates (5a-h in Figure 4); these reactions are all variations of the 5a to 7a conversion described above in Figure 2. These allowed us to somewhat systematically probe the effect, primarily, of modification in the alkyl group R at the terminus of the 1,3-diyne, which proffers its C–H bond(s) to the carbenic center. In three cases two products were isolated (see boxes). Collectively, the products in Figure 4 represent all that were formed to the extent of >~5% in any of the reactions.
Figure 4.
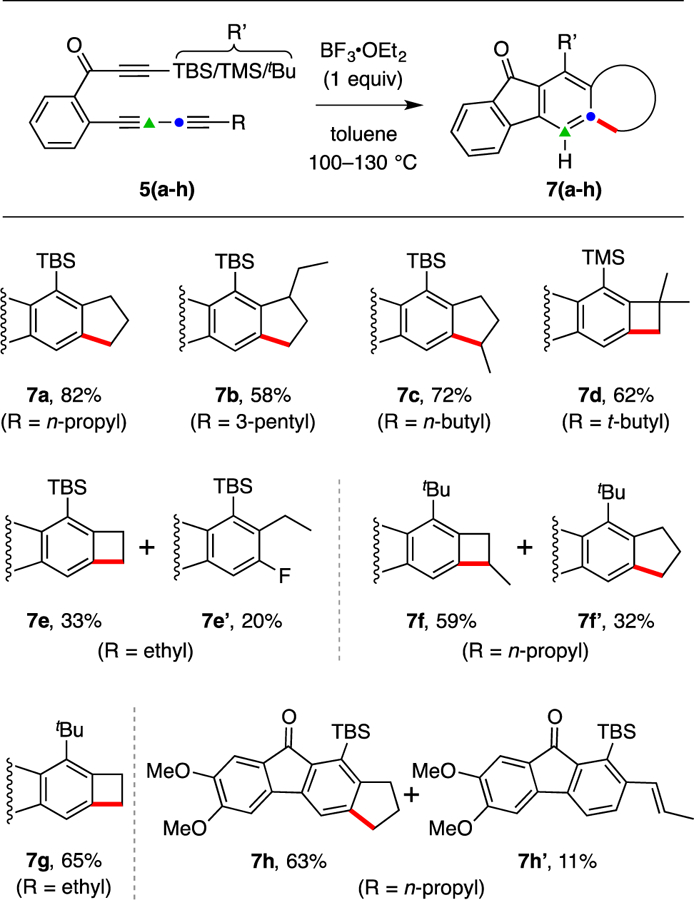
The major products (7a-h) isolated from C-H insertion experiments using triyne precursors 5a-h. Yields are for isolated, purified (SiO2) material. The nature of the R and R’ groups present in each precursor 5 can be deduced from the structure of each resultant product 7. TMS = trimethylsilyl; tBu = tertiary-butyl.
The reaction to produce 7b (the non-deuterated analog of 13, Figure 3c) behaves analogously to that giving 7a. The formation of 7c indicates that insertion into a methylene with concomitant formation of a five-membered ring is significantly favored over insertion into a stronger methyl C–H bond with accompanying formation of a six-membered ring.7 The R group in 5d is t-butyl; C–H insertion still occurs even though the product, 7d, is a benzocyclobutene. This supports the view that the insertion step passes through an early transition state geometry wherein low portion of the strong primary C–H bond cleavage and only a small fraction of the eventual ring strain has evolved. When R is an ethyl rather than t-butyl group as in 5e, the unsubstituted benzocyclobutene product 7e is still formed, but with diminished efficiency relative to that of 7d, reflecting the lower probability of the carbene carbon to encounter an insertable C–H bond (three vs. nine methyl hydrogens to give 7e vs. 7d, respectively) as well as, perhaps, a Thorpe-Ingold effect that leads the methyl C–H of 5d to more intimately sample the orbital space of the carbene center. Product 7e is accompanied by the formation of the HF adduct 7e’. Although we do not know the mechanism of this reaction,8 it is presumably slower than the insertion processes. With the exception of the reaction giving 7b (where a trace of an HF adduct was detected by GC-MS analysis), this pathway was not observed for any of the other reactions.
Reaction of substrate 5f proceeds via a benzyne containing a t-butyl instead of TBS ring-substituent. In contrast to the clean formation of 7a for the latter, the major product from 5f was now the methylated benzocyclobutene derivative 7f rather than the isomeric cyclopentane 7f’. It is possible that the more compact steric bulk of the t-butyl group has compressed the Ar–CH2–CH2Me bond angle, leading to closer proximity of the weaker methylene C–H bond to the reaction center. Consistent with this explanation, the reaction of 5g proceeded to give the benzocyclobutene 7g more efficiently than that of 5e to 7e, and no trace of the (more slowly formed) HF adduct analogous to 7e’ was detected. Finally, substrate 5h, an analog of 5a now bearing 4,5-dimethoxy groups, is also a competent substrate for the benzyne C–H insertion reaction. The principal product, 7h, is analogous to 7a, but a second product, the alkene 7h’, was also isolated.9
We then explored five other HDDA polyyne substrates that differ primarily in atoms ABC (10a-e, Figure 5) that compose the tether between the diyne and diynophile. All give at least some amount of insertion product(s) 11, demonstrating that the process is not unique to the phenylketone linker in substrates 5. Moreover, products 11d and 11e show that a conjugated carbonyl group within the tether is not a prerequisite. We attribute the lower yield in these last two cases to the fact that the 1-pentynyl substituent is thin and that the benzyne arising from each of the substrates 10d and 10e each experience reduced compression between a propyl C–H bond and the carbene carbon. This slows the rate of the insertion reaction, allowing more time for competing, benzyne-consuming reactions (such as reaction with another molecule of polyyne substrate10) to ensue. Substrate 10b contains a tBu group on the terminus of the diynophile and, like tBu-containing triynes 5f and 5g, gave a preponderance of a benzocyclobutene product, the lactam 11b. Finally, the TBS containing analog 10c gave only the cyclopentane-containing compounds 11c/c’/c’’. The second of these products reveals an interesting fluorodemethylation process. A control experiment (GCMS) demonstrated that isolated 11c was converted to 11c’ when heated in toluene in the presence of BF3•OEt2. Coordination of the ortho TBS and ketone groups to reversibly form a zwitterionic silicate complex would open a path for methyl group removal either by BF3 or an adventitious, Lewis acid-activated, proton source such as water.
Figure 5.
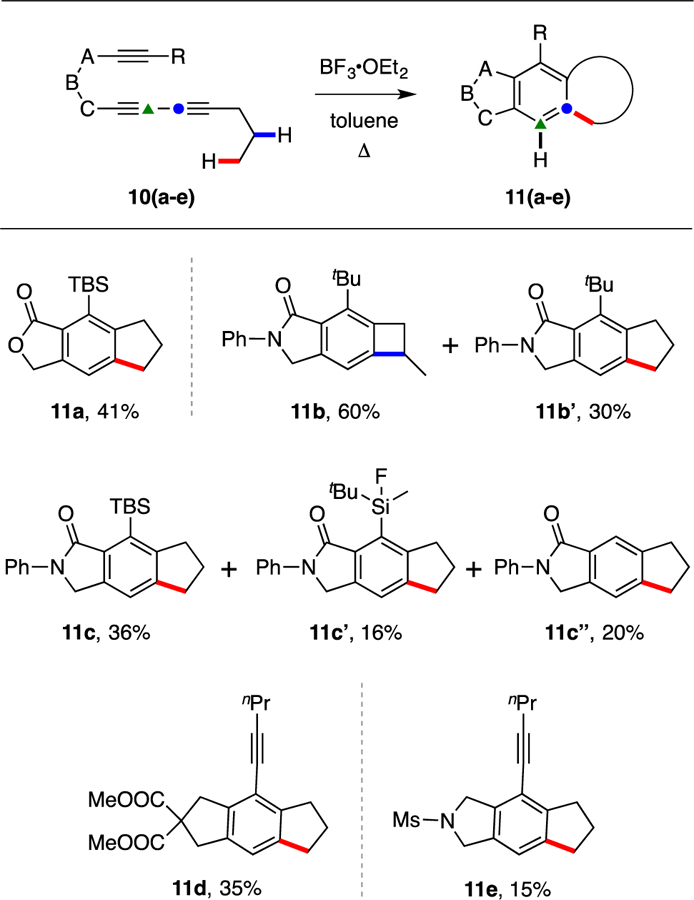
The major products (11a-e) isolated from experiments using triyne and tetrayne precursors 10a-e, each having a different linker structure (ABC) from that in 5. Yields are for isolated, purified (SiO2) material. The nature of the ABC linker and the R group present in each precursor 10 can be deduced from the structure of each resultant product 11.
In conclusion, we have described an unprecedented mode of activation of an aryne. Namely, the Lewis acid BF3 engages benzynes to promote carbene-like reactivity at the adjacent benzyne carbon atom. This work serves as another example11 in which benzynes derived from the thermal HDDA cycloisomerization reaction and, therefore, in the absence of any other benzyne-generating reagents or byproducts, has allowed a fundamentally new type of transformation to be discovered.
Supplementary Material
ACKNOWLEDGMENT
Support for this work was provided by the Institute of General Medical Sciences of the U.S. Department of Health and Human Services (R01 GM65597, then R35 GM127097) and the National Science Foundation (CHE-1665389). A portion of the NMR data were obtained with an instrument purchased with funds from the NIH Shared Instrumentation Grant program (S10OD011952). Computations were performed with resources made available by the University of Minnesota Supercomputing Institute (MSI).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures for all reactions; spectroscopic characterization data for all new compounds; details of computational methods; copies of 1H and 13C NMR spectra.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Laird DW; Gilbert JC Norbornyne: A Cycloalkyne Reacting Like a Dicarbene. J. Am. Chem. Soc 2001, 123, 6704–6705 [DOI] [PubMed] [Google Scholar]; (b) Bachrach SM; Gilbert JC; Laird DW DFT Study of the Cycloaddition Reactions of Strained Alkynes. J. Am. Chem. Soc 2001, 123, 6706–6707 [DOI] [PubMed] [Google Scholar]; (c) Zeidan T; Kovalenko SV; Manoharan M; Clark RJ; Ghiviriga I; Alabugin IV Triplet Acetylenes as Synthetic Equivalents of 1,2-Bicarbenes: Phantom n,π* State Controls Reactivity in Triplet Photocycloaddition. J. Am. Chem. Soc 2005, 127, 4270–4285 [DOI] [PubMed] [Google Scholar]; (d) Alabugin IV; Gold B “Two Functional Groups in One Package”: Using Both Alkyne π-Bonds in Cascade Transformations. J. Org. Chem 2013, 78, 7777–7784. [DOI] [PubMed] [Google Scholar]
- (2).For example: (a) Yasufuku K; Yamazaki H Chemistry of Mixed Transition Metal Complexes of New Complexes III*. Preparation and Acetylenes Coordinated on Iron and Nickel. J. Organometal. Chem 1972, 35, 367–373 [Google Scholar]; (b) Leutzsch M; Wolf LM; Gupta P; Fuchs M; Thiel W; Farès C; Fürstner A Formation of Ruthenium Carbenes by gem-Hydrogen Transfer to Internal Alkynes: Implications for Alkyne trans-Hydrogenation. Angew. Chem. Int. Ed 2015, 54, 12431–12436 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kelch H; Kachel S; Celik MA; Schäfer M; Wennemann B; Radacki K; Petrov AR; Tamm M; Braunschweig H Elucidating the Reactivity of Vicinal Dicarbenoids: From Lewis Adduct Formation to B–C Bond Activation. Chem. Eur. J 2016, 22, 13815–13818. [DOI] [PubMed] [Google Scholar]
- (3).Hoffman RW Dehydrobenzene and Cycloalkynes; Academic: New York, 1967. [Google Scholar]
- (4).(a) Yun SY; Wang K-P; Lee N-K; Mamidipalli P; Lee D Alkane C–H Insertion by Aryne Intermediates with a Silver Catalyst. J. Am. Chem. Soc 2013, 135, 4668–4671 [DOI] [PubMed] [Google Scholar]; (b) Mamidipalli P; Yun SY; Wang K-P; Zhou T; Xia Y; Lee D Formal Hydrogenation of Arynes with Silyl Cβ–H Bonds as an Active Hydride Source. Chem. Sci 2014, 5, 2362–2367 [Google Scholar]; (c) Karmakar R; Lee D Reactions of Arynes Promoted by Silver Ions. Chem. Soc. Rev 2016, 45, 4459–4470. [DOI] [PubMed] [Google Scholar]; (d) Karmakar R; Le A; Xie P; Xia Y; Lee D Reactivity of Arynes for Arene Dearomatization. Org. Lett 2018, 20, 4168–4172. [DOI] [PubMed] [Google Scholar]
- (5).(a) Majer JR; Capey WD; Robb JC Isotope Effect in Radical Reactions. Nature 1964, 294–295; (b) More O’Ferall RA Model Calculations of Hydrogen Isotope Effects for Non-linear Transition States. J. Chem. Soc. (B) 1970, 785–790; (c) Baldwin JE; Andrist AH Mechanism and Inverse Primary Kinetic Isotope Effect in the Reaction of Fluorenylidene with 3-Deuteriocyclohexene. Chem. Comm 1971, 1512–1513; (d) Churchill DG; Janak KE; Wittenberg JS; Parkin G Normal and Inverse Primary Kinetic Deuterium Isotope Effects for C-H Bond Reductive Elimination and Oxidative Addition Reactions of Molybdenocene and Tungstenocene Complexes: Evidence for Benzene σ-Complex Intermediates. J. Am. Chem. Soc 2003, 125, 1403–1420 [DOI] [PubMed] [Google Scholar]; (e) Horino Y; Yamamoto T; Ueda K; Kuroda S; Toste FD Au(I)-Catalyzed Cycloisomerizations Terminated by sp3 C-H Bond Insertion. J. Am. Chem. Soc 2009, 131, 2809–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Mascarelli L Contributo Alla Conoscenza Del Bifenile E Dei Suoi Derivati.—Nota XV. Passaggio Dal Sistema Bifenilico a Quello Fluorenico. Gazz. Chim. Ital 1936, 66, 843–850 [Google Scholar]; (b) Cohen T; Lipowitz J The Question of a Benzene Cation Insertion Reaction. A Novel Intramolecular Electrophilic Substitution. J. Am. Chem. Soc 1964, 86, 2515–2516 [Google Scholar]; (c) Allemann O; Duttwyler S; Romanato P Baldridge KK; Siegel JS Proton-Catalyzed, Silane-Fueled Friedel-Crafts Coupling of Fluoroarenes. Science 2011, 332, 574–577 [DOI] [PubMed] [Google Scholar]; (d) Shao B; Bagdasarian AL; Popov S; Nelson HM Arylation of Hydrocarbons Enabled by Organosilicon Reagents and Weakly Coordinating Anions. Science 2017, 355, 1403–1407; [DOI] [PubMed] [Google Scholar]
- (7).For an early study demonstrating the strong preference for (rhodium) carbenoid insertions to produce five-rather than six-membered rings, see: (a) Taber DF; Petty EH General Route to Highly Functionalized Cyclopentane Derivatives by Intramolecular C-H Insertion. J. Org. Chem 1982, 47, 4808–4809 [Google Scholar]; (b) Taber DF; Ruckle RE Cyclopentane Construction by Dirhodium Tetraacetate-Mediated Intramolecular C-H Insertion: Steric and Electronic Effects. J. Am. Chem. Soc 1986, 108, 7686–7693. [DOI] [PubMed] [Google Scholar]; (c) For selective reviews that include carbene C-H insertion reactions see:Maas G Transition-metal Catalyzed Decomposition of Aliphatic Diazo Compounds–New Results and Applications in Organic Synthesis. Top. Curr. Chem 1987, 137, 75–253 [Google Scholar]; (d) Doyle MP; Forbes DC Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev 1998, 98, 911–935 [DOI] [PubMed] [Google Scholar]; (e) Sulikowski GA; Cha KL; Sulikowski MM Stereoselective Intramolecular Carbon–Hydrogen Insertion Reactions of Metal Carbenes. Tetrahedron: Asymmetry 1998, 9, 3145–3169 [Google Scholar]; (f) Davies HML; Beckwith REJ Catalytic Enantioselective C–H Activation by Means of Metal–Carbenoid-Induced C–H Insertion. Chem. Rev 2003, 103, 2861–2904 [DOI] [PubMed] [Google Scholar]; (g) Doyle MP; Duffy R; Ratnikov M; Zhou L Catalytic Carbene Insertion into C–H Bonds. Chem. Rev 2010, 110, 704–724. [DOI] [PubMed] [Google Scholar]
- (8).Using HBF4 instead of BF3•OEt2 under otherwise similar conditions, we observed formation of a larger amount of 7e’ compared to 7e, implying that a Brønsted acid is playing a role in the net HF addition process.
-
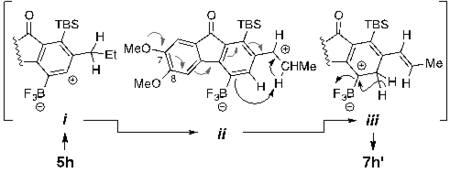
(9).An alkene product
was only observed in the case of the reaction of the
4,5-dimethoxyphenone derivative 5h. We suggest that the
derived benzyne•BF3 adduct i can give
rise to the benzylic carbenium ion ii, the formation of
which (more than one route can be envisioned for this
transformation) is enabled by the electron donating ability of the
7-OMe substituent (cf. gray arrows). Intramolecular proton transfer
restores boron carbenoid character in iii, which,
finally, aromatizes to 7h’ via 1,2-hydride
migration (cf. 3 to 4, Fig.
1).
- (10).Xiao X; Woods BP; Xiu W; Hoye TR Benzocyclobutadienes: An Unusual Mode of Access Reveals Unusual Modes of Reactivity. Angew. Chem. Int. Ed 2018, 57, 9901–9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Diamond OJ; Marder TB Methodology and Applications of the Hexadehydro-Diels–Alder (HDDA) Reaction. Org. Chem. Front 2017, 4, 891–910. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.