

CS6, a prevalent surface antigen expressed in nearly 20% of clinical enterotoxigenic Escherichia coli (ETEC) isolates, is comprised of two major subunit proteins, CssA and CssB. Using donor strand complementation, we constructed a panel of recombinant proteins of 1 to 3 subunits that contained combinations of CssA and/or CssB subunits and a donor strand, a C-terminal extension of 16 amino acids that was derived from the N terminus of either CssA or CssB.
KEYWORDS: CS6, enterotoxigenic Escherichia coli, mice, vaccines
ABSTRACT
CS6, a prevalent surface antigen expressed in nearly 20% of clinical enterotoxigenic Escherichia coli (ETEC) isolates, is comprised of two major subunit proteins, CssA and CssB. Using donor strand complementation, we constructed a panel of recombinant proteins of 1 to 3 subunits that contained combinations of CssA and/or CssB subunits and a donor strand, a C-terminal extension of 16 amino acids that was derived from the N terminus of either CssA or CssB. While the entire panel of recombinant proteins could be obtained as soluble, folded proteins, it was observed that the proteins possessing a heterologous donor strand, derived from the CS6 subunit different from the C-terminal subunit, had the highest degree of physical and thermal stability. Immunological characterization of the proteins, using a murine model, demonstrated that robust anti-CS6 immune responses were generated from fusions containing both CssA and CssB. Proteins containing only CssA were weakly immunogenic. Heterodimers, i.e., CssBA and CssAB, were sufficient to recapitulate the anti-CS6 immune response elicited by immunization with CS6, including the generation of functional neutralizing antibodies, as no further enhancement of the response was obtained with the addition of a third CS6 subunit. Our findings here demonstrate the feasibility of including a recombinant CS6 subunit protein in a subunit vaccine strategy against ETEC.
INTRODUCTION
On a global scale, enterotoxigenic Escherichia coli (ETEC) is one of the leading bacterial causes of acute diarrhea in children in developing countries as well as in travelers to these areas (1–3). Although recent global estimates are imprecise, ETEC is estimated to cause roughly 74,000 deaths per year (4). The pathogenicity of ETEC strains is associated with the production of colonization factors (CFs), polymeric protein structures expressed on the surface of the bacterial cell that facilitate adherence to the small intestine, and diarrheagenic enterotoxins, heat-labile (LT) and/or heat-stable (ST) toxins (5, 6). Thus, a CF/enterotoxin-based approach is the primary strategy of many of the current ETEC vaccines in development (7), and clinical studies have demonstrated that antibodies (Abs) generated against CFs, as well as against subunits of CFs, are protective against ETEC-induced diarrhea (8–10). A hurdle to vaccine development is the variety of CFs, with over 25 different ETEC CFs identified (5, 11). Additionally, a significant proportion of clinically isolated ETEC strains possess no detectable CFs, but it is unclear whether this is due to a true lack of CFs, the expression of unknown CFs, or shortcomings in detection methodologies. Seven CFs, CFA/I and CS1 to CS6, are more prevalent in clinical isolates, and a vaccine comprised of these CFs and an LT toxin component could potentially provide coverage against 80% of global ETEC strains (12). Of the seven above-mentioned CFs, CS6 is an attractive vaccine target, as it is highly prevalent, expressed alone or with additional CFs in approximately 20% of clinical isolates globally (12–14). However, past efforts to develop a vaccine using purified, recombinant CS6 antigen (Ag) administered via the transcutaneous route or microencapsulated and administered via the oral route have been unsuccessful (15–17; D. Tribble, unpublished data).
Our efforts have been directed toward developing a multivalent subunit vaccine against ETEC. Initially, we focused on the tip adhesins of the class 5 fimbriae expressed by pathogenic ETEC strains, with the intent to disrupt initial intestinal binding by the bacteria, thus preventing colonization and abrogating disease (10, 18). However, the structure of CS6 is distinct from that of the rod-like class 5 fimbriae, which have a repeating structural subunit making up the length of the structure and a tip adhesin subunit that aids in intestinal binding (19, 20). Instead, CS6 is afimbrial in structure, associating closely with the bacterial cell surface instead of extending from the surface as is typical of the fimbrial CFs (11). Furthermore, it is made up of two structural subunits, CssA and CssB, in a 1:1 ratio (21). The bioassembly of CS6 is encoded by a plasmid-associated operon consisting of four genes (cssABCD) that is transcribed as a single transcript. In addition to the two structural subunits, the CssC and CssD proteins have been ascribed chaperone and usher roles, respectively (22–24). The intestinal binding moiety of CS6 has not been specifically determined, but binding functionalities have been ascribed to both CssA and CssB, which bind host cell fibronectin and sulfatide, respectively (24, 25). Thus, a representative CS6-derived subunit vaccine antigen would likely need to incorporate a combination of one or more of the CssA and CssB protein units, especially considering that multiple alleles of both proteins have been identified (26). Although distinct in structure and sequence from the class 5 CFs, it has been shown that much like the rigid class 5 fimbrial structures (27) and that of the Yersinia pestis F1 antigen (28), CS6 forms from the donor strand complementation of the two adjacent structural subunits (29).
Here, we describe the engineering of a panel of donor strand-complemented fusions of CssA and CssB subunits, in which the fold is completed by an in cis fusion of the N-terminal donor β-strand from either CssA or CssB to its C terminus. These vaccine candidates were characterized immunologically in BALB/c mice in order to select the ideal antigen that would induce a robust serum immune response against CS6 while satisfying the minimal production requirements in purity (>90%) and yield (1 mg purified protein/g of cell paste) for future evaluation in a protection study.
RESULTS
Expression, purification, and characterization of “homologous donor strand-complemented” CS6-derived recombinant proteins.
It has been observed that purified CS6 (21) as well as its CssA and CssB subunits (S. J. Savarino, unpublished data) form oligomeric complexes in solution. This has also been described for the class 5 major fimbrial subunit CfaB, which multimerizes via donor strand interactions between two subunits (27). Thus, in order to develop stable CS6-derived vaccine candidates, we applied donor strand complementation technology previously described for CfaE (27) and FimH (30) in the design of an initial panel of 11 His-tagged, homologous donor strand-complemented CS6 subunit proteins (Fig. 1A). The dimers or trimers contained various combinations of the CssA and CssB proteins of CS6 and an in cis donor strand fused to the C terminus. For “homologous donor strand complementation,” the C-terminal protein also served as the source of the donor strand. For instance, in the dscBCssAB fusion, CssA is the N-terminal protein, CssB is the C-terminal protein, and the donor strand used to complement the C-terminal CssB protein originated from the N terminus of CssB. The donor strand complementation occurring between proteins within the fusion are dictated by the protein fused to its C terminus. Thus, in the same dscBCssAB fusion, the fold of the N-terminal CssA subunit is completed by the donated donor strand from the C-terminally fused CssB unit, resulting in an internal “heterologous donor strand” between the CssA and CssB subunits. Thus, while it was possible for a given fusion to contain both heterologous and homologous donor strand interactions, for “homologous fusions,” the C-terminal donor strand was always homologous to the C-terminal protein. The pattern of donor strand complementation for each construct is schematically represented in Fig. 1A.
FIG 1.

Design of recombinant CS6 subunit monomers and fusion proteins. (A) Initial CS6 subunit fusions with homologous donor strand complementation, the donor strand fused to the C terminus derived from the C-terminal protein. The CssA protein is denoted in solid blue shading, the CssB protein is in solid red shading, the donor strand of CssA is in blue diagonal shading, the donor strand of CssB is in red diagonal shading, the DNKQ linker sequence is in black shading, and the hexahistidine tag is in green shading. (B and C) SDS-PAGE (B) and anti-CS6 Western blot (C) analyses of homologous donor strand-complemented CssA and CssB monomer and multimeric fusions. Lane 1, dscACssA; lane 2, dscBCssB; lane 3, dscACssAA; lane 4, dscBCssBB; lane 5, dscBCssAB; lane 6, dscACssBA; lane 7, dscACssABA; lane 8, dscBCssABB; lane 9, dscACssBBA; lane 10, dscBCssBAB; lane 11, dscACssBAA. Anti-CS6 sera were used at a concentration of 1:106, with the exception of dscACssA, which was blotted with an antibody concentration of 1:105. Abbreviations: ntd, deletion of the N-terminal donor strand; dsc, donor strand complementation of the C-terminal protein with the donor strand from either CssA (dscA) or CssB (dscB); MW, molecular weight.
Using standard column chromatographic techniques, the 11 recombinant proteins diagrammed in Fig. 1A were purified to >85% purity with low endotoxin content (<8 endotoxin units per 25 μg) (Table 1 and Fig. 1B). Additionally, each fusion reacted with rabbit antiserum raised against recombinant CS6, here referred to as CS6 (Fig. 1C), as well as antiserum raised against the component subunits, namely, CssB and/or CssA (data not shown).
TABLE 1.
Physicochemical properties of CS6 subunit fusions
| Protein | Theoretical mol wta (kDa) | Purityb (%) | Yieldc (mg/g) | Tm (°C) | HPLC-SEC |
||
|---|---|---|---|---|---|---|---|
| Peak retention time(s) (min) | Mol wt (kDa)d | Oligomeric state | |||||
| CssA fusions | |||||||
| dscACssA | 18.5 | 95 | 1.3 | 55 | 10.4 | 20.9 | Monomeric |
| dscACssAA | 34.0 | 95 | 1.3 | 49.9, 75.3 | 9.3 | 34.2 | Monomeric |
| dscBCssAB | 35.0 | 98 | 8.9 | 58.8, 74.5 | 7.5, 8.0, 9.2 | 45.7, 94.6 | Multimeric |
| dscACssABA | 50.4 | 95 | 2.1 | 54.3, 75 | 8.5 | 59.2 | Monomeric |
| dscBCssABB | 51.3 | 94 | 4.0 | 59.5, 74 | 7.2, 7.5, 8.5 | 67.4, 132 | Multimeric |
| Refined CssAB fusions | |||||||
| dscACssAB | 34.8 | 97 | 4.9 | 70 | 9.2 | 35.5 | Monomeric |
| ntd_dscBCssAB | 33.3 | 99 | 3.8 | 59, 74.6 | 9.3 | 34.1 | Monomeric |
| ntd_dscACssAB | 33.2 | 97 | 3.7 | 69.7, 76.1 | 9.4 | 33.9 | Monomeric |
| CssB fusions | |||||||
| dscBCssB | 19.4 | 91 | 3.1 | 60.1 | 10.4 | 22.5 | Monomeric |
| dscBCssBB | 35.8 | 100 | 2.7 | 58, 92.6 | 8.1, 9.2 | 37.5, 94 | Multimeric |
| dscACssBA | 34.8 | 91 | 4.3 | 69.2, 74.7 | 7.7, 8.2, 9.5 | 39.8, 93 | Multimeric |
| dscACssBAA | 50.4 | 90 | 1.0 | 71.8, 77.6 | 7.1, 8.0 | 141 | Multimeric |
| dscBCssBAB | 51.3 | 89 | 1.2 | 59.7, 75.5 | 7.6, 8.0, 8.6 | 63.1 | Multimeric |
| dscACssBBA | 51.2 | 91 | 0.5 | 59, 71.4, 76 | 7.2, 7.9, 8.9 | 65.4, 122.7 | Multimeric |
| Refined CssBA fusions | |||||||
| dscBCssBA | 35.0 | 97 | 8.2 | 74.1 | 9.4 | 37.3 | Monomeric |
| ntd_dscACssBA | 33.2 | 97 | 0.5 | 76.1 | 9.4 | 33.7 | Monomeric |
| ntd_dscBCssBA | 33.3 | 98 | 3.0 | 74.7 | 9.3 | 33.9 | Monomeric |
The theoretical molecular weight of each protein was determined from the primary amino acid sequence.
Protein purity was determined by densitometric measurements of the protein separated on an SDS-PAGE gel.
Milligrams of protein obtained per gram of wet bacterial cell paste lysed.
The experimental molecular weight was determined using the refractive index of molecules eluting from the HPLC-SEC column.
While analysis by SDS-PAGE (Fig. 1B) showed homogenous purified proteins, size exclusion high-performance liquid chromatography (HPLC-SEC) of the CS6-derived fusion proteins determined that some contained multiple species, manifesting as one or more additional peaks on the HPLC-SEC chromatograms (Fig. 2A and B). In contrast, the singular dscACssA and dscBCssB proteins both eluted as one peak, migrating as monomeric proteins.
FIG 2.
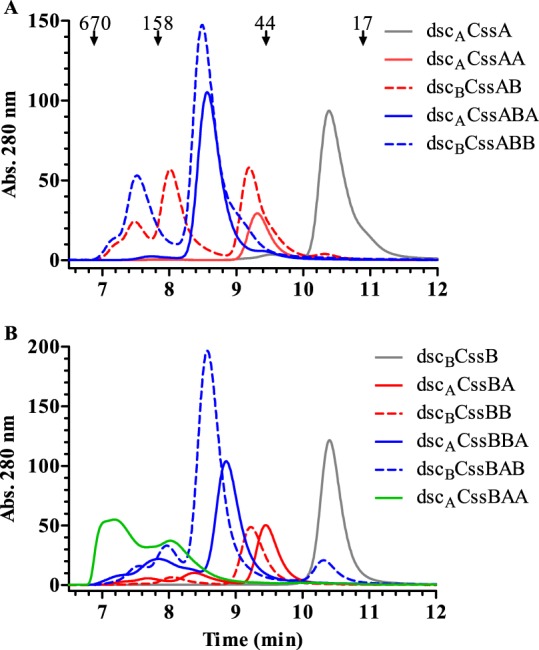
Size exclusion HPLC analysis of donor strand-complemented CssA (A) and CssB (B) monomer and multimeric fusions. Abs., absorbance.
Proteins extended from an N-terminal CssA unit exhibited various HPLC-SEC profiles (Fig. 2A). Higher-molecular-weight species, indicating possible multimer formation or aggregation, were observed for the dscBCssAB and dscBCssABB fusions, while proteins containing a C-terminal CssA protein (dscACssA, dscACssAA, and dscACssABA) eluted as a single peak. Interestingly, the two proteins forming higher-molecular-weight species both possess a C-terminal CssB protein complemented with a homologous donor strand and a potential heterologous donor strand present in the N-terminal CssA protein. The experimental masses of the species eluting during HPLC-SEC analysis argue against nonspecific aggregation, indicating that dscBCssAB and dscBCssABB are associating into dimers and trimers, respectively. Furthermore, these two proteins were the ones with the highest yields among the CssA fusions. Fusions with a C-terminal CssA donor strand, dscACssAA and dscACssABA, which have identical N-terminal and C-terminal donor strands, did not form multimers. These findings suggested that the donor strand in the N-terminal CssA protein from a second fusion may be outcompeting the internal C-terminal CssB donor strand, resulting in a more stable interaction being formed between two or more protein fusions.
Proteins extended from an N-terminal CssB unit, with the exception of dscBCssB, all exhibited some propensity to form higher-molecular-weight complexes, with dscACssBAA existing primarily as a multimer (Fig. 2B). However, unlike the protein fusions containing an N-terminal CssA donor strand, the N-terminal CssB donor strand in these fusions could interchange with the homologous C-terminal donor strand, whether from CssB or CssA, allowing for the formation of multimers. While the majority of these fusions had initial melting temperature (Tm) values around or below 60°C, those for dscACssBA and dscACssBAA fusions were higher, near 70°C (Table 1), possibly due to thermal stability conferred to the proteins due to multimer formation. With regard to protein recovery, of the CssB fusions, the dscACssBA fusion yielded the most material. Overall, the biochemical characterization of these CS6 subunit fusions indicated that complementation of the C terminus of these proteins with a homologous donor strand may not be the ideal fit for stabilizing the fold of the C-terminal protein unit, resulting in a degree of instability.
Expression, purification, and characterization of refined CS6-derived fusions.
After the initial evaluation of the CS6-derived protein fusions, we believed that the presence of an N-terminal donor strand, as well as a suboptimal donor strand, was resulting in instability in the recombinant fusions. To rectify this, we designed CS6 subunit fusions where the N-terminal donor strand (“ntd,” for N-terminal deletion) was removed, where the C-terminal protein was complemented with a donor strand derived from the heterologous “sister” subunit (e.g., complementation of CssA with a donor strand from CssB), and where both modifications were made. We then determined whether these modifications resulted in more-stable, monomeric fusion proteins. As we had selected the CssAB and CssBA protein fusions for further study based on their immunological properties (see below), we generated additional fusions for both CssAB and CssBA. For CssAB, these were ntd_dscBCssAB, dscACssAB (CssA-derived donor strand complementing C-terminal CssB), and ntd_dscACssAB (containing both modifications). For CssBA, ntd_dscACssBA, dscBCssBA (CssB-derived donor strand complementing C-terminal CssA), and ntd_dscBCssBA (containing both modifications) were constructed (Fig. 3A).
FIG 3.

(A) Refinement of dscACssBA and dscBCssAB fusions through deletion of the N-terminal donor strand and/or incorporation of heterologous donor strand complementation of the C-terminal protein. The CssA protein is denoted in solid blue shading, the CssB protein is in solid red shading, the donor strand of CssA is in blue diagonal shading, the donor strand of CssB is in red diagonal shading, the DNKQ linker sequence is in black shading, and the hexahistidine tag is in green shading. (B and C) SDS-PAGE (B) and anti-CS6 Western blot (C) analyses of refined CssAB and CssBA fusion proteins. Lane 1, dscBCssAB; lane 2, dscACssAB; lane 3, ntd_dscBCssAB; lane 4, ntd_dscACssAB; lane 5, dscACssBA; lane 6, dscBCssBA; lane 7, ntd_dscBCssBA; lane 8, ntd_dscACssBA. Anti-CS6 sera were used at a concentration of 1:106.
As with the initial panel of fusions, the refined CssAB and CssBA fusions were produced at high purity and with low endotoxin content (Table 1 and Fig. 3B). Additionally, all of the fusions reacted with rabbit antiserum against CS6 (Fig. 3C). HPLC-SEC analyses demonstrated that all of the refined CssAB and CssBA fusions, whether they contained a heterologous donor strand, the deletion of the donor strand of the N-terminal protein, or both modifications, did not form high-molecular-weight species, eluting as a single peak on the chromatogram (Fig. 4A and B). However, the fusions with heterologous donor strand complementation had the highest observed Tm values. For example, the ntd_dscBCssAB fusion, which had its N-terminal donor strand deleted and a homologous C-terminal donor strand, was monomeric but still had two melting transitions, one being below 60°C (Table 1). However, when the source of the C-terminal donor strand was changed from CssB to CssA (ntd_dscACssAB), only one Tm, above 70°C, was observed. While modification of this CssAB fusion through the deletion of the N-terminal donor strand did not appear to increase thermal stability, the lack of the N-terminal donor strand prevents any possibility of its participation in intermolecular interactions. The refined fusions, with the exception of ntd_dscACssBA, had yields at or above 3 mg of protein per g of wet cell paste lysed.
FIG 4.
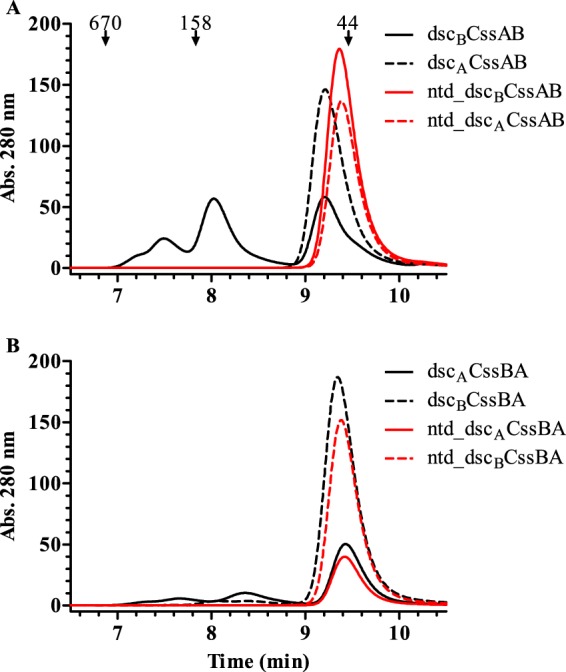
Size exclusion HPLC analysis of refined dscBCssAB (A) and dscACssBA (B) fusions.
Immunogenicity of CS6-derived recombinant proteins.
The first mouse experiment evaluated the immunogenicity of the monomeric and dimeric CS6 subunits in comparison to CS6 protein in the presence or absence of the adjuvant LTR192G when administered by the intradermal (i.d.) route. An enzyme-linked immunosorbent assay (ELISA) was used to measure the levels of anti-CS6, anti-CssB, and anti-CssA IgG antibodies (Abs) as well as serum anti-CS6 IgA Abs. Mice did not have detectable Ab titers against CS6, CssB, or CssA at baseline, i.e., before the first immunization (data not shown). After three immunizations, on day 42, the monomer dscBCssB and the dimers dscBCssBB, dscACssBA, and dscBCssAB, all homologous donor strand-complemented, elicited levels of serum anti-CS6 IgG Ab comparable to those elicited by CS6 (Fig. 5A). The monomer of CssA (dscACssA) failed to elicit anti-CS6 Abs, while the dimer (dscACssAA) induced only modest levels, which were significantly lower than those elicited by CS6, dscACssBA, or dscBCssAB (P < 0.05). Similar antibody responses against CssB were observed, with the exception that no IgG anti-CssB response was detected for either the monomer or dimer of CssA (Fig. 5B). Evaluation of anti-CssA Ab responses revealed that only the heterodimers dscACssBA and dscBCssAB were able to elicit anti-CssA IgG Ab levels comparable to those observed with CS6, while dscACssAA promoted only a modest response (Fig. 5C). Anti-CS6 IgA Ab levels were similar among the groups immunized with CS6, dscBCssB, dscACssBA, and dscBCssBA (Fig. 5D), while responses were below the limit of detection (LOD) for groups immunized with dscACssA and dscACssAA. For all parameters, the addition of the adjuvant LTR192G significantly increased the levels of antibodies observed (P < 0.05). Of note, we have not observed CF-specific antibody responses in animals immunized by the intradermal route with LTR192G only (data not shown).
FIG 5.

Immunogenicity of CS6 subunit-based monomers and dimers. Groups of 10 BALB/c mice were immunized three times, 2 weeks apart, by the i.d. route with 25 μg of each protein (CS6 or monomers or dimers of CS6 subunits CssA and/or CssB) with or without LTR192G as an adjuvant. Data are shown as means and SD of the log10-transformed titers for each group, on day 42, 2 weeks after the last immunization. Baseline levels, before the first immunization, were below the limit of detection (LOD) for the assays for all mice and are not shown in the graph. The dotted horizontal line indicates the LOD (1:50 or 101.7). (A) Serum anti-CS6 IgG Ab titers; (B) serum anti-CssB IgG Ab titers; (C) serum anti-CssA IgG Ab titers; (D) serum anti-CS6 IgA Ab titers. Black columns, protein plus LTR192G; white columns, protein alone. *, P < 0.05.
FIG 6.

Immunogenicity of CS6 subunit-based trimers. Groups of 10 BALB/c mice were immunized three times, 2 weeks apart, by the i.d. route with 25 μg of each protein (CS6 or dimers or trimers of CS6 subunits CssA and CssB) plus LTR192G or PBS. Data are shown as means and SD of the log10-transformed titers for each group on day 42. Baseline levels, before the first immunization, were below the LOD for the assays for all mice and are not shown. The dotted horizontal line indicates the LOD for the assay (1:50 or 101.7). (A) Serum anti-CS6 IgG Ab titers; (B) serum anti-CssB IgG Ab titers; (C) serum anti-CssA IgG Ab titers; (D) serum anti-CS6 IgA Ab titers. *, P < 0.05.
A second mouse study focused on evaluating the heterotrimeric CS6 subunits. The homologous donor strand-complemented heterodimer dscBCssAB, which promoted high levels of anti-CS6, -CssB, and -CssA IgG Abs in the first experiment, was used as a positive control, along with the CS6 protein. In addition, due to the significant increase in antibody titers observed in the previous study with the use of LTR192G, all proteins were coadministered by the i.d. route with this adjuvant in order to maximize the responses. All of the trimers, as well as the dscBCssAB control, promoted levels of serum anti-CS6 IgG Abs comparable to those observed with CS6 (Fig. 6A). Moreover, anti-CS6 antibody responses were comparable among the trimers, except for dscBCssBAB, which elicited anti-CS6 IgG levels significantly lower than those elicited by dscACssABA (P < 0.05). Most trimers also elicited an anti-CssB IgG Ab response that was comparable to the CS6 response, except for dscBCssABB, which elicited a significantly higher response (P < 0.05) (Fig. 6B). Anti-CssA Ab responses were more variable. Specifically, dscBCssBAB and dscACssBBA induced levels of anti-CssA IgG Ab that were significantly lower than those observed with the CS6 protein (P < 0.05) (Fig. 6C). All proteins tested induced similar serum anti-CS6 IgA Ab levels (Fig. 6D). Consistent with data from the first study, the heterodimer dscBCssAB (positive control) elicited responses comparable to those observed with CS6 across all parameters.
A third mouse study assessed the immunogenicity of the refined CS6 subunit dimers. For this study, animals were immunized intradermally with 10 μg of each protein, since parallel experiments demonstrated that doses of 10 and 25 μg gave comparable results (data not shown). This study incorporated the two modifications delineated above: (i) the molecules were stabilized by a heterologous donor strand, and (ii) the N-terminal chain was deleted to avoid intermolecular multimerization of the dimers. Animals immunized with CS6, dscBCssAB, and dscACssBA were included as controls. In comparison to the first and second mouse studies, where immunization with CS6 protein elicited anti-CS6 IgG Ab titers (log10) of 5.0 ± 0.15 (Fig. 5A) and 5.5 ± 0.32 (Fig. 6A) (means ± standard deviations [SD]), respectively, here we observed slightly reduced antibody levels, i.e., 4.4 ± 0.19 (Fig. 7A). In the same three studies, dscBCssAB reproducibly induced similar levels of anti-CS6 IgG Ab: 5.37 ± 0.24, 5.67 ± 0.21, and 5.15 ± 0.24. Hence, likely due to the weaker performance of CS6 in this study, the anti-CS6 IgG Ab titers promoted by all of the dimers are significantly higher than those observed with CS6 (P < 0.05) (Fig. 7A). Compared among themselves, all dimers elicited similar anti-CS6 IgG Ab titers. In addition, all of them induced high anti-CssB and anti-CssA IgG Ab titers (Fig. 7B) and promoted modest serum anti-CS6 IgA Ab titers (Fig. 7C). Overall, the serological response induced by the CS6 subunit trimers did not outperform the response observed with the CssAB and CssBA dimers, suggesting that the latter is sufficient in inducing a robust anti-CS6 response.
FIG 7.

Immunogenicity of refined CS6 subunit-based dimeric proteins. Groups of eight BALB/c mice were immunized three times, 2 weeks apart, by the i.d. route with 10 μg of each protein (CS6 or dimers of CS6 subunits CssA and/or CssB) plus LTR192G or PBS. Assays were performed with day 42 sera. The dotted horizontal line indicates the LOD for the assay (1:50 or 101.7). (A) Serum anti-CS6 IgG Ab titers. Data are shown as means and SD of the log10-transformed titers for each group. (B) Serum anti-CssB IgG Ab titers. Groups were assayed in pools. (C) Serum anti-CssA IgA Ab titers. Groups were assayed in pools. *, P < 0.05.
Evaluation of anti-CS6 functional neutralizing antibodies.
Pools of sera from mice immunized with CS6-based subunit vaccine candidates were assayed for their capacity to inhibit the binding of a CS6-positive (CS6+) ETEC strain to HT-29 cells in vitro as a measure of functional Abs. The results clearly show that immunization with the CS6 protein elicited anti-CS6 Abs that were able to neutralize bacterial adherence to the HT-29 cell line (average, 91.6%) (Fig. 8). Moreover, any recombinant protein, or fusion, containing the CssB subunit also elicited comparable high levels of anti-CS6 neutralizing Abs (86.8 to 92.9% inhibition). In contrast, the monomer or homodimer of CssA (i.e., CssA and CssAA) generated significantly lower neutralizing Ab responses than any of the groups immunized with the CS6 protein (20.9% and 38.0%, respectively).
FIG 8.

Serum-mediated inhibition of bacterial adherence. Pools of sera from mice immunized with CS6-based vaccine candidates (n = 8 to 10/pool) were assayed for the quantification of anti-CS6 functional neutralizing antibodies in an HT-29–CS6+ ETEC in vitro assay. Pools were prepared with day 42 sera from mice immunized with CS6 protein and monomers and dimers of CS6 subunits (evaluated in the first mouse experiment), CS6 protein and trimers of the CS6 subunits (evaluated in the second mouse experiment), and redesigned dimers of CS6 subunits (evaluated in the third mouse experiment). In the same assay, a pool of sera from PBS-immunized mice was also assayed, and the results from that pool were used to normalize the results as 100% binding (“zero” inhibition; 1.81 × 106 ± 0.54 × 106 CFU bacteria recovered [means ± SD]). Bars indicate intraexperimental means ± SD of data from evaluations performed in triplicate. *, P < 0.05 (compared to CS6 [first study]).
DISCUSSION
CS6 is a prevalent CF expressed by ETEC strains and is of special interest for a multivalent ETEC vaccine approach, as it is found globally in approximately 20% of clinically isolated ETEC strains (13, 22). CS6 is a polymeric structure consisting of two major structural subunits, CssA and CssB, with both having potential host intestinal binding activities (24, 25). Thus, the incorporation of both proteins into an adhesin-based ETEC vaccine may be required. Therefore, using various combinations and formulations of CssA and/or CssB, we aimed to maximize antigenicity, stability, and yield in order to identify an optimal composition for a CS6-derived vaccine candidate that could be incorporated into a multivalent subunit vaccine strategy targeting ETEC.
Adapting a similar donor strand complementation strategy that we used previously in designing a stable recombinant form of the ETEC CFA/I adhesin CfaE (27), we produced a panel of various in cis donor strand-complemented CS6 dimeric and trimeric subunit fusions constructed from combinations of the CssA and CssB subunits. The most stable fusions were those in which the C-terminal CS6 subunit was complemented with a heterologous donor strand, derived from the sister subunit, as opposed to one derived from itself.
Concurrent with our efforts, Roy et al. demonstrated, both experimentally and through structural determination, that heterologous donor strand complementation of CS6 subunits is chemically and energetically preferred and that CS6 is a polymer consisting of alternating CssA and CssB subunits (29). The results presented in this study lend further support to these findings.
The immunological downselection of our proteins was based on two criteria: (i) the robustness of the anti-CS6 IgG and IgA response induced in the serum of immunized mice and (ii) the generation of significant serum immune responses to both CssA and CssB, as both proteins may play a role in adhesion to host intestinal cells. We observed that while the monomer and dimer of CssB were able to elicit anti-CS6 IgG levels comparable to those observed with CS6, the CssA monomer failed to induce a detectable anti-CS6 response, and dimeric CssAA induced only a mediocre response. Both heterodimers, CssAB and CssBA, elicited anti-CS6 IgG levels comparable to or higher than those observed with CS6. All of the proteins studied, with the exception of CssA and the CssAA dimer, elicited comparable serum antibody titers against CssB. Finally, only CS6 and the heterodimers (CssAB and CssBA) were able to elicit high anti-CssA responses. Compared to the heterodimers, the incorporation of more than two CS6 subunits into a fusion protein did not lead to an increase in serum antibody responses against CS6, CssB, or CssA. Taken together, our results show that although CssB and CssBB elicited a robust anti-CS6 serum response, only the heterodimers (CssAB and CssBA) were able to generate strong responses against CS6, CssA, and CssB.
BLAST(P) analysis of the amino acid sequences of native CssA and CssB from ETEC strain E8775 shows that they share only limited sequence similarity (∼30% identity); hence, it is likely that they elicit a different repertoire of unique antibodies. This could explain why dscBCssB (and dscBCssBB) induced anti-CssB responses but not anti-CssA responses. In contrast, the monomer dscACssA failed to generate serum antibody responses against itself, and only modest antibody levels were observed with dimeric dscACssAA. These observations could be explained by (i) misfolding and/or (ii) an inherent low immunogenicity of the dscACssA protein. Our biochemical characterization supports that the proteins, including dscACssA, were appropriately folded. Moreover, high levels of responses were measured against the dscACssA protein using antisera generated against CS6, suggesting that the protein was folded with immunogenic epitopes properly displayed. Thus, it appears that the weak immunogenicity of the dscACssA protein in mice is an inherent property of the protein. Of note, in silico predictions for CssA and CssB proteins indicate that both proteins display similar numbers of possible B cell epitopes, providing no structural rationale as to why CssA is less immunogenic than CssB (see Fig. S1 in the supplemental material). In addition, dimeric dscACssAA was able to elicit modest anti-CssA titers, indicating that the anti-CssA immunogenicity is partially dependent on the association with another subunit, which has been suggested previously (23). This hypothesis is further supported by the fact that CssAB and CssBA heterodimers were able to elicit high levels of anti-CssA antibodies.
To determine whether the extension of the heterodimers would enhance immune responses, we constructed and evaluated CS6 subunit heterotrimers. Several iterations of CssA and CssB trimers were manufactured and used to immunize mice i.d. with LTR192G as an adjuvant. In general, the anti-CS6 IgG titers elicited by the trimers were comparable to those elicited by the CS6 protein and by the CssAB and CssBA heterodimers. Similar immune responses against CssB and CssA were also observed. Moreover, comparable anti-CS6 IgA titers were observed with all proteins tested. These results indicated that the inclusion of a third subunit, either CssA or CssB, did not enhance the immune response against CS6. Trimers, and possibly tetramers, of CssA and CssB as anti-CS6 vaccine candidates could be interesting approaches if the allelic variations identified in CS6 proteins (26) need to be incorporated into the vaccine.
Our final evaluations readdressed heterodimers (dscCssBA/dscCssAB) as the best candidates to elicit anti-CS6, anti-CssB, and anti-CssA responses. We compared the original heterodimers to the redesigned proteins where the homologous donor strand was replaced by a heterologous version, which increased stability. Our evaluations showed that all heterodimers tested were able to elicit high levels of anti-CS6 IgG antibodies as well as anti-CssA and -CssB responses. These results demonstrated that although the heterologous donor strand improved stability, it did not lead to discernible immunogenic differences compared to the original homologous donor strand versions.
Cell-based methodologies have been explored for the evaluation of functional anti-CS6 Abs (24, 31). We employed a recently developed cell binding assay to investigate the capacity of the proteins to generate anti-CS6 Abs that could inhibit the binding of a CS6+ ETEC strain (32) to the HT-29 cell line in vitro. Immunization with the monomer or homodimer of CssA (i.e., dscCssA and dscCssAA) elicited only marginal levels of inhibition, which seem to correlate with the weak immunogenicity of the protein and the low anti-CS6 IgG Ab titers observed by ELISAs. In contrast, all candidates with at least one copy of CssB, including the monomer dscCssB, generated high levels of anti-CS6 neutralizing Abs, comparable to those elicited by the CS6 protein and resembling the high anti-CS6 IgG levels measured by ELISAs. Previously, Tobias et al. (23) and Jansson et al. (25) implicated CssB in the binding mechanism of CS6, while Ghosal et al. (24) proposed that CssA may play a role in adherence. However, each of those investigations employed different experimental systems (i.e., different substrates), which could have led to contrasting conclusions. Additionally, it is plausible that both subunits may be involved in adherence to host intestinal epithelial cells. Here, we show that serum antibodies against CssB are significantly more effective than those against CssA in preventing the in vitro binding of a CS6+ ETEC strain to a cell substrate, likely through binding and interfering with functional sites on the CssB protein. This could also be explained by the lower anti-CS6 antibody titers induced by CssA immunization. However, immunization with CssB is sufficient in generating a functional anti-CS6 response similar to that induced by CS6, implying that CssB is critical to adherence and antigenicity. Overall, we observed that a CS6-derived fusion containing the CssB protein induced an immune response that resulted in a 10-fold reduction in the number of bacteria adhering to the HT-29 cell line, for example, for ntd_dscACssBA (1.62 × 105 CFU) in comparison to the phosphate-buffered saline (PBS) control (1.81 × 106 CFU). This indicates that an anti-CS6 response can impair the in vitro binding of a CS6+ ETEC strain to a surrogate cell line for the human intestine. The degree to which this impairment will translate into protection from diarrhea in immunized human volunteers will need to be assessed in vaccination-challenge clinical trials.
We evaluated numerous iterations of CS6 subunit recombinant proteins and determined that the heterologous donor strand-complemented CssBA and CssAB fusions have the most favorable biochemical profile for development as vaccine antigens. Both antigens are thermally stable and can be purified to homogeneity. The yields for these proteins, which were at or above 3 mg of protein per g of bacterial cells lysed, were also acceptable for proceeding into clinical-grade manufacturing. The assessments of immunogenicity by an ELISA and functional neutralizing Abs by a cell assay support the choice of these proteins for further development, as they were able to elicit responses comparable to those elicited by the CS6 protein. Recently, the efficacy of one of these proteins, ntd_dscBCssBA, was tested against oral challenge with CS6+ ETEC (B7A strain) in nonhuman primates, i.e., Aotus nancymaae monkeys. The animals responded with high serum anti-CS6 IgG Ab titers after intradermal immunization with ntd_dscBCssBA plus LTR192G/L211A (dmLT) as the adjuvant and were protected from diarrhea (unpublished data). This promising result led to a phase 1 safety-and-immunogenicity clinical trial, which is currently ongoing (ClinicalTrials.gov registration no. NCT03404674).
MATERIALS AND METHODS
Plasmid construction.
The genes for the donor strand-complemented CS6 subunit monomeric and fusion proteins were constructed with a hexahistidine (His) tag in the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible expression vector pET24a(+) (Novagen) as described in Tables S1 and S2 in the supplemental material. Proteins were expressed in E. coli BL21(DE3). The design of the recombinant proteins as well as the nomenclature used to identify them are summarized in Fig. 1A and B. The native leader sequences for these proteins were not included, which allowed for the cytoplasmic expression of the proteins. The CS6 operon from ETEC strain E8775 (GenBank accession no. U04846.1) was used as the source for the cssA and cssB structural genes. For simplicity, the names for the fusion proteins were condensed. For example, CssA-dscBCssB, the fusion of dscBCssB (CssB with the first 16 N-terminal amino acids of CssB [dscB] fused to its C terminus) to the C terminus of CssA, is abbreviated to dscBCssAB. A DNKQ tetrapeptide linker provided a bridge between subunit-subunit and subunit-donor strand interfaces.
Expression and purification of recombinant proteins.
BL21(DE3) expression clones were grown at 37°C with agitation at 200 rpm in 2.8-liter baffled Fernbach flasks containing 1 liter of Select APS Superbroth (Difco) and 50 μg/ml kanamycin sulfate. After reaching an optical density at 600 nm (OD600) of approximately 0.8, protein expression was induced by the addition of 1 mM IPTG. Cells were harvested after 3 h of induction by centrifugation. Cell pellets were resuspended in ∼100 ml of binding buffer (20 mM Tris-HCl [pH 7.9], 5 mM imidazole, and 0.5 M sodium chloride [NaCl]) and lysed by microfluidization (model M-110Y apparatus; Microfluidic Corp., Newton, MA). Cell lysates were applied to a 2-ml column packed with His-Bind resin (Novagen) using gravity flow. Proteins were eluted from the column with a buffer containing 20 mM Tris-HCl (pH 7.9), 0.5 M NaCl, and 300 mM imidazole. Fractions containing the protein were pooled and diluted 20-fold with 20 mM Tris (pH 8.0) before being loaded onto either a 1-ml or a 2-ml Q-Sepharose Fast Flow column (GE Healthcare). The Q column was washed with a solution containing 20 mM Tris (pH 8.0) and 50 mM NaCl and eluted stepwise with increasing salt concentrations (100 mM, 200 mM, and 300 mM). The dscACssA and dscACssAA proteins were eluted in the 50 mM NaCl wash step and in the flowthrough of the Q column. For these proteins, the flowthrough and wash fractions were pooled. All other proteins were eluted from the column using either 100 or 200 mM NaCl, and fractions containing the proteins were pooled. Finally, all proteins were concentrated and passed through a 0.22-μm filter. Endotoxin levels in protein preparations were determined by the Limulus amebocyte lysate (LAL) gel clot assay using Pyrotell lysate (Associates of Cape Cod Inc., E. Falmouth, MA) following manufacturer protocol.
Recombinant CS6 (CS6) prepared from an E. coli expression strain containing the CS6 operon from ETEC strain E8775 was kindly provided by the Walter Reed Army Institute of Research (WRAIR) (17).
SDS-PAGE and immunodetection of CS6-derived recombinant proteins.
For each recombinant protein preparation, 3 to 5 μg of protein was separated on a 15% SDS-PAGE gel along with a Precision Plus protein dual-color standard (Bio-Rad, Hercules, CA) and stained using GelCode blue stain reagent (Thermo Fisher Scientific, Waltham, MA). The purity of each protein was determined by densitometric analysis of the protein samples separated on the SDS-PAGE gel. The reactivity of proteins to anti-CS6 antibodies was analyzed by Western blotting using antisera at either a 1:105 or 1:106 dilution. Rabbit polyclonal antiserum was previously prepared against CS6, and rabbit immunizations and antiserum collection were performed by Harlan Bioproducts for Science, Inc. (Indianapolis, IN).
High-performance size exclusion chromatography.
A TSKgel G2000SWxl size exclusion column (Tosoh Bioscience, King of Prussia, PA) was equilibrated with a solution containing 0.1 M sodium phosphate and 0.1 M sodium sulfate (pH 6.7) at a flow rate of 0.5 ml/min. Purified recombinant proteins and a gel filtration standard (Bio-Rad, Hercules, CA) were applied to the column and eluted under the same conditions. Eluted proteins were observed by monitoring the absorbance of the eluate at 280 nm.
Molecular weight determination by HPLC-SEC analysis was performed on a Viscotek GPC/SEC system consisting of a GPCmax integrated autosampler-pump-degasser module, a VE2501 UV detector, and a TDA 302-050 triple-detector array comprising two laser light scattering detectors (right-angle and low-angle laser light scattering) and a viscometer in a series (Malvern Instruments, Worcestershire, UK). A Sup-Rs Stabitherm column oven (Prolabo, France) was used to maintain column temperature. OmniSEC 4.1 software (Malvern Instruments) was used for the acquisition and analysis of SEC data. The system was equipped with a TSKgel G3000 PWXL column (5 μm, 7.8-mm internal diameter by 30-cm length; Tosoh Biosciences). The mobile phase consisted of phosphate-buffered saline (PBS) (pH 7.4). The flow rate was 0.6 ml/min at a column temperature of 30°C. Samples were injected at a theoretical concentration of 500 μg/ml under a volume of 100 μl. The different detectors were calibrated by injecting pullulan standards of a defined molecular weight (Mw) and of a known concentration to calculate the mass constant for the refractometer, the constant for the viscometer, and constants for each of the two light scattering detectors. Column void volume (V0) and total column volume (Vt) were determined by injecting DNA and sucrose standards, respectively. Molecular weights at the peak and weight-average molecular weights of the pullulan standards were provided by the supplier, Malvern Instruments. The Mws of the polymer samples were calculated from the light scattering intensity signal (LS) according to the equation LS = KLS (dn/dc)2 Mw C, where KLS is the instrument calibration constant, C is the solution concentration, and dn/dc is the refractive index increment. The dn/dc value was taken from the literature.
Differential scanning calorimetry.
Differential scanning calorimetry (DSC) measurements were performed using a VP-DSC microcalorimeter (MicroCal, Inc., Piscataway, NJ). DSC thermograms of each protein were recorded in PBS (pH 7.4) from 5°C to 110°C at a scan rate of 200°C per h (filtering period of 10 s, prescan thermostat of 5 min, and postscan thermostat of 0 min). Baseline correction was performed by subtracting the buffer thermograms obtained under identical conditions. The data were analyzed using MicroCal Origin 7.0 software (OriginLab Corp., Northampton, MA), assuming a non-two-state unfolding model.
Mouse immunization and sample collection.
Animal studies involving mice were reviewed and approved by the Walter Reed Army Institute of Research/Naval Medical Research Center Institutional Animal Care and Use Committee in compliance with all applicable Federal regulations governing the protection of animals in research. Female BALB/c mice (8 to 10/group), aged 6 to 8 weeks (The Jackson Laboratory, Bar Harbor, ME), were housed in laminar flow cages for 7 days before use. Food and water were provided ad libitum. Three experiments were performed to evaluate the serological responses to immunization with CS6 or iterations of CS6 subunits (monomers, homo- and heterodimers, and trimers described above). Ten to twenty-five micrograms of each protein was administered via the intradermal (i.d.) route, with or without LTR192G as an adjuvant (100 ng per dose). Immunizations were performed on day 0, day 14, and day 28. One day prior to immunization, mice were restrained, and the dorsal surface of each animal was shaved using an electric clipper. i.d. immunizations were delivered in 20 μl/dose using a 31-gauge needle fitted to a 1-ml syringe inserted almost parallel to the skin. After delivery, the syringe was removed quickly; a bleb at the site of injection indicated a proper i.d. delivery of the formulation. Each of three doses was administered in a different quadrant on the animal’s dorsum. Blood was sampled prior to the first immunization (day 0) and approximately 2 weeks after each immunization (day 14, day 28, and day 42). On day 42, animals were anesthetized with ketamine-xylazine (Phoenix Scientific, Inc., St. Josephs, MO), exsanguinated by cardiac puncture with a 25-gauge needle fitted to a 1-ml syringe, and euthanized by cervical dislocation. Blood was centrifuged at 400 × g at 4°C for 15 min, and the separated serum was stored at −20°C until use.
Evaluation of the serum antibody response by an ELISA.
Mouse sera were evaluated for IgG and IgA anti-CS6, -CssB, and -CssA antibody (Ab) titers by ELISAs. Duplicate assays were performed with individual samples except where otherwise indicated (i.e., in pools). Antigen (Ag)-specific IgG and IgA ELISAs were performed on Nunc MicroWell and Nunc MicroWell MaxiSorp 96-well plates, respectively (Thermo Scientific, Rochester, NY). Plates were coated with CS6, dscBCssB, or dscACssA at 1.0 μg/ml (IgG) or 2.0 μg/ml (IgA) in carbonate buffer. Plates were coated with a volume of 100 μl/well for 1 h at 37°C, followed by overnight incubation at +4°C for IgG assays or overnight incubation at 37°C for IgA assays. After three washes with PBS-T (0.05% Tween 20 [Sigma-Aldrich]–PBS), all plates were blocked with 200 μl/well of 5% nonfat milk (Sigma-Aldrich) in PBS-T for 1 h at 37°C in a humidified chamber. After three washes with PBS-T, serum samples were added at a starting dilution of 1:50 in 1% nonfat milk–PBS-T, followed by a 3-fold serial dilution, and incubated for 1.5 h at room temperature (RT). Plates were washed 5 times with PBS-T, followed by the addition of peroxidase-conjugated goat anti-mouse IgG(H+L) (KPL, Gaithersburg, MD) at a 1:1,000 dilution or biotin-labeled goat anti-mouse IgA (KPL) at a 1:1,000 dilution in 1% nonfat milk–PBS-T for 1.5 h at 37°C in a humidified chamber. IgG plates were developed by the addition of orthophenylenediamine (OPD) substrate for 20 min at RT, while IgA assays were developed with 1-Step Ultra TMB (3,3′,5,5′-tetramethylbenzidine; Fisher Scientific, Waltham, MA) for 30 min at RT and stopped according to the manufacturer’s directions. The OD was measured at 450 nm using a Multiskan EX ELISA reader with Ascent software (Thermo Scientific). The cutoff for each plate was set at 0.4 U above the averaged background OD. A linear regression was fitted to the experimental data. The endpoint titer was determined as the reciprocal of the interpolated sample dilution that intersected with the cutoff and was log10 transformed. The average from duplicate assays was calculated as the final result. Based on the first serum dilution, the limit of detection (LOD) for all assays was established as 1:50 (indicated in the graphs by a horizontal line at 1.7). For computational purposes, serum samples with maximal ODs below the cutoff were assigned a value of one-half of the lowest dilution (i.e., 1:25) and are shown in the graphs as 1.4.
Cell binding inhibition assay.
Pools of sera from selected groups immunized with CS6-based vaccine candidates previously evaluated by ELISAs were prepared and assayed for the quantification of anti-CS6 functional neutralizing antibodies (see Fig. 8 for sample details) in a recently developed cell assay, as follows. HT-29 (ATCC HTB-38) cells, propagated in Dulbecco’s modified Eagle’s medium, were seeded into 48-well plates at 4 × 105 cells/well and grown overnight at 37°C with 5% CO2. Cells should be at approximately 95% confluence by the time of the assay. For the assay, the bacterial broth culture of CS6+ ETEC strain 214-4 (32), grown statically for 24 h, was diluted to 2 × 108 bacteria/ml. A volume of 800 μl of the diluted bacteria was incubated with 80 μl of each mouse test serum as well as control serum (1:10 dilution of serum), with rotation for 1 h at 37°C. After incubation, 250 μl of the bacterium-antibody mix was added to each of 3 wells of HT-29 cells and incubated at 37°C with 5% CO2 for 1.5 h. Given that HT-29 cells grow by about 85% after overnight incubation (95% confluence), the multiplicity of infection (MOI) by the time of the assay is approximately 67:1. The cells were washed with 500 μl per well of 1× DPBS (Dulbecco’s phosphate-buffered saline) (Corning) three times with vigorous shaking (200 rpm) for 5 min. The cells were lysed with 1% Triton X-100, serially diluted, and spot plated to enumerate attached bacteria. The percentage of bacterial binding inhibition for each pool sample was calculated using the average of bacteria enumerated in PBS control groups as 100% binding (i.e., “zero” inhibition), as follows: 100% − [(no. of bacteriasample/no. of bacteriaPBS) × 100%].
Statistical analysis.
Comparisons between animal groups were analyzed by one-way analysis of variance (ANOVA) with Bonferroni’s multiple-comparison test, and significance was assessed at P values of <0.05 throughout the study. All statistical analyses were performed using GraphPad Prism version 6 (GraphPad Software, San Diego, CA).
Supplementary Material
ACKNOWLEDGMENTS
We thank the Walter Reed Army Institute of Research for the provision of recombinant CS6. Additionally, we are thankful for the technical support from Glomil Corbin, William Hulsey, Diana Zhang, Patrick Bonifassi, and Laurence Fourrichon. We also acknowledge Julianne Rollenhagen for her assistance in reviewing the manuscript and Claudia Costabile for her assistance with image formatting.
This research was supported by U.S. Army Military Infectious Diseases Research Program Work Unit no. U.S. Navy 6000.RAD1.DA2.A0307, the Henry M. Jackson Foundation for the Advancement of Military Medicine, and Sanofi Pasteur (NCRADA-NMR-09-3183).
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, or the U.S. Government.
S.J.S. and M.G.P. served as military service members over the course of this work, which was prepared as part of their official duties. Title 17 USC §105 provides that “copyright protection under this title is not available for any work of the United States Government.” Title 17 USC §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties. P.D. and G.R-M. are employees of Sanofi Pasteur. S.J.S. served as the principal investigator for the NMRC and USUHS at the time of the study and is now an employee of Sanofi Pasteur. All other authors declare that there are no financial, institutional, or other relationships that might lead to bias or a conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00788-18.
REFERENCES
- 1.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, et al. 2012. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O’Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 3.Steffen R, Hill DR, DuPont HL. 2015. Traveler’s diarrhea: a clinical review. JAMA 313:71–80. doi: 10.1001/jama.2014.17006. [DOI] [PubMed] [Google Scholar]
- 4.Hosangadi D, Smith PG, Giersing BK. 11 October 2017. Considerations for using ETEC and Shigella disease burden estimates to guide vaccine development strategy. Vaccine doi: 10.1016/j.vaccine.2017.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qadri F, Svennerholm AM, Faruque AS, Sack RB. 2005. Enterotoxigenic Escherichia coli in developing countries: epidemiology, microbiology, clinical features, treatment, and prevention. Clin Microbiol Rev 18:465–483. doi: 10.1128/CMR.18.3.465-483.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans DG, Silver RP, Evans DJ Jr, Chase DG, Gorbach SL. 1975. Plasmid-controlled colonization factor associated with virulence in Escherichia coli enterotoxigenic for humans. Infect Immun 12:656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Svennerholm AM, Tobias J. 2008. Vaccines against enterotoxigenic Escherichia coli. Expert Rev Vaccines 7:795–804. doi: 10.1586/14760584.7.6.795. [DOI] [PubMed] [Google Scholar]
- 8.Freedman DJ, Tacket CO, Delehanty A, Maneval DR, Nataro J, Crabb JH. 1998. Milk immunoglobulin with specific activity against purified colonization factor antigens can protect against oral challenge with enterotoxigenic Escherichia coli. J Infect Dis 177:662–667. doi: 10.1086/514227. [DOI] [PubMed] [Google Scholar]
- 9.Tacket CO, Losonsky G, Link H, Hoang Y, Guesry P, Hilpert H, Levine MM. 1988. Protection by milk immunoglobulin concentrate against oral challenge with enterotoxigenic Escherichia coli. N Engl J Med 318:1240–1243. doi: 10.1056/NEJM198805123181904. [DOI] [PubMed] [Google Scholar]
- 10.Savarino SJ, McKenzie R, Tribble DR, Porter CK, O’Dowd A, Cantrell JA, Sincock SA, Poole ST, DeNearing B, Woods CM, Kim H, Grahek SL, Brinkley C, Crabb JH, Bourgeois AL. 2017. Prophylactic efficacy of hyperimmune bovine colostral antiadhesin antibodies against enterotoxigenic Escherichia coli diarrhea: a randomized, double-blind, placebo-controlled, phase 1 trial. J Infect Dis 216:7–13. doi: 10.1093/infdis/jix144. [DOI] [PubMed] [Google Scholar]
- 11.Gaastra W, Svennerholm AM. 1996. Colonization factors of human enterotoxigenic Escherichia coli (ETEC). Trends Microbiol 4:444–452. doi: 10.1016/0966-842X(96)10068-8. [DOI] [PubMed] [Google Scholar]
- 12.Isidean SD, Riddle MS, Savarino SJ, Porter CK. 2011. A systematic review of ETEC epidemiology focusing on colonization factor and toxin expression. Vaccine 29:6167–6178. doi: 10.1016/j.vaccine.2011.06.084. [DOI] [PubMed] [Google Scholar]
- 13.Kharat VB, Ahmed M, Jiang ZD, Riddle MS, DuPont HL. 2017. Colonization factors in enterotoxigenic Escherichia coli strains in travelers to Mexico, Guatemala, and India compared with children in Houston, Texas. Am J Trop Med Hyg 96:83–87. doi: 10.4269/ajtmh.16-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolf MK. 1997. Occurrence, distribution, and associations of O and H serogroups, colonization factor antigens, and toxins of enterotoxigenic Escherichia coli. Clin Microbiol Rev 10:569–584. doi: 10.1128/CMR.10.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lapa JA, Sincock SA, Ananthakrishnan M, Porter CK, Cassels FJ, Brinkley C, Hall ER, van Hamont J, Gramling JD, Carpenter CM, Baqar S, Tribble DR. 2008. Randomized clinical trial assessing the safety and immunogenicity of oral microencapsulated enterotoxigenic Escherichia coli surface antigen 6 with or without heat-labile enterotoxin with mutation R192G. Clin Vaccine Immunol 15:1222–1228. doi: 10.1128/CVI.00491-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katz DE, DeLorimier AJ, Wolf MK, Hall ER, Cassels FJ, van Hamont JE, Newcomer RL, Davachi MA, Taylor DN, McQueen CE. 2003. Oral immunization of adult volunteers with microencapsulated enterotoxigenic Escherichia coli (ETEC) CS6 antigen. Vaccine 21:341–346. doi: 10.1016/S0264-410X(02)00613-8. [DOI] [PubMed] [Google Scholar]
- 17.Guerena-Burgueno F, Hall ER, Taylor DN, Cassels FJ, Scott DA, Wolf MK, Roberts ZJ, Nesterova GV, Alving CR, Glenn GM. 2002. Safety and immunogenicity of a prototype enterotoxigenic Escherichia coli vaccine administered transcutaneously. Infect Immun 70:1874–1880. doi: 10.1128/IAI.70.4.1874-1880.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sincock SA, Hall ER, Woods CM, O’Dowd A, Poole ST, McVeigh AL, Nunez G, Espinoza N, Miller M, Savarino SJ. 2016. Immunogenicity of a prototype enterotoxigenic Escherichia coli adhesin vaccine in mice and nonhuman primates. Vaccine 34:284–291. doi: 10.1016/j.vaccine.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 19.Sakellaris H, Scott JR. 1998. New tools in an old trade: CS1 pilus morphogenesis. Mol Microbiol 30:681–687. doi: 10.1046/j.1365-2958.1998.01088.x. [DOI] [PubMed] [Google Scholar]
- 20.Baker KK, Levine MM, Morison J, Phillips A, Barry EM. 2009. CfaE tip mutations in enterotoxigenic Escherichia coli CFA/I fimbriae define critical human intestinal binding sites. Cell Microbiol 11:742–754. doi: 10.1111/j.1462-5822.2009.01287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabui S, Debnath A, Ghosal A, Wajima T, Hamabata T, Ramamurthy T, Ghosh AN, Basak S, Chatterjee NS. 2016. Characterization of oligomeric assembly of colonization factor CS6 from enterotoxigenic Escherichia coli. Microbiology 162:72–83. doi: 10.1099/mic.0.000180. [DOI] [PubMed] [Google Scholar]
- 22.Wolf MK, de Haan LA, Cassels FJ, Willshaw GA, Warren R, Boedeker EC, Gaastra W. 1997. The CS6 colonization factor of human enterotoxigenic Escherichia coli contains two heterologous major subunits. FEMS Microbiol Lett 148:35–42. doi: 10.1111/j.1574-6968.1997.tb10263.x. [DOI] [PubMed] [Google Scholar]
- 23.Tobias J, Lebens M, Kallgard S, Nicklasson M, Svennerholm AM. 2008. Role of different genes in the CS6 operon for surface expression of enterotoxigenic Escherichia coli colonization factor CS6. Vaccine 26:5373–5380. doi: 10.1016/j.vaccine.2008.07.091. [DOI] [PubMed] [Google Scholar]
- 24.Ghosal A, Bhowmick R, Banerjee R, Ganguly S, Yamasaki S, Ramamurthy T, Hamabata T, Chatterjee NS. 2009. Characterization and studies of the cellular interaction of native colonization factor CS6 purified from a clinical isolate of enterotoxigenic Escherichia coli. Infect Immun 77:2125–2135. doi: 10.1128/IAI.01397-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jansson L, Tobias J, Jarefjall C, Lebens M, Svennerholm AM, Teneberg S. 2009. Sulfatide recognition by colonization factor antigen CS6 from enterotoxigenic Escherichia coli. PLoS One 4:e4487. doi: 10.1371/journal.pone.0004487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabui S, Ghosal A, Dutta S, Ghosh A, Ramamurthy T, Nataro JP, Hamabata T, Chatterjee NS. 2010. Allelic variation in colonization factor CS6 of enterotoxigenic Escherichia coli isolated from patients with acute diarrhoea and controls. J Med Microbiol 59:770–779. doi: 10.1099/jmm.0.017582-0. [DOI] [PubMed] [Google Scholar]
- 27.Poole ST, McVeigh AL, Anantha RP, Lee LH, Akay YM, Pontzer EA, Scott DA, Bullitt E, Savarino SJ. 2007. Donor strand complementation governs intersubunit interaction of fimbriae of the alternate chaperone pathway. Mol Microbiol 63:1372–1384. doi: 10.1111/j.1365-2958.2007.05612.x. [DOI] [PubMed] [Google Scholar]
- 28.Zavialov AV, Kersley J, Korpela T, Zav’yalov VP, MacIntyre S, Knight SD. 2002. Donor strand complementation mechanism in the biogenesis of non-pilus systems. Mol Microbiol 45:983–995. doi: 10.1046/j.1365-2958.2002.03066.x. [DOI] [PubMed] [Google Scholar]
- 29.Roy SP, Rahman MM, Yu XD, Tuittila M, Knight SD, Zavialov AV. 2012. Crystal structure of enterotoxigenic Escherichia coli colonization factor CS6 reveals a novel type of functional assembly. Mol Microbiol 86:1100–1115. doi: 10.1111/mmi.12044. [DOI] [PubMed] [Google Scholar]
- 30.Barnhart MM, Pinkner JS, Soto GE, Sauer FG, Langermann S, Waksman G, Frieden C, Hultgren SJ. 2000. PapD-like chaperones provide the missing information for folding of pilin proteins. Proc Natl Acad Sci U S A 97:7709–7714. doi: 10.1073/pnas.130183897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viboud GI, McConnell MM, Helander A, Svennerholm AM. 1996. Binding of enterotoxigenic Escherichia coli expressing different colonization factors to tissue-cultured Caco-2 cells and to isolated human enterocytes. Microb Pathog 21:139–147. doi: 10.1006/mpat.1996.0049. [DOI] [PubMed] [Google Scholar]
- 32.Levine MM, Caplan ES, Waterman D, Cash RA, Hornick RB, Snyder MJ. 1977. Diarrhea caused by Escherichia coli that produce only heat-stable enterotoxin. Infect Immun 17:78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.