

Pathogenic Yersinia species deliver Yop effector proteins through a type III secretion system into host cells. Among these effectors, YopE and YopT are Rho-modifying toxins, which function to modulate host cell physiology and evade immune responses.
KEYWORDS: inflammasome, macrophages, pyrin, Yersinia
ABSTRACT
Pathogenic Yersinia species deliver Yop effector proteins through a type III secretion system into host cells. Among these effectors, YopE and YopT are Rho-modifying toxins, which function to modulate host cell physiology and evade immune responses. YopE is a GTPase-activating protein (GAP) while YopT is a protease, and they inhibit RhoA by different modes of action. Modifications to RhoA are sensed by pyrin, which, once activated, assembles a caspase-1 inflammasome, which generates cytokines such as interleukin-1β (IL-1β) and cell death by pyroptosis. In Yersinia-infected macrophages, YopE or YopT triggers inflammasome assembly only in the absence of another effector, YopM, which counteracts pyrin by keeping it inactive. The glucosyltransferase TcdB from Clostridium difficile, a well-studied RhoA-inactivating toxin, triggers activation of murine pyrin by dephosphorylation of Ser205 and Ser241. To determine if YopE or YopT triggers pyrin dephosphorylation, we infected lipopolysaccharide (LPS)-primed murine macrophages with ΔyopM Yersinia pseudotuberculosis strains expressing wild-type (wt) or YopE mutant variants or YopT. By immunoblotting pyrin after infection, we observed that wt YopE triggered dephosphorylation of Ser205 and inflammasome activation. Pyrin dephosphorylation was reduced if a YopE variant had a defect in stability or RhoA specificity but not membrane localization. We also observed that wt YopT triggered pyrin dephosphorylation but more slowly than YopE, suggesting that YopE is dominant in this process. Our findings provide evidence that RhoA-modifying toxins trigger activation of pyrin by a conserved dephosphorylation mechanism. In addition, by characterization of YopE and YopT, we show that different features of effectors, such as RhoA specificity, affect the efficiency of pyrin dephosphorylation.
INTRODUCTION
Protein toxins can function as potent virulence factors and play a very important role in host-pathogen interactions and the outcome of infections (1, 2). There are several types of toxins, which can vary in structure, modes of action, and targets and how they are delivered into the host (2, 3). An important class of toxins consists of those that modify the Rho family of small GTPases (4). Rho GTPases have essential functions in the host cells and are known as “switch proteins” due to their ability to cycle between an inactive and active state (4, 5). When active, GTPases are bound to GTP and can control stress fiber formation, regulate actin cytoskeleton and cell shape, control progression of cell division, coordinate cell migration and phagocytosis, and regulate adhesion to endothelial and epithelial tissues (5–8). This process is tightly regulated by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). GEFs can catalyze the dissociation of GDP, leading to the binding of GTP and activation of the GTPase, while GAPs can facilitate the hydrolysis of GTP into GDP, inactivating the GTPase (9). Therefore, GEFs and GAPs are essential to control the activity cycle of the small GTPases such as RhoA, Rac1, and Cdc42 (8, 9). Since Rho GTPases are such important proteins, inactivating them can generate major effects in cell homeostasis, and some pathogens have evolved toxins that affect this pathway with different strategies in order to evade host defense mechanisms (4).
The genus Yersinia consists of 17 species, in which three are well-known human pathogens: Yersinia enterocolitica, Yersinia pestis, and Yersinia pseudotuberculosis (10, 11). Among many virulence factors, pathogenic Yersinia species share the presence of a 70-kb plasmid that encodes a conserved type III secretion system (T3SS) and a variety of effector proteins named Yersinia outer proteins (Yops) (12, 13). Upon infection, Yersinia can assemble the T3SS and through a contact-dependent mechanism can deliver several Yops into the host cell cytosol (14–16). Among them, two are known Rho GTPase-inactivating effectors: YopE, that mimics the host GAP and facilitates the hydrolysis of GTP into GDP, inactivating the Rho GTPase, and YopT, a cysteine protease that cleaves the C terminus of Rho GTPases, leading to its release from the membrane and, consequently, its inactivation (15–18). Although YopE and YopT have different Rho GTPase inactivation mechanisms, they generate similar detrimental effects in the host cells, such as disruption of the actin cytoskeleton leading to changes in cell morphology and blockage of phagocytosis (19).
Toxins that inactivate RhoA such as Clostridium difficile TcdB, YopE, and YopT can also trigger the pyrin inflammasome, a compensatory innate immune response inside host cells (15, 20, 21). Pyrin is a pattern recognition receptor (PRR) and inflammasome sensor (15, 22) that is preferentially expressed in activated macrophages, cytokine-activated monocytes, and granulocytes but can also be found in serosal and synovial fibroblasts (23). In nonintoxicated cells pyrin is phosphorylated on two serine residues and bound to a dimer of protein 14-3-3, locking this PRR in the inactive state (22). Studies with covalently modifying toxins such as TcdB indicate that upon inactivation of RhoA, pyrin is dephosphorylated, and 14-3-3 is released (22, 24). Consequently, pyrin becomes active and can bind to the adaptor protein ASC, which recruits pro-caspase-1, forming the multiprotein inflammasome (25). This assembly activates caspase-1, an important cysteine protease that can generate mature interleukin-1β (IL-1β) and IL-18 cytokines and can cleave gasdermin D (GSDMD) (26). The generated GSDMD N-terminal fragments relocalize to the plasma membrane, where they oligomerize to form small pores. These pores allow the release of mature IL-1β and IL-18 and lead to a unique cell death, known as pyroptosis (27, 28).
Pyroptosis can restrict intracellular bacteria from growing and spreading to counteract infection (29). In addition, the secretion of IL-1β and IL-18 leads to recruitment of leukocytes to the site of the infection and the activation of these cells, which can help in the clearance of pathogens (30). Triggering of the pyrin inflammasome by YopE and YopT results in a protective immune response mediated by IL-1β and IL-18 against systemic Yersinia infection (15, 21, 31). A third Yersinia effector, YopM, inhibits pyrin to limit inflammasome-mediated immunity against systemic infection (15, 21).
It is not understood how inactivation of RhoA by bacterial toxins and effectors leads to activation of pyrin. Although toxins such as TcdB that covalently modify switch I of RhoA GTPase cause dephosphorylation of serines 205 and 241 in murine pyrin (22, 24), it is not clear if other mechanisms of RhoA inactivation lead to the same mechanism of pyrin activation. Here, we determined that YopE, which noncovalently inactivates RhoA via GAP activity (18), and YopT, which cleaves the C terminus of RhoA (17), also trigger dephosphorylation of pyrin. In addition, we determined that YopE and YopT trigger dephosphorylation of pyrin at different rates in order to shed light on the previous observation that these two effectors cause IL-1β to be released at different levels from Yersinia-infected macrophages (15).
We also further investigated the mechanism of YopE-triggered pyrin activation by taking advantage of existing YopE variants since target specificity, toxin stability, and membrane localization had been shown previously to be important features for the GAP activity of YopE against RhoA and macrophage killing of the Y. pseudotuberculosis IP2666 strain (16). YopEL109A has an amino acid change in the predicted binding pocket that interacts with switch I and II regions of Rho GTPases and consequently possesses less substrate specificity toward RhoA (70% of wild-type [wt] level) (16, 32); YopER62K is an unstable naturally occurring variant of YopE, which is targeted for ubiquitination and therefore degradation (33); and YopE3N is a mutant that presents three amino acid modifications (leucine to asparagine) in its membrane localization domain (MDL), leading to the disruption of its ability to localize to the membrane (34). These three variants had their abilities of triggering the pyrin inflammasome compared to the ability of the wild-type YopE. We observed that target specificity and toxin stability were critical factors to determine the YopE ability to activate pyrin, suggesting that the Rho GAP activity of YopE toward RhoA is what specifically leads to the pyrin inflammasome formation. Thus, altogether our data suggest that RhoA inactivation by YopE and YopT leads to the same conserved response by the host immune system.
RESULTS
YopE triggers pyrin Ser205 dephosphorylation in the absence of YopM.
To determine if YopE triggers pyrin dephosphorylation leading to assembly of the pyrin inflammasome, infections with a Y. pseudotuberculosis yopE yopM mutant strain IP6ΔM expressing YopE from a plasmid vector under the control of its native promoter (pYopE) or carrying the empty vector (Table 1) were carried out. Bone marrow-derived macrophage (BMDMs) primed with Escherichia coli lipopolysaccharide (LPS) were infected with the above strains at a multiplicity of infection (MOI) of 30 or treated with the RhoA-modifying toxin TcdB as a positive control. After 90 min, we observed the cells through phase-contrast microscopy to detect YopE cytotoxicity, represented by the BMDM cell rounding and detachment (see Fig. S1D in the supplemental material). A similar rounding effect was observed upon TcdB intoxication (Fig. S1C) but not in BMDMs infected with the empty vector strain (Fig. S1B) or left uninfected (Fig. S1A). Infection supernatants were also collected for quantification of IL-1β secretion and lactate dehydrogenase (LDH) release (a measure of pyroptosis). As shown in Fig. 1A, in the presence of YopE or TcdB, BMDMs release a larger amount of IL-1β than under the empty vector condition. The same trend was observed for LDH release (Fig. 1B). Thus, the data suggest that in the presence of the IP6ΔM strain containing pYopE, the pyrin inflammasome is being triggered, while in the infection with the empty vector, the inflammasome is not being assembled.
TABLE 1.
Y. pseudotuberculosis strains and plasmids used in this work
| Strain | Relevant characteristic(s) | Reference or source |
|---|---|---|
| IP6 | Serogroup O3, pYV+, yopE mutant | 18 |
| IP6ΔM harboring an empty vector | pYV+, yopE yopM mutant, naturally yopT mutant; harboring pMMB67HE, tac promoter | This study |
| IP6ΔM/pYopE | IP6ΔM (pPEYopE), yopE wt controlled by native promoter | This study |
| IP6ΔM/pYopE3N | IP6ΔM (pPEYopE L55N I59N L63N), yopE variant controlled by yopE native promoter | This study |
| IP6ΔM/pYopER62K | IP6ΔM (pPEYopER62K) yopE variant controlled by yopE native promoter | This study |
| IP6ΔM/pYopEL109A | IP6ΔM (pPEYopEL109A) yopE variant controlled by yopE native promoter | This study |
| IP6ΔMmJ harboring an empty vector | IP6ΔM yopJC172A, harboring pMMB67HE, tac promoter | This study |
| IP6ΔMmJ/pYopE | IP6ΔMmJ (pPEYopE), yopE controlled by native promoter | This study |
| IP6ΔMmJ/pYopE3N | IP6ΔMmJ (pPEYopE L55N I59N L63N) yopE variant controlled by yopE native promoter | This study |
| IP6ΔMmJ/pYopER62K | IP6ΔMmJ (pPEYopER62K) yopE variant controlled by yopE native promoter | This study |
| IP6ΔMmJ/pYopEL109A | IP6ΔMmJ (pPEYopEL109A) yopE variant controlled by yopE native promoter | This study |
| 32777 ΔyopM | Serogroup O1, pYV+ yopE+ yopT+ yopM mutant | 39 |
| yopER144A strain | 32777 ΔyopM yopER144A | 15 |
| yopTC139A strain | 32777 ΔyopM yopTC139A | 15 |
| yopER144A yopTC139A strain | 32777 ΔyopM yopER144A yopTC139A | 15 |
FIG 1.
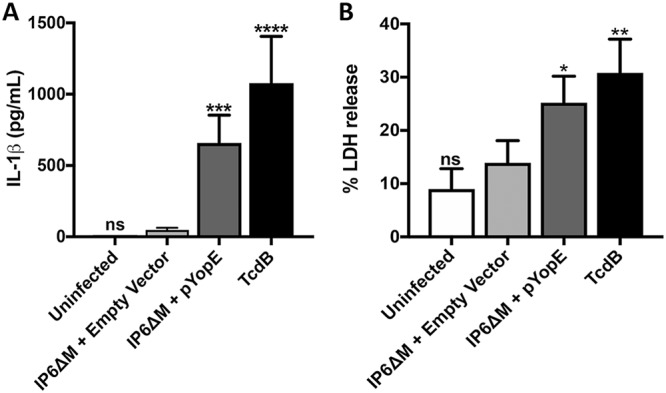
Analysis of IL-1β release and pyroptosis in BMDMs. LPS-primed BMDMs were left uninfected or infected with Y. pseudotuberculosis strain IP6ΔM harboring an empty vector or IP6ΔM/pYopE for 90 min at an MOI of 30. As a positive control, TcdB was used to intoxicate BMDMs for 90 min at a concentration of 0.2 μg/ml. Supernatants were collected and analyzed by ELISA for released IL-1β (A) or by measurement of LDH release for pyroptosis (B). Data represent average values ± the standard errors of the means from three independent experiments. Significant differences compared to results for BMDMs infected with IP6ΔM harboring the empty vector were determined by one-way ANOVA. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Next, to investigate the phosphorylation status of pyrin, lysates of the BMDMs treated as above were analyzed by immunoblotting using antibodies that recognize phosphorylated Ser205 (24) or total pyrin. Immunoblotting with antibody to actin was used to control for loading. To generate lysates, a nondenaturing NP-40 lysis buffer was used, and the lysate was separated into soluble and insoluble fractions, which were analyzed separately. Serine 205-phosphorylated pyrin and total pyrin were mostly detected in the soluble fractions of the BMDMs left uninfected or infected with the strain harboring the empty vector (Fig. 2A and B, lanes 1 and 2, respectively). After infection with the strain harboring pYopE, total pyrin was detected at higher levels in the insoluble fraction, and S205-phosphorylated pyrin was not detected in either fraction (Fig. 2A and B, lanes 3). Together, these data suggest that in the presence of YopE, a movement of pyrin from a soluble to insoluble form occurs and that this is associated with its dephosphorylation.
FIG 2.

Western blot analysis of pyrin S205 dephosphorylation in BMDMs. LPS-primed BMDMs were left uninfected, infected with Y. pseudotuberculosis strain IP6ΔM harboring an empty vector or IP6ΔM/pYopE or intoxicated with TcdB for 90 min. (A and B) Lysates were processed with nondenaturing NP-40 lysis buffer and separated into soluble and insoluble fractions by centrifugation. (C) Lysates were processed with denaturing RIPA lysis buffer, and the soluble fractions were collected. Samples were subjected to Western blotting using antibodies to pSer205 of pyrin, total pyrin, or β-actin.
To further investigate pyrin dephosphorylation triggered by YopE, denaturing radioimmunoprecipitation assay (RIPA) lysis buffer was used to generate new BMDM cell lysates after infection. Lysates from TcdB-intoxicated BMDMs were also obtained as a positive control. Soluble samples of the RIPA cell lysates were immunoblotted as previously described. As shown in Fig. 2C, dephosphorylation of pyrin was observed in the presence of YopE or TcdB (Fig. 2C, lanes 3 and 4). Altogether, these data demonstrate that, similar to TcdB, YopE triggers dephosphorylation of pyrin on serine 205.
Ubiquitination or RhoA specificity variants of YopE trigger reduced pyrin dephosphorylation.
Amino acid variants of YopE have previously been generated to determine if features such as membrane localization, stability, and Rho GTPase specificity are important for its GAP function (16, 32–34). To examine several of these variants for their abilities to trigger pyrin dephosphorylation, we constructed Y. pseudotuberculosis IP6ΔM strains harboring the following plasmids: pYopEL109A, encoding a variant with reduced GAP activity toward RhoA and Rac1; pYopER62K, encoding a variant that is ubiquitinated and therefore, less stable, or pYopE3N, encoding a variant defective for membrane localization (Table 1). SDS-PAGE was used to detect the YopE variants secreted in vitro by these strains in comparison to results with the previously used IP6ΔM strain harboring pYopE or empty vector (Fig. S2). Importantly, the different YopE variants were secreted at similar or higher levels than YopE (Fig. S2, lanes 2, 4, 5, and 6).
Next, the above strains were used to infect BMDMs as previously described. After 90 min of infection, supernatants were analyzed for the presence of IL-1β. Compared to levels with the YopE strain, all three YopE variants caused reduced secretion of this cytokine. However, only in the case of YopER62K was there a significant change in the IL-1β secretion level (Fig. S3).
To increase the IL-1β secretion sensitivity, the empty vector and plasmids encoding the different YopE proteins were introduced into the strain IP6ΔMmJ lacking YopJ activity as well as YopM (Table 1). By inactivating YopJ, which also inhibits caspase-1-dependent responses in LPS-primed BMDMs (35), we could better observe the effects of YopE. After the above strains were used to infect BMDMs, supernatants were analyzed. We observed that YopE triggered higher IL-1β release when the BMDMs were infected with the IP6ΔMmJ background strain than with the IP6ΔM background strain (Fig. 3). In addition, YopER62K and YopEL109A but not YopE3N strains triggered significantly reduced IL-1β release in the IP6ΔMmJ background compared to the level with YopE (Fig. 3).
FIG 3.
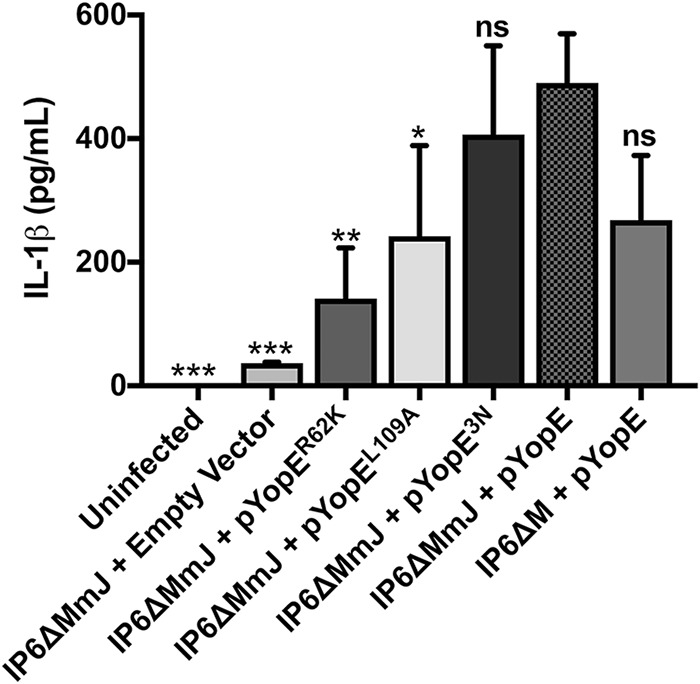
Analysis of IL-1β release in BMDMs. LPS-primed BMDMs were left uninfected or infected with Y. pseudotuberculosis IP6ΔM/pYopE or IP6ΔMmJ mutants harboring empty vector, pYopER62K, pYopEL109A, pYopE3N, or pYopE for 90 min. After infection, supernatants were collected and analyzed by ELISA to detect the release of IL-1β. Data represent average values ± the standard errors of the means from three independent experiments. Significant differences compared to results with IP6ΔMmJ/pYopE-infected BMDMs were determined by one-way ANOVA. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
RIPA lysates from BMDMs infected with the above strains were also analyzed for the presence of pSer205 pyrin through immunoblotting. As shown in Fig. 4, YopER62K and YopEL109A strains were defective for triggering pyrin dephosphorylation (lanes 5 and 6) compared to results with YopE3N (lane 7) or YopE (lane 4). There was no difference observed in levels of pyrin dephosphorylation triggered by YopE between the strains IP6ΔM and IP6ΔMmJ (Fig. 4, lanes 3 and 4). Together, these results suggest that avoidance of ubiquitination and RhoA target specificity in YopE are critical for it to trigger pyrin Ser205 dephosphorylation and inflammasome assembly.
FIG 4.
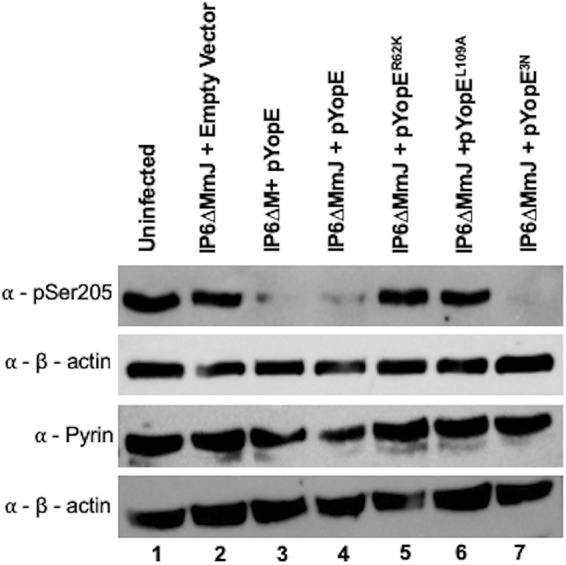
Western blot analysis of pyrin S205 dephosphorylation in BMDMs. LPS-primed BMDMs were left uninfected or infected with the indicated Y. pseudotuberculosis IP6ΔM or IP6ΔMmJ mutants for 90 min at an MOI of 30. After infection, lysates were processed with denaturing RIPA lysis buffer, collected, and subjected to Western blotting using antibodies to pSer205 of pyrin, total pyrin, or β-actin.
YopT triggers slower pyrin dephosphorylation than YopE.
To determine if the other RhoA-modifying effector YopT triggers pyrin dephosphorylation leading to assembly of the pyrin inflammasome, infections with Y. pseudotuberculosis 32777 ΔyopM strains expressing wt YopE and YopT or only active YopT (yopER144A [the yopE gene encoding an R-to-A change at position 144]) or YopE (yopTC139A) or inactive YopT and YopE (yopER144A yopTC139A) (Table 1) were carried out. LPS-primed BMDMs were infected as previously described. After 90 min, YopE- or YopT-induced cell rounding was observed through phase-contrast microscopy. Although both mutants generated cell rounding and detachment, in the presence of YopT (yopER144A) these effects were more prominent than those in the mutant expressing wt YopE (yopTC139A) (Fig. S4B and C). The cytotoxic effects were not observed in BMDM infections in which YopE and YopT were both catalytically dead (yopER144A yopTC139A) or in the uninfected BMDMs (Fig. S4A and D).
BMDM infection supernatants were also collected for quantification of IL-1β secretion and LDH release, as previously described. Mutants lacking active YopE or YopT or both caused less IL-1β release than 32777 ΔyopM, but the decrease was significant only for yopER144A and yopER144A yopTC139A strains (Fig. 5A), as expected from our previous publication (15). Thus, YopE has a dominant role in triggering IL-1β release, and the effect of YopT is significant only in the absence of YopE (Fig. 5A). Interestingly, the reduced IL-1β release triggered by YopT compared to that of YopE after 90 min did not correspond with the cell-rounding effects of these effectors as observed by microscopy (Fig. S4B and C). Although LDH release caused by the mutants followed the same trends as for IL-1β, the differences were not significant (Fig. 5B).
FIG 5.
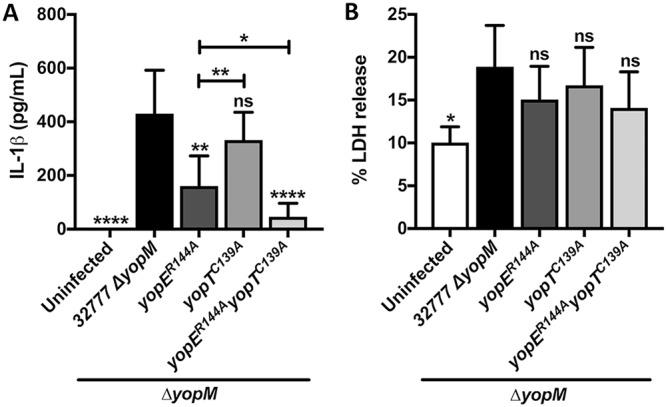
Analysis of IL-1β release and pyroptosis in BMDMs. LPS-primed BMDMs were left uninfected or infected with a Y. pseudotuberculosis 32777 ΔyopM, yopER144A, yopTC139A, or yopER144A yopTC139A strain for 90 min. Supernatants were collected and analyzed by ELISA for release of IL-1β (A) or by measurement of LDH release for pyroptosis (B). Data represent average values ± the standard errors of the means from three independent experiments. Significant differences compared to results for the 32777 ΔyopM or yopER144A mutant-infected BMDMs were determined by one-way ANOVA. ns, not significant; *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Infected BMDM RIPA lysates were used to analyze the pyrin phosphorylation status by immunoblotting with the previously used antibodies. Our results indicated that YopT could trigger pyrin dephosphorylation. However, as observed for IL-1β release, YopE was dominant in this activity, and the effect of YopT was observed only in the mutant lacking YopE (Fig. 6, lanes 3 to 5). Altogether, these results showed that when the two effector activities were compared in isolation at 90 min, YopT generated more cell rounding in BMDMs but less triggering of pyrin dephosphorylation than YopE. Similarly, Ratner et al. found that YopE was dominant over YopT in triggering IL-1β release in BMDMs infected with Y. pestis (21).
FIG 6.
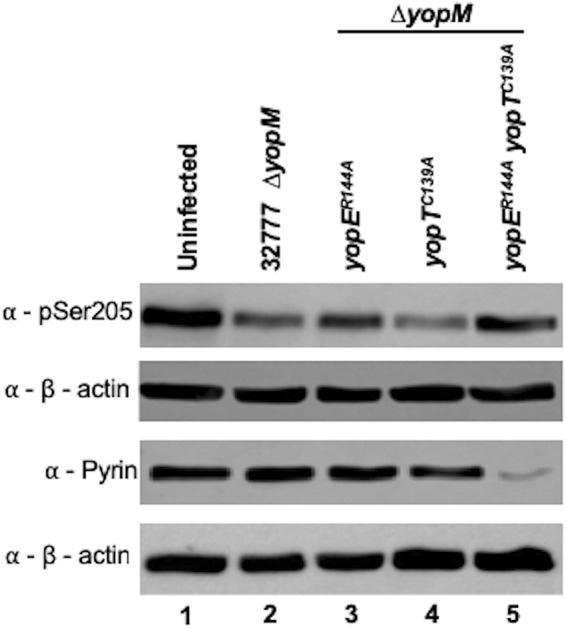
Western blot analysis of pyrin S205 dephosphorylation in BMDMs. LPS-primed BMDMs were left uninfected or infected with the indicated Y. pseudotuberculosis 32777 ΔyopM strain for 90 min. After infection, lysates were processed with denaturing RIPA lysis buffer, collected, and subjected to Western blotting using antibodies to pSer205 of pyrin, total pyrin, or β-actin.
To further investigate the role of YopT in triggering pyrin dephosphorylation, 180-min infections with the previously described strains at an MOI of 30 were performed and analyzed by phase-contrast microscopy, IL-1β enzyme-linked immunosorbent assay (ELISA), and pyrin immunoblotting. By microscopy, no obvious differences were observed at 180 min compared to observations of the 90-min infections, with the exceptions that over time the numbers of bacteria increased, and there was a subtle increase in cell rounding in BMDMs infected with the strain expressing inactive YopE and YopT (yopER144A yopTC139A strain) (Fig. S4B to D and F to H).
At 180 min, the mutants expressing active YopE or YopT exhibited increased release of IL-1β compared to that at 90 min (Fig. 7). Interestingly, the magnitude of the increase over time was greater for YopT than for YopE (Fig. 7). In addition, when the yopER144A and yopER144A yopTC139A strain infections were compared, YopT was responsible for a significantly higher level of IL-1β secretion at 180 but not at 90 min (Fig. 7). Although YopT can induce higher levels of IL-1β secretion at 180 min, YopE also showed a higher induction at the same time point, which confirms its dominance during the infection (Fig. 7).
FIG 7.
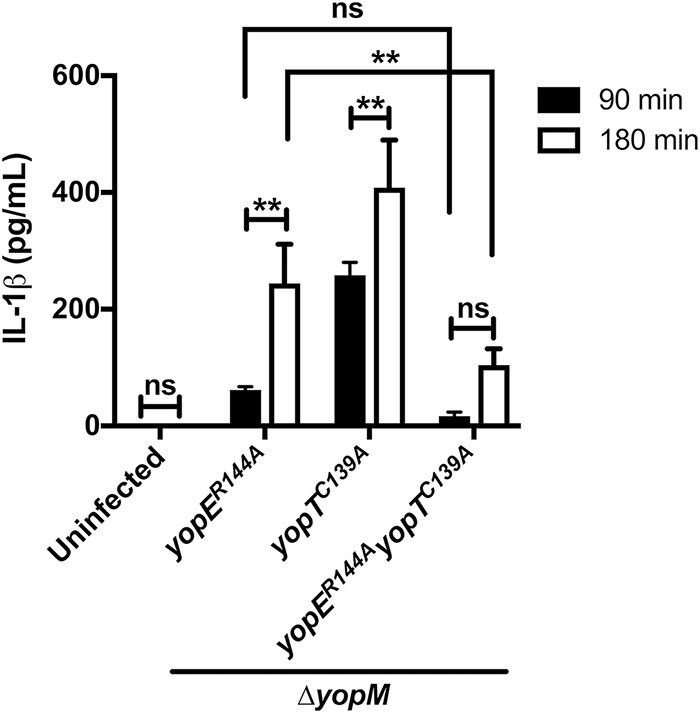
Analysis of IL-1β release in BMDMs. LPS-primed BMDMs were left uninfected or infected with indicated Y. pseudotuberculosis 32777 ΔyopM strains for 90 min or 180 min. Supernatants were collected and analyzed by ELISA to detect the release of IL-1β. Data represent average values ± the standard errors of the means from three independent experiments. Significant differences comparing the results of the same strains or different strains at the two time points were determined by two-way ANOVA. ns, not significant; **, P < 0.01.
Immunoblotting showed that effector-triggered pyrin dephosphorylation was mostly completed by 90 min with YopE but not until 180 min with YopT (Fig. 8, compare lanes 2 and 3 and lanes 6 and 7). Interestingly, the mutant strain lacking both active YopE and YopT caused dephosphorylation of pyrin at 180 min, suggesting that pyrin might be activated in this case also (Fig. 8, compare lanes 4 and 8). This result also corresponds with the IL-1β release observed in the presence of this strain at 180 min, which was increased (although not significantly) compared to the level of the same strain at 90 min (Fig. 7). Altogether, these results suggest that while YopT causes faster cell rounding, it triggers slower pyrin dephosphorylation, inflammasome assembly, and IL-1β release than YopE.
FIG 8.
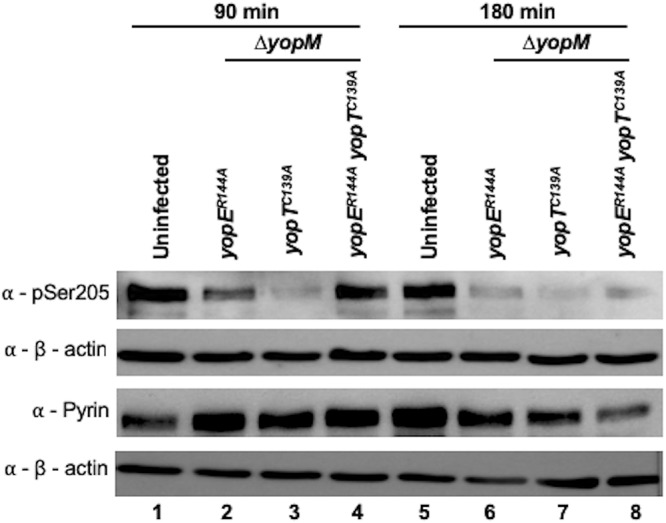
Western blot analysis of pyrin S205 dephosphorylation in BMDMs. LPS-primed BMDMs were left uninfected or infected with the indicated Y. pseudotuberculosis 32777 ΔyopM strain for 90 or 180 min. After infection, lysates were processed with denaturing RIPA lysis buffer, collected, and subjected to Western blotting using antibodies to pSer205 of pyrin, total pyrin, or β-actin.
DISCUSSION
In this study, we aimed to determine if and how the Yersinia sp. effectors YopE and YopT trigger inflammasome assembly by a conserved mechanism of pyrin dephosphorylation. It is crucial to understand how RhoA-modifying toxins interact with the host in order to promote pathogenesis and how the host can sense the effects generated by this class of toxins. YopE is a GAP that inactivates RhoA in a noncovalent way by facilitating the hydrolysis of the GTP into GDP. YopT is a cysteine protease that inactivates RhoA covalently by cleaving it from the membrane, leading to its mislocalization. Both modifications, although enzymatically different, can, in the absence of YopM, trigger assembly of the pyrin inflammasome (15). Other RhoA-modifying toxins, such as C3 from Clostridium botulinum and TcdB from C. difficile were also demonstrated to trigger assembly of the pyrin inflammasome, which further confirms that pyrin can sense disturbances in RhoA generated by different toxins and effectors (20).
Since the sensing of modifications in RhoA seems to be a conserved mechanism in the host immune system, we hypothesized that the mechanism by which pyrin is being activated is also conserved. Recently, TcdB was shown to trigger activation of mouse pyrin by the dephosphorylation of serines 205 and 241 (24). Here, we show that in the absence of YopM, YopE triggers activation of pyrin through the dephosphorylation of serine 205, and we assume that serine 241 is dephosphorylated as well. As in previous infection experiments performed under similar conditions (15), we observed a decrease in the amount of pyrin in BMDM cell supernatants generated with the nonionic detergent NP-40. Here, we demonstrate that loss of active pyrin from NP-40 supernatants is a result of the protein becoming insoluble. Using the more denaturing RIPA lysis buffer, we could recover dephosphorylated pyrin in the soluble phase. These results suggest that when pyrin is dephosphorylated and binds to ASC and pro-caspase-1 to form inflammasomes, the resulting multiprotein complexes are insoluble in nonionic detergent.
To understand the features of YopE that are important for pyrin inflammasome assembly, we used Y. pseudotuberculosis strains harboring different YopE variants. The variants are less stable (YopER62K), have reduced affinity for RhoA (YopEL109A) or are defective for membrane localization (YopE3N). All three variants were shown previously to be less efficient than wild-type YopE in triggering the killing of intracellular Yersinia by murine BMDMs (16). Our results showed that stability and RhoA target specificity in YopE were important for inflammasome activation since the corresponding variants (YopER62K and YopEL109A) had reduced ability to trigger dephosphorylation of pyrin and release of IL-1β.
It is important to highlight that Y. enterocolitica serotype O:8 naturally harbors a YopE protein with a lysine at position 62 (33). It was shown that in this strain YopE is targeted for ubiquitination, making this effector an unstable molecule. The degradation of YopE was shown to contribute to the pathogenesis of this specific Y. enterocolitica strain by increasing the translocation of other Yop proteins into host cells (33). Our results suggest that YopE with a lysine at position 62 is a more pathogenic variant because it not only fine-tunes Yop translocation (33) but also triggers less pyrin inflammasome assembly (this study) and reduced macrophage killing of intracellular IP2666 bacteria (16).
YopEL109A also triggered reduced assembly of the pyrin inflammasome. This variant was shown in previous work to have a reduced specificity for RhoA and Rac2, demonstrated by the reduced ability to generate cell rounding and reduced ability to inhibit reactive oxygen species (ROS) production, respectively (32). We have also observed that the GAP domain from SptP of Salmonella enterica serovar Typhimurium, which is specific for Rac1 and Cdc42 but not RhoA (32), does not trigger pyrin inflammasome assembly (unpublished data). Together, these data confirm that RhoA specificity is an important feature for the ability of toxins and effectors to trigger the assembly of the pyrin inflammasome (20).
YopE3N was not significantly different from wt YopE in its ability to trigger pyrin inflammasome assembly, suggesting that the localization of YopE to the plasma membrane is not necessary for RhoA inactivation. Interestingly, the YopE3N variant did display a significant defect in triggering killing of IP2666 in BMDMs, which was suggested to require inactivation of Rac1 (16). Altogether, these data suggest that membrane localization is less important for YopE to deactivate RhoA than to deactivate Rac1.
We showed that YopT, in addition to YopE, was also able to trigger inflammasome assembly through the dephosphorylation of serine 205 on pyrin. However, YopT seemed not to be as efficient as YopE in this process since YopT triggered less dephosphorylation of pyrin and less IL-1β release at 90 or 180 min when the two effectors were compared individually. On the other hand, YopT had faster cell-rounding activity than YopE as determined by phase-contrast microscopy. Differences in mechanisms of action or target specificity could explain why YopT and YopE trigger pyrin dephosphorylation at different rates. Unlike YopE, YopT directly cleaves and detaches Rho GTPases from the membrane (17). In the case of Rac1, after being cleaved by YopT, the GTP-bound form of the protein remains in an active form and can be translocated to the nucleus (36). It is possible that cleaved active RhoA does not efficiently trigger the assembly of the pyrin inflammasome. The other possibility is that YopT has a preference for inactivating another Rho GTPase, such as Rac1, over RhoA, while YopE preferentially targets RhoA. The inactivation of Rac1 may be generating a more drastic change to cell morphology by YopT, but the more efficient YopE activity as a RhoA inactivator leads to faster inflammasome formation.
Mutational inactivation of YopE and YopT in Yersinia can increase translocation of other effectors (18), increase phagocytosis (18), and increase survival of the bacteria inside macrophages (16). We acknowledge that changes in these features of the Yersinia-macrophage interaction could impact the outcome of our assays used to investigate the roles of YopE and YopT in triggering pyrin activation. For example, inactivation of YopE would be expected to increase translocation of YopT, and as a result we may be overestimating the ability of YopT translocated at native levels to trigger the pyrin inflammasome.
We also observed pyrin dephosphorylation and IL-1β release at 180 min of infection in the absence of both YopE and YopT. Yersinia strains are able to translocate other effectors, such as YpkA, into host cells (19). YpkA is a Rho-modifying effector with two different domains: a serine/threonine kinase that phosphorylates G proteins and a Rho guanine nucleotide dissociation inhibitor (GDI)-like domain with affinity for RhoA and Rac1 (37). As a Rho GDI, YpkA can associate with RhoA-GDP, inhibiting the transition of this GTPase into its GTP-bound form and, consequently, keeping RhoA inactive (38). It will be important to determine if YpkA is responsible for pyrin dephosphorylation and IL-1β release detected at 180 min of infection in the absence of both YopE and YopT.
Our work showed how the RhoA-modifying toxins YopE and YopT can trigger the pyrin inflammasome through the same mechanism as described for TcdB (24). Other RhoA-modifying toxins still need further investigation to prove that they also trigger the pyrin inflammasome through the dephosphorylation of specific serines in pyrin. It would not be surprising if the same mechanism is used since it has been shown that the pyrin inflammasome is a conserved response to RhoA inactivation. Further studies are also necessary to understand if any additional proteins play a role in the process of pyrin activation. Studying RhoA-inactivating toxins can lead to a better understanding of how the immune system recognizes and responds to certain bacterial infections.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All Y. pseudotuberculosis strains and plasmids used in this study are listed in Table 1. Y. pseudotuberculosis was grown at 28°C on LB agar plates or with shaking in LB broth, supplemented with antibiotics when necessary. To generate IP6ΔM, an unmarked deletion of the yopM open reading frame was introduced into the IP6 strain (18) using the vector pSB890 and an allelic exchange method, as previously described (39). To generate IP6ΔMmJ, the C172A codon change was introduced into the yopJ gene of IP6ΔM by allelic exchange as described previously (40). In order to create Y. pseudotuberculosis strains expressing the different YopE variants, E. coli S17-1 donor strains (16) containing plasmid pPEYopE, pPEYopER62K, pPEYopEL109A, pPEYopE3N, or pMMB67HE (the empty vector) were conjugated with IP6ΔM or IP6ΔMmJ recipient strains, as described previously (16, 18). The 32777ΔM (39), yopER144A, yopTC139A, and yopER144A yopTC139A strains were generated and described previously (15).
Cell culture.
Bone marrow-derived macrophages (BMDMs) were obtained from femurs of 8-week-old wt C57BL/6 mice (Jackson Laboratories) and cultured as previously described (15, 41). On day 7, macrophages were seeded in MGM 10/10 medium which consists of Dulbecco’s modified Eagle Medium (DMEM) plus GlutaMAX (Gibco) containing 10% fetal bovine serum (FBS) (GE Health Care), 10% L929 cell-conditioned medium, 1 mM sodium pyruvate (Gibco), 10 mM HEPES (Gibco), and 100 ng/ml O26:B6 Escherichia coli LPS (Sigma) and divided into six-well plates at a density of 0.8 × 106 cells/well in a total volume of 3 ml.
Y. pseudotuberculosis Yop secretion assay.
Y. pseudotuberculosis IP6ΔM strains harboring yopER62K, yopEL109A, or yopE3N were tested for the ability to secrete the different YopE variants. The above strains and control strain IP6ΔM harboring the empty vector or wt yopE were grown overnight (ON) at 28°C with shaking in LB broth in the presence of antibiotics. On the following day, a 1:40 dilution of the ON culture was inoculated in 10 ml of antibiotic-free LB broth and containing 20 mM sodium oxalate and 20 mM MgCl2. The cultures were grown with shaking for 2 h at 28°C and then shifted to 37°C for 4 h to allow production of the T3SS and secretion of the Yops. The bacterial culture (1.5 ml) was centrifuged at room temperature for 5 min at 21,592 × g, and 1 ml of the obtained supernatant was incubated ON with shaking at 4°C with trichloroacetic acid (TCA). Proteins were pelleted the next day by centrifugation at 4°C for 30 min at 21,592 × g. Supernatants were removed, and the pellets were washed by centrifugation at 4°C for 2 min at 21,592 × g with 1 ml of cold acetone. The resulting pellets were dried for 10 min in a SpeedVac concentrator (Savant) and resuspended in 100 μl of 1× SDS-PAGE protein loading buffer containing 0.1 M dithiothreitol (DTT). The mixture was boiled for 5 min and cooled down in ice for 5 min, and 5 μl of secreted Yops was resolved by SDS-PAGE using 10% gels. Protein bands in gels derived from secreted Yops were stained with Gel Code Blue Stain Reagent (ThermoFisher Scientific) according to the manufacturer’s instructions. For long-term storage, stained gels were incubated in 10 ml of a mixture of 10% glycerol and 20% ethanol for 30 min. Last, the gel was clipped with a gel frame between two cellophane sheets and allowed to dry overnight.
Macrophage infections.
Y. pseudotuberculosis strains were grown ON at 28°C in LB broth, supplemented with antibiotics when necessary. On the day of infection, a 1:40 dilution of the ON culture was inoculated in 10 ml of antibiotic-free LB broth containing 20 mM sodium oxalate and 20 mM MgCl2. The cultures were grown for 1 h at 28°C and then shifted to 37°C for 2 h. Bacteria were diluted into serum-free MGM 10/10 medium at 8 × 106 CFU/ml. LPS-primed BMDMs were washed once with warm 1× PBS and serum-free MGM 10/10 containing no bacteria or bacteria at a multiplicity of infection (MOI) of 30 was added to the wells. The six-well plates were centrifuged for 5 min at 112 × g to facilitate the contact between the Y. pseudotuberculosis and macrophages. Plates were incubated at 37°C with 5% CO2. At 90 min postinfection, phase-contrast microscopy was performed. In addition, cell supernatants were collected and processed for IL-1β detection by ELISA and for pyroptosis detection by lactate dehydrogenase (LDH) release assay (see below). Cell lysates were obtained and processed for Western blot analysis to detect host proteins (see below).
Phase-contrast microscopy.
After infection, six-well plates were used for BMDM analysis using phase-contrast microscopy. At least two pictures of each well were taken from random fields containing both macrophages and bacteria at a ×10 magnification. Images were taken with a SPOT camera connected to an Axiovert S100 microscope (Zeiss).
Protein analysis by SDS-PAGE and Western blotting.
To obtain cell lysates, BMDMs were lysed using nondenaturing NP-40 lysis buffer (1% NP-40, 150 mM NaCl, and 50 mM Tris-HCl, pH 8.0) or denaturing RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid [DOC], and 0.1% SDS) with protease inhibitor cocktail (Roche). Total protein (3 to 20 μg) from cell lysates was resolved by SDS-PAGE using 10% gels. Proteins in gels derived from cell lysates were transferred to a polyvinylidene difluoride (PVDF) membrane and were subjected to Western blotting with rabbit monoclonal antibodies against mouse pyrin (ab195975; Abcam) or against mouse S205-phosphorylated pyrin (ab201784; Abcam). For a loading control, membranes were probed with rabbit polyclonal antibody against β-actin (Cell signaling). Anti-rabbit secondary antibody conjugated to horseradish peroxidase (HRP) (Jackson laboratory) was used as a secondary reagent. Proteins were visualized using chemiluminescent detection reagent (GE Healthcare). Images of chemiluminescence signals on Western blots were obtained with Amersham Imager 600 (GE Health Care), and images of stained gels were obtained with a GS-800 calibrated densitometer (Bio-Rad).
IL-1β detection and LDH release assays.
Levels of IL-1β and LDH released into BMDM supernatants were assessed using commercially available kits (R&D Systems and Promega, respectively) according to the companies’ instructions.
Statistical analysis.
Experimental data from at least three independent experiments were analyzed for significance using GraphPad Prism. The P values for IL-1β and LDH graphs were calculated using one-way analysis of variance (ANOVA) or grouped in two-way ANOVA.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jose Delgado, Amrapali Gosh, and Nicole Loeven for editing the manuscript and Lawton Chung for construction of Y. pseudotuberculosis strains.
The preparation of this publication was supported by the NIH under award R01AI099222 (J.B.B.) and by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazil, under grant 3830 - Full PhD Abroad Program (N.P.M.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00822-18.
REFERENCES
- 1.do Vale A, Cabanes D, Sousa S. 2016. Bacterial toxins as pathogen weapons against phagocytes. Front Microbiol 7:42. doi: 10.3389/fmicb.2016.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemichez E, Barbieri JT. 2013. General aspects and recent advances on bacterial protein toxins. Cold Spring Harb Perspect Med 3:a013573. doi: 10.1101/cshperspect.a013573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dean P. 2011. Functional domains and motifs of bacterial type III effector proteins and their roles in infection. FEMS Microbiol Rev 35:1100–1125. doi: 10.1111/j.1574-6976.2011.00271.x. [DOI] [PubMed] [Google Scholar]
- 4.Aktories K. 2015. Rho-modifying bacterial protein toxins. Patho Dis 73:ftv091. doi: 10.1093/femspd/ftv091. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt A, Hall A. 2002. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev 16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 6.Jaffe AB, Hall A. 2005. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 7.Etienne-Manneville S, Hall A. 2002. Rho GTPases in cell biology. Nature 420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 8.Visvikis O, Maddugoda MP, Lemichez E. 2010. Direct modifications of Rho proteins: deconstructing GTPase regulation. Biol Cell 102:377–389. doi: 10.1042/BC20090151. [DOI] [PubMed] [Google Scholar]
- 9.Bos JL, Rehmann H, Wittinghofer A. 2007. GEFs and GAPs: critical elements in the control of small G proteins. Cell 129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 10.Duan R, Liang J, Shi G, Cui Z, Hai R, Wang P, Xiao Y, Li K, Qiu H, Gu W, Du X, Jing H, Wang X. 2014. Homology analysis of pathogenic Yersinia species Yersinia enterocolitica, Yersinia pseudotuberculosis, and Yersinia pestis based on multilocus sequence typing. J Clin Microbiol 52:20–29. doi: 10.1128/JCM.02185-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pha K, Navarro L. 2016. Yersinia type III effectors perturb host innate immune responses. World J Biol Chem 7:1–13. doi: 10.4331/wjbc.v7.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cornelis GR. 2002. Yersinia type III secretion: send in the effectors. J Cell Biol 158:401–408. doi: 10.1083/jcb.200205077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plano GV, Schesser K. 2013. The Yersinia pestis type III secretion system: expression, assembly and role in the evasion of host defenses. Immunol Res 57:237–245. doi: 10.1007/s12026-013-8454-3. [DOI] [PubMed] [Google Scholar]
- 14.Cornelis GR. 2006. The type III secretion injectisome. Nat Rev Microbiol 4:811. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- 15.Chung LK, Park YH, Zheng Y, Brodsky IE, Hearing P, Kastner DL, Chae JJ, Bliska JB. 2016. The Yersinia virulence factor YopM hijacks host kinases to inhibit type III effector-triggered activation of the pyrin inflammasome. Cell Host Microbe 20:296–306. doi: 10.1016/j.chom.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Parashar K, Sitaram A, Bliska JB. 2014. The GAP activity of type III effector YopE triggers killing of Yersinia in macrophages. PLoS Pathog 10:e1004346. doi: 10.1371/journal.ppat.1004346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao F, Vacratsis PO, Bao Z, Bowers KE, Fierke CA, Dixon JE. 2003. Biochemical characterization of the Yersinia YopT protease: Cleavage site and recognition elements in Rho GTPases. Proc Natl Acad Sci U S A 100:904–909. doi: 10.1073/pnas.252770599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Black DS, Bliska JB. 2000. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol Microbiol 37:515–527. [DOI] [PubMed] [Google Scholar]
- 19.Viboud GI, Bliska JB. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- 20.Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong YN, Peng X, Xi JJ, Chen S, Wang F, Shao F. 2014. Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature 513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 21.Ratner D, Orning MPA, Proulx MK, Wang D, Gavrilin MA, Wewers MD, Alnemri ES, Johnson PF, Lee B, Mecsas J, Kayagaki N, Goguen JD, Lien E. 2016. The Yersinia pestis effector YopM inhibits pyrin inflammasome activation. PLoS Pathog 12:e1006035. doi: 10.1371/journal.ppat.1006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park YH, Wood G, Kastner DL, Chae JJ. 2016. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 17:914–921. doi: 10.1038/ni.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, Kastner DL. 2006. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A 103:9982–9987. doi: 10.1073/pnas.0602081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao W, Yang J, Liu W, Wang Y, Shao F. 2016. Site-specific phosphorylation and microtubule dynamics control pyrin inflammasome activation. Proc Natl Acad Sci U S A 113:E4857–E4866. doi: 10.1073/pnas.1601700113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Torre-Minguela C, Mesa del Castillo P, Pelegrín P. 2017. The NLRP3 and pyrin inflammasomes: implications in the pathophysiology of autoinflammatory diseases. Front Immunol 8:43. doi: 10.3389/fimmu.2017.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma D, Kanneganti T-D. 2016. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol 213:617–629. doi: 10.1083/jcb.201602089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. 2015. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kovacs SB, Miao EA. 2017. Gasdermins: effectors of pyroptosis. Trends Cell Biol 27:673–684. doi: 10.1016/j.tcb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahoo M, Ceballos-Olvera I, del Barrio L, Re F. 2011. Role of the inflammasome, IL-1β, and IL-18 in bacterial infections. ScientificWorldJournal 11:2037–2050. doi: 10.1100/2011/212680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratner D, Orning MPA, Starheim KK, Marty-Roix R, Proulx MK, Goguen JD, Lien E. 2016. Manipulation of Interleukin-1β and Interleukin-18 production by Yersinia pestis effectors YopJ and YopM and redundant impact on virulence. J Biol Chem 291:9894–9905. doi: 10.1074/jbc.M115.697698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Songsungthong W, Higgins MC, Rolán HG, Murphy JL, Mecsas J. 2010. ROS-inhibitory activity of YopE is required for full virulence of Yersinia in mice. Cell Microbiol 12:988–1001. doi: 10.1111/j.1462-5822.2010.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaus K, Hentschke M, Czymmeck N, Novikova L, Trülzsch K, Valentin-Weigand P, Aepfelbacher M, Ruckdeschel K. 2011. Destabilization of YopE by the ubiquitin-proteasome pathway fine-tunes Yop delivery into host cells and facilitates systemic spread of Yersinia enterocolitica in host lymphoid tissue. Infect Immun 79:1166–1175. doi: 10.1128/IAI.00694-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Barbieri JT. 2005. A leucine-rich motif targets Pseudomonas aeruginosa ExoS within mammalian cells. Infect Immun 73:7938–7945. doi: 10.1128/IAI.73.12.7938-7945.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schoberle TJ, Chung LK, McPhee JB, Bogin B, Bliska JB. 2016. Uncovering an important role for YopJ in the Inhibition of caspase-1 in activated macrophages and promoting Yersinia pseudotuberculosis virulence. Infect Immun 84:1062–1072. doi: 10.1128/IAI.00843-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong K-W, Isberg RR. 2005. Yersinia pseudotuberculosis spatially controls activation and misregulation of host cell Rac1. PLoS Pathog 1:e16. doi: 10.1371/journal.ppat.0010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pha K, Wright ME, Barr TM, Eigenheer RA, Navarro L. 2014. Regulation of Yersinia protein kinase A (YpkA) kinase activity by multisite autophosphorylation and identification of an N-terminal substrate-binding domain in YpkA. J Biol Chem 289:26167–26177. doi: 10.1074/jbc.M114.601153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prehna G, Ivanov MI, Bliska JB, Stebbins CE. 2006. Yersinia virulence depends on mimicry of host Rho-family nucleotide dissociation inhibitors. Cell 126:869–880. doi: 10.1016/j.cell.2006.06.056. [DOI] [PubMed] [Google Scholar]
- 39.McPhee JB, Mena P, Bliska JB. 2010. Delineation of regions of the Yersinia YopM protein required for interaction with the RSK1 and PRK2 host kinases and their requirement for interleukin-10 production and virulence. Infect Immun 78:3529–3539. doi: 10.1128/IAI.00269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Murtha J, Roberts MA, Siegel RM, Bliska JB. 2008. Type III secretion decreases bacterial and host survival following phagocytosis of Yersinia pseudotuberculosis by macrophages. Infect Immun 76:4299–4310. doi: 10.1128/IAI.00183-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. 2010. A Yersinia secreted effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7:376–387. doi: 10.1016/j.chom.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.