

Abstract
Target-specific imaging probes represent a promising tool in the molecular imaging of human cancer. Fluorescently-labeled target-specific probes are useful in imaging cancers because of their ability to bind a target receptor with high sensitivity and specificity. The development of probes relies upon preclinical testing to validate the sensitivity and specificity of these agents in animal models. However, this process involves both conventional histology and immunohistochemistry, which require large numbers of animals and samples with costly handling. In this chapter, we describe a novel validation tool that takes advantage of genetic engineering technology, whereby cell lines are transfected with genes that induce the target cell to produce fluorescent proteins with characteristic emission spectra, thus enabling their easy identification as cancer cells in vivo. Combined with multicolor fluorescence imaging, this can provide rapid validation of newly-developed exogenous probes that fluoresce at different wavelengths. For example, the plasmid containing the gene encoding red fluorescent protein (RFP) was transfected into cell lines previously developed to either express or not express specific cell surface receptors. Various antibody-based or ligand-based optical-contrast agents, with green fluorophores were developed to concurrently target cancer cells and validate their positive and negative controls, such as the β-D-galactose receptor, HER1, and HER2 in a single animal/organ. Spectrally-resolved multicolor fluorescence imaging was used to detect separate fluorescence emission spectra from the exogenous green fluorophore and RFP. Here, we describe the use of “co-staining” (matching the exogenous fluorophore and the endogenous fluorescent protein to the positive control cell line) and “counter-staining” (matching the exogenous fluorophore to the positive control and the endogenous fluorescent protein to the negative control cell line) to validate the sensitivity and specificity of target-specific probes. Using these in vivo imaging techniques, we are able to determine the sensitivity and specificity of target-specific optical contrast agents in several distinct animal models of cancer in vivo, thus exemplifying the versatility of our technique, while reducing the number of animals needed to conduct these experiments.
Keywords: Cancer, Spectral fluorescence imaging, Fluorescent probe, Fluorescent protein, Sensitivity, Specificity, Molecular imaging, Molecular targeting
1. Introduction
As more novel imaging probes are developed, it is necessary to optimize a method to efficiently assess their effectiveness at identifying pathological processes in vivo. The utilization of optically-detectable agents is promising for clinical applications because of their high sensitivity and specificity. Fluorescently-labeled probes provide the advantage of high signal-to-background ratios, the ability to utilize multiple fluorophores to differentiate even subtle cellular differences, and decreased exposure to ionizing radiation. The application of using fluorescently-labeled probes in the identification and clinical monitoring of cancers shows promise for the future. However, novel probes must undergo sufficient preclinical assessment before these probes can be utilized clinically. The ideal probe binds to its target receptor with high affinity resulting in high sensitivity and specificity, has high signal-to-background ratio, and any probe that remains unbound is quickly removed from the body with low systemic toxicity. Therefore, a preclinical model must be able to monitor binding efficiency, clearance, sensitivity, and specificity efficiently.
An example of the kind of target-specific optical imaging agent that may play a role in disease in the future is one designed to detect ovarian cancer in the peritoneum as an aid to surgery. Using fluorescence imaging in a mouse model with disseminated peritoneal cancer, the agent was able to localize tumor nodules as small as 100 in diameter, which were invisible to the naked eye (1). In order to validate new imaging probes such as this, however, it is important to document that the areas of fluorescence actually correspond to sites of disease. Both the “true positives” and the “true negatives” need to be assessed to determine the sensitivity and specificity of the probe. The traditional method of doing this is to use large numbers of animals, painstakingly remove tumor implants, as well as normal regions and correlate these with the images. In addition to being time consuming, expensive, and necessitating large numbers of animals, the analysis is incomplete because it is difficult to sample more than a small percentage of the total lesions. There are also always uncertainties regarding the reliability of the co-localization of the histologic specimen to the image. When the tumor nodules are too small and too closely clustered to be individually defined by direct visualization, it is almost impossible to correlate each positive nodule in the fluorescence image to its respective histologic counter part. In order to overcome this problem, we developed an alternative approach using genetically-engineered probes that constitutively express fluorescent proteins in the cancer cells (2).
In this chapter, we describe a method to assess the sensitivity and specificity of novel exogenous imaging probes by comparing them with endogenously-expressed fluorescent proteins. We used “co-staining” (matching the exogenous fluorophore and the endogenous fluorescent protein to the positive control cell line) and “counter-staining” (matching the exogenous fluorophore to the positive control and the endogenous fluorescent protein to the negative control cell line) to validate the sensitivity and specificity of target-specific probes.
2. Materials
2.1. Plasmid
The RFP (DsRed2)-expressing plasmid was purchased from Clontech Laboratories, Inc. (Mountain View, CA).
2.2. Antibodies and Ligands
Trastuzumab was purchased from Genentech, Inc. (San Francisco, CA).
Daclizumab was purchased from Hoffmann-La Roche, Inc. (Nutley, NJ).
Galactosamine serum albumin (GmSA) was purchased from Sigma Chemical (St. Louis, MO).
2.3. Fluorophores
Rhodamine Green (RhodG) NHS was purchased from Invitrogen Corporation (Carlsbad, CA).
ICG-sulfo-OSu was purchased from Dojindo Molecular Technologies (Rockville, MD).
Alexa680-NHS was purchased from Invitrogen Corporation (Carlsbad, CA).
2.4. Molecular Probes
2.5. In Vivo Imaging
Female nude mice (National Cancer Institute Animal Production Facility, Frederick, MD, USA).
Maestro In Vivo Imaging System (CRi Inc., Woburn, MA, USA).
FluorVivo real-time color fluorescence imaging system (INDEC Biosystems, Santa Clara, CA).
3. Methods
3.1. Co-staining Method
3.1.1. Concept
In co-staining, a target cell line is labeled with an endogenous fluorescent protein. Next, the probe of interest is conjugated to an exogenous fluorophore that is optically distinguishable from the transfected fluorescent protein, and then injected into an animal bearing tumors derived from a fluorescent-protein-labeled cancer cell line. Since multicolor spectral fluorescence imaging enables the detection of these two distinct fluorescent wave lengths, two fluorescent images, one from the endogenous fluorescent protein and another from the exogenous fluorophore, can be obtained and analyzed (see Fig. 1) (see Note 1). For analysis, true-positive results are tumor foci that show two distinct fluorescence-emission patterns. False positives are those tumor foci that demonstrate exogenous fluorescence only, but not endogenous fluorescence. False negatives are tumor foci that have endogenous fluorescence only, but no exogenous fluorescence. To determine the true negative value, however, it is necessary to randomly choose regions of interest (ROIs) over normal tissues negative for endogenous fluorescence emission and determine whether exogenous fluorescence is present. Once these values are determined, it is possible to determine sensitivity and specificity of a given probe. However, this method is most accurate at determining sensitivity, rather than specificity. This is because specificity estimation requires the knowledge of the true-negative value, and the requirement to place ROIs over “normal” tissue is sometimes difficult because normal tissue is often not validated and this may lead to inaccurate determination of specificity.
Fig. 1.
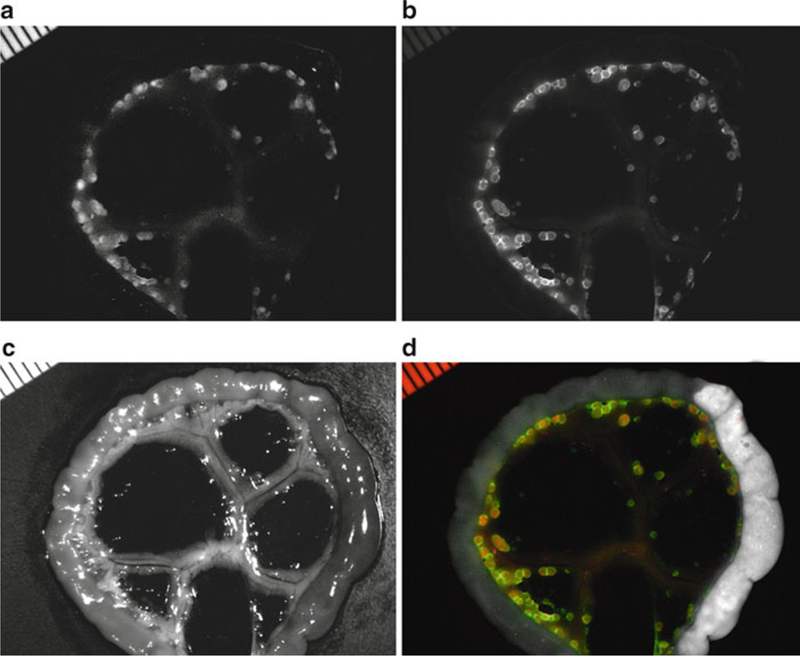
Co-staining in a peritoneal-dissemination model of ovarian cancer. Spectral fluorescence images of a SHIN3-RFP tumor-bearing mouse which was injected with GmSA-RhodG. Unmixed spectral fluorescence images display (a) SHIN3-RFP tumors in the RFP spectrum and (b) the tested imaging probe (GmSA-RhodG) in the RhodG spectrum. (c) White light image of gut and mesentery. (d) Overlay of spectral images illustrates the co-localization of both the RFP and RhodG fluorescence.
3.1.2. Tumor Model and Probe (Peritoneal Dissemination Model)
Here, we describe one method of co-staining using a β-D-galactose receptor-positive cell line (SHIN3) and a newly-developed β-D-galactose receptor-targeted optical probe GmSA-RhodG in a peritoneal-dissemination model of ovarian cancer (3).
The red fluorescent protein (RFP:DsRed2) plasmid was transfected into SHIN3 cells (human ovarian cancer cell line) which is positive for the β-D-galactose receptor, and then cloned to establish a cell line stably expressing RFP (SHIN3-RFP).
The tested imaging probe, D-galactosamine serum albumin (GmSA) which binds to the β-D-galactose receptor, is conjugated with a green fluorophore, rhodamine green (GmSA- RhodG).
An intraperitoneal-dissemination model of ovarian cancer is established 14 days after intraperitoneal injection of 2 × 106 SHIN3-RFP cells, suspended in 200 μL PBS, into female nude mice.
Three hours after intraperitoneal injection of 500 pmol GmSA- RhodG diluted in 300 μL PBS, mice are sacrificed with carbon dioxide and then subjected to fluorescence imaging.
3.1.3. In Vivo Fluorescence Imaging (Spectral Fluorescence Imaging)
Spectral fluorescence imaging is a method that enables the unmixing of two or more fluorescent signals by their distinct spectral fluorescence patterns. This has several advantages over conventional fluorescence imaging. First, it can separate and eliminate autofluorescence from the target fluorescence signal efficiently by differentiating their distinct spectral fluorescence patterns. Second, fluorophores/fluorescent proteins having similar, but distinct, emission spectra can be unmixed. Thus, multiplexed use of several fluorophore/fluorescent proteins is feasible with this method. Also, low-intensity target fluorescence can be detected and enhanced after spectral unmixing. Therefore, the brightness of target fluorescence matters less than conventional fluorescence imaging. The procedure using spectral fluorescence imaging, with co-staining to validate GmSA as a probe to identify SHIN3 tumors, in the intraperitoneal-dissemination model, is described below.
After euthanasia, the abdominal cavity is exposed, and the mesentery is spread out on a nonfl uorescent plate.
Close-up spectral fl uorescence imaging of the mesentery is performed with an in vivo spectral-fl uorescence imager (Maestro, CRi, Waltham, MA). Spectral-fl uorescence image data are obtained in which each pixel has its own distinct spectral pattern in a range of acquisition wavelengths. In this particular experiment (RFP and RhodG), parameters of acquisition are as follows: a band-pass fi lter from 445 to 490 nm and a long pass fi lter over 515 nm were used for excitation and emission light, respectively. The tunable fi lter was automatically stepped in 10 nm increments from 500 to 800 nm while the camera captured images at each wavelength interval with constant exposure. Note that these parameters need to be optimized depending on the combination of fluorescent proteins and fluorophores used.
To unmix spectral fluorescence data into its component spectral fluorescences, it is necessary to first create a standard library that represents each fluorophore of interest and a library that represents autofluorescence (see Note 2). In our sample, an RFP spectrum is obtained using RFP transfected SHIN3 cells resuspended in solution, a RhodG spectrum is established using GmSA-RhodG in solution, and an autofluorescence spectrum of the gut is created using a nude mouse without any fluorophore.
All spectral-fluorescence data are unmixed with this spectral library, and finally unmixed fluorescence images of RhodG and RFP are created and then subjected to image analysis.
3.1.4. Image Analysis
ROIs were drawn on two unmixed fluorescence images of either endogenous fluorescent protein or exogenous fluorophore, to determine sensitivity and specificity as described in Subheading 3.1.1. In this experiment, ROIs are drawn over both the lesions, depicted by RFP spectral fluorescence (standard reference for cancer foci) and in the surrounding adjacent areas (standard reference for non-cancerous foci). The average fluorescence intensity of each ROI is calculated, both on the RFP and the RhodG spectral unmixed images. Then, additional ROIs are drawn on the nodules depicted only by the RhodG spectral fluorescence to count the number of false-positive lesions. The average fluorescence intensities of false-positive foci are calculated, both on the RFP and the RhodG unmixed images. The number of foci positive for both RhodG and RFP, negative for both RhodG and RFP, and positive only for RhodG or RFP, are counted. Finally, sensitivity and specificity are calculated based on these values, as described in Subheading 3.1.1.
3.2. Counter-Staining Method
3.2.1. Concept
In counter-staining, two or more cancer cells lines are utilized to determine sensitivity and specificity of a given probe. One cell line must express the target receptor of interest that will bind to a testing probe and the other cell line must not express the receptor of interest. The cell line that does not express the target receptor of interest is labeled with an endogenous fluorescent protein. Then, the probe of interest is conjugated with an exogenous fluorophore that is optically distinguishable from the transfected fluorescent protein. The conjugated probe is then injected into an animal bearing tumors derived from both cell lines. Next, spectral fluorescence imaging is used to distinguish the endogenous fluorescent protein and the exogenous probe (see Fig. 2). For analysis, true positive results are tumor foci that fluoresce at the spectra of the exogenous probe. False positives are the tumor foci that fluoresce at both the exogenous-probe and the endogenous-protein fluorescence spectra. True negatives are tumor foci that fluoresce at only the spectra that represent the endogenous fluorescent protein. False negatives are represented by tumor foci that form nodules visible with white light imaging or microscopic imaging but have no exogenous or endogenous fluorescence emission. In contrast to co-staining, this method is most accurate in determining specificity, because this can determine the true-negative value more clearly than the co-staining method. However, to determine the false- negative value, white light images serve as the gold standard and small tumors may be invisible on white light images leading to an overestimation of specificity.
Fig. 2.
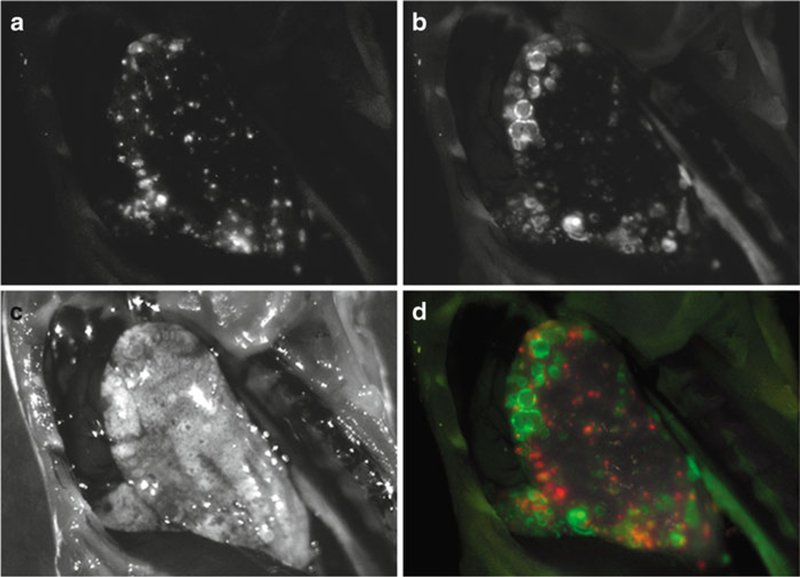
Counter staining in a model of lung metastasis. Spectral-fluorescence images of a mouse bearing 3T3-HER2 (HER2+) and Balb3T3-RFP (HER2-) tumors in a lung metastasis model that was injected with RhodG-conjugated trastuzumab (anti-HER2). (a) Unmixed RFP spectral image localizes RFP tumors. (b) Unmixed RhodG spectral image identifies 3T3-HER2 tumors labeled with trastuzumab-RhodG. (c) White light images of the left lung. (d) Two-color spectral-fluorescence overlay localizes HER2+ and HER2- tumors which are clearly differentiated from each other and do not co-localize.
3.2.2. Tumor Model and Probe (Metastatic Lung Cancer Model)
Here, we demonstrate a counter-staining method using a HER-2 receptor-positive cell line (NIH3T3 cells transfected with the HER-2 gene) and a HER-2 receptor-negative cell line (Balb3T3) in a lung metastasis model (7).
NIH3T3 cells were transfected with the HER-2 gene and cloned to establish a cell line stably over-expressing HER-2 receptors (3T3-HER2). The red fluorescent protein (RFP:DsRed2) plasmid was transfected into Balb3T3 cells, which are negative for the HER-2 receptor and cloned to establish a cell line stably expressing RFP (Balb3T3-RFP).
The imaging probe to be tested, trastuzumab which binds to the HER2 receptor, is conjugated with a green fluorophore, rhodamine green (trastuzumab-RhodG).
A metastatic lung tumor model is established 12 days after intravenous injection of 2 × 106 Balb3T3/RFP cells and 19 days post-injection of 2 × 106 3T3/HER-2+ cells suspended in 200-μL PBS into female nude mice.
24 h after tail vein injection of 50 μg trastuzumab-RhodG, diluted in 200 μL PBS in mice with lung metastatic tumors, the mice are sacrificed with carbon dioxide and then subjected to fluorescence imaging.
3.2.3. In Vivo Fluorescence Imaging
Immediately after sacrifice, the pleural cavity is exposed, and the mouse is placed on a nonfluorescent plate.
Spectral-fluorescence images were obtained using an in vivo spectral-fluorescence imager as described in Subheading 3.1.3. Then fluorescence images of RhodG and RFP are unmixed based on the respective emission spectra of RhodG and RFP.
3.2.4. Image Analysis
Two unmixed fluorescent images are obtained representing either the endogenous RFP or exogenous RhodG. Detection of RhodG-positive tumors, RFP-positive tumors, and double-negative tumors are performed using magnified fluorescence composite images and stereoscopic microscope images. Finally, sensitivity and specificity were determined as described above in Subheading 3.2.1.
3.3. Multi-excitation
In the method described above, we utilized a single excitation method for in vivo spectral fluorescence imaging. Using this method, we are limited by the range of excitation light. By using a blue excitation light (445–490 nm), we are limited to fluorophores that are excitable in that range. When a green fluorophore and RFP are used, although both fluorophores are excitable by blue light, the RFP is excited less efficiently than the green fluorophore, increasing “false positive” tumor nodules in the co-staining analysis. For instance, when calculating the sensitivity of GmSA-RhodG using co-staining, three foci positive for GmSA-RhodG were not confirmed by RFP and were determined to be false positives (3). It is difficult to determine, but this may have resulted from RhodG being excited with better efficiency than RFP by the blue light (see Fig. 3) (see Note 3).
Fig. 3.
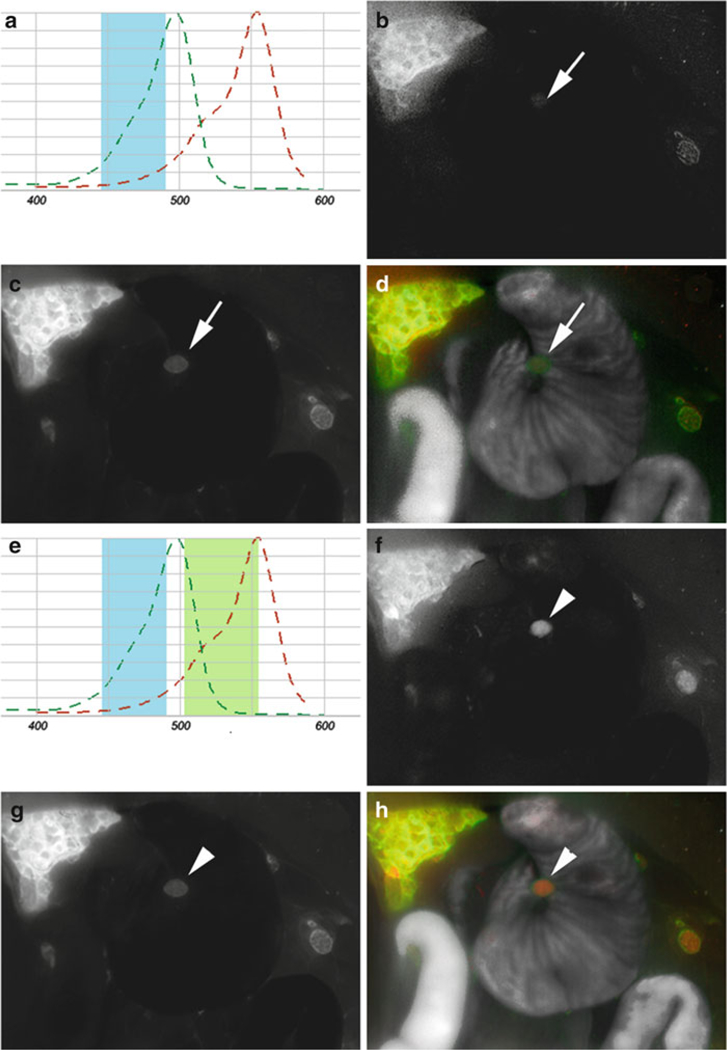
Comparison of single- and multi-excitation in a peritoneal-dissemination model of ovarian cancer. (a) Schematic of RFP (red) and RhodG (green) absorbance curves, and range of single blue excitation light (blue). The blue light adequately excites RhodG, but only suboptimally excites RFP. (b-d) Spectral fluorescence images taken with a single excitation blue light using a co-staining method in a SHIN3-RFP-tumor-bearing mouse receiving GmSA-RhodG. (b) RFP spectrum identifies endogenous expression of RFP by SHIN3 cells but is unable to recognize tumor nodules (arrowS) that are visible on the RhodG spectrum (c). (d) Two-color overlay using single excitation light. (e) Schematic of multi-excitation (blue and green) light on the absorbance curves of RFP (red) and RhodG (green) demonstrating more efficient excitation of RFP by green light. (f-h) Spectral-fluorescence images, taken with multiple-excitation filters in the same mouse, demonstrates the ability to identify the previously-invisible tumor nodules on the RFP spectrum (f, arrowhead) that are still present on the RhodG spectrum (g). (h) Two-color overlay using multiple excitation filters.
Ideally, we would like to be able to excite each fluorophore with different wavelengths of light that excite each fluorophore with equal efficiency, resulting in the most intense emission possible for each fluorophore. Multiple excitation methods enable us to use many more fluorophores that span a wider emission spectra. Using this method, the fluorescent signals from each fluorophore/ fluorescent protein increase, leading to a higher signal-to-noise ratio on unmixed images. Furthermore, since each pixel has two or more spectral patterns excited by multiple excitation lights, this method can unmix each fluorophore’s spectrum more efficiently (8). The procedure for multiple-excitation image acquisition is essentially the same as the single excitation method, except the filter settings are changed during acquisition of fluorescence imaging. Currently, several filter settings can be used for excitation during one image acquisition with the Maestro spectral-fluorescence imager (CRi) (e.g., blue, green, and red excitation).
3.4. Other Tumor Models
3.4.1. Subcutaneous Transplant Model
The strategies described above may also be used in subcutaneous- xenograft models of tumors. For example, we applied the counter- staining method to a subcutaneous-transplant model with interleukin-2α-receptor (IL-2Rα)-positive tumors (ATAC4 cells) and IL-2Rα-negative tumors (A431 cells) in the same mice (5). A431 cells were labeled with an endogenous fluorophore (RFP) and cloned to establish stable expression. ATAC4 cells and A431- RFP cells were injected subcutaneously in the left and right dorsum of female nude mice, respectively. After intravenous injection of an exogenous probe, daclizumab conjugated to ICG, ATAC4 tumors were depicted by only ICG spectral-fluorescence. A431- RFP was depicted with only RFP spectral fluorescence (counter- staining) (see Fig. 4).
Fig. 4.
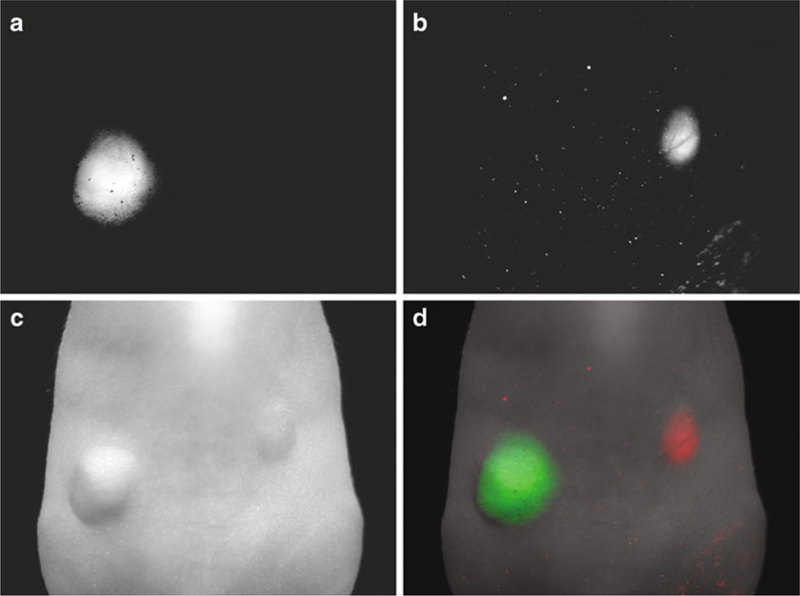
Subcutaneous-xenograft model identified with counter-staining. Spectral-fluorescence images of a mouse bearing ATAC4 (IL-2Rα+) and A431-RFP (IL-2Rα-) tumors receiving ICG-conjugated daclizumab (anti-IL-2Rα). Unmixed spectral fluorescence images illustrate (a) the ICG spectrum demonstrating the localization of ATAC4 tumors labeled with ICG conjugated daclizumab, (b) the RFP spectrum localizing A431 cells endogenously expressing RFP. (c) White light image, and (d) two-color overlay demonstrates that ATAC4 tumor (IL-2Rα+) is depicted only in the ICG spectrum (green), while a A431-RFP tumor is depicted only in the RFP spectrum (red) and does not show any fluorescence in ICG spectrum.
3.4.2. Two Tumor Peritoneal Dissemination Model
Co-staining and counter-staining methods can be performed simultaneously in a single mouse model. This has been demonstrated using our animal model of “co-incident” peritoneal cancer implants ( 6). Using two cancer cell lines, SHIN3 and SKOV3, which both express the β-D-galactose receptor (D-galR), but differ in their expression of another receptor (HER-2), we were able to specifically distinguish between the two cell populations in a single mouse. SHIN3 cells were transfected with a plasmid expressing RFP (D-galR+, HER2-, RFP+). SKOV3 cells (D-galR+, HER2+, RFP-) and SHIN3 cells were co-injected into nude mice to establish a coincident model of intraperitoneal dissemination. After establishing the tumor model, galactosyl serum albumin (GSA) conjugated to RhodG, which targets D-galR, and trastuzumab conjugated to Alexa680, which targets HER-2, were co-injected into the tumor-bearing mice. In vivo multi-excitation spectral imaging was carried out and each fluorescence signal was unmixed. SHIN3 cells were identified by fluorescence in the RFP and RhodG spectrum (co-staining), but no emission in the Alexa680 spectrum. SKOV3 cells were identified by fluorescence in the RhodG and Alexa680 spectrum, but no emission in the RFP spectrum (counter-staining). With this method, we were able to verify the multiplexed use of probes to distinguish between subpopulations of cancer cells in vivo. Theoretically, this method can be applied to other mouse models, including subcutaneous-xenograft and lung metastasis models.
3.5. Other Fluorescence Imaging Methods
The in vivo fluorescence imaging methods described above are performed using a spectral-fluorescence imager (Maestro, CRi). Theoretically, these strategies can be applied to other fluorescence imaging devices. For example, counter-staining was demonstrated with an in vivo real-time color fluorescence imager (FluorVivo, Indec) in a co-incident two-tumor peritoneal-dissemination model (SHIN3-RFP and SKOV3) (4). In this study, trastuzumab- RhodG was injected into the co-incident two-tumor model. Fluorescence-guided surgery was then performed. SHIN3-RFP tumors were depicted by their red fluorescence, while SKOV3 tumors were depicted by green fluorescence (labeled fluorescence) in real-time (counter-staining). Furthermore, if emission wavelengths of two fluorescence spectra can be distinguished, it may be possible to perform co-staining or counter-staining with conventional fluorescence imagers which can separate two fluorescent signals only by filter settings. However, it may require higher emission light yields and lower autofluorescence than are currently attainable to assess each tumor nodule accurately.
Footnotes
Notes
1.Selection of fluorescent proteins
In using single-excitation filter settings for co-staining and counter-staining methods, there are several characteristics to consider when choosing a combination of fluorescent proteins and fluorophores. First, both the fluorescent protein and fluorophore must be sufficiently excited by the chosen excitation light. Second, it is ideal to choose a fluorescent protein and fluorophore with emission spectra that are close enough to one another that their spectra will both fall into the predetermined acquisition spectrum. However, it should be possible to differentiate emission signals when both fluorophores have well-characterized emission spectra that are distinct from one another. Spectral fluorescence imaging is also effective at resolving two fluorophores with very similar peak emission spectra that have distinct spectral widths (9). When combining a fluorophore with a narrow spectral width with a fluorophore with a wide spectral width, even if both have the same peak emission wavelength, spectral imaging can be used to effectively differentiate between the two fluorophores.
This differs from real-time fluorescence imaging where the ideal fluorophores have distinct emission spectra (i.e., colors) with as little overlap as possible, but are still simultaneously excited by a single excitation filter. As fluorophores commonly exhibit considerable spectral overlap, it becomes more difficult to optically differentiate between a fluorescent protein and a fluorophore.
When using multiple excitation methods, the limitations in fluorophore selection can be reduced ( 8). However, one must still be careful that unmixed signals do not overlap one another, since this could yield confounding data due to crosstalk between fluorophores.
2.Unmixing procedure
In the unmixing process, it is necessary to compare an acquired signal relative to a reference spectral library that represents the “pure” fluorophore of interest. However, difficulty in unmixing two target fluorescence spectra correctly is occasionally encountered with spectral libraries, as described above. Fluorophores may undergo processing if taken into cells after binding their target. This may expose the fluorophores to different pH levels or enzymatic changes that may affect the fluorophore’s spectral map. A fluorophore may be exposed to different levels of tissue-dependent absorption or scatter that may result in a change in spectral map (10). Thus, it may sometimes be necessary to create a spectral library for a given fluorophore after it is already in the tissue of interest. In that case, it is acceptable to obtain a reference spectral library from an excised tumor nodule labeled with the fluorophore of interest.
After determining a spectral library, it is also important to confirm that there is no crosstalk between endogenous fluorescent proteins and exogenous fluorophores. In the above-described SHIN3-RFP and GmSA-RhodG experiment, mice bearing SHIN3-RFP tumors need to be sacrificed and imaged without addition of the exogenous fluorophore conjugated probe, and confirmation that a lack of fluorescence signal on RhodG spectral unmixed images must be made. Also, mice bearing SHIN3 (no RFP) tumors also need to be sacrificed and imaged after GmSA-RhodG administration, in order to confirm lack of fluorescence signal in RFP spectral unmixed images.
3.Weak fluorescence from fluorescent protein-labeled tumor nodules
Another possibility that may lead to a “false positive” is the unstable expression of fluorescent proteins in a transfected cell population. This causes weak fluorescence from tumor nodules, especially small tumor nodules that cannot be detected by spectral-fluorescence imaging. This may be an unavoidable limitation when using an endogenously-expressed fluorescent protein. However, using brighter fluorescent proteins and/or obtaining a new clone expressing a fluorescent protein more stably can minimize this limitation. Furthermore, in some circumstance, it is also acceptable to disregard very tiny tumor nodules from image analysis, although it depends on the purpose of the experiment. Even though cancer cells may express fluorescent proteins heterogeneously, larger tumor nodules containing exponentially more cancer cells, will naturally have enough fluorescent protein expression to be detected by spectral- fluorescence imaging.
References
- 1.Hama Y, Urano Y, Koyama Y, Kamiya M, Bernardo M, Paik RS, et al. (2006) In vivo spectral fluorescence imaging of submillimeter peritoneal cancer implants using a lectin- targeted optical agent. Neoplasia 8(7), 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kobayashi K, Hama Y, Koyama Y, Barrett T, Urano Y, Choyke P (2007) Whole-body multicolor spectrally resolved fluorescence imaging for development of target-specific optical contrast agents using genetically engineered probes. Proc SPIE 6449, 644914. [Google Scholar]
- 3.Hama Y, Urano Y, Koyama Y, Choyke PL, Kobayashi H (2007) d-galactose receptor-targeted in vivo spectral fluorescence imaging of peritoneal metastasis using galactosamine-conjugated serum albumin-rhodamine green. JBiomed Opt 12(5), 051501. [DOI] [PubMed] [Google Scholar]
- 4.Longmire M, Kosaka N, Ogawa M, Choyke PL, Kobayashi H (2009) Multicolor in vivo targeted imaging to guide real-time surgery of HER2-positive micrometastases in a two-tumor coincident model of ovarian cancer. Cancer Sci 100(6), 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogawa M, Kosaka N, Choyke PL, Kobayashi H (2009) In vivo molecular imaging of cancer with a quenching near-infrared fluorescent probe using conjugates of monoclonal antibodies and indocyanine green. Cancer Res 69(4), 1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosaka N, Ogawa M, Longmire MR, Choyke PL, Kobayashi H (2009) Multitargeted multi-color in vivo optical imaging in a model of disseminated peritoneal ovarian cancer. J Biomed Opt 14(1), 014023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koyama Y, Hama Y, Urano Y, Nguyen DM, Choyke PL, Kobayashi H (2007) Spectral fluorescence molecular imaging of lung metastases targeting HER2/neu. Clin Cancer Res 13(10), 2936–2945. [DOI] [PubMed] [Google Scholar]
- 8.Koyama Y, Barrett T, Hama Y, Ravizzini G, Choyke PL, Kobayashi H (2007) In vivo molecular imaging to diagnose and subtype tumors through receptor-targeted optically labeled monoclonal antibodies. Neoplasia 9(12), 1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neher R, Neher E (2004) Optimizing imaging parameters for the separation of multiple labels in a fluorescence image. J Microsc 213(Pt 1), 46–62. [DOI] [PubMed] [Google Scholar]
- 10.Mansfield JR, Gossage KW, Hoyt CC, Levenson RM (2005) Autofluorescence removal, multiplexing, and automated analysis methods for in-vivo fluorescence imaging. J Biomed Opt 10(4), 41207. [DOI] [PubMed] [Google Scholar]