

The W-Beijing strain family is globally distributed and is associated with multidrug-resistant tuberculosis (TB) and treatment failure. Therefore, in this study, we examined the contribution of Toll-like receptor 2 (TLR2) to host resistance against Mycobacterium tuberculosis HN878, a clinical isolate belonging to the W-Beijing family.
KEYWORDS: CXCL5, Mycobacterium tuberculosis, TLR2, immunopathology, inflammation, neutrophils
ABSTRACT
The W-Beijing strain family is globally distributed and is associated with multidrug-resistant tuberculosis (TB) and treatment failure. Therefore, in this study, we examined the contribution of Toll-like receptor 2 (TLR2) to host resistance against Mycobacterium tuberculosis HN878, a clinical isolate belonging to the W-Beijing family. We show that TLR2 knockout (TLR2KO) mice infected with M. tuberculosis HN878 exhibit increased bacterial burden and are unable to control tissue-damaging, pulmonary neutrophilic inflammation. Consistent with a critical role for CXCL5 in regulating neutrophil influx, expression of epithelial cell-derived CXCL5 is significantly enhanced in TLR2KO mice prior to their divergence from wild-type (WT) mice in M. tuberculosis replication and neutrophilic inflammation. Depletion of neutrophils in TLR2KO mice by targeting Ly6G reverts lung inflammation and bacterial burden to levels comparable to those of WT mice. Together, the results establish that TLR2 controls neutrophil-driven immunopathology during infection with M. tuberculosis HN878 infection, likely by curtailing CXCL5 production.
INTRODUCTION
Several groups have reported that Toll-like receptor 2 (TLR2) does not contribute to host resistance against laboratory-derived Mycobacterium tuberculosis H37Rv (1–3). Yet other studies have found that TLR2 knockout (TLR2KO) mice are susceptible to M. tuberculosis H37Rv (4–6). Consistent with the latter studies, we found that TLR2KO mice are also susceptible to M. tuberculosis Erdman, another lab-derived virulent strain (7). These mice developed exaggerated immunopathology characterized by pneumonitis and enhanced cellular infiltration (7), indicating that in vivo TLR2 functioned mainly as an immunoregulator. Since the role of TLR2 so far had been studied only with laboratory-derived M. tuberculosis strains, we argued that it was essential to investigate whether TLR2 contributed to host resistance against highly virulent clinical strains of M. tuberculosis. The association of TLR2 polymorphisms with increased susceptibility to pulmonary tuberculosis (TB) in humans with TB disease (8–10) makes it all the more critical to study whether TLR2 influences disease outcome following infection with clinical strains of M. tuberculosis.
Several M. tuberculosis genotypes exist and have been extensively studied to understand the impact of the genetic differences on phenotypic outcomes (11). The W-Beijing genotype has expanded more rapidly than other lineages, and a high incidence of this genotype has been observed among isolates of multidrug-resistant M. tuberculosis (12) and with treatment relapse (13–15). W-Beijing strain HN878 has caused several outbreaks of TB worldwide (reviewed in reference 12) and in animal models is more virulent than strains from other lineages (16). There is now accumulating evidence that the M. tuberculosis lineage influences innate immune responses and virulence (17–19), and this is associated with distinct cell envelope lipid profiles (20). Increased virulence in mice infected with M. tuberculosis HN878 has also been linked to downregulation of Th1 responses, possibly influenced by the increase in type 1 interferons (IFNs) and a concomitant decrease in tumor necrosis factor (TNF) and interleukin-12 (IL-12) by infected macrophages (16, 19). Subsequent studies proposed that the downregulation of the Th1 response and reduced survival of M. tuberculosis HN878-infected animals was possibly due to the rapid surge in T regulatory (Treg) cells that followed the early Th1 response (21, 22). In this study, we sought to understand the requirement for TLR2 in regulating immunopathology and in mediating host resistance during infection with the clinical strain M. tuberculosis HN878.
RESULTS
Loss of early mycobacterial containment in the lungs of M. tuberculosis HN878-infected TLR2KO mice.
At 2 weeks following M. tuberculosis HN878 infection, there was no difference in bacterial replication levels between wild-type (WT) and TLR2KO mice. By 4 weeks, however, we observed a 3-log increase in CFU counts in the lungs of TLR2KO mice compared to the level in WT mice (Fig. 1A). By 8 weeks, although the bacterial numbers began to stabilize, there was still a marked difference in the lung bacterial burdens between the two groups of mice. Similarly, increased bacterial burden was also seen in the spleen of TLR2KO mice at both 4 and 8 weeks of infection compared to levels in WT mice (Fig. 1B). These data demonstrate that TLR2 is required early during acute M. tuberculosis HN878 infection to restrict bacterial replication in the lung. The increased bacterial burden in the spleen indicates that TLR2 either limits dissemination to the spleen or regulates bacterial survival in the spleen.
FIG 1.

Loss of early mycobacterial containment in the lungs of M. tuberculosis HN878-infected TLR2KO mice. WT and TLR2KO mice were aerosol infected with ∼20 CFU of M. tuberculosis HN878, and at indicated time points bacterial burden was determined in lung (A) and spleen (B). Five mice were included for each time point for WT and TLR2KO groups. Data are presented as means ± standard errors of the means and are representative of one of two individual experiments. Statistical significance was calculated using two-way ANOVA with Bonferroni’s correction. Mtb, M. tuberculosis; wk, weeks. ***, P < 0.001.
Absence of TLR2 leads to enhanced pulmonary inflammation during M. tuberculosis HN878 infection.
Next, lung pathological disease was evaluated through high-resolution scanning of hematoxylin and eosin (H&E)-stained lung sections. At 4 weeks following infection, the lungs of WT mice exhibited small and compact granulomas whereas the granulomas in the lungs of TLR2KO mice at this time point appeared larger and less compact (Fig. 2A). Also, a significant part of the lung had clear alveolar spaces in the WT compared to findings in the TLR2KO mice (Fig. 2A). Visualization of the lungs at higher magnification showed the characteristic compact aggregation of lymphocytic clusters (L) within the granuloma in WT mice, but in the lungs of TLR2KO small lymphocytic clusters were found scattered throughout the granulomatous lesion that also contained large numbers of foamy macrophage (FM) (see Fig. S1 in the supplemental material). By 8 weeks postinfection, the WT mice had compact granulomas, and, in contrast, the entire lung of TLR2KO mice presented with excessive inflammation (Fig. 2A). There was a marked loss of clear alveolar spaces in the TLR2KO compared to tissue of WT mice (Fig. 2A). We next examined pulmonary inflammation in lung parenchyma, outside granulomatous tissue, in WT and TLR2KO mice. Stark differences in parenchymal inflammation were noticeable upon closer examination of the parenchyma. The alveoli in the WT mice displayed single layers of epithelial (EL) cells indicative of healthier lung tissue, and, in contrast, TLR2KO lung parenchyma displayed increased cellular infiltration (CI) and marked thickening of the epithelial cell layer (Fig. S1).
FIG 2.

Increased pulmonary inflammation in HN878-infected TLR2KO mice. (A) Formalin-fixed, paraffin-embedded lung tissue sections were H&E stained, and scanned images were obtained. Pictures are presented at a magnification of ×2 of WT and TLR2KO lungs at 4 and 8 weeks of infection. (B) The percent area of lung involved in inflammation was evaluated at 4 weeks and 8 weeks using ImageProJ software from Nikon. (C) The trypan blue exclusion method was used to calculate the total number of live cells in the lungs of WT and TLR2KO mice at weeks 2, 4, and 8 weeks post-M. tuberculosis HN878 infection. For each time point, 5 WT and TLR2KO mice were evaluated. Data are representative of one of two individual experiments. Data represented as means ± standard errors of the means. Statistical significance was calculated using a two-way ANOVA with Bonferroni’s correction. ***, P < 0.001; **, P < 0.01.
The total area of lung involved in inflammation as calculated using ImageProJ software was significantly higher in the lungs of TLR2KO mice than in those of WT mice (Fig. 2B). Of note, while the granulomatous inflammation began to stabilize in the WT mice between 4 and 8 weeks (∼40% to ∼45%), it significantly increased in the TLR2KO mice (∼55% to ∼80%) (Fig. 2B). Together, these data suggest that disease progression during M. tuberculosis HN878 infection in TLR2KO mice, unlike that in WT mice, does not stabilize with resolution of inflammation.
Consistent with enhanced inflammation, enumeration of total lung infiltrates at 4 weeks of infection showed an increase in the number of live cells in M. tuberculosis HN878-infected TLR2KO mice compared to the number in WT mice (Fig. 2C). Even at 8 weeks of infection, the cell numbers in the lungs of TLR2KO mice were higher than those of WT mice (Fig. 2C). The composition of inflammatory cells recruited to the lungs following infection was evaluated by flow cytometry to establish the immune cell types contributing to the increased inflammatory pathology in the TLR2KO mice. The gating strategy is provided in Fig. S3. There was no difference in the total numbers of B220+ B cells, CD11b− CD11c+ alveolar macrophages and CD11b+ CD11c+Ly6C- dendritic cells between WT and TLR2KO mice (Fig. 3). However, CD4+ T cells, CD8+ T cells, CD11b+ CD11c− Ly6C− recruited macrophages, CD11b+ CD11c− Ly6C+ inflammatory macrophages, and CD11bhi Ly6G+ (expressing high levels of CD11b) neutrophils were significantly higher in the TLR2KO mice (Fig. 3). Compared to other myeloid subsets, CD11bhi Ly6G+ neutrophil numbers were markedly elevated in the lungs of TLR2KO mice (Fig. 3). Consistent with flow cytometry data, immunohistochemical staining of lung sections showed the presence of large numbers of Ly6G+ neutrophils in the granulomatous lesions of 4-week-infected TLR2KO mice compared with numbers in WT mice (Fig. S2). The neutrophils appeared karyorrhectic, characteristic of necrotic lesions (Fig. S2). Previously, we had reported that TLR2KO mice infected with M. tuberculosis Erdman had significantly reduced accumulation of FoxP3+ T regulatory (Treg) cells in the lungs compared to levels in WT mice. Interestingly, we did not see differences in Treg cell accumulations between M. tuberculosis HN878-infected WT and TLR2KO mice (Fig. S4).
FIG 3.

Increased recruitment of immune cells to the lungs of 4-week-infected TLR2KO mice. Lung single-cell suspensions were prepared from lungs of mice at 4 weeks postinfection and were surface stained with directly conjugated antibodies to CD4 CD8, B220, CD11b, CD11c, Ly6G and Ly6C to determine the absolute number of specific cell types infiltrating the lungs of WT and TLR2KO mice. Cells were appropriately gated to enumerate the following cell subsets: B220+ (B cells), CD4+ T cells, CD8+ T cells, CD11bhi Ly6G+ neutrophils; CD11b− CD11c+ (CD11b−c+)alveolar macrophages, CD11b+ CD11c− (CD11b+c−) Ly6C+ inflammatory monocytes, CD11b+ CD11c− Ly6C− recruited macrophages, and CD11b+ CD11c+ Ly6C− dendritic cells. Data are represented as means ± standard errors of the means. Statistical significance was calculated using a two-way ANOVA with Bonferroni’s correction. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
Increased epithelial cell-derived CXCL5 expression in TLR2KO mice.
To gain better insight into early events that could be driving increased neutrophil influx and bacterial replication in TLR2KO mice, we evaluated the expression of CXCL5 in the lungs of animals infected for 2 weeks, a time point when the bacterial burdens are still comparable between the two groups. We found that CXCL5 was significantly increased in TLR2KO mice (Fig. 4A). Epithelial cell-derived CXCL5 is critical for recruiting neutrophils to M. tuberculosis-infected lungs (23), and therefore the cellular source of the increased CXCL5 in TLR2KO mice was next investigated. Epithelial cell adhesion molecule-positive (EpCAM+) cells, CD11b+ CD11c+ dendritic cells (DCs), CD11b− CD11c+ alveolar macrophages, CD11b+ CD11c− recruited macrophages, and CD11bhi Ly6G+ neutrophils were purified from 2-week-infected lungs by fluorescence-activated cell sorting (FACS). We found significantly increased expression of Cxcl5 in EpCAM+ cells from TLR2KO mice in comparison to the levels in cells purified from WT mice. Alveolar macrophage was the only other subset that expressed Cxcl5, but the levels were significantly lower than those of EpCAM+ in both groups of mice (Fig. 4B). Together, these data indicate that the exuberant neutrophilic inflammation in TLR2KO mice could be caused by increased epithelial cell-derived CXCL5.
FIG 4.

TLR2 restricts CXCL5 expression. (A) Five WT and TLR2KO mice were infected with M. tuberculosis HN878, and 2 weeks following infection, CXCL5 protein level in lung lysates was measured by ELISA. (B) Single-cell suspensions from 2-week-infected WT and TLR2KO were pooled. Cells were sorted by flow cytometry for EpCAM+ cells and the different myeloid subsets. RNA was extracted from the sorted cells, and CXCL5 gene expression was measured in each of the sorted subsets by real-time PCR using TaqMan probes (Life Technologies). Total RNA from uninfected lung was used as calibrators. Relative gene expression was expressed as 2−ΔΔCT. Data are representative of one of two individual experiments. Statistical significance was calculated using an unpaired t test. ***, P < 0.001; *, P < 0.05.
Depletion of neutrophils early in infection reduces pathology and bacterial load in M. tuberculosis HN878-infected TLR2KO mice.
Since we saw a significant increase in neutrophils, cells that are associated with pathological disease in TB, and CXCL5 that drives neutrophil accumulation, we next determined whether blocking neutrophil recruitment would improve disease outcome in TLR2KO mice. M. tuberculosis HN878-infected TLR2KO mice were treated with an anti-Ly6G antibody or an isotype control at 17, 19, 21, and 24 days following infection and at day 27 evaluated for cellular recruitment, histopathological response, and bacterial burden in the lung. WT mice treated with isotype were included as controls. As expected, TLR2KO mice treated with isotype had significantly increased neutrophil infiltration compared to the level in WT mice treated with the same antibody, whereas TLR2KO mice treated with anti-Ly6G antibody had significantly reduced neutrophils (Fig. 5A and S5). Interestingly, total cell numbers were also reduced in anti-Ly6G-treated TLR2KO animals, indicating that neutrophil blockade resulted in an overall reduction in cellular infiltration (Fig. 5B). The lungs of isotype-treated WT mice manifested with small granulomas and minimal inflammation, and the majority of the lung presented with clear alveolar spaces (Fig. 5D). In contrast, the lungs of the TLR2KO mice treated with isotype were highly inflamed with large granulomatous lesions and very few clear alveoli spaces (Fig. 5D). The lungs of anti-Ly6G-treated TLR2KO mice appeared less inflamed, with more compact granulomas, than isotype-treated animals, similar to observations in the WT mice (Fig. 5D). The percent area of lung involved in inflammation was quantitated and was consistent with the microscopic examination; TLR2KO mice treated with isotype had an increased percentage of inflamed area (Fig. 5C).
FIG 5.

Neutrophil blockade reduces the pulmonary pathology of M. tuberculosis HN878-infected TLR2KO mice to WT level. WT and TLR2KO mice were aerosol infected with ∼100 CFU of M. tuberculosis HN878, and at days 17, 19, 21, and 24 following infection, mice were injected intraperitoneally with either anti-mouse Ly6G antibody or an isotype control. Mice were euthanized on day 27, and lungs from WT mice receiving control isotype antibody, TLR2KO mice receiving control isotype antibody, and TLR2KO mice receiving anti-Ly6G antibody, as indicated, were evaluated for neutrophil recruitment (A) and total cellular number (B). The percent lung area involved in inflammation was calculated for all three groups of mice using the ImageProJ software from Nikon (C). Scanned images of lung tissue were obtained by using Act-1 software from Nikon (D). Data are represented as means ± standard errors of the means. Statistical significance was calculated using a one-way ANOVA with Bonferroni’s correction. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
We next examined the effect of neutrophil depletion on bacterial containment in the lungs. At 2 weeks postinfection, prior to anti-Ly6G treatment, the bacterial burdens in the lungs of WT and TLR2KO mice were similar (data not shown), confirming the previous finding shown in Fig. 1. Following neutrophil depletion, we observed that the bacterial burden in the lungs of isotype-treated TLR2KO mice at 27 days following infection was significantly higher than that in the WT mice, again consistent with data presented in Fig. 1. Remarkably, anti-Ly6G treatment of TLR2KO mice reduced the bacterial burden to levels present in WT mice (Fig. 6). Together, these data establish that TLR2 signals limit pathology and restrict M. tuberculosis replication by restricting neutrophil recruitment to the lungs.
FIG 6.
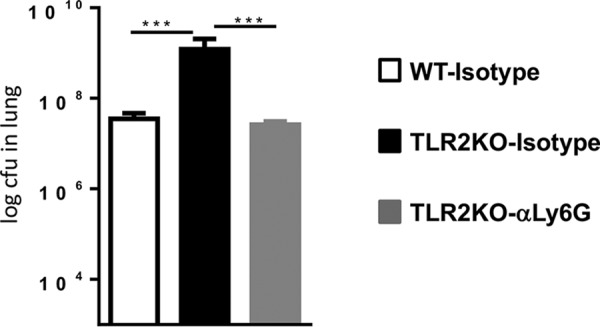
Neutrophil blockade reduces bacterial burden in M. tuberculosis HN878-infected TLR2KO mice to WT levels. Lungs obtained from mice described in the legend of Fig. 5 were evaluated for CFU counts. Data are represented as means ± standard errors of the means. Statistical significance was calculated using one-way ANOVA with Bonferroni’s correction. ***, P < 0.001.
DISCUSSION
Increased pathology and enhanced susceptibility to M. tuberculosis infection are consistently associated with dysregulated neutrophil recruitment to the lungs (24–28). Human TB is also associated with a neutrophil-driven type 1 interferon signature (29), and neutrophils are the predominant infected cells in the airways of patients with active TB (30). Thus, it is reasonable to have several nonredundant mechanisms in place in the host to restrain neutrophil influx. The present study has uncovered that during M. tuberculosis HN878 infection, TLR2 plays a critical role in preventing excessive neutrophil influx into the lungs and the associated increase in inflammatory pathology and bacterial replication. The recruited neutrophils could directly enhance inflammatory pathology or, alternatively, promote an environment that favors extravasation of inflammatory monocytes (iMs) (31, 32).
Previous studies have reported that IFN-γ (33) and nitric oxide (NO) (34) signaling pathways also control inflammation by restricting recruitment of tissue-damaging neutrophils. Depletion of neutrophils in Nos2−/− mice led to decreased pathology and improved control of M. tuberculosis replication while in Ifnγr−/− mice, survival was prolonged but bacterial control was not improved. Mechanistically, NO inhibited inflammation by repressing the caspase 1-dependent processing of pro-IL-1β (35) while IFN-γR signaling suppressed the generation of Th17 cells (33). The findings from the current study suggest that TLR2 also contributes to the control of neutrophil influx into the lung during M. tuberculosis infection by restricting the production of CXCL5 that causes destructive neutrophilic inflammation (23). Whether there is a link between TLR2-regulated CXCL5 to the nitric oxide (NO)- and IFN-γ-mediated suppression of neutrophil recruitment needs to be further investigated. M. tuberculosis infection of diversity outbred (DO) mice resulted in infection outcomes ranging from resistant to susceptible to supersusceptible, and disease severity strongly correlated with neutrophil numbers and CXCL1 expression (27). Clearly, there are several pathways regulating neutrophil influx into the lung to prevent pathological damage.
In our study, depletion of neutrophils in TLR2KO mice not only resulted in reduced pathological inflammation but also restored bacterial numbers to WT levels. Nos2−/− and C3HeB/FeJ mice are highly susceptible to M. tuberculosis infection and present with concomitant accumulation of large numbers of neutrophils in the lung (34). Similar to results in TLR2KO mice, neutrophil depletion in the Nos2−/− and C3HeB/FeJ susceptible mice also reduced bacterial burden in the lung (34), indicating that the recruited neutrophils were creating a growth-permissive environment for M. tuberculosis in the lesions. The uptake of apoptotic Mycobacterium bovis BCG-infected neutrophils by macrophages leads to lipid body formation (36), a microenvironment that is conducive to M. tuberculosis replication. Neutrophil activation also leads to the release of neutrophil extracellular traps (NETs) (37) that contribute to host defense (38), but dysregulated production of NETs (NETosis) can cause tissue damage and destruction (39, 40) that promotes a growth-permissive environment for M. tuberculosis. Other intracellular pathogens also exploit the host neutrophilic inflammatory response for their growth. For example, Salmonella enterica serovar Typhimurium can use the abundantly generated by-products of neutrophil activation to engage in anaerobic respiration and thereby find a means to outgrow the normal bacterial communities present in the gut (41). Further studies are needed to determine the exact mechanism of neutrophil-mediated damage in M. tuberculosis infection.
It is intriguing that the absence of TLR2 did not impact infection with M. tuberculosis H37Rv in some studies (1–3) while in other studies TLR2KO mice were found to be moderately susceptible to infection with low-dose M. tuberculosis H37Rv infection (6), and high-dose infection led to severe disease (4, 5). Our previous study with low-dose M. tuberculosis Erdman infection (7) and the current study with M. tuberculosis HN878 infections show that absence of TLR2 severely impacts host resistance. Also notable is that although TLR2 restricts inflammation in both Erdman and HN878 infections, the mechanisms employed to achieve this are different. During chronic M. tuberculosis Erdman infection, TLR2 signaling on macrophages is critical for the recruitment of Treg cells to the granuloma, which in turn restrains excessive inflammation (7). Surprisingly, the course of M. tuberculosis HN878 infection was not influenced by Treg cell-mediated immunoregulation. In M. tuberculosis HN878 infection, TLR2’s immunoregulatory functions were engaged much earlier to put a break on pathogenic neutrophil influx. Given that there is cross-regulation between Th17 and Treg cells (42), it is possible that HN878 infection induces an early Th17 response that limits Treg cell expansion. The early Th17 response may influence CXCL5 expression (43), and the role for TLR2, therefore, is to regulate this early inflammatory response during HN878 infection. The temporal and functional differences in TLR2-mediated immunoregulation in M. tuberculosis HN878 and Erdman infections underscore the importance of studying the contribution of host immune factors essential in mediating protection against clinical strains of M. tuberculosis. The requirement and the timing of TLR2’s immunoregulatory functions may depend on the extent of the inflammatory response induced in the host by a particular M. tuberculosis strain. This argument is supported by the finding that a heterogeneous inflammatory response is induced from macrophages when they are infected with different clinical isolates that have distinct cell envelop lipid profiles (20).
In conclusion, the new finding from this study is that TLR2 is a key pathway that the host employs to control neutrophil-mediated immunopathology. Pathological damage to lungs not only increases M. tuberculosis replication but can also have significant repercussions, including the greater risk of TB treatment failure, increased transmission, and long-term pulmonary dysfunction even after cure (44–47). Thus, future studies investigating the role of TLR2 and its polymorphic variants in regulating immunopathology in human TB are warranted.
MATERIALS AND METHODS
Ethics statement.
The studies with mice described here conform to the Rutgers-New Jersey Medical School (NJMS) Institutional Animal Care and Use Committee (IACUC) Guidelines, and NIH and USDA policies on the care and use of animals in research. Animal protocols pertaining to the experiments described below were approved by the Rutgers IACUC (assurance number A3158-01). Efforts were taken to ensure minimal animal pain and suffering, and, when applicable, approved anesthesia methods were employed for the same.
Mice.
C57BL/6 WT mice were purchased from Charles River/National Cancer Institute (NCI). TLR2KO mice were bred under pathogen-free conditions in a biosafety level 2 (BSL2) animal facility at Rutgers-New Jersey Medical School, Newark, NJ. M. tuberculosis infections were performed in a BSL3 facility, and infected mice were housed in a BSL3 facility for the duration of the study.
Aerosol infections and tissue harvests.
Female mice (aged 6 to 10 weeks) were infected via aerosol. At 24 h after aerosol infection, 4 to 5 mice were euthanized, and total lung homogenates were plated on 7H11 selective medium to determine the infection dose per experiment. At appropriate time intervals following aerosol infection, mice were euthanized by cervical dislocation, and lungs and spleen were harvested.
Single-cell preparation and flow cytometry.
Lung lobes were digested with 20 μg/ml of collagenase D at 37°C for 30 min, and the reaction was stopped by addition of 5 mM EDTA. Digested lung tissue was mashed through a 40-μm-pore-size nylon membrane filter. Red blood cells were lysed by treating with ACK (ammonium-chloride-potassium) lysing buffer. Single-cell suspensions of lung cells were surface stained with directly conjugated fluorochrome-labeled anti-mouse CD4-V450 (BD Horizon), anti-mouse CD8-Alexa Fluor 488 (AF488) (BD Pharmingen), anti-mouse B220-PECF594 (where PE is phycoerythrin) (BD Pharmingen), anti-mouse CD11b-PE (BD Pharmingen), anti-mouse Ly6G-PECy7 (BD Pharmingen), anti-mouse CD11c-AF700 (BD Pharmingen), and anti-mouse Ly6c-PerCP5.5 (where PerCP is peridinin chlorophyll protein) (eBiosciences). The gating strategy was as follows. Live/Dead cell viability staining was used to gate the fluorescence-negative population as total live cells in the lung single cell suspension. From this, the CD4+ CD8+ and the double-negative (CD4− CD8−) cell populations were separated. Of the CD4− CD8− population, the B220+ cells were gated out based on low side scatter (SSC) as B cells. The remaining B220− population was gated for CD11b and Ly6G. The cell population with high expression for CD11b and Ly6G was gated out as the neutrophils, and the remaining population of cells was identified as CD11b− and CD11b+. The CD11b− population was then gated for expression of CD11c, and the CD11b− CD11c+ population was identified as the alveolar macrophages. This population was negative for Ly6C. The CD11b+ macrophages were further gated for CD11c and Ly6C to get the following populations of immune cells: CD11b+ CD11c+ Ly6C− myeloid dendritic cells (mDCs), CD11b+ CD11c− Ly6C+ inflammatory monocytes (iMs), and CD11b+ CD11c− Ly6C− resident macrophages (RMs).
Histopathological analysis of lung tissue.
Lungs of M. tuberculosis-infected mice were perfused with sterile phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 7 days, followed by paraffin embedding. For histopathological analysis, 5- to 7-μm sections were cut and stained using a standard H&E protocol. A Leica SCN400 F whole-slide scanner (Experimental Pathology Research Lab, NYU Langone Health) was used for scanning histological sections, and images were analyzed using an Aperio ImageScope. Stereoscopic images were obtained using Act-1 software from Nikon. For quantitation of granulomatous inflammation in the lung section, Image-Pro Discovery software was used to create a grid overlay onto each photomicrograph of H&E-stained lung section, and the numbers of points hitting areas of granulomatous infiltration were counted. Paraffin-embedded serial sections of lung tissue were prepared for Foxp3 immunohistochemical staining as previously described (7). Ly6G staining was performed similar to that for Foxp3 using the anti-Ly6G antibody (clone 1A8; Biolegend).
In vivo blockade of neutrophils.
C57BL/6 WT and TLR2KO mice were aerosol infected with a low dose of M. tuberculosis HN878. At days 17, 19, 21, and 24 following infection, mice were injected intraperitoneally with 0.5 mg of either anti-mouse Ly6G antibody (clone 1A8, BioXCell) or isotype control (rat IgG2a, clone 2A3; BioXCell) in 200 μl of PBS. The treated mice were euthanized on day 27 and evaluated for cellular recruitment, neutrophil numbers, bacterial burden, and histopathological response.
Cell sorting for EpCAM+ cells.
Lungs of 2-week-infected mice were perfused with 10 ml of PBS, and lung tissues were collected in PBS containing collagenase D (Roche) and Miltenyi Biotech lung dissociation enzymes and then incubated for 30 min at 37°C. After incubation, 30 μl of 10 mM EDTA was added to stop the reaction. Digested lung tissues were mechanically dissociated and passed through a 70-μm-pore-size cell strainer. Single-cell suspensions were collected and centrifuged at 1,200 rpm for 10 min. Following this, red blood cell (RBC) lysis was performed, and, finally, cell pellets were resuspended in 1 ml of FACS buffer (PBS with 2% fetal calf serum [FCS] and 0.02% sodium azide). Based on viable cell numbers, suspensions from 3 to 4 mice from each genotype were pooled to get a final cell density of 8 × 106 to 10 × 106 cells/ml. The following cell surface marker antibodies were used to sort the respective cellular populations: anti-mouse CD45.2 PerCpCy5.5 (or V450), GR1-allophycocyanin (APC), CD3-fluorescein isothiocyanate (FITC), CD11b-APCCy7, CD11c-AF700, Ly6G-PECy7 (all, BD Biosciences), and EpCAM-PE (Biolegend). These pooled single-cell suspensions were stained using a FACS surface staining protocol and later sorted on a BD FACSAria in the BSL3 facility. The gating strategy was as follows. Live/Dead cell viability staining was used to gate the fluorescence-negative population as total live cells in the lung single-cell suspension. From this, cells were first gated separately on EpCAM+ and CD45+, and subsequently the CD45+ cells were gated on CD3+ and CD11b+ cells. The CD11b+ cells were further gated on Ly6G and CD11c. After collection, live sorted cells were processed for real-time PCR to measure gene expression.
Gene expression by real-time PCR.
Total RNA was extracted using an RNeasy column (Qiagen) with the flow cytometry-sorted cells. cDNA was prepared from the total RNA by reverse transcription using Superscript II (Invitrogen) or a High Capacity RNA to c-DNA kit (Applied Biosystems). Real-time PCR was performed using TaqMan probes (Life Technologies). β-Actin was used as the endogenous control. Total RNA from uninfected naive lung was used as calibrators. Relative gene expression was expressed as 2−ΔΔCT (where CT is threshold cycle).
Statistical analysis.
All statistical analyses were performed using GraphPad Prism software. For analysis of two groups, an unpaired t test was used. For more than two groups, one-way or two-way analysis of variance (ANOVA) with Bonferroni’s correction was used. In all cases, a P value of <0.05 was considered to be statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the NIH (AI0848221 and 1S10RR025465) to P.S.
We thank the NJMS Flow Cytometry and Immunology Core Laboratory for excellent assistance with the flow sorting and Selvakumar Subbian for providing M. tuberculosis HN878.
A.G., J.D., S.V., and M.B. performed the experiments. A.G., J.D., and P.S. designed the study. A.G., J.D., and P.S. wrote the manuscript. All authors have reviewed the manuscript and agree with its content.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00760-18.
REFERENCES
- 1.Holscher C, Reiling N, Schaible UE, Holscher A, Bathmann C, Korbel D, Lenz I, Sonntag T, Kroger S, Akira S, Mossmann H, Kirschning CJ, Wagner H, Freudenberg M, Ehlers S. 2008. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, -4 and -9. Eur J Immunol 38:680–694. doi: 10.1002/eji.200736458. [DOI] [PubMed] [Google Scholar]
- 2.Reiling N, Ehlers S, Holscher C. 2008. MyDths and un-TOLLed truths: sensor, instructive and effector immunity to tuberculosis. Immunol Lett 116:15–23. doi: 10.1016/j.imlet.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 3.Rath P, Huang C, Wang T, Wang T, Li H, Prados-Rosales R, Elemento O, Casadevall A, Nathan CF. 2013. Genetic regulation of vesiculogenesis and immunomodulation in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 110:E4790–E4797. doi: 10.1073/pnas.1320118110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning CJ, Goyert S, Ehlers S. 2002. Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol 169:3480–3484. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 5.Drennan MB, Nicolle D, Quesniaux VJ, Jacobs M, Allie N, Mpagi J, Fremond C, Wagnet H, Kirschning CJ, Ryffel B. 2004. Toll-Like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am J Pathol 164:49–57. doi: 10.1016/S0002-9440(10)63095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. 2005. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med 202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McBride A, Konowich J, Salgame P. 2013. Host Defense and Recruitment of Foxp3(+) T regulatory cells to the lungs in chronic Mycobacterium tuberculosis infection requires Toll-like receptor 2. PLoS Pathog 9:e1003397. doi: 10.1371/journal.ppat.1003397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Jiang T, Yang X, Xue Y, Wang C, Liu J, Zhang X, Chen Z, Zhao M, Li JC. 2013. Toll-like receptor -1, -2, and -6 polymorphisms and pulmonary tuberculosis susceptibility: a systematic review and meta-analysis. PLoS One 8:e63357. doi: 10.1371/journal.pone.0063357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang JJ, Xia X, Tang SD, Wang J, Deng XZ, Zhang Y, Yue M. 2013. Meta-analysis on the associations of TLR2 gene polymorphisms with pulmonary tuberculosis susceptibility among Asian populations. PLoS One 8:e75090. doi: 10.1371/journal.pone.0075090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schurz H, Daya M, Moller M, Hoal EG, Salie M. 2015. TLR1, 2, 4, 6 and 9 variants associated with tuberculosis susceptibility: a systematic review and meta-analysis. PLoS One 10:e0139711. doi: 10.1371/journal.pone.0139711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coscolla M, Gagneux S. 2014. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol 26:431–444. doi: 10.1016/j.smim.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanekom M, Gey van Pittius NC, McEvoy C, Victor TC, Van Helden PD, Warren RM. 2011. Mycobacterium tuberculosis Beijing genotype: a template for success. Tuberculosis (Edinb) 91:510–523. doi: 10.1016/j.tube.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Sun YJ, Lee AS, Wong SY, Paton NI. 2006. Association of Mycobacterium tuberculosis Beijing genotype with tuberculosis relapse in Singapore. Epidemiol Infect 134:329–332. doi: 10.1017/S095026880500525X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huyen MN, Buu TN, Tiemersma E, Lan NT, Dung NH, Kremer K, Soolingen DV, Cobelens FG. 2013. Tuberculosis relapse in Vietnam is significantly associated with Mycobacterium tuberculosis Beijing genotype infections. J Infect Dis 207:1516–1524. doi: 10.1093/infdis/jit048. [DOI] [PubMed] [Google Scholar]
- 15.Parwati I, Alisjahbana B, Apriani L, Soetikno RD, Ottenhoff TH, van der Zanden AG, van der Meer J, van Soolingen D, van Crevel R. 2010. Mycobacterium tuberculosis Beijing genotype is an independent risk factor for tuberculosis treatment failure in Indonesia. J Infect Dis 201:553–557. doi: 10.1086/650311. [DOI] [PubMed] [Google Scholar]
- 16.Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE, Freedman VH, Kaplan G. 2001. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci U S A 98:5752–5757. doi: 10.1073/pnas.091096998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez B, Aguilar D, Orozco H, Burger M, Espitia C, Ritacco V, Barrera L, Kremer K, Hernandez-Pando R, Huygen K, van Soolingen D. 2003. A marked difference in pathogenesis and immune response induced by different Mycobacterium tuberculosis genotypes. Clin Exp Immunol 133:30–37. doi: 10.1046/j.1365-2249.2003.02171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reed MB, Domenech P, Manca C, Su H, Barczak AK, Kreiswirth BN, Kaplan G, Barry CE. 2004. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431:84–87. doi: 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- 19.Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, Barry C, Kaplan G. 2005. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res 25:694–701. doi: 10.1089/jir.2005.25.694. [DOI] [PubMed] [Google Scholar]
- 20.Krishnan N, Malaga W, Constant P, Caws M, Tran TH, Salmons J, Nguyen TN, Nguyen DB, Daffe M, Young DB, Robertson BD, Guilhot C, Thwaites GE. 2011. Mycobacterium tuberculosis lineage influences innate immune response and virulence and is associated with distinct cell envelope lipid profiles. PLoS One 6:e23870. doi: 10.1371/journal.pone.0023870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shang S, Harton M, Tamayo MH, Shanley C, Palanisamy GS, Caraway M, Chan ED, Basaraba RJ, Orme IM, Ordway DJ. 2011. Increased Foxp3 expression in guinea pigs infected with W-Beijing strains of M. tuberculosis. Tuberculosis (Edinb) 91:378–385. doi: 10.1016/j.tube.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C, Basaraba RJ, Orme IM. 2007. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol 179:522–531. doi: 10.4049/jimmunol.179.1.522. [DOI] [PubMed] [Google Scholar]
- 23.Nouailles G, Dorhoi A, Koch M, Zerrahn J, Weiner J, Faé KC, Arrey F, Kuhlmann S, Bandermann S, Loewe D, Mollenkopf H-J, Vogelzang A, Meyer-Schwesinger C, Mittrücker H-W, McEwen G, Kaufmann SHE. 2014. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J Clin Invest 124:1268–1282. doi: 10.1172/JCI72030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almeida FM, Ventura TL, Amaral EP, Ribeiro SC, Calixto SD, Manhaes MR, Rezende AL, Souzal GS, de Carvalho IS, Silva EC, Silva JA, Carvalho EC, Kritski AL, Lasunskaia EB. 2017. Hypervirulent Mycobacterium tuberculosis strain triggers necrotic lung pathology associated with enhanced recruitment of neutrophils in resistant C57BL/6 mice. PLoS One 12:e0173715. doi: 10.1371/journal.pone.0173715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S. 2006. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect Immun 74:4295–4309. doi: 10.1128/IAI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. 2015. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528:565–569. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niazi MK, Dhulekar N, Schmidt D, Major S, Cooper R, Abeijon C, Gatti DM, Kramnik I, Yener B, Gurcan M, Beamer G. 2015. Lung necrosis and neutrophils reflect common pathways of susceptibility to Mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis Model Mech 8:1141–1153. doi: 10.1242/dmm.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marzo E, Vilaplana C, Tapia G, Diaz J, Garcia V, Cardona PJ. 2014. Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberculosis (Edinb) 94:55–64. doi: 10.1016/j.tube.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eum S-Y, Kong J-H, Hong M-S, Lee Y-J, Kim J-H, Hwang S-H, Cho S-N, Via LE, Barry CE. 2010. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 137:122–128. doi: 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soehnlein O, Lindbom L, Weber C. 2009. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood 114:4613–4623. doi: 10.1182/blood-2009-06-221630. [DOI] [PubMed] [Google Scholar]
- 32.Soehnlein O, Zernecke A, Weber C. 2009. Neutrophils launch monocyte extravasation by release of granule proteins. Thromb Haemost 102:198–205. doi: 10.1160/TH08-11-0720. [DOI] [PubMed] [Google Scholar]
- 33.Nandi B, Behar SM. 2011. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med 208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, Smith CM, Phuah JY, Long JE, Dubuke ML, Palace SG, Goguen JD, Baker RE, Nambi S, Mishra R, Booty MG, Baer CE, Shaffer SA, Dartois V, McCormick BA, Chen X, Sassetti CM. 2017. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol 2:17072. doi: 10.1038/nmicrobiol.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, Sassetti CM. 2013. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol 14:52–60. doi: 10.1038/ni.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D'Avila H, Roque NR, Cardoso RM, Castro-Faria-Neto HC, Melo RC, Bozza PT. 2008. Neutrophils recruited to the site of Mycobacterium bovis BCG infection undergo apoptosis and modulate lipid body biogenesis and prostaglandin E production by macrophages. Cell Microbiol 10:2589–2604. doi: 10.1111/j.1462-5822.2008.01233.x. [DOI] [PubMed] [Google Scholar]
- 37.Almyroudis NG, Grimm MJ, Davidson BA, Rohm M, Urban CF, Segal BH. 2013. NETosis and NADPH oxidase: at the intersection of host defense, inflammation, and injury. Front Immunol 4:45. doi: 10.3389/fimmu.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papayannopoulos V. 2017. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18:134–147. doi: 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 39.Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT. 2012. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One 7:e32366. doi: 10.1371/journal.pone.0032366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, Solis AG, Bielecki P, Mohanty S, Trentalange M, Homer RJ, Flavell RA, Wagner DD, Montgomery RR, Shaw AC, Staeheli P, Iwasaki A. 2016. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science 352:463–466. doi: 10.1126/science.aaf3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winter SE, Lopez CA, Baumler AJ. 2013. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep 14:319–327. doi: 10.1038/embor.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sehrawat S, Rouse BT. 2017. Interplay of regulatory T Cell and Th17 cells during infectious diseases in humans and animals. Front Immunol 8:341. doi: 10.3389/fimmu.2017.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen K, Eddens T, Trevejo-Nunez G, Way EE, Elsegeiny W, Ricks DM, Garg AV, Erb CJ, Bo M, Wang T, Chen W, Lee JS, Gaffen SL, Kolls JK. 2016. IL-17 receptor signaling in the lung epithelium is required for mucosal chemokine gradients and pulmonary host defense against K. pneumoniae. Cell Host Microbe 20:596–605. doi: 10.1016/j.chom.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jung JW, Choi JC, Shin JW, Kim JY, Choi BW, Park IW. 2015. Pulmonary impairment in tuberculosis survivors: the Korean National Health and Nutrition Examination Survey 2008–2012. PLoS One 10:e0141230. doi: 10.1371/journal.pone.0141230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ralph AP, Kenangalem E, Waramori G, Pontororing GJ, Sandjaja, Tjitra E, Maguire GP, Kelly PM, Anstey NM. 2013. High morbidity during treatment and residual pulmonary disability in pulmonary tuberculosis: under-recognised phenomena. PLoS One 8:e80302. doi: 10.1371/journal.pone.0080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willcox PA, Ferguson AD. 1989. Chronic obstructive airways disease following treated pulmonary tuberculosis. Respir Med 83:195–198. doi: 10.1016/S0954-6111(89)80031-9. [DOI] [PubMed] [Google Scholar]
- 47.Ravimohan S, Tamuhla N, Kung SJ, Nfanyana K, Steenhoff AP, Gross R, Weissman D, Bisson GP. 2016. matrix metalloproteinases in tuberculosis-immune reconstitution inflammatory syndrome and impaired lung function among advanced HIV/TB co-infected patients initiating antiretroviral therapy. EBioMedicine 3:100–107. doi: 10.1016/j.ebiom.2015.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.