

Abstract
Melanoma is one of the most highly mutated cancer types. To identify functional drivers of melanoma, we searched for cross-species conserved mutations utilizing a mouse melanoma model driven by loss of PTEN and CDKN2A, and identified mutations in Kras, Erbb3, and Ptpn11. PTPN11 encodes the SHP2 protein tyrosine phosphatase (PTP) that activates the RAS/RAF/MAPK pathway. Although PTPN11 is an oncogene in leukemia, lung, and breast cancers, its roles in melanoma are not clear. In this study, we found that PTPN11 is frequently activated in human melanoma specimens and cell lines and is required for full RAS/RAF/MAPK signaling activation in BRAF wild-type (either NRAS mutant or wild-type) melanoma cells. PTPN11 played oncogenic roles in melanoma by driving anchorage-independent colony formation and tumor growth. In Pten and Cdkn2a null mice, tet-inducible and melanocyte-specific PTPN11 E76K expression significantly enhanced melanoma tumorigenesis. Melanoma cells derived from this mouse model showed doxycycline-dependent tumor growth in nude mice. Silencing PTPN11 E76K expression by doxycycline withdrawal caused regression of established tumors by induction of apoptosis and senescence, and suppression of proliferation. Moreover, the PTPN11 inhibitor (SHP099) also caused regression of NRAS Q61K-mutant melanoma. Using a quantitative tyrosine phosphoproteomics approach, we identified GSK3α/β as one of the key substrates that were differentially tyrosine-phosphorylated in these experiments modulating PTPN11. This study demonstrates that PTPN11 plays oncogenic roles in melanoma and regulates RAS and GSK3β signaling pathways.
Keywords: melanoma, PTPN11, SHP2, SHP099, GSK3α/β
Introduction
Genetically engineered mouse models have been utilized to mine complex human genomic data to identify novel human cancer genes. Integration of high-resolution copy number profiles generated using mouse and human tumor genomes identified cross-species conserved genomic alterations, which led to the identification of cIAP and YAP1 as driver oncogenes in liver cancer (1) and NEDD9 in melanoma (2). Whole genome sequencing (WGS) analysis of a tumor developed in an acute promyelocytic leukemia (APL) mouse model uncovered recurrent somatic mutations in Jak1, pointing to the importance of the JAK/STAT pathway in pathogenesis of human APL (3). These studies have demonstrated that mice and humans sustain syntenic genetic events in the development of cancer, that mouse-human comparative oncogenomics can facilitate the discovery of new cancer genes that were not immediately obvious from the human cancer genome alone, and that the mouse models may be utilized to gain insights into the function of these new cancer genes.
Melanoma displays frequent activation of RAS/RAF/MAPK and PI3K/AKT signaling pathways, as well as inactivation of PTEN and CDKN2A (INK4A/ARF) tumor suppressors via genetic and epigenetic alterations. The high frequency of activating mutations in BRAF (40~60%) and NRAS (15~25%) support the importance of the RAS/RAF/MEK/ERK pathway in melanoma (4,5). Several recent genomic studies have identified mutations in genes regulating the RAS pathway such as RasGAPs (NF1, RASA1, RASA2) and PTPN11 in melanoma (4,6–10). These Ras regulators have the potential of being exploited as therapeutic targets.
Protein-Tyrosine Phosphatase (PTP), Non-Receptor Type 11 (PTPN11, also known as SHP2) is ubiquitously expressed in various tissues and cell types, and activates the Ras signaling pathway as downstream of most, if not all, receptor tyrosine kinases (RTKs). PTPN11 contains two SH2 domains and a PTP domain. PTPN11 is auto-inhibited via a SH2-PTP domain interaction and is activated by binding to tyrosine-phosphorylated RTKs, immune inhibitory receptors, or docking proteins (such as Gab1/Gab2, IRS, FRS) and by point mutations that disrupt the SH2-PTP domain interaction. RTKs, such as EPHA2, phosphorylate Y542 and Y580 on PTPN11, which prolongs ERK activation by maintaining PTPN11’s open conformation (11). Mutations in PTPN11 have been linked to Noonan and LEOPARD syndromes and pathogenesis of multiple cancer types. Oncogenic roles of PTPN11 in leukemia, lung, and breast cancers have been established with PTPN11 regulating invasion, metastasis, apoptosis, senescence, DNA damage, cell proliferation, cell cycle progression, and drug resistance (12,13). However, PTPN11 has tumor suppressive roles in liver cancer (14) and in bone/cartilage cancer (15), supporting PTPN11’s cell context dependent effects.
PTPN11 protein tyrosine phosphatase (PTP) activity regulates several molecules involved in Ras signaling (16). Specifically, PTPN11 negatively regulates RasGAP recruitment by dephosphorylating RTKs (e.g., EGFR and PDGFRβ), leading to activation of Ras signaling. PTPN11 also dephosphorylates PAG (phosphoprotein associated with glycosphingolipid-enriched membrane microdomains) and paxillin, resulting in the release of CSK (C-terminal Src kinase) and subsequent activation of Src family kinases (SFKs). A recent study by Bunda et al. showed dephosphorylation and activation of Ras by PTPN11 (17). Additional targets of PTPN11 include Sprouty, signal regulatory protein-α (SRPα), and protein zero-related (PZR). Although the list of PTPN11 substrates continues to grow, it is possible that PTPN11 dephosphorylates differential sets of substrates depending on the cell context in each cancer type.
In this study, to identify functional driver mutations of melanoma, we analyzed melanoma genomes from a mouse model driven by loss of CDKN2A (INK4A/ARF) and PTEN (IP model), commonly observed alterations in human melanoma patients, by whole exome sequencing (WES). This study identified several cross-species orthologous mutations, including those in Kras and Erbb3 that are implicated in melanomagenesis. Notably, we also identified S506P mutation in Ptpn11, which is orthologous to an activating S502P mutation in human PTPN11. Although implicated, the role of PTPN11 in melanoma tumorigenesis and its utility as a therapeutic target has not been fully addressed in melanoma. Our work indicates that PTPN11 is frequently activated in human melanoma specimens, plays oncogenic roles in melanoma, is required for full activation of the RAS/RAF/MAPK signaling pathway in BRAF wt melanoma, and is a potential therapeutic target. We also identified GSK3α/β as one of the key targets of PTPN11 that regulates β-catenin, cyclin D1, and others.
Materials and methods
Detailed materials and methods can be found in Supplementary Information.
Mouse models and allograft studies
PA662T cells (containing vector control, PTPN11 wt, or E76K, 2.3 million cells/injection), W331 (1.5 million cells), 5037 (2 million cells, all on doxy) were inoculated subcutaneously into seven-week-old female nude mice (Crl:NU-Foxn1 Nu/Nu). For 5037, when tumors reached 200 mm3, mice were randomly enrolled onto vehicle control (1% CMC (carboxymethyl cellulose)/0.5% Tween-80), MEK162 (25mg/kg, p.o., b.i.d.), or SHP099 (100mg/kg, p.o., q.d.) treatment groups. All animals were maintained according to the guidelines of the Comparative Medicine Department of the University of South Florida (M4473 and R3312).
Whole exome sequencing analysis of mouse melanomas with matched normal tissue
Genomic DNA from 3 melanomas developed in IP mice with matched normal tissue (kidney or tail) were subjected to whole exome capture and paired end sequencing on Illumina Hi-Seq 2000 sequencer by Beckman Coulter Genomics, Inc. Overall average coverage of 63.5X was achieved. Sequence reads obtained were aligned to the mm10 mouse reference sequence (C57BL/6J strain) using the Burrows-Wheeler Alignment tool (BWA) (18). Known SNPs of FVB/N mouse strain (strain background of the IP model) from v.3 of the Mouse Genomes Project (19) and variants mapped outside of targeted region were removed. Utilizing Strelka (20), somatic single nucleotide variants (SNVs) and small indels were identified from the aligned sequencing reads of matched tumor-normal samples.
Phosphoproteomic analysis
Tumors were homogenized in denaturing buffer containing 8M urea and clarified by centrifugation. Proteins were reduced, alkylated, and digested overnight with trypsin (Worthington). After buffer exchange, tyrosine phosphorylated peptides were immunoprecipitated using anti-phosphotyrosine antibody beads (p-Tyr-100), followed by analysis on liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) peptide sequencing (RSLC-QExactive Plus, Thermo), as previously described (21). Label free quantification by MaxQuant (22) produced 245 molecularly defined tyrosine phosphorylation sites with relative quantification for the levels of phosphorylation. LIMMA analysis was then performed to identify differentially phosphorylated peptides and proteins (23).
Patient samples
This study utilized archived metastatic melanoma samples that were collected under the Total Cancer Care (TCC) protocol from the consented patients at Moffitt Cancer Center. Frozen tissues (BRAF/MEK targeted therapy naïve) were obtained following de-identification (n=38). This study received expedited approval by the Institutional Review Board at the University of South Florida: Category 5 (45CFR46.110 and 21 CFR 56.110).
Result
Whole-exome sequencing analysis of murine melanomas identified cross-species conserved alterations
Loss of Cdkn2a and Pten are commonly observed alterations in human melanomas. To identify evolutionarily conserved alterations driving melanoma, we analyzed melanomas that developed in IP mice (Cdkn2a(Ink4a/Arf) L/L; PtenL/L; Tyr-CreERT2). We confirmed deletion of Cdkn2a (Ink4a/Arf) (24) and Pten (25) upon 4-OHT (4-hydroxytamoxifen) topical application by the well-characterized Tyr-CreERT2 allele (26) (Fig. 1). These mice developed cutaneous and ocular melanomas following 4-OHT treatment with long latency (median melanoma free survival: 425 days) and low penetrance (n=4/28), suggesting that the acquisition of the additional genetic alterations is required to drive melanoma formation in these mice.
Figure 1. Identification of oncogenic mutations arising spontaneously in Cdkn2a (Ink4a/Arf) and Pten null (IP) tumors.
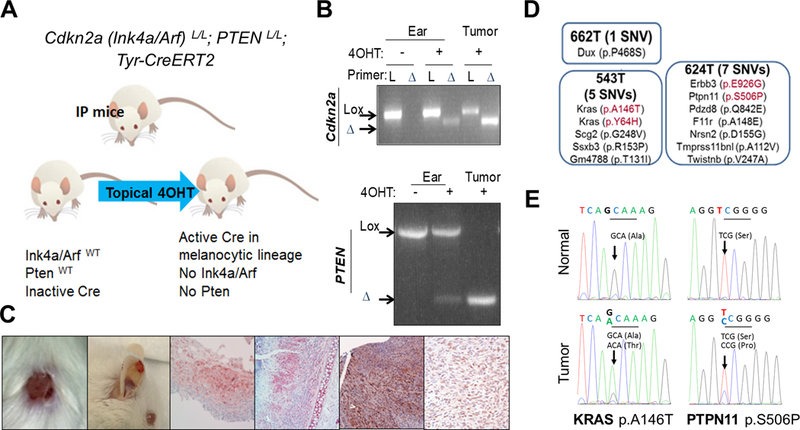
An illustration of the IP mouse model (A) where topical 4OHT treatment induces Cre activation within melanocytes and subsequent Ink4a/Arf and Pten deletion (B: PCR analysis of ear and tumor DNA). Gross and histological representations of melanomas arising in IP mice (C). Results of whole exome sequencing performed on melanoma and matched normal tissue samples from IP mice (D). KRAS A146T and Ptpn11 S506P mutation is confirmed by Sanger sequencing in melanoma, but not in matched normal DNA of PA543 and PA624 mice, respectively (E).
Whole exome sequencing analyses of cutaneous (ear, back skin) and genital melanomas (PA662T, PA543T, and PA624T) along with matched normal tissues identified 16 nonsynonymous single nucleotide variants and 1 frame shift mutation that caused coding changes in 16 genes (Supplementary Fig. S1 and Supplementary table 1). This list includes several orthologous mutations observed in human tumors (Fig. 1D). For example, Kras A146T and Y64H are orthologous to KRAS A146T and Y64H, Erbb3 E926G to ERBB3 E928G, and Ptpn11 S506P to PTPN11 S502P. Functionally, KRAS A146T was shown to increase active Ras-GTP compared to wild type KRAS (27) and ERBB3 E928G was shown to transform colonic and breast epithelial cells in a ligand-independent manner in collaboration with ERBB2 (28). KRAS A146 and Y64 mutations, ERBB3 E928 mutation, and PTPN11 S502 mutation are recurring mutations in many large-scale integrative cancer studies (29,30). These mutations were confirmed by Sanger sequencing (Fig. 1E for Kras and Ptpn11); since KRAS and ERBB3 have already been implicated in melanoma tumorigenesis, we focused on PTPN11.
PTPN11 is frequently activated in melanoma and activates ERK in BRAF wt melanomas
PTPN11 is frequently mutated in human cancers with mutational hot spots such as E76, S502, G503, and Q510 (Fig. 2A). S502 in the PTP domain interacts with E76 in the SH2 domain (Supplementary Fig. S2C), leading to auto inhibition of PTPN11, and mutations in S502 or E76 were shown to activate PTPN11 (13,31). PTPN11 showed a 2~3% mutation rate in melanoma. PTPN11 mutations identified in human melanoma in multiple cancer studies (4,7,10,32) are listed in Supplementary Fig. S2A. Although the mutation rate is low across melanoma samples, PTPN11 can be activated by RTKs or docking proteins (such as Gab1/Gab2). To address the activation status of PTPN11 in melanoma, we tested the expression level and phosphorylation of PTPN11 in metastatic melanoma specimens and cell lines. We observed phosphorylation on Tyr 542 of PTPN11 in 40% (n=15/38, Fig. 2B) of melanoma specimens and the majority of human melanoma cell lines (Fig. 2C), supporting frequent activation of PTPN11 in human melanoma. Immunohistochemical (IHC) staining of human melanoma tissues with pPTPN11 (Tyr542) showed the staining in the tumor cells (Supplementary Fig. S3). Reduced PTPN11 expression via siRNAs (A and B) decreased ERK activation (Fig. 2D) in NRAS mutant (WM1361A, WM1366, WM1346) and BRAF/NRAS wt (MeWo, WM3211, CHL1) melanoma cells, but not in BRAF mutant (1205Lu, IGR1, WM983C) cells. These results support that PTPN11 is required for full ERK activation in BRAF wild-type melanoma cells (including NRAS mutant cells), possibly due to its roles in Ras activation downstream of activated RTKs. We also observed suppression of NRAS activation by PTPN11 knock-down (KD) in NRAS mutant WM1366 and WM1361A (Fig. 2E).
Figure 2. PTPN11 is activated in human melanoma and regulates ERK phosphorylation in BRAF wt melanoma cell lines.
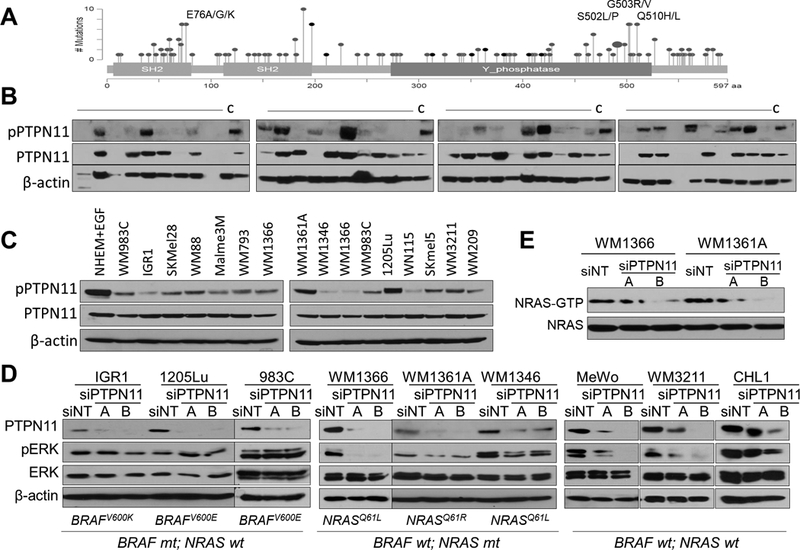
Mutations identified in human cancers (adapted from cBioPortal): SH2 and Y phosphatase domains; Gray: missense mutation; Black: in frame deletion (A). Western blot analysis of human metastatic melanoma specimens and WM793 cell lysates (“C”) as a control (B) and melanoma cell lines (C) with pPTPN11 (Y542), PTPN11, and β-actin antibodies. The effect of PTPN11 KD with siRNAs (A & B) on ERK activation in BRAF or NRAS mutant and BRAF/NRAS wt melanoma cells (D) and NRAS activation ((RAS-GTP pull down detected with NRAS antibody) in NRAS mutant cells (E).
PTPN11 plays oncogenic roles in BRAF wt melanomas
Functionally, knock-down of Ptpn11 via shRNAs in PA624T (murine melanomas cells from IP mice) harboring the Ptpn11S506P mutant reduced and ectopic expression of activated mutant PTPN11 (E76K, G503V, S502P) in melanoma cells derived from PA662T (murine melanomas cells from IP mice) increased anchorage-independent growth and ERK activation (Fig. 3A&B and Supplementary Fig. S4). This ERK activation by PTPN11E76K expression was associated with increased activation of Ras, c-Raf, and MEK (Supplementary Fig. S5). PTPN11 knock-down also suppressed anchorage-independent growth and ERK activation in WM3211 and MeWo human melanoma cell lines (Supplementary Fig. S6). Moreover, PTPN11E76K expression (n=14/14) in PA662T melanoma cells increased subcutaneous tumor growth in nude mice compared to control (n=0/12) or PTPN11 wt (n=4/12) cells (Fig. 3C). Although none of the control mice developed tumors by 52 days, control animals kept alive developed small tumors after an additional 2 months (day 112, n=8/12). PTPN11 E76K expression (and wt PTPN11 expression with milder effect) increased pERK and proliferation (p-Histone 3 (pH3)) and decreased apoptosis (cleaved caspase 3) (Fig. 3D). These data support the oncogenic roles of PTPN11 in melanoma through RAS activation leading to RAF, MEK, and ERK signaling.
Figure 3. PTPN11 expression regulates growth of cells in soft agar and as xenografts.
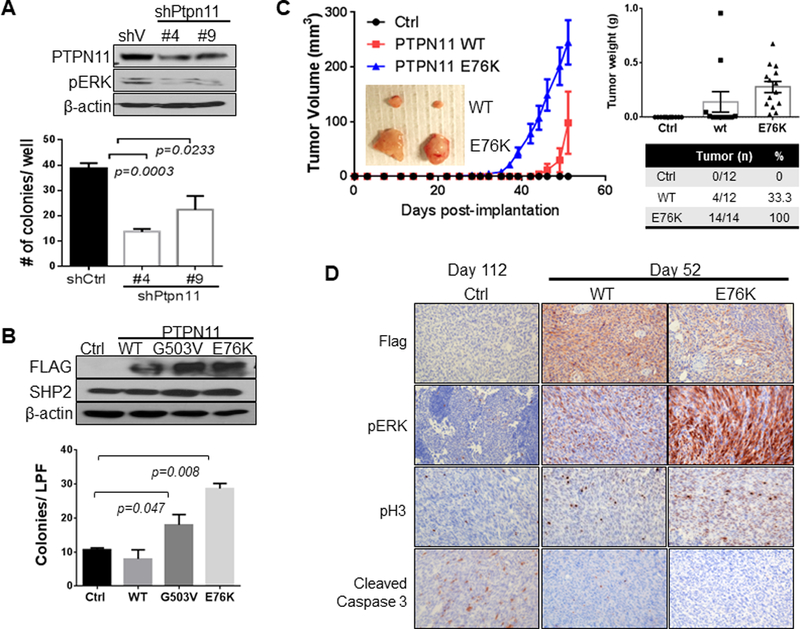
PA624T and PA662T cells are murine melanomas cells from Cdkn2a(Ink4a/Arf) L/L; PtenL/L; Tyr-CreERT2 mice. Western analysis of PA624T with vector control or shRNAs (#4 and #9) targeting Ptpn11 (A top) and PA662T with vector control, wt, G503V, or E76K mutant PTPN11 expression (B top). Number of colonies grown in soft agar (A&B, bottom, seeded in triplicate) is shown graphically as mean +/− SD (p-value: two-tailed T-test). Subcutaneous tumor growth in nude mice implanted with PA662T cells expressing control, wt, or E76K PTPN11 over the time course (mean tumor volume +/− SEM, C). Significant difference (p<0.0001) is determined by two-way ANOVA comparing E76K with both control and wt on day 42 and comparing wt and control on day 52. Immunohistochemical (IHC) analysis of tumors for PTPN11 (flag Ab), pHistone H3 (pH3), pERK, and cleaved caspase 3 (D).
PTPN11E76K expression is required for melanoma initiation and maintenance in Pten and Cdkn2a (Ink4a/Arf) null mice
To test whether PTPN11 activation can drive melanoma tumorigenesis, bitransgenic (BT: Tyr-rtTA; tetO-PTPN11E76K) and control monotransgenic (MT: Tyr-rtTA)) mice were generated in a Cdkn2a(Ink4a/Arf)−/−; PtenL/L; Tyr-CreERT2 genetic context (Fig. 4). When treated with 4-OHT and fed with doxycycline (hereinafter doxy), 5 out of 7 BT mice (median melanoma free survival: 183 days) developed melanomas compared to 1 out of 9 MT mice within ~200 days (p=0.009, Log-rank test). These melanomas are positive for S100, Flag (PTPN11 E76K), pPTPN11 (Y542), and pERK, supporting the role of PTPN11 activation in melanoma tumorigenesis (Fig. 4B). Similarly, PTPN11 E76K transgene expression in IP mice led to development of melanoma (n=7/41) when placed on doxy with longer latency, possibly due to incomplete deletion of Cdkn2a (Fig. 1B). However, none of the IP mice without the PTPN11 E76K transgene on doxy nor any mice on normal chow developed melanoma (p=0.0016, Log-rank test) (Supplementary Fig. S7A).
Figure 4. PTPN11 E76K is required for growth and maintenance of tumors in mice.
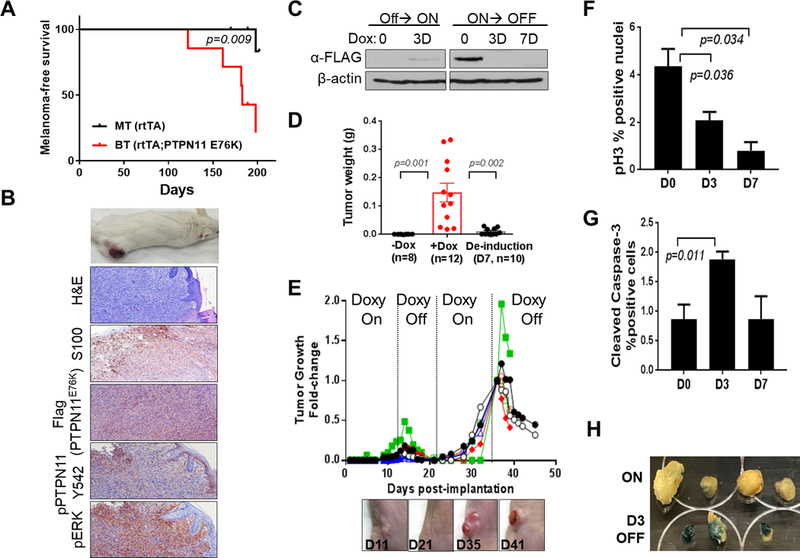
Kaplan-Meier melanoma free survival curves of BT (PTPN11 E76K positive) and MT (rtTA only) mice on doxy (A). Representative images of a cutaneous melanoma developed in a BT mouse, H&E, and IHC staining for S100, Flag, pPTPN11 Y542 and pERK (B). Western blot analysis of W331 cells derived from a BT melanoma (Tyr-rtTA; tetO-PTPN11 E76K;Cdkn2a(Ink4a/Arf) L/L; PtenL/L; Tyr-CreERT2) (C). Weights of W331 allograft tumors (mean tumor weight (g) +/− SEM) grown in nude mice fed with (+Dox) or without (-Dox) doxy on day 11 following implantation (p=0.001; +Dox vs. -Dox group) or deinduced for 7 days following growth on doxy for 11 days (p=0.002; De-induction vs. +Dox group) (D). Repeated cycles of doxy induction/de-induction (PTPN11 E76K expression On/Off) drive growth and regression of s.c. tumors (E). Quantification of p-Histone H3 (F) and cleaved caspase 3 (G) by IHC analysis on tumor sections following PTPN11 de-induction on D0, D3, and D7. Senescence associated β-galactosidase staining (blue) of tumors on D0 and D3 (H). p-value is calculated using two-tailed t-test for D, F, and G.
To test whether PTPN11 can serve as a therapeutic target for melanoma patients, we addressed the continued requirement of PTPN11 activity in established tumors. We generated a cell line (W331) from a melanoma developed in a Tyr-rtTA; tetO-PTPN11E76K; Cdkn2a(Ink4a/Arf) L/L; PtenL/L; Tyr-CreERT2 mouse (Fig. 4C). When implanted subcutaneously into nude mice, all mice fed with doxy showed tumor growth (n=12), while none of the mice on normal diet developed tumors (n=10) on Day 11 following implantation. Moreover, withdrawal of doxy and subsequent extinction of PTPN11 E76K expression led to regression of tumors (n=10, de-induced for 7 days following 11 days of induction, Fig. 4D). Complete regression of tumors was observed in additional mice kept off doxy (n=10) on day 10 following doxy withdrawal (Fig. 4E). Re-induction of PTPN11 E76K expression by doxy re-administration caused growth of the tumor on the same spot (n=8), which showed regression following doxy withdrawal (Fig. 4E, n=6). Tumor regression was associated with ERK inactivation, decreased proliferation, increased apoptosis, and increased senescence (Fig. 4F–H and Supplementary Fig. S8A&B). Doxy withdrawal caused regression of a primary tumor developed in BT mice as well (Supplementary Fig. S7B). Therefore, activated PTPN11 E76K drives melanoma growth and is required for maintenance of established tumors.
Phosphoproteomics approach identifies peptides differentially tyrosine phosphorylated by PTPN11
To identify downstream targets of PTPN11, we analyzed protein lysates from allograft tumors generated with PA662T expressing vector control, wt, or E76K PTPN11 (Fig. 3C) and with W331 on (D0) or 3 Day off (D3 de-induced) doxy (Fig. 4). Tyrosine (tyr) phosphorylated peptides were immunoprecipitated and analyzed with liquid chromatography-tandem mass spectrometry (LC-MS/MS) peptide sequencing and quantified using the peaks heights of each peptide ion signal (MaxQuant). Overlapping alterations were observed both in PA662 (E76K vs. ctrl, y axis) and W331 (Dox vs. de-induced, x axis) (Fig. 5A), many of which were also regulated by wt PTPN11 (Supplementary Table 2 and Supplementary Fig. S9B). Total 13 of 245 peptides were identified as differentially phosphorylated by PTPN11 (p<0.05), including Dyrk2/4, Cltc, Prap, Nck2, and Ptprc as decreased and Gab1 as increased peptides (Supplementary Fig. S9C). Supplementary Fig. S9D shows peptides with the highest fold changes (ex, Prpf4b, Gsk3α/β, Dyrk1a/1b, Grlf1, Pik3r1/r3, and Dyrk2/4).
Figure 5. Phosphotyrosine proteomic analysis of PTPN11 expressing tumors identifies candidate substrates of PTPN11, including GSK3α/β.
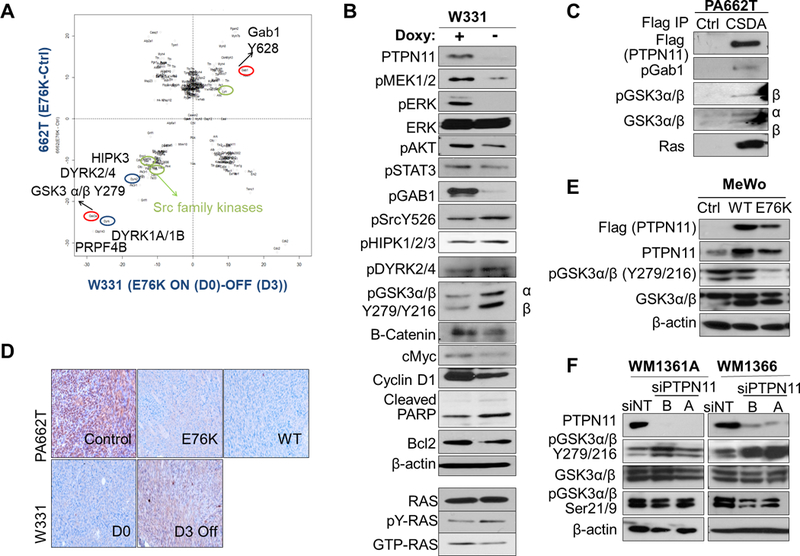
A. Normalized scatter plot of the log2 values of the median (E76K/ctrl) ratios for individual phosphosites. Peptides changed in PA662T (Cdkn2aL/L; PtenL/L;Tyr-CreERT2) transduced with E76K vs. vector ctrl are shown along the y axis and ones altered in W331 (Tyr-rtTA;tetO-PTPN11E76K; Cdkn2aL/L; PtenL/L;Tyr-CreERT2) on doxy (D0, E76K+) vs. de-induced (D3, E76K-) are on the x axis. B. Western blot analysis of W331 cells with or without doxy with indicated antibodies. Immunoblotting with Ras antibody detected the level of total, activated (Ras-GTP pull-down), and tyr phosphorylated (pTyr pull down) Ras in W331. C. Co-immunoprecipitation using anti-FLAG beads in control or CSDA-PTPN11-Flag (trapping mutant) expressing 662T cells were immunoblotted with antibodies against pGab1, pY-GSK3, total GSK3 and Ras. D. IHC staining of pY-GSK3 in 662T control, E76K, and wt PTPN11 expressing tumors and in W331 tumors (D0 and D3). Western analysis of human melanoma cell lines MeWo (E, BRAF/NRAS wt) with PTPN11 (wt or E76K mutant) and WM1361A and 1366 (F, NRAS mutant) with PTPN11 KD (siRNAs A, B) compared to controls.
While pGab1 activates PTPN11, PTPN11 was shown to modulate a positive feedback loop and activate Gab1 to regulate the PI3K/AKT pathway (33). In this murine model, we also observed increased pGab1 (Y628 (Y627 in human)) by PTPN11 in W331. Reduced tyrosine phosphorylation of pGSK3α/β (Y279/Y216, hereinafter pY-GSK3), pSrc (Y526), pDYRK2/4 (Y380/Y389 (Y382/Y264 in human)), and pHIPK3 (Y359) by PTPN11 expression was confirmed by Western blot in W331 cells (Fig. 5B). To identify substrates of PTPN11, we utilized the PTPN11 CSDA mutant, which is PTP defective and traps substrates. In PA662T cells, PTPN11 CSDA mutant physically interacted with GSK3α/β, pGab1, and Ras (Fig. 5C and Supplementary Fig. S9E).
Phosphorylation of GSK3α/β (hereinafter GSK3) at Ser 9 and Thr 43 inactivates, while Tyr279/216 phosphorylation activates, this kinase. Decreased pY-GSK3 by PTPN11 was confirmed on W331 and PA662T tumors (Fig. 5D). Moreover, wt or E76K mutant PTPN11 expression in a human melanoma cell line, MeWo, decreased, while PTPN11 knock-down in WM1366 and WM1361A increased, pY-GSK3 (Fig. 5E&F), supporting GSK3 as a downstream target of PTPN11. DYRKs and HIPKs are known to serve as priming kinases for substrates of GSK3 such as c-Myc, cyclin D1, and c-Jun, regulating their protein levels. When PTPN11 is de-induced in W331 cells, protein levels of c-Myc, cyclin D1, and β-catenin, known downstream targets of GSK3, were decreased (Fig. 5B). Thus, PTPN11 dephosphorylates and inactivates GSK3, which may contribute to the increased level of β–catenin, c-Myc, and cyclin D1.
PTPN11 inhibition led to regression of NRAS Q61K; Cdkn2a−/− melanoma, increased pY-GSK3, and decreased pERK and cyclin D1
We tested the effect of pharmacological inhibition of PTPN11 in melanoma. PTPN11 inhibitor (SHP099 (34)) treatment significantly reduced cell viability of 5037 and 2187 cells in a dose-dependent manner; these mouse melanoma cells are derived from NRAS Q61K; Cdkn2a−/− mice (35) (Fig. 6A). In addition, when treated with SHP099 (100 mg/kg), the majority of subcutaneous tumors generated with 5037 cells (n=16/18) demonstrated a significant reduction in tumor volume (average D3/D0 ratio=0.52+/−0.04 SEM) on day 3 followed by a cytostatic response (D8/D0 ratio=0.46+/−0.05), while tumor volume continuously increased in the vehicle control group (n=16, 2.1 fold+/− 0.22 on D3 and 4.2+/−0.48 on D8) (Fig. 6B and Supplementary Fig. S10A). Tumor weight was significantly lower in the SHP099 treated group (n=8) compared to the vehicle treated group (n=7) on day 8 (0.12 +/−0.03 g vs. 0.74 +/−0.08 g, p=0.0003) (Fig. 6C). This rapid tumor regression was associated with decreased ERK activation and proliferation as well as increased apoptosis on day 3 followed by a cytostatic response associated with partial re-activation of ERK on day 8 (Fig. 6E and Supplementary Fig. S10B). Two out of 18 tumors treated with SHP099 showed tumor stasis without regression and eventually started to grow on treatment (PR: partial-responder, Fig. 6D and Supplementary Fig. S10A). PR tumors showed higher level of pERK and proliferation compared to responder tumors.
Figure 6. Pharmacological inhibition of PTPN11 in mouse melanoma cells with NRAS Q61K induces tumor regression in part through regulation of GSK3 and cyclin D1.
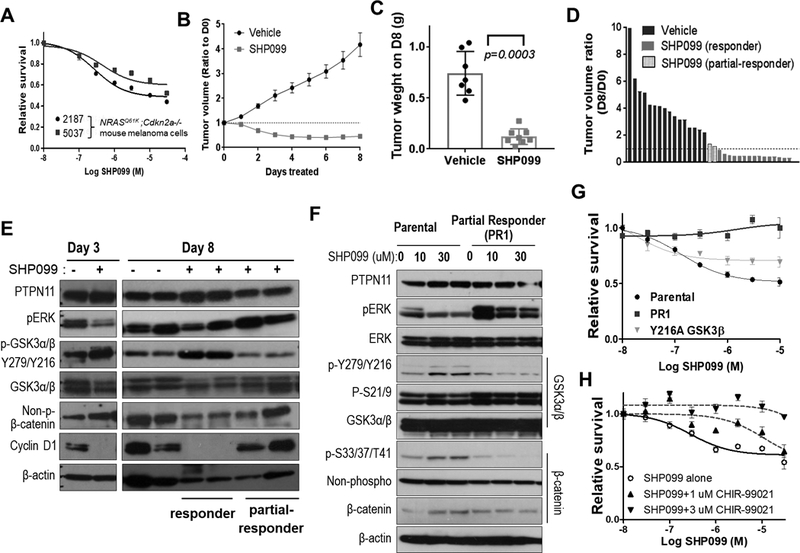
Survival analysis of 2187 and 5037 (mouse melanoma cells with Tyr-rtTA; tetO-NRAS Q61K; Cdkn2a (Ink4a/Arf) −/−) following treatment with increasing concentrations of SHP099 at 72 hours (A). Decreased tumor volume following treatment of established subcutaneous allograft 5037 tumors with SHP099 (PTPN11 inhibitor, 100 mg/kg; po qd) compared to treatment with vehicle alone. Data shown as mean tumor volume ratio to day 0 +/− SEM (B) and final tumor weight at harvest (p=0.0003; two-tailed t-test) on day 8 (C). Waterfall plot demonstrating ratio of D8 to D0 tumor volume for tumors treated with vehicle or SHP099. SHP099 treated tumor segregated into (red) responders and partial-responders (blue) with and without regression, respectively (D). Western analysis of 5037 subcutaneous tumors with (+) or without (–) SHP099 treatment for 3 or 8 days (E). Western analysis of parental 5037 and a cell line established from a partial responding tumor (PR1; blue in panel D) treated with 0, 10, or 30 uM SHP099 for 3 hrs (F). Cell survival was assessed at 72 hrs of post-treatment as the mean +/−SD survival (relative to DMSO or GSK3β inhibitor CHIR-99021 alone) (G&H). The response of 5037 with vector control or GSK3β-Y216A mutant and PR1 cells to SHP099 (G) and that of 5037 parental cells to increasing concentrations of SHP099 alone or in combination with 1μM or 3μM CHIR-99021 (H) were shown.
To address molecular mechanisms, we compared signaling responses to SHP099 in responder vs. partial responder (PR) as well as parental vs. PR1 cells that were derived from PR. In addition to a decline of pERK, PTPN11 inhibition with SHP099 increased pY-GSK3 and decreased non-phospho β-catenin (stabilized form) and cyclin D1 in responder tumors compared to vehicle control tumors, but not in PR on day 8 (Fig. 6E). SHP099 did not cause cell death or increase pY-GSK3 and p-S33/37/T41-β–catenin in PR1 cells; although, SHP099 did partially decrease pERK (Fig. 6F&G). GSK3 is known to phosphorylate Ser33, Ser37, and Thr41 on β–catenin (36). To test the functional impact of Y216 phosphorylation of GSK3β, we expressed phosphorylation defective Y216A or kinase-dead K85A mutant forms of GSK3β in 5037 and W331 cells and observed decreased p-S33/37/T41-β–catenin and increased pERK (Supplementary Fig. S11). These mutant forms of GSK3β increased anchorage-independent colony growth in soft agar independent of PTPN11 induction in W331 cells (Supplementary Fig. S11B). Thus, GSK3 inactivation by loss of kinase activity or dephosphorylation by PTPN11 promotes colony growth. Moreover, expression of GSK3β Y216A mutant in 5037 cells partially suppressed response to SHP099 (Fig. 6G and Supplementary Fig. S11A&C), increased cyclin D1, and decreased p-S33/37/T41-β–catenin. Inhibition of GSK3 with CHIR-99021 suppressed response to SHP099 in a dose-dependent manner (Fig. 6H). Up-regulation of pY-GSK3 and p-S33/37/T41-β–catenin upon SHP099 treatment was abrogated by CHIR-99021, suggesting the requirement of GSK3 function for response to SHP099 (Supplementary Fig. S11D). Inhibition of MEK (MEK162, 50mg/kg, twice daily, Supplementary Fig. S12) in tumors generated with 5037 cells elicited only a cytostatic effect. Although MEK162 also suppressed pERK, it failed to increase pY-GSK3 or decrease cyclin D1, suggesting the importance of cyclin D1 regulation by GSK3α/β. These data combined support PTPN11 as an important regulator of the RAS/RAF/MAPK and GSK3/ β-catenin/cyclinD1 pathways.
Discussion
Identification of genes and pathways targeted both in mouse and human cancers support their possible key roles in tumorigenesis. Our study identified cross-species conserved orthologous mutations in Kras, Erbb3, and Ptpn11. While implicated, the role of PTPN11 in melanoma tumorigenesis and its utility as a therapeutic target has not been fully addressed in melanoma. Here, we have shown the frequent activation of PTPN11 in human melanoma cell lines and tumor specimens and the oncogenic roles of PTPN11 in RAS/RAF/MAPK signaling activation in BRAF wt melanoma (either NRAS mutant or wt). We generated a tet-inducible, melanocyte-specific, PTPN11 E76K transgenic mouse model in a PTEN and CDKN2A null background and have shown that PTPN11 activation drives melanoma formation and is required for tumor maintenance. These findings establish oncogenic roles for PTPN11 in melanoma tumorigenesis and support its value as a therapeutic target.
Several studies established the role of PTPN11 in both intrinsic and acquired vemurafenib resistance and the requirement of PTPN11 in cancers with activated RTKs, but not in the context of treatment-naive BRAF mutant or KRAS mutant cancers (34,37,38). However, our study supports the possible therapeutic effect of PTPN11 inhibition on NRAS mutant, in addition to NRAS/BRAF wt, melanomas. Additionally, recent studies showed that activated KRAS mutant tumors depend on PTPN11, and its inhibition suppressed GTP loading on Ras and suppressed tumor growth (39,40). Either wt or mutant forms of Ras are shown to be phosphorylated on conserved Y32 by SFKs, which interrupts its interaction with RAF and promotes GTP hydrolysis (41), and its dephosphorylation by PTPN11 is required for the re-activation of Ras (17). We also observed co-immunoprecipitation of RAS with PTPN11 and decreased tyrosine phosphorylation and activation of RAS in W331 cells with PTPN11 expression (Fig. 5). NRAS-GTP was suppressed by PTPN11 knock-down in NRAS mutant cells (Fig. 2E), supporting the roles of PTPN11 in regulating Ras activation in melanoma.
Efforts are underway to develop PTPN11 inhibitors as novel targeted therapy drugs. PTPN11 active site catalytic inhibitors often have poor bioavailability and low selectivity. Circumventing these problems, several allosteric PTPN11 inhibitors, which stabilize PTPN11 in an auto-inhibited confirmation, have been developed including SHP099 (34). A phase I clinical trial with an allosteric PTPN11 inhibitor (TNO155) is currently ongoing for patients with advanced EGFR-mutant non-small cell lung cancer, head-and-neck squamous cell cancer, or RAS/RAF wt other solid tumors. Based on the recent findings, PTPN11 targeted therapy could be expected to include Ras mutant tumors in the future. In addition, it will be necessary to identify which drug combination would elicit clinical efficacy by reducing adaptive feedback in response to inhibition and to delay or suppress emergence of resistance. MEK inhibitors were shown to activate RTKs and thus PTPN11 due to a feedback loop, providing a strong rationale for dual MEK/PTPN11 targeting. In fact, combined inhibition of MEK and PTPN11 induced regression of KRAS mutant (39,40) or KRAS amplified tumors (42). We also observed that SHP099 sensitized NRAS mutant WM1366 cells to MEK inhibitor, eliciting stronger ERK suppression and cell death both in 2D (at 72 hours) and 3D spheroids (Supplementary Fig S13). Further study will establish the possible benefit of a MEK/PTPN11 co-targeting strategy in melanoma.
The signaling mechanisms and PTP substrates of PTPN11 in melanoma are not well understood. This understanding is particularly important to predict response and resistance to PTPN11 inhibitors and to develop rational co-targeting strategies. In this study, we identified pY-GSK3 as novel downstream targets of PTPN11, of which dephosphorylation by PTPN11 mediates growth on soft agar and survival. Some studies suggest that GSK3 is constitutively autophosphorylated at Tyr 279/216 (43), while others show that apoptotic stimuli induce pY-GSK3 phosphorylation within the nucleus by Fyn and PYK2 (36,44). Our study shows that PTPN11 dephosphorylates Y279/216 on GSK3. Although these cells show high levels of pSer21/9- GSK3 due to loss of PTEN or NRAS activation, tyrosine dephosphorylation of GSK3, which may affect its subcellular localization and protein complex formation, suppresses its activities and provides growth and survival benefits.
The glycogen synthase kinase-3, GSK3, gene family consists of two ubiquitously expressed and highly conserved members, GSK3α and GSK3β. GSK3 is a serine/threonine kinase involved in control of metabolism, growth, development, and oncogenesis by regulating Wnt/β-catenin, PI3K/PTEN/AKT, RAS/RAF/MAPK, hedgehog, Notch and other signaling pathways. Thus, aberrant activities of GSK3 have been implicated in multiple diseases including cancer, Alzheimer’s, Parkinson’s disease, diabetes, and obesity (36). GSK3 is reported to have both tumor promoting (glioblastoma, pancreatic, ovarian, and blood cancers) and tumor suppressive (breast and skin cancers) roles (45). GSK3 regulates expression or stability of anti-apoptotic Bcl2, Bcl2L12A, Mcl-1, and VEGF, promoting tumors (36,45). On the other hand, GSK3 phosphorylates and destabilizes β-catenin leading to downregulation of c-Myc and cyclin D1 in breast and skin tumors. GSK3 also phosphorylates T286 on cyclin D1, leading to its nuclear export and degradation (46). Additionally, GSK3 functions as a negative regulator of ERK1/2 (47), promotes p53 translation by inactivating RNPC1 (48), and facilitates p53 Lys382 acetylation activating senescence (49). In melanoma, GSK3 inhibition was shown to suppress both N-Cadherin expression and Focal adhesion kinase phosphorylation, modulating the motile and invasive behavior (50). Therefore, more studies are needed to understand the multifaceted roles of GSK3.
Our study shows that PTPN11 suppresses pY-GSK3 and p-β-catenin and increases pERK and cyclin D1. Previously, Kwong et al. reported that genetic extinction of oncogenic NRAS suppressed MEK and CDK4 leading to tumor regression associated with decreased proliferation and apoptosis, while MEK inhibition alone caused tumor stasis and activated apoptosis, but not cell-cycle arrest (35). Consistently, our study also showed that MEK inhibition (MEK162) alone failed to regress tumors, while genetic or pharmacological PTPN11 inhibition caused tumor regression. Here, PTPN11 inhibition elicited not only suppression of MEK/ERK but also activation of GSK3 and suppression of cyclin D1. When this response is lost, tumor stasis instead of regression was observed following PTPN11 inhibition. This suggests the possible importance of cyclin D1/CDK4 downregulation to cause tumor regression upon PTPN11 inhibition.
In summary, we have shown that PTPN11 plays oncogenic roles in melanoma driving melanoma tumorigenesis, and genetic or pharmacological inhibition of PTPN11 causes regression of fully established tumors. In addition to activation of the RAS/RAF/MAPK pathway, PTPN11 suppresses pY-GSK3, regulating β-catenin and cyclin D1. Furthermore, the loss GSK3 signaling suppresses response to PTPN11 inhibition. Our findings suggest that PTPN11 can serve as a therapeutic target for BRAF wt (either NRAS mutant or wt) melanoma.
Supplementary Material
Implications:
This study identifies PTPN11 as an oncogenic driver and a novel and actionable therapeutic target for BRAF wild-type melanoma.
Acknowledgments
We are grateful to Drs. Lynda Chin and Lawrence Kwong for providing NRASQ61K mutant mouse melanoma cell lines; Lynda Chin and Meenhard Herlyn for human melanoma cell lines; Lynda Chin and Ronald A. DePinho for mouse alleles; Jeffrey Weber for the critical reading of the manuscript. This work was supported by research grants from the American Cancer Society (Research Scholar Grant,TBG-122705 (MK)); from the Florida Department of Health (Bankhead-Coley Cancer Research Program, 5BC03 (MK)); from the Veteran’s Administration (VA Merit Review, I BX001792 (CEC) and a Research Career Scientist Award, 13F-RCS-002 (CEC)); from the National Institutes of Health via HL125353 (CEC), HD087198 (CEC), RR031535 (CEC), AI139072 (CEC). This work has been supported in part by the Molecular Genomics, Proteomics, and Tissue Core Facility at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292).The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006;125:1253–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell 2006;125:1269–81 [DOI] [PubMed] [Google Scholar]
- 3.Wartman LD, Larson DE, Xiang Z, Ding L, Chen K, Lin L, et al. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J Clin Invest 2011;121:1445–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsao H, Chin L, Garraway LA, Fisher DE. Melanoma: from mutations to medicine. Genes Dev 2012;26:1131–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding L, Kim M, Kanchi KL, Dees ND, Lu C, Griffith M, et al. Clonal architectures and driver mutations in metastatic melanomas. PLoS One 2014;9:e111153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sung H, Kanchi KL, Wang X, Hill KS, Messina JL, Lee JH, et al. Inactivation of RASA1 promotes melanoma tumorigenesis via R-Ras activation. Oncotarget 2016;7:23885–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arafeh R, Qutob N, Emmanuel R, Keren-Paz A, Madore J, Elkahloun A, et al. Recurrent inactivating RASA2 mutations in melanoma. Nat Genet 2015;47:1408–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet 2015;47:996–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas N Genomic Classification of Cutaneous Melanoma. Cell 2015;161:1681–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miura K, Wakayama Y, Tanino M, Orba Y, Sawa H, Hatakeyama M, et al. Involvement of EphA2-mediated tyrosine phosphorylation of Shp2 in Shp2-regulated activation of extracellular signal-regulated kinase. Oncogene 2013;32:5292–301 [DOI] [PubMed] [Google Scholar]
- 12.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev 2008;27:179–92 [DOI] [PubMed] [Google Scholar]
- 13.Chan RJ, Feng GS. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 2007;109:862–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell;19:629–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 2013;499:491–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tiganis T, Bennett AM. Protein tyrosine phosphatase function: the substrate perspective. Biochem J 2007;402:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y, et al. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun 2015;6:8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 2011;477:289–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–7 [DOI] [PubMed] [Google Scholar]
- 21.Li J, Rix U, Fang B, Bai Y, Edwards A, Colinge J, et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol 2010;6:291–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26:1367–72 [DOI] [PubMed] [Google Scholar]
- 23.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004;3:Article3 [DOI] [PubMed] [Google Scholar]
- 24.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003;17:3112–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008;455:1129–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosenberg M, Muthusamy V, Curley DP, Wang Z, Hobbs C, Nelson B, et al. Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis 2006;44:262–7 [DOI] [PubMed] [Google Scholar]
- 27.Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res 2010;70:5901–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell 2013;23:603–17 [DOI] [PubMed] [Google Scholar]
- 29.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LaRochelle JR, Fodor M, Xu X, Durzynska I, Fan L, Stams T, et al. Structural and Functional Consequences of Three Cancer-Associated Mutations of the Oncogenic Phosphatase SHP2. Biochemistry 2016;55:2269–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneeberger VE, Ren Y, Luetteke N, Huang Q, Chen L, Lawrence HR, et al. Inhibition of Shp2 suppresses mutant EGFR-induced lung tumors in transgenic mouse model of lung adenocarcinoma. Oncotarget 2015;6:6191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016;535:148–52 [DOI] [PubMed] [Google Scholar]
- 35.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med 2012;18:1503–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014;5:2881–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prahallad A, Heynen GJ, Germano G, Willems SM, Evers B, Vecchione L, et al. PTPN11 Is a Central Node in Intrinsic and Acquired Resistance to Targeted Cancer Drugs. Cell Rep 2015;12:1978–85 [DOI] [PubMed] [Google Scholar]
- 38.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell 2017;170:564–76 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med 2018 [DOI] [PubMed] [Google Scholar]
- 40.Mainardi S, Mulero-Sanchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med 2018 [DOI] [PubMed] [Google Scholar]
- 41.Bunda S, Heir P, Srikumar T, Cook JD, Burrell K, Kano Y, et al. Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc Natl Acad Sci U S A 2014;111:E3785–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghon V. A chaperone-dependent GSK3beta transitional intermediate mediates activation-loop autophosphorylation. Mol Cell 2006;24:627–33 [DOI] [PubMed] [Google Scholar]
- 44.Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, et al. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci U S A 2000;97:11074–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol 2006;79:173–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer 2007;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Q, Zhou Y, Wang X, Evers BM. Glycogen synthase kinase-3 is a negative regulator of extracellular signal-regulated kinase. Oncogene 2006;25:43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang M, Zhang J, Chen X, Cho SJ, Chen X. Glycogen synthase kinase 3 promotes p53 mRNA translation via phosphorylation of RNPC1. Genes Dev 2013;27:2246–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eom TY, Jope RS. GSK3 beta N-terminus binding to p53 promotes its acetylation. Mol Cancer 2009;8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.John JK, Paraiso KH, Rebecca VW, Cantini LP, Abel EV, Pagano N, et al. GSK3beta inhibition blocks melanoma cell/host interactions by downregulating N-cadherin expression and decreasing FAK phosphorylation. J Invest Dermatol 2012;132:2818–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.