

Summary
Synthetic peptidomimetics of antimicrobial peptides (AMPs) are promising antimicrobial drug candidates because they promote membrane disruption and exhibit greater structural and proteolytic stability than natural AMPs. We previously reported selective antifungal 14-helical β-peptides, but the mechanism of antifungal toxicity of β-peptides remains unknown. To provide insight into the mechanism, we studied antifungal β-peptide binding to artificial membranes and living Candida albicans cells. We investigated the ability of β-peptides to interact with and permeate small unilamellar vesicle models of fungal membranes. The partition coefficient supported a pore-mediated mechanism characterized by the existence of a critical β-peptide concentration separating low and high partition coefficient regimes. Live cell intracellular tracking of β-peptides showed that β-peptides translocated into the cytoplasm, and then disrupted the nucleus and vacuole sequentially, leading to cell death. This understanding of the mechanisms of antifungal activity will facilitate design and development of peptidomimetic AMPs, including 14-helical β-peptides, for antifungal applications.
Keywords: antifungal β-peptide, peptidomimetics, mechanism of antifungal peptides, 14-helical β-peptide, intracellular target, Candida albicans, small unilamellar vesicles, partition coefficients
Graphical Abstract
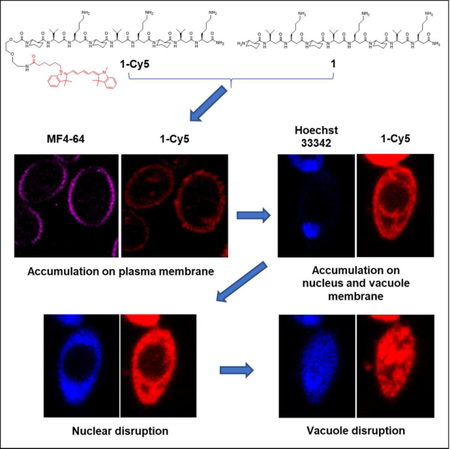
eTOC
Lee et al. elucidate how antifungal 14-helical β-peptides interact with model membrane vesicles and plasma membranes of living Candida albicans cells. They show that β-peptide binding to these membranes is consistent with pore formation. After permeabilizing the plasma membrane, β-peptides accumulate on intracellular membranes then disrupt the nucleus and vacuole.
Introduction
Antimicrobial peptides (AMPs) are components of innate immune systems found in a wide variety of invertebrates and vertebrates (Beutler, 2004; Gallo and Nizet, 2003; Ganz, 2003), and have demonstrated broad spectrum activity against bacteria, fungi, viruses, and cancer cells (Brogden et al., 2003; Hancock and Lehrer, 1998; Zasloff, 2002). Even though some instances of resistance to naturally-occurring AMPs have been reported (Andersson and Hughes, 2010; Fjell et al., 2012; Lai and Gallo, 2009; Mahlapuu et al., 2016; Nizet, 2006; Peschel and Sahl, 2006), AMPs are, as a class, less susceptible than conventional antibiotics to the emergence of microbial resistance because of their lack of a single target (Mahlapuu et al., 2016). Thus, there is significant interest in developing AMPs as drugs against infectious agents since many existing antimicrobials are plagued by the emergence of resistant strains. Unfortunately, the viability of natural AMPs as therapeutics is reduced by their low structural stability and activity in physiologic environments (Chu et al., 2013; Goldman et al., 1997; Huang et al., 2011; Smith et al., 1996) and their susceptibility to proteolytic cleavage (Schmidtchen et al., 2002).
A variety of synthetic or non-natural peptidomimetic compounds have been developed to exploit the antimicrobial properties of AMPs and to address their low stability (Godballe et al., 2011; Lienkamp et al., 2013). One such class of compounds, β-peptides, is composed of β-amino acids and can fold into protein secondary structures found in AMPs, including the α-helix (Appella et al., 1997; Seebach et al., 1997; Seebach and Matthews, 1997), β-sheet (Chung et al., 2000; Krauthauser et al., 1997), and γ-turn (Chung et al., 1998). Structure-functional analyses of β-peptides have demonstrated that these compounds can be designed to exhibit antibacterial (Arvidsson et al., 2001; Epand et al., 2003; Hamuro et al., 1999; Porter et al., 2000; Raguse et al., 2002) and antifungal (Karlsson et al., 2009; Raman et al., 2015) activities at concentrations that induce little hemolysis in mammalian red blood cells. In prior work, we demonstrated that fungicidal activity of 14-helical β-peptides against the opportunistic pathogen Candida albicans requires a globally-amphiphilic structure, composed of hydrophobic and cationic faces, with approximately three helical turns (Lee et al., 2014). We observed that both antifungal and hemolytic activities increased as β-peptides became more hydrophobic, and we identified a hydrophobicity window that resulted in high antifungal activity but low hemolysis. The addition of helix-stabilizing residues, such as aminocyclohexane carboxylic acid (ACHC), increased the activity and specificity of these 14-helical β-peptides against C. albicans.
While prior studies have shown that β-peptides and other synthetic AMP peptidomimetics have promise as antimicrobial therapeutics, little is known about their mechanisms of action compared to those of natural AMPs. The canonical mechanism of AMP-mediated toxicity involves permeabilization of the target cell membrane, a process that depends on both the physicochemical properties of the AMP and those of the target cell membrane. Several models have been proposed to describe the mechanisms of AMP permeabilization of cell membranes (Brogden, 2005; Huang, 2000; Shai, 1999; Yeaman and Yount, 2003). In the “barrel-stave model”, AMP monomers bind to the cell membrane and then oligomerize and form structured pores with the hydrophobic region of the peptide oriented toward the membrane lipids and the hydrophilic region comprising the pore channel. For example, aggregation of four to six alamethicin molecules in a cell membrane has been shown to form voltage-dependent ion channels (Boheim, 1974; Gordon and Haydon, 1972, 1975). In contrast, in the “toroidal model”, peptides insert into the cell membrane and associate with lipid headgroups to induce lipid bending. Thus, the pore lining is composed of both the peptides and membrane lipids. Magainin 2 (Matsuzaki et al., 1995; Yang et al., 1998), protegrin-1 (Lazaridis et al., 2013; Yamaguchi et al., 2002), melittin (Yang et al., 2001; Zhou et al., 2014), LL-37 (Lee et al., 2011), MSI-78 (Hallock et al., 2003), and other AMPs have been shown to act via the toroidal model. Finally, in the “carpet model”, AMPs first interact with the target membrane via electrostatic interactions, and then they effectively solubilize and disrupt the membrane. Dermaceptin S (Pouny et al., 1992), cecropin (Gazit et al., 1996), caerin 1.1 (Pukala et al., 2004), ovispirin (Yamaguchi et al., 2001), and other AMPs have been suggested to follow the carpet model. Little is known about how antifungal peptidomimetic 14-helical β-peptides may induce membrane lysis and promote cell death in C. albicans.
In addition to permeabilizing the cell membrane, some AMPs act on intracellular targets to induce microbial death. Modes of intracellular action include binding nucleic acids (e.g. buforin II (Park et al., 1998) and tachyplesin (Yonezawa et al., 1992)) and inhibiting cell-wall synthesis (mersacidin (Brotz et al., 1995)), nucleic acid and protein synthesis (pleurocidin (Patrzykat et al., 2002), PR-39 (Boman et al., 1993), and HNP-1 (Lehrer et al., 1989)), and enzymatic activity (histatins (Nishikata et al., 1991; Puri and Edgerton, 2014), pyrrhocoricin (Gagnon et al., 2016; Otvos et al., 2000), and drosocin (Otvos et al., 2000)).
Prior studies have focused on interaction of AMPs with the cell membrane, but their effects on intracellular compartments, such as the nucleus, vacuole, and mitochondria have not been studied. In addition, there is little understanding of membrane disrupting mechanism of peptidomimetic β-peptides and the fates of these compounds inside the target cell. Here, we studied interactions between 14-helical β-peptides and small unilamellar vesicles (SUVs) to elicit the membrane disrupting mechanism and tracked fluorescently-labeled β-peptide to understand the interactions between peptide and intracellular organelles.
To elucidate mechanisms of β-peptide interactions with fungal cells, we first employed SUVs to investigate how membrane composition influences β-peptide adhesion to and the subsequent permeabilization of the vesicles. Next, we used confocal laser scanning microscopy (CLSM) and super resolution structure illumination microscopy (SR-SIM) to validate that cooperative binding of β-peptides observed in SUVs also occurs in C. albicans cells. Finally, time lapse microscopy tracking of fluorescently labelled β-peptides demonstrated that, after permeabilizing the cell membranes, the β-peptides lysed the nucleus and vacuole, resulting in cell death. Our results are consistent with pore-mediated mechanisms of membrane permeabilization and suggest that disruption of intracellular organelles also plays a role in the fungicidal activity of β-peptides. The improved understanding of mechanisms of β-peptide interactions with the cell membrane and organelles will enhance efforts to further develop these compounds as active and selective antifungal agents.
RESULTS
Hydrophobicity of β-peptide regulates the antifungal activity.
To study the potential mechanisms of antifungal β-peptide toxicity, we synthesized β-peptides 1 (NH2-(ACHC-β3hVal-β3hLys)3-CONH2), 2 (NH2-(ACHC-β3hVal-β3hArg)3-CONH2) and 3 (NH2- (ACHC-β3hAal-β3hLys)3-CONH2) (Fig. 1). We previously demonstrated that β-peptides 1, 2, and 3 share structural properties important in antifungal activity, including charge, number of β-amino acid residues, amphiphilicity, and helical stability (Lee et al., 2014; Raman et al., 2015). However, the hydrophobicity of these β-peptides (as determined by RP-HPLC retention time) (Browne et al., 1982; Jiang et al., 2008; Kondejewski et al., 1999; Wilson et al., 1981), which also regulates the antifungal activity, varies substantially. The more hydrophobic β-peptides 1 and 2 exhibit a MIC against C. albicans of 8 µg/mL, while the MIC of less hydrophobic β-peptide 3 is 512 µg/mL (Table S1). We also synthesized NBD labeled analogues of β-peptides 1, 2, and 3 (1-NBD, 2-NBD, and 3-NBD) and Cy5-labeled β-peptide 1 (1-Cy5) (Fig. 1) (NBD = nitrobenzoxadiazole) to facilitate quantification of β-peptide partition coefficients into synthetic lipid vesicle membranes and to visualize the interactions between the β-peptides and live C. albicans cells, respectively. NBD is a small, polar fluorophore with a low dipole moment (Gazit et al., 1994; Pouny et al., 1992; Pouny and Shai, 1992; Rapaport and Shai, 1991). After NBD-labeling of β-peptides 1, 2, and 3, peptide retention times increased, indicating that the NBD label increased the hydrophobicity of these β-peptides (Table S1, Fig. S1). The MICs of β-peptides 1-NBD and 2-NBD were within a factor of 2 of the MICs of the corresponding unlabeled β-peptides (Table S1, Fig. S2) while the MIC of 3-NBD was 8-fold lower than the MIC of β-peptide 3 (Table S1, Fig. S2), likely resulting from the increase in β-peptide hydrophobicity upon adding the NBD label. β-Peptide 3-NBD still exhibited a 4- and 8-fold greater MIC than 1-NBD and 2-NBD, respectively. While NBD labeling had little effect on β-peptide hydrophobicity and activity, we are unable to rule out possible influence of the NBD label on mechanism of action. To characterize mechanisms of antifungal β-peptide toxicity, we investigated biophysical interactions of fluorescently-labelled β-peptides with model phospholipid membranes and C. albicans cell membranes, as well as the subsequent localization of β-peptides to intracellular compartments.
Figure 1.
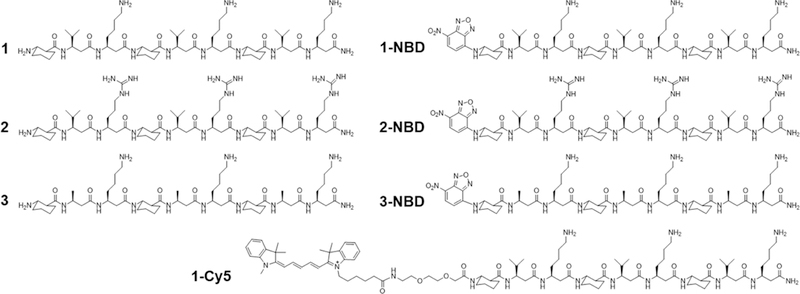
Structures of β-peptides used in this study. See also Figures S1 and S2, and Table S1.
Permeabilization of synthetic vesicles by β-peptides depends upon membrane composition
We used the leakage of calcein from synthetic small unilamellar vesicles (SUVs) to assess the specificity of β-peptide disruption of phospholipid membranes composed of phosphatidylglycerol (PG, anionic phospholipid), phosphatidylcholine (PC, neutral phospholipid), and phosphatidylethanolamine (PE, neutral phospholipid). We prepared SUVs with different phospholipid compositions to characterize the influence of β-peptide on the disruption of SUVs containing zwitterionic (PC:PE) and anionic (PC:PG) phospholipids. After preparation, the SUV size and phospholipid ratio were quantified using dynamic light scattering and 31P nuclear magnetic resonance (Table S2). Magainin 2, a pore-forming antibacterial peptide, and melittin, a pore-forming non-selective AMP, were used as controls for membrane lysis. Magainin 2 induced substantial calcein leakage only from the model bacterial membrane vesicles (PC:PG 60:40) (Fig. 2, and S3a–c). In contrast, melittin induced calcein leakage from all SUVs tested (Fig. 2, and S3a–c), as expected given its broad activity against bacterial, fungal, and mammalian cells. The phospholipid selectivities of magainin 2 and melittin for SUVs are consistent with prior reports of PG selectivity of magainin 2 (Gregory et al., 2009; Wieprecht et al., 1996) and broad phospholipid targeting of melittin (Magzoub et al., 2005) against large unilamellar vesicles (LUVs).
Figure 2.
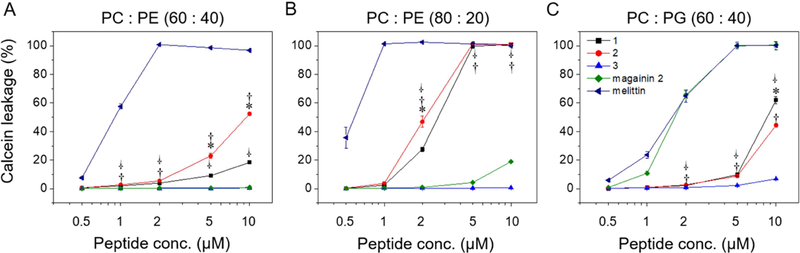
β-Peptide concentration-dependent calcein leakage from SUVs of the indicated phospholipid concentrations. Calcein leakage was monitored 10 min after incubation of β-peptides and SUVs (total [lipid] = 130 µM). To quantify calcein leakage, the fluorescence intensity at 515 nm was measured and normalized to the intensity obtained after adding 2% Triton X-100, corresponding to 100% calcein leakage. Error bars denote standard deviation (n = 3). Statistical analysis was performed using one-way ANOVA for β-peptide 1 vs. 2 (*), β-peptide 1 vs. 3 (†), and β-peptide 2 vs. 3 ( · ) (*, †, and · p ≤ 0.01). See also Figure S3 and Table S2.
The disruption of SUVs by 14-helical β-peptides depended on the SUV and β-peptide compositions. The active antifungal β-peptides 1 and 2 resulted in about 10–20% calcein leakage from PC:PE (60:40) SUVs and 100% calcein leakage from PC:PE (80:20) SUVs after 10 min exposure to the β-peptides at the antifungal MIC (5 µM, Table S1) (Fig. 2, Fig. S3a–b). Hydrophobic β-peptides 1 and 2 induced greater calcein leakage from PC:PE (60:40) SUVs than did the more hydrophilic β-peptide 3. β-Peptide 2 induced greater calcein leakage than β-peptide 1, which may be due to increased hydrophobicity of β-peptide 2. This difference could also result, at least in part, from differences in β-peptide head group composition (Lee et al., 2014). The inactive β-peptide 3 resulted in less than 2% calcein leakage even after 1 hr from neutral SUVs (Fig. 2, Fig. S3a–b).
We also quantified calcein leakage from negatively-charged PC:PG (60:40) SUVs to investigate the effects of electrostatic interactions on phospholipid membrane permeabilization by 14-helical β-peptides. β-peptides 1 and 2 lysed PC:PG (60:40) SUVs to a substantially greater extent than β-peptide 3 at and above 5 µM (Fig. 2c). However, β-peptides 1 and 2 elicited similar calcein leakage in PC:PG (60:40) SUVs as PC:PE (60:40) SUVs at 10 µM. β-peptide 3 caused more than ten-fold greater calcein leakage after 1 h from PC:PG (60:40) SUVs than from PC:PE (60:40) SUVs (Fig. S3c). The activity of hydrophobic β-peptides 1 and 2 appeared to be less sensitive to electrostatic interactions than the less hydrophobic β-peptide 3. Together, these results demonstrate that the ability of 14-helical β-peptides to lyse model microbial cell membranes correlates with their antifungal activity, and suggest a potential mechanism of antifungal activity that involves cell membrane disruption.
14-Helical β-peptides lyse SUV membranes via pore formation
To characterize mechanisms of membrane disruption and distinguish between pore-forming and carpet mechanisms, we quantified the partition coefficients of NBD-labeled β-peptides in model SUV membranes. Carpet formation is characterized by a monotonic correlation between peptide concentration and membrane binding (Rapaport and Shai, 1991). In contrast, a hallmark of pore formation is two regimes of peptide binding to the membrane (Huang, 2000; Jackman et al., 2016). Below the critical concentration, a low partition coefficient results from peptide interactions with the membrane surface, while above the critical concentration a higher partition coefficient results from cooperative insertion of the peptides into the membrane as pores are formed (Brogden, 2005; Huang, 2000; Shai, 1999; Yeaman and Yount, 2003).
We measured partition coefficients of 1-NBD, 2-NBD and 3-NBD in PC:PE (60:40), PC:PG (60:40), and PC:PE:ergosterol (50:30:20) SUVs (Table S4, and S3, and Fig. 3, S4, and S5a–f). Partition coefficients were obtained by plotting Xb*, the corrected molar ratio of bound peptide per total outer leaflet lipid (assumed to be 60% of the total lipid) (Michaelson et al., 1974) versus Cf, the equilibrium concentration of free peptide in solution. The partition coefficient was calculated from the slope of this curve (Fig. 3, and S5a–f). 1-NBD and 2-NBD, which exhibit high antifungal activity (Table S1), demonstrated greater partition coefficients, 23.6- and 35-fold higher respectively, than the less active and more hydrophilic 3-NBD in PC:PE (60:40) SUVs (Table S4, Fig. 3). The partition coefficients of 1-NBD and 2-NBD varied by 2-fold or less between PC:PG SUVs, while 3-NBD exhibited a 16-fold greater partition coefficient in PC:PG SUVs than PC:PE SUVs, indicating a greater dependence of partition coefficient on electrostatic interactions for 3-NBD (Table S4, Fig. 3). As anticipated, the partition coefficients for NBD-labeled β-peptides correlated with the extent of calcein leakage caused by the unlabeled β-peptides (Fig. 2). Critical concentrations of 13.7 and 17.8 nM were identified for 1-NBD and 2-NBD, respectively, for PC:PE (60:40) SUVs, but 3-NBD did not exhibit a critical concentration against PC:PE SUVs, consistent with a mechanism of pore formation by 1-NBD and 2-NBD but not 3-NBD. All three NBD-labeled β-peptides exhibited critical concentrations between 11 and 16 nM against PC:PG (60:40) SUVs, suggesting that all three NBD-labeled β-peptides formed pores in PC:PG (60:40) SUVs (Table S4).
Figure 3.
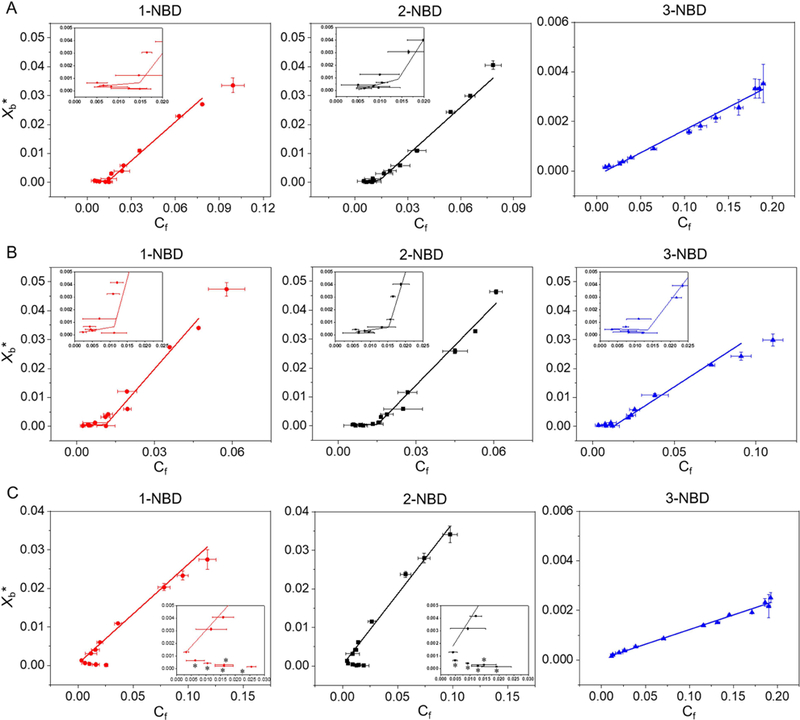
Binding isotherms of NBD-labeled β-peptides (0.2 µM) to (A) PC:PE (60:40), (B) PC:PG (60:40) and (C) PC:PE:ergosterol (50:30:20) SUVs ([lipid] = 5 to 2000 µM) with the indicated compositions after 2 min incubation. Xb* represents the molar ratio of bound peptide per 60% of total lipid and Cf is the equilibrium concentration of free peptide in solution. The partition coefficient is the slope of the binding isotherm and the critical concentration is the intersection of the low-slope and high-slope isotherms, shown in the insets. The placement of asterisk (*) near a data point indicates conditions at which vesicle aggregation was observed visually (see text). Data points represent the averages of three independent experiments and error bars denote standard deviation (n = 3). Lines in A, B, and C show linear and non-linear (piecewise fit lines by piecewise fitting function (pwl2s) consisting of two linear segments) data fits. The statistical data for each fit are: red line in A (non-linear fit, p = 0.0014, R2 = 0.85, intercept = 14.8 nM), black line in A (non-linear fit, p = 7.82 × 10−5, R2 = 0.87, intercept = 14.3 nM), blue line in A (linear fit, p = 1.71 × 10−11, R2 = 0.98); red line in B (non-linear fit, p = 1.51 x= 10−6, R2 = 0.96, intercept = 11.3 nM), black line in B (non-linear fit, p = 1.13 × 10−5, R2 = 0.91, intercept = 14.1 nM), blue line in B (non-linear fit, p = 5.89 × 10−4, R2 = 0.80, intercept = 13.0 nM); red line in C (linear fit, p = 9.33 × 10−7, R2 = 0.98), black line in C (linear fit, p = 5.83 × 10−8, R2 = 0.98), blue line in C (linear fit, p = 3.00 × 10−13, R2 = 0.99). See also Figures S4 and S5, and Tables S3 and S4.
We also investigated the effects of ergosterol on β-peptide partition coefficients in PC:PE:ergosterol (50:30:20) SUVs (Fig. 3c). The ratio of ergosterol was chosen to mimic that found in the C. albicans cell membrane (Warschawski et al., 2011). SUV aggregation was visually observed at high [lipid[]/[peptide] ratios, corresponding to low Cf in Fig. 3c. Since SUV aggregation occurred, we were unable to ascertain whether two distinct binding regimes existed. However, SUVs were stable at higher Cf values, permitting measurement of a partition coefficient. The partition coefficients of all NBD-labeled β-peptides in PC:PE:ergosterol (50:30:20) SUVs were lower than in PC:PE (60:40) SUVs (Fig. 3c, Table S4). The membrane sterols may inhibit the β-peptide adhesion to the membrane, as previously suggested (Bennett et al., 2009; Mason et al., 2007). While ergosterol inhibits β-peptide association with SUVs, it is not clear whether a critical concentration for pore formation exists in ergosterol-containing SUVs.
14-Helical β-peptides are cooperatively inserted into the plasma membrane of C. albicans
The calcein leakage and partition coefficient experiments suggest that antifungal β-peptides permeate synthetic phospholipid membranes by binding to and forming pores in the membranes. To explore mechanisms of β-peptide permeation of C. albicans cell membranes, we used confocal laser scanning microscopy (CLSM) and image analysis to measure relative binding of β-peptides to live C. albicans cells as a function of β-peptide concentration. In these experiments, the concentration of fluorescently-labeled β-peptide 1-Cy5 was fixed at 0.5 µM and unlabeled β-peptide 1 was added to vary the total β-peptide concentration from 0.5 to 7.0 µM. The cell-associated fluorescence signal was significantly greater above the MIC of β-peptide 1 (5.0 µM) than below the MIC (Fig. 4 and Fig. S6), suggesting cooperative binding of the β-peptide to the C. albicans cell membrane. These results are consistent with a pore-forming mechanism of fungal cell toxicity and suggest a similar mechanism of poration of SUVs and living C. albicans cell membranes.
Figure 4.
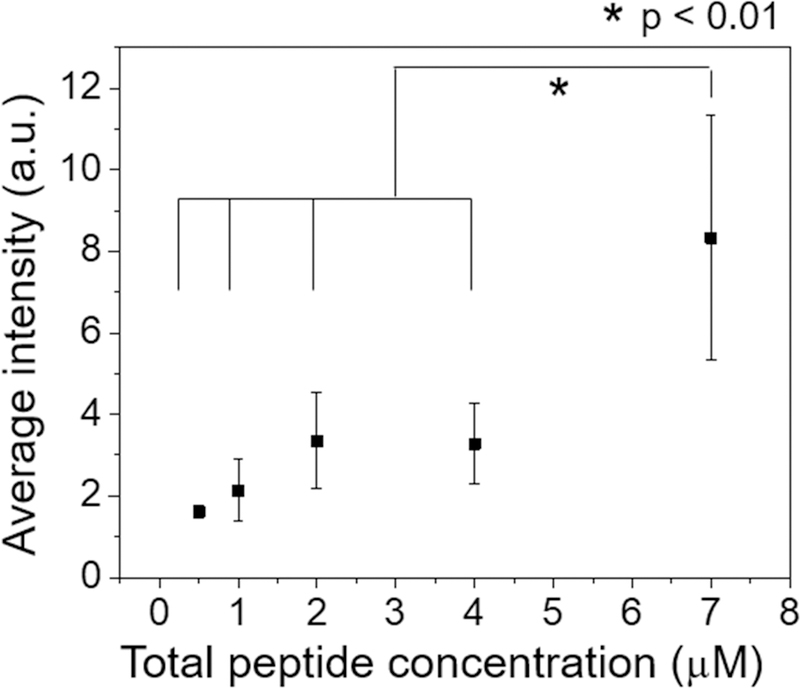
Cooperative insertion of β-peptide 1-Cy5 in the C. albicans plasma membrane. Cy5 fluorescence intensity associated with each cell was measured by fluorescence microscopy after 3 min incubation with a solution of 0.5 µM β-peptide 1-Cy5 and 0 – 6.5 µM β-peptide 1 in DPBS. The fluorescence intensity was measured for individual cells and then background intensity subtracted (Fig. S6). The images was acquired using same detector gain, amplifier gain, and pinhole settings. Data points are the averages of three independent experiments with approximately 20 cells/experiment, and error bars denote standard deviation (n = 3). Statistical analysis was performed using one-way ANOVA. See also Figure. S6.
We next used super-resolution structure illumination microscopy (SR-SIM) to visualize β-peptide distribution on the C. albicans membranes. A sub-MIC concentration of β-peptide 1-Cy5 (2 µM) was incubated with live C. albicans for 3 min then imaged. Fig. 5 demonstrates that the β-peptides congregated into punctate structures around 300 nm diameter on the cell surface (Fig. 5). This result suggests rapid adhesion and aggregation of β-peptides on the plasma membrane. We speculate that the concentrated β-peptides in these structures could facilitate the supramolecular aggregation between β-peptide and lipids on the plasma membrane surface, which enables cooperative insertion into the plasma membrane to form pores in the C. albicans cell membrane.
Figure 5.
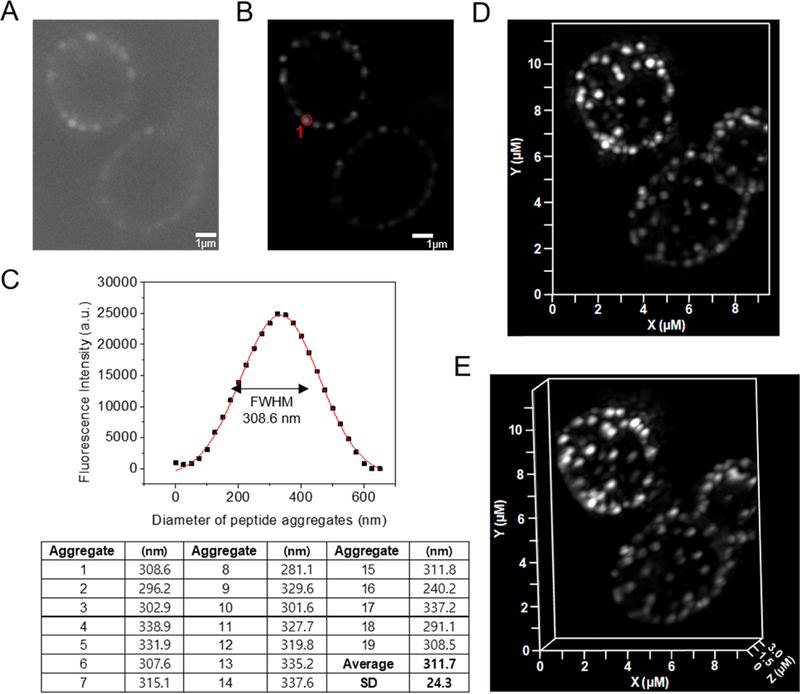
SR-SIM of β-peptide aggregation on the plasma membrane of C. albicans. 2 µM β-peptide 1-Cy5 was added to C. albicans cells and cells were imaged after 3 min incubation. Panel (A) shows a sample raw z-section image while panel (B) shows the same image processed using Zeiss ZEN software. (C) Fluorescence intensity distribution of the peptide aggregate circled in red in (B). To measure the size of peptide aggregate, the fluorescence intensity vs diameter plot was fit to a Gaussian distribution (red line). The full width at half maximum intensity illustrates the aggregate size distribution. Average peptide aggregate size was calculated from 19 aggregates from 5 different cells. SD denotes standard deviation (n = 19). Panels (d) and (e) show 3D-SR SIM image projections of stacked of Z-sections. Scale bar indicates 1 µm in (A) and (B).
14-Helical β-peptides form PI-permeable pores on live C. albicans
Data in Figures 3–5 are consistent with β-peptide pore formation in model phospholipid membranes and living C. albicans cell membranes. Next, we investigated whether these pores facilitate molecular translocation across the cell membrane and into the cytoplasm using the membrane-impermeable DNA-binding fluorescent dye propidium iodide (PI) as a tracer. We incubated C. albicans cells in sub-MIC (0–3 µM) concentrations of β-peptide 1 in the presence of 10 µg/mL PI and monitored PI accumulation inside the cell for 45 min using CLSM. At 2–3 µM β-peptide 1, some cells were lysed, indicated by high intensity PI staining (red cells in Fig. 6a and S7). Quantification of PI staining in the living cells demonstrated that as β-peptide concentration increased, the PI uptake into living cells increased throughout the incubation period (Fig. 6b). These results suggest that β-peptides can form pores in the plasma membrane that permit molecular translocation at concentrations below the MIC.
Figure 6.
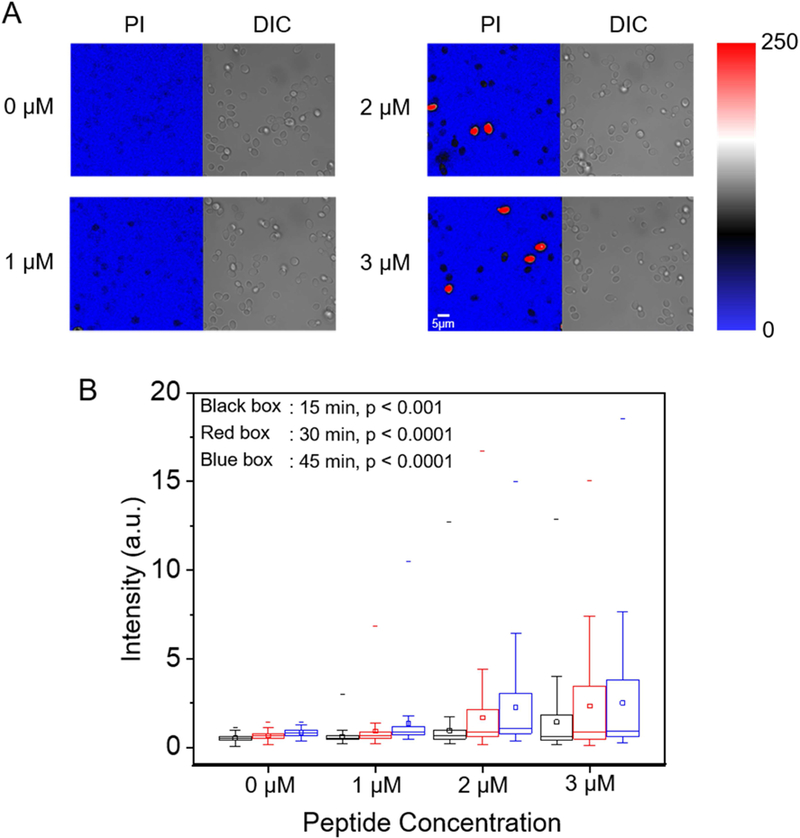
PI uptake into live C. albicans. Cells were incubated with 10 µg/mL PI and the indicated sub-MIC concentration of β-peptide 1 (1, 2, or 3 µM) in DPBS. (A) CLSM and corresponding DIC images of PI uptake into C. albicans after 45 min incubation in β-peptide solutions of the indicated concentrations. Color represents fluorescence imaging pixel values. Red cells are lysed cells that were excluded from quantitative analysis. Scale bar indicates 5 µm. (B) Box chart of the distribution of PI uptake in live C. albicans as a function of β-peptide concentration and incubation time. Black, red, and blue bars represent 15 min, 30 min, and 45 min incubation times, respectively. The box chart shows median intensity values (solid horizontal line in the box), average intensity (open square in box), 50th percentile values (box outline), 10th and 90th percentile values (whiskers), and minimum and maximum intensities (−). Statistical analysis was performed using one-way ANOVA. p-Values compare cells exposed to β-peptides to no-peptide controls. See also Figure S7.
Nuclear and vacuolar membranes are intracellular targets of 14-helical β-peptides
Because β-peptides form pores in the C. albicans membrane that mediate molecular translocation into the cytoplasm, it is possible that β-peptides could also cross the cell membrane and act on intracellular targets to promote cell death. To track β-peptide localization inside the cell, we monitored the co-localization of β-peptide 1-Cy5 with organelle-specific dyes. C. albicans cells were incubated with organelle-specific dyes, a mixture of β-peptides 1-Cy5 and 1, and PI. Specifically, FM 4–64 was used to stain the plasma membrane, and SYTO 13 and Mitotracker Green were pre-incubated to stain the nucleus and mitochondria, respectively. MDY-64 was added after removing the β-peptide solution to stain the vacuolar membrane. Fig. 7a shows that β-peptide co-localized with FM 4–64, consistent with prior findings of β-peptide localization to the cell membrane (Walther and Wendland, 2004). Following adhesion to the cell membrane, the β-peptide next translocated into the cytoplasm and associated with the nuclear membrane and the vacuolar membrane. Fig. 7b shows the β-peptide interacting with the nuclear membrane, while Fig. 7c and 7e show β-peptide association with the vacuole. However, β-peptide did not interact with all intracellular membranes. For example, β-peptide did not co-localize with mitochondria (Fig. 7d). These colocalization studies suggest that a mechanism involving lysis of nuclear and/or vacuolar membranes may contribute to the antifungal activity of β-peptides.
Figure 7.
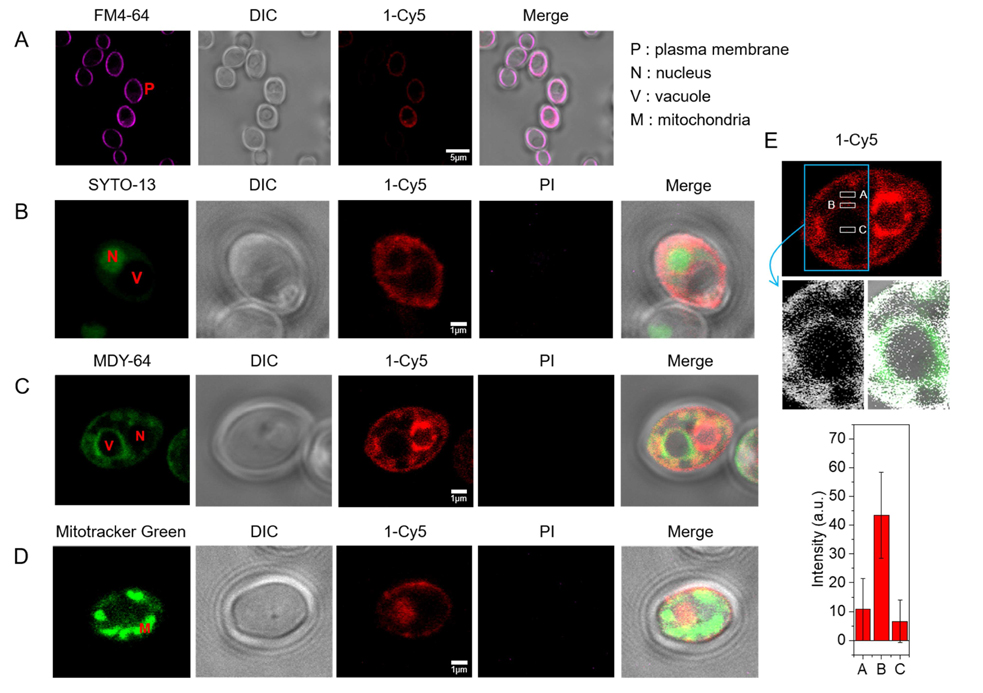
Subcellular localization of β-peptide 1-Cy5. 2 µM β-peptide 1-Cy5, 3 µM unlabeled β-peptide 1 and an organelle specific dye (1 µg/mL FM 4–64 for plasma membrane, 2.5 µM SYTO 13 for nucleus, 0.5 µM Mitotracker Green for mitochondria, and 5 µM MDY 64 for intracellular membranes, and 1 µg/mL PI for dead cell) were incubated with C. albicans cells for 5 – 30 min, followed by β-peptide incubation, and then imaged. (A) C. albicans cells were incubated with FM 4–64 for 5 min (34 of 34 cells in three experiments exhibited plasma membrane localization of β-peptide). (B) Cells were incubated with SYTO 13 for 20 min (10 of 21 cells in three experiments exhibited nuclear localization). (C) Cells were incubated with MDY 64 for 5 min (11 of 25 cells in three experiments exhibited vacuolar localization). (D) Cells were incubated with Mitotracker green for 30 min (0 of 50 cells in three experiments exhibited mitochondrial localization). (E) Quantitation of 1-Cy5 fluorescence in three areas near the vacuole: A (cytosol), B (vacuole membrane), and C (center of vacuole). Top image shows the whole cell with regions used for fluorescence quantitation outlined in white. Blue indicates the cropped vacuole region (below, white represents 1-Cy5, and green represents MDY-64). The plot in the bottom of the panel shows average fluorescence intensities in regions A, B, and C taken from measurements on images of 11 different cells similar to the representative images shown here. Scale bars indicates 5 µm for (A) and 1 µm for (B), (C), and (D). P labels the plasma membrane, N the nucleus, V the vacuole, and M the mitochondria. See also Movie S1.
To determine whether β-peptides also lyse the nucleus and vacuole, we used CLSM to monitor the kinetics of association of β-peptide 1-Cy5 with intracellular organelles. Movie S1 shows that the β-peptides rapidly adhered to the C. albicans cell membrane. Next, the cell appeared to shrink, likely as a result of leakage of cytoplasmic contents following poration of the cell membrane. The β-peptide then translocated into the cytoplasm where it adhered to the nuclear and vacuolar membranes. The nuclear membrane was then disrupted, resulting in DNA dispersal throughout the cytoplasm and then the vacuole expanded until the vacuolar membrane disrupted. This sequence of events occurred over 15 min and was observed in all C. albicans cells examined. Together, these results establish the plasma membrane, nuclear membrane, and vacuole membrane as targets for amphiphilic 14-helical β-peptides that exhibit antifungal activity.
DISCUSSION
The mode of action of helical antimicrobial peptides involves electrostatic interactions between a randomly-oriented amphiphilic peptide and the outer membrane of a target cell at the adhesion stage. Upon binding, AMPs adopt a stable and primarily helical secondary structure and self-associate on the outer membrane until a threshold concentration is reached, resulting in cell membrane disruption, either by formation of structured pores or less-organized carpets (Shai, 1999; Yeaman and Yount, 2003). Here, we demonstrate that peptidomimetic 14-helical β-peptides that display antifungal activity lyse the cell membrane by pore formation, allowing the β-peptides to translocate into the cytoplasm where they disrupt the nuclear and vacuolar membranes, resulting in cell death. First, we demonstrated that β-peptide permeabilization of SUVs depends upon SUV composition as well as β-peptide hydrophobicity. Based on prior work showing that β-peptide activity depends on hydrophobicity, charge, and helical stability, we predicted that these interactions would also influence β-peptide permeation of SUVs (Lee et al., 2014; Raman et al., 2015). Indeed, calcein leakage from SUVs correlated with β-peptide hydrophobicity. Neutral charged PC:PE (60:40) SUVs exhibited greater calcein leakage than negatively charged PC:PG (60:40) SUVs after exposure to cationic β-peptides. Hydrophobic β-peptides 1 and 2 induced greater calcein leakage than the more hydrophilic β-peptide 3. Thus, β-peptide hydrophobicity appears have a more significant influence on the ability to disrupt neutral SUVs than anionic SUVs. Hydrophobic β-peptide 2 induced similar calcein leakage from both neutral and negatively charged SUVs at 10 µg/mL peptide concentration, while less hydrophobic β-peptides 1 and 3 resulted in greater calcein leakage from negatively-charged SUVs than neutral SUVs.
The ability of β-peptides to permeabilize SUVs correlated with binding to the cell membrane of live C. albicans and to antifungal activity. Wieprecht et al. showed that hydrophobic derivatives of magainin 2a caused greater calcein leakage from POPC/POPG LUVs than native magainin 2a (Wieprecht et al., 1997). Jin et al. demonstrated that the antimicrobial activities of sheet-structured 18-mer and helical 21-mer oligopeptides correlated with calcein leakage from POPC/POPG and POPE/POPG LUVs (Jin et al., 2005). Similarly, our results illustrate that the more active antifungal β-peptides 1 and 2 induce significantly greater calcein leakage from PC:PE(60:40) and PC:PE(80:20) SUVs than the less active β-peptide 3.
SUV studies also demonstrated cooperative binding of antifungal 14-helical β-peptides in a manner consistent with pore formation. In the rapid accumulation stage, the β-peptides adhere to the cell membrane with a low partition coefficient. After reaching a threshold concentration, the β-peptides insert into the core of lipid bilayer and permeabilize the membrane in a cooperative manner, resulting in a higher partition coefficient (Huang, 2000; Shai, 1999). Rapaport et al. reported a critical concentration of 35 nM for the pore-forming pardaxin derivative (C1-NBD-par) against PC SUVs (Rapaport and Shai, 1991). Gazit et al. demonstrated that two helical peptides derived from the pore-forming Bacillus thuringiensis serotype israelensis (Bti) toxin exhibited similar critical concentrations and partition coefficients to the full length Bti toxin (Gazit et al., 1994). The NBD-labeled 14-helical β-peptides used in this study exhibited high partition coefficients and low critical concentrations against model membranes (Table S4).
Our live C. albicans studies also support a pore-forming mechanism. CLSM measurement of fluorescence accumulation using mixtures of β-peptides 1-Cy5 and 1 showed cooperative binding to the plasma membrane of live C. albicans near the MIC of β-peptide 1. This cooperative adhesion is likely related to a transition from the low to the high partition coefficient observed in the SUVs. Interestingly, β-peptide 1 permitted PI translocation across the cell membrane in live C. albicans at concentrations below the MIC. The intracellular concentration of PI depended on the β-peptide concentration and incubation time, but some membrane permeabilization was observed without resulting in cell death. Conversely, β-peptide concentrations above the MIC resulted in rapid entry of high concentrations of PI. Together, these synthetic SUV and live cell studies are consistent with a pore-forming mechanism of β-peptide permeabilization of the C. albicans cell membrane.
Mechanisms involving both plasma membrane permeation and intracellular targets have been reported. For example, buforin 2 forms toroidal pores in bacterial cell membranes, then translocates into the cytoplasm and binds to DNA and RNA (Kobayashi et al., 2004; Park et al., 1998; Takeshima et al., 2003). Once inside the cell, 14-helical β-peptides can disrupt the nuclear and vacuole membranes, as determined by time-lapse co-localization of Cy5-labeled β-peptides with organelle-specific fluorescent dyes. After translocating through the cell membrane, the β-peptides bind and permeabilize nuclear and vacuole membranes, sequentially (Movie S1). We show that β-peptide-induced disruption of cellular membranes, including the plasma membrane, nucleus, and vacuole, results in cell death. We predict that the potential of these compounds to disrupt multiple cell membranes could help impede development of resistance mechanisms involving changes in membrane composition.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sean Palecek (sppalecek@wisc.edu)
Experimental Model and Subject Details
SUVs were used to study calcein leakage and measure β-peptide partition coefficients. C. albicans strain SC5314 was used to study β-peptide localization in living cells.
Details of C. albicans cells
-
o
Genotype : Wild-type
-
o
Species/strain : C. albicans / SC5314 (ATCC MYA-2876)
-
o
Culture conditions: Frozen stocks were streaked on YPD agar plates and grown at 30 °C for 24 h. A single colony of C. albicans (SC5314) was suspended in 5 mL YPD medium and grown overnight at 32.5 °C. The cells were collected by centrifugation (2000 rpm, 5 min) and washed with DPBS buffer. The cells (106 - 107 cells/mL in 400 µL) were seeded on ConA-coated plates and allowed to attach for 30 min prior to CLSM and SR-SIM microscopy.
Method Details
Synthesis of fluorescently-labeled β-peptides
β-peptides were synthesized as previously reported by microwave-assisted protocols of solid-phase peptide synthesis(Lee et al., 2014). NDB and Cy5 labeling were performed after coupling and deprotection of the last β-amino acid. For NBD labeling, NBD-Cl (3 eq), and DIEA (6 eq) were premixed in DMF and then mixed with the β-peptide resin. After stirring overnight at room temperature, the resin was washed with DMF (5 times), CH2Cl2 (5 times), and DMF (5 times). Prior to Cy5 labeling, β-peptides were coupled with an ethoxy-containing tether, (2-(2-(Fmoc-amino)ethoxy)-ethoxy)acetic acid (3 eq), and then the resin was washed with DMF (5 times), CH2Cl2 (5 times), and DMF (5 times). Cy5-NHS (2 eq), and DIEA (4 eq) were dissolved in DMF and then mixed with the β-peptide resin. After stirring overnight at room temperature, the resin was washed with DMF (5 times), CH2Cl2 (5 times) and DMF (5 times). After the NBD and Cy5 labeling, the resin was washed with CH2Cl2 and then the peptide was cleaved from the resin by addition of TFA containing H2O (2.5%) and triisopropylsilane (2.5%) for 1 to 2 h. The crude products of dye-labeled β-peptides were purified by preparative RP-HPLC with a gradient of 25–73% CH3CN in water containing 0.1% TFA over 35 min. The peptides were validated using mass spectrometry. 1-NBD: HRMS (m/z, ESI) calcd for C66H112N16O12 [M+2H]2+ 661.4396, measured 661.4417, 2-NBD: HRMS (m/z, ESI) calcd for C66H112N22O12 [M+TFA+2H]2+ 760.4452, measured 760.4482, 3-NBD: HRMS (m/z, ESI) calcd for C60H100N16O12 [M+2H]2+ 619.3926, measured 619.3947, 1-Cy5: MALDI-TOF (m/z) calcd for C98H159N16O13 [M]+ 1768.2, measured 1768.5.
Measurement of Minimum Inhibitory Concentration (MIC) of Antifungal Peptides
The MICs of fluorophore-labeled β-peptides (1-NBD, 2-NBD, 3-NBD, and 1-Cy5), magainin 2, and melittin against C. albicans (SC5314) were determined in accordance with the broth microdilution assay suggested by the Clinical and Laboratory Standards Institute, including quantitative XTT cell metabolic assessment (Karlsson et al., 2009). MIC was calculated as the minimum concentration that resulted in at least 90% decrease in cell viability. Peptides were assayed in RPMI medium containing 1% DMSO.
Preparation of Small Unilamellar Vesicles
The total phospholipid was maintained at 36 µmol to prepare calcein-encapsulating small unilamellar vesicles (SUVs): PC/PE (21.6/14.4 µmol, 60:40), PC/PE (28.8/7.2 µmol, 80:20), PC/PG (21.6/14.4 µmol, 60:40). The desired amounts of phospholipids were dissolved in chloroform (5 mL) in a round bottom flask and then chloroform was evaporated to form multilamellar layers of phospholipids. After drying the round bottom flask under vacuum for 1 h, 70 mM calcein in TBS buffer (10 mM Tris.HCl, 150 mM NaCl, 0.1 mM EDTA, pH 7.4, 4 mL) and 10 N NaOH (3 eq) was added into the flask and then the solution was vortexed to form a suspension of multilamellar vesicles. This suspended solution was sonicated for 30 to 45 min in an ice bath using an ultrasonicator (550 Sonic Dismembrator, Fisher Scientific). Unilamellar vesicles were purified by Sephadex G-50 size exclusion chromatography (L/D = 26) under gravity. After purification, the vesicle composition was measured by 31P NMR and the effective vesicle size was quantified by dynamic light scattering (DLS). The DLS of a 1 mL purified SUV solution was measured at 25 °C in a glass cell in a decahydronaphthalene environment. Light scattering was measured at a 90° angle using a 100 mW, 532 nm laser (Coherent Compass 315M-100) and BI-9000AT digital autocorrelator (Brookhaven Laser Light Scattering System, Brookhaven Instruments Corporation). The effective vesicle size was measured between 50 – 90 nm (Table S2). To measure β-peptide partition coefficients, unilamellar vesicles were prepared as described for calcein-encapsulating vesicles. PC/PE (60:40) and PC/PG (60:40) vesicles were prepared at a 36 µmol scale in TBS buffer (4 mL). PC/PE/ergosterol (22.5/13.5/9 µmol, 50:30:20) vesicles were prepared at a 45 µmol scale in TBS buffer (9 mL). After purification, the vesicle composition and phospholipid concentration were measured by 31P NMR and 1H NMR (Fig. S4) and the effective size of vesicles was measured by DLS. The effective vesicle size was measured between 40 – 80 nm (Table S3).
Calcein leakage
The desired concentrations of β-peptides (1 - 3), magainin 2, and melittin in TBS buffer were added to a 96 well plate (75 µL) with positive (2% Triton X100 in TBS buffer) and negative (TBS buffer) lysis controls. SUVs (75 µL, 260 µM) were added into the 96 wells of the plate and the fluorescence intensity was recorded (ext. 495 / em. 515 nm) over 1 h at 80 s intervals using a FL800 Universal Microplate Reader) (Bio-Tek Instruments). The calcein leakage (%) was calculated as
Where and are the average fluorescence intensities at 515 nm of SUVs treated with peptides, TBS, and Triton X100, respectively. Duplicate samples were analyzed for experiments repeated on at least three different days.
Partition coefficient measurement
All NBD labeled β-peptide stock solutions were prepared in DMSO (20 µM). SUV solution (792 µL) and peptide stock solution (8 µL) were mixed in a fluorescence cuvette and then reacted for 2 min before the recording fluorescence (ext. 468) spectrum from 480 – 600 nm (F-4500 Fluorescence Spectrometer, Hitachi). Experiments were performed in triplicate using freshly-prepared SUVs on different days. After recording fluorescence spectra, the partition coefficients were calculated as reported previously (Pouny et al., 1992; Rapaport and Shai, 1991). Briefly, the binding isotherms were assessed as partition equilibria using the following equation:
where Xb, Kp and Cf are the molar ratio of bound peptide per total lipid, the partition coefficient, and the equilibrium concentration of free peptide in the solution, respectively. In order to calculate Xb, F∞ (the estimated maximum fluorescence intensity when vesicles are saturated by peptide) was calculated from the peptide titration curve. The fb (fraction of bound peptide) was calculated from the following equation:
Where F, and F0 are the fluorescence intensity of bound and unbound NBD-peptide, respectively. Xb equations were corrected by the fraction of phospholipid accessible to the β- peptide (60% of total lipid). Thus, the binding isotherm was represented as:
where and are the molar ratio of bound peptide per 60% of total lipid and the estimated surface partition constant, respectively.
Adhesion of C. albicans to glass plates for microscopy
A single colony of C. albicans (SC5314) was suspended in 5 mL YPD medium and grown overnight at 32.5 °C. The cells were collected by centrifugation (2000 rpm, 5 min) and washed with DPBS buffer. ConA (2 mg/mL in DPBS, 500 µL) was adsorbed to the glass bottomed cell culture dish for 24 h at 4 °C. After incubation, the dish was rinsed with DPBS. The C. albicans cells (106 - 107 cells/mL in 400 µL) were seeded on ConA-coated plates and allowed to attach for 30 min prior to CLSM and SR-SIM microscopy.
Confocal laser scanning microscopy
C. albicans cells adhered to a glass-bottomed dish were incubated with 1-Cy5 (0.5 µM) and varying concentrations of β-peptide 1 (0, 0.5, 1.5, 3.5, 6.5 µM) in DPBS (300 µL). After 3 min incubation, cells were imaged by CLSM (Zeiss LSM 510) with a 100x objective. The excitation and emission filter were selected, excitation filter: 633 nm and emission filter: BP 650–710 IR, respectively. Images were processed using Zeiss LSM image browser.
Super resolution structure illumination microscopy (SR-SIM)
C. albicans cells adhered to a glass-bottomed dish were incubated with 2 µM 1-Cy5 in DPBS (300 µL) for 3 min and then cells were imaged by CLSM (Zeiss LSM 780) with an alpha plan-apochromat 100x/1.46 oil objective equipped with a 642 nm laser. Immersion oil was applied on the objective to minimize refractive index mismatch and spherical aberration. 3D SR-SIM image stacks of 3.84 µm thickness were acquired with 0.133 µm z-steps and 15 images (5 rotations per angle) per z-section. Images were processed using Zeiss ZEN Software.
Characterization of PI uptake by cells
10 µg/mL PI and β-peptide 1 (1, 2, or 3 µM) in DPBS (300 µL) were added to cells adhered to a glass-bottomed dish. Images were acquired for 45 min with 3 min intervals by CLSM (Zeiss LSM 780) with a 100x objective. The excitation and emission filter were selected, excitation filter: 561 nm and emission filter: 575–600 nm bandpass filter, respectively. Images were processed using Zeiss ZEN software.
Characterization of co-localization of β-peptide with cellular organelles
C. albicans cells were adhered to a glass-bottomed dish as described above. For FM 4–64/1-Cy5: A solution of 1 µg/mL FM 4–64, 2.0 µM β-peptide 1-Cy5, and 3.0 µM β-peptide 1 in DPBS (300 µL) were added to the cells and incubated for 5 min. Then, the confocal images were acquired. For SYTO 13/1-Cy5: A solution of 2.5 µM SYTO 13 in DPBS (300 µL) was added to the cells and incubated for 20 min. After incubation, the SYTO 13 solution was removed from the dish and then a solution of 2.5 µM SYTO 13, 2.0 µM β-peptide 1-Cy5, 3.0 µM β-peptide 1, and 1 µg/mL PI in DPBS (300 µL) was added to the dish. After 5 – 10 min, confocal images were acquired. For MDY-64/1-Cy5: A solution of 2.0 µM β-peptide 1-Cy5 and 3.0 µM β-peptide 1 in DPBS (300 µL) was added to the cells and incubated for 10 min. After incubation, the peptide solution was removed from the dish and the cells were washed 2 times with DPBS. A solution of 5 µM MDY-64 and 1 µg/mL PI in 10 mM HEPES containing 5% glucose, pH 7.4 (300 µL) was added to the dish. After 5 min, confocal images were acquired. For MitoTracker Green/1-Cy5: A solution of 0.5 µM MitoTracker in DPBS (300 µL) was added to the cells and incubated for 30 min. After incubation, the MitoTracker solution was removed from the dish and then a solution of 2.0 µM β-peptide 1-Cy5, 3.0 µM β-peptide 1, and 1 µg/mL PI in DPBS (300 µL) was added to the dish. After 5 – 10 min, confocal images were acquired. Images were acquired by CLSM (Zeiss LSM 510) using a 100x objective with 3x zoom. The excitation wavelengths and emission filters were selected as followed: SYTO 13 (ext. 488 nm, filter: BP 500–530 IR), Cy-5 (ext. 633 nm, filter: BP 650–710 IR), FM 4–64 and PI (ext. 561 nm, filter: BP 575–615 IR), MDY-64 and MitoTracker Green (ext. 458 nm, filter: BP 480–520 IR). Images were processed using Zeiss LSM image browser.
Time lapse tracking of β-peptide
A solution of 1 µg/mL Hoechst in DPBS (300 µL) was added to cells adhered to a glass-bottomed dish and incubated for 30 min. After incubation, the Hoechst solution was replaced with the solution of 1 µg/mL Hoechst and 0.5 µM Mitotracker green in DPBS (300 µL) and incubated for an additional 20 min. Then a solution of 1 µg/mL PI, 2.0 µM β-peptide 1-Cy5, and 3.0 µM β-peptide 1 in DPBS (300 µL) was added to the dish. Confocal images were acquired for Hoechst, Mitotracker green, PI and Cy5 every 40 s. Cells were imaged using CLSM (LSM 700) with a 60x objective. The excitation wavelength and emission wavelength filters were selected as followed: Hoechst (ext. 405 nm, filter EM 500–530 nm), Cy5 (ext. 639 nm, filter EM 650–710 nm), Mitotracker green (ext. 488 nm, filter EM 480–520 nm), and PI (ext. 561 nm, filter EM 575–615 nm). Images were processed using Zeiss ZEN software.
Quantification and Statistical Analysis
Partition coefficients were calculated by nonlinear (piecewise fitting function (pwl2s) consisting of two linear segments) and linear data fits using OriginPro (version 2018b, Northampton, MA) (n=3). Statistical analysis was performed using one-way ANOVA in OriginPro (n=3). p<0.05 was considered statistically significant.
Data and Software Availability
CLSM and SR-SIM Images were processed using Zeiss ZEN software (https://www.zeiss.com/).
Supplementary Material
Key Resource Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fmoc-L-β-homoalanine | Chem-Impex International | Cat # 12776 |
| Fmoc-L-β-homovaline | Chem-Impex International | Cat # 15225 |
| Fmoc-(1S,2S)-2-aminocyclohexane carboxylic acid | Chem-Impex International | Cat # 15082 |
| Nβ-Fmoc-Nω-Boc-L-β-homolysine | Chem-Impex International | Cat # 12769 |
| Fmoc-Nω-(2,2,5,7,8-pentamethyl-chromane-6-sulfonyl)-L-β-homoarginine | Chem-Impex International | Cat # 12775 |
| HBTU (O-(benzotriaole-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophos-phate | Advanced ChemTech | Cat # RC8106 |
| HOBt.H2O (N-hydroxybenzotrizole monohydrate) | Advanced ChemTech | Cat # RC8201 |
| Calcein | Sigma-Aldrich | Cat # C0875 |
| Triethylphosphine oxide | Sigma-Aldrich | Cat # 562394 |
| (2-(2-(Fmoc-amino)-ethoxy)ethoxy)acetic acid | Sigma-Aldrich | Cat # 95003 |
| Ergosterol | Acros | Cat # 117810050 |
| 4-chloro-7-nitrobenzofurazan | Sigma-Aldrich | Cat # 25455 |
| Cyanine5 NHS ester | Lumiprobe | Cat # 53020 |
| Magainin 2 | American Peptide Company | Cat # 72-2-26 |
| Melittin | American Peptide Company | Cat # 86-4-10 |
| TentaGel S RAM Fmoc | Advanced ChemTech | Cat # SJ5030 |
| Sephadex G-50 | Sigma-Aldrich | Cat # G50150 |
| Concanavalin A (ConA) | Sigma-Aldrich | Cat # C2010 |
| 1,2-dipalmitoyl-sn-glycero-3-phosphocholine | Sigma-Aldrich | Cat # P0763 |
| L-α-Phosphatidylglycerol (Egg, Chicken) | Avanti | Cat # 841138 |
| L-α-phosphatidylethanolamine (Egg, Chicken) | Avanti | Cat # 840021 |
| L-α-phosphatidylcholine (Egg, Chicken) | Avanti | Cat # 840051 |
| SYTO 13 | Life Technologies | Cat # S7575 |
| MDY-64 | Life Technologies | Cat # Y7536 |
| FM 4–64 | Life Technologies | Cat # T13320 |
| MitoTracker Green | Life Technologies | Cat # M7514 |
| Experimental Models: Organisms/Strains | ||
| C. albicans: SC5314 | ATCC | MYA-2876 |
| Software and Algorithms | ||
| pwl2s; piecewise fitting function consisting of two linear segments | OriginPro 2018b | https://www.originlab.com/doc/Tutorials/Fitting-Piecewise-Linear |
| Zeiss ZEN | Zeiss | https://www.zeiss.com/ |
| Other | ||
| Glass bottom cell culture plates | Cellvis | Cat # D35–20-1.5-N |
| CELLVIEW cell culture dishes | Greiner Bio-One | Cat # 627861 |
Highlights.
14-Helical β-peptides can permeabilize synthetic lipid vesicles.
β-peptide binding to synthetic vesicles is consistent with a pore formation model.
Antifungal β-peptides permeabilize the plasma membrane of Candida albicans.
β-Peptides disrupt the nuclear membrane and vacuole to kill fungal cells.
Significance.
Peptidomimetic AMPs have tremendous potential as antimicrobial agents. However, the mechanisms of their activity and selectively are not well-understood, limiting efforts to design more effective peptidomimetic AMPs. Our results establish that 14-helical β-peptides disrupt artificial membranes and live C. albicans cell membranes. β-Peptides bind to membranes via hydrophobic and electrostatic interactions, and upon reaching a critical threshold concentration insert into and permeabilize the cell membrane in a cooperative manner. Once inside C. albicans cells, β-peptides bind and permeabilize nuclear and vacuolar membranes sequentially, leading to cell death. This understanding of the mechanisms of antifungal activity will facilitate design and development of AMP peptidomimetics, including 14-helical β-peptides, for antifungal applications.
Acknowledgements
The authors thank Sarah Swanson at the UW-Madison Newcomb Imaging Center and Julie Last and the UW-Madison Materials Science Center for imaging assistance. This research was supported by NIH grants RO1AI092225 and R21AI127442. The ESI mass spectrometry for peptide identification was supported by NIH grant 1S10OD020022-1.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersson DI, and Hughes D (2010). Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8, 260–271. [DOI] [PubMed] [Google Scholar]
- Appella DH, Christianson LA, Klein DA, Powell DR, Huang XL, Barchi JJ, and Gellman SH (1997). Residue-based control of helix shape in beta-peptide oligomers. Nature 387, 381–384. [DOI] [PubMed] [Google Scholar]
- Arvidsson PI, Frackenpohl J, Ryder NS, Liechty B, Petersen F, Zimmermann H, Camenisch GP, Woessner R, and Seebach D (2001). On the antimicrobial and hemolytic activities of amphiphilic beta-peptides. Chembiochem 2, 771–773. [DOI] [PubMed] [Google Scholar]
- Bennett WFD, MacCallum JL, and Tieleman DP (2009). Thermodynamic Analysis of the Effect of Cholesterol on Dipalmitoylphosphatidylcholine Lipid Membranes. J Am Chem Soc 131, 1972–1978. [DOI] [PubMed] [Google Scholar]
- Beutler B (2004). Innate immunity: an overview. Mol Immunol 40, 845–859. [DOI] [PubMed] [Google Scholar]
- Boheim G (1974). Statistical analysis of alamethicin channels in black lipid membranes. J Membr Biol 19, 277–303. [DOI] [PubMed] [Google Scholar]
- Boman HG, Agerberth B, and Boman A (1993). Mechanisms of action on Escherichia coli of cecropin P1 and PR-39, two antibacterial peptides from pig intestine. Infect Immun 61, 2978–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogden KA (2005). Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3, 238–250. [DOI] [PubMed] [Google Scholar]
- Brogden KA, Ackermann M, McCray PB, and Tack BF (2003). Antimicrobial peptides in animals and their role in host defences. Int J Antimicrob Agents 22, 465–478. [DOI] [PubMed] [Google Scholar]
- Brotz H, Bierbaum G, Markus A, Molitor E, and Sahl HG (1995). Mode of action of the lantibiotic mersacidin - inhibition of peptidoglycan biosynthesis via a novel mechanism. Antimicrob Agents Chemother 39, 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne CA, Bennett HPJ, and Solomon S (1982). The Isolation of Peptides by High-Performance Liquid Chromatograpy Using Predicted Elution Positions. Anal Biochem 124, 201–208. [DOI] [PubMed] [Google Scholar]
- Chu HL, Yu HY, Yip BS, Chih YH, Liang CW, Cheng HT, and Cheng JW (2013). Boosting Salt Resistance of Short Antimicrobial Peptides. Antimicrob Agents Chemother 57, 4050–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YJ, Christianson LA, Stanger HE, Powell DR, and Gellman SH (1998). A beta-peptide reverse turn that promotes hairpin formation. J Am Chem Soc 120, 10555–10556. [Google Scholar]
- Chung YJ, Huck BR, Christianson LA, Stanger HE, Krauthauser S, Powell DR, and Gellman SH (2000). Stereochemical control of hairpin formation in beta-peptides containing dinipecotic acid reverse turn segments. J Am Chem Soc 122, 3995–4004. [Google Scholar]
- Epand RF, Umezawa N, Porter EA, Gellman SH, and Epand RM (2003). Interactions of the antimicrobial beta-peptide beta-17 with phospholipid vesicles differ from membrane interactions of magainins. Eur J Biochem 270, 1240–1248. [DOI] [PubMed] [Google Scholar]
- Fjell CD, Hiss JA, Hancock REW, and Schneider G (2012). Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov 11, 37–51. [DOI] [PubMed] [Google Scholar]
- Gagnon MG, Roy RN, Lomakin IB, Florin T, Mankin AS, and Steitz TA (2016). Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition. Nucleic Acids Res 44, 2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo RL, and Nizet V (2003). Endogenous production of antimicrobial peptides in innate immunity and human disease. Curr Allergy Asthma Rep 3, 402–409. [DOI] [PubMed] [Google Scholar]
- Ganz T (2003). Defensins: Antimicrobial peptides of innate immunity. Nat Rev Immunol 3, 710–720. [DOI] [PubMed] [Google Scholar]
- Gazit E, Lee WJ, Brey PT, and Shai YC (1994). Mode of action of the antibacterial cecropin B2 - a spectrofluorometic study. Biochemistry 33, 10681–10692. [DOI] [PubMed] [Google Scholar]
- Gazit E, Miller IR, Biggin PC, Sansom MSP, and Shai Y (1996). Structure and orientation of the mammalian antibacterial peptide cecropin P1 within phospholipid membranes. J Mol Biol 258, 860–870. [DOI] [PubMed] [Google Scholar]
- Godballe T, Nilsson LL, Petersen PD, and Jenssen H (2011). Antimicrobial beta-Peptides and alpha-Peptoids. Chem Biol Drug Des 77, 107–116. [DOI] [PubMed] [Google Scholar]
- Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, and Wilson JM (1997). Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 88, 553–560. [DOI] [PubMed] [Google Scholar]
- Gordon LGM, and Haydon DA (1972). The unit conductance channel of alamethicin. Biochimica Et Biophysica Acta 255, 1014–1018. [DOI] [PubMed] [Google Scholar]
- Gordon LGM, and Haydon DA (1975). Potential-dependent conductances in lipid membranes containing alamethicin. Philos Trans R Soc Lond Ser B-Biol Sci 270, 433–447. [DOI] [PubMed] [Google Scholar]
- Gregory SM, Pokorny A, and Almeida PFF (2009). Magainin 2 Revisited: A Test of the Quantitative Model for the All-or-None Permeabilization of Phospholipid Vesicles. Biophys J 96, 116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallock KJ, Lee DK, and Ramamoorthy A (2003). MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys J 84, 3052–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y, Schneider JP, and DeGrado WF (1999). De novo design of antibacterial beta-peptides. J Am Chem Soc 121, 12200–12201. [Google Scholar]
- Hancock REW, and Lehrer R (1998). Cationic peptides: a new source of antibiotics. Trends Biotechnol 16, 82–88. [DOI] [PubMed] [Google Scholar]
- Huang HW (2000). Action of antimicrobial peptides: Two-state model. Biochemistry 39, 8347–8352. [DOI] [PubMed] [Google Scholar]
- Huang JF, Hao DM, Chen Y, Xu YM, Tan JJ, Huang YB, Li F, and Chen YX (2011). Inhibitory effects and mechanisms of physiological conditions on the activity of enantiomeric forms of an alpha-helical antibacterial peptide against bacteria. Peptides 32, 1488–1495. [DOI] [PubMed] [Google Scholar]
- Jackman JA, Goh HZ, Zhdanov VP, Knoll W, and Cho NJ (2016). Deciphering How Pore Formation Causes Strain-Induced Membrane Lysis of Lipid Vesicles. J Am Chem Soc 138, 1406–1413. [DOI] [PubMed] [Google Scholar]
- Jiang ZQ, Kullberg BJ, van der Lee H, Vasil AI, Hale JD, Mant CT, Hancock REW, Vasil ML, Netea MG, and Hodges RS (2008). Effects of Hydrophobicity on the Antifungal Activity of alpha-Helical Antimicrobial Peptides. Chem Biol Drug Des 72, 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Hammer J, Pate M, Zhang Y, Zhu F, Zmuda E, and Blazyk J (2005). Antimicrobial activities and structures of two linear cationic peptide families with various amphipathic beta-sheet and alpha-helical potentials. Antimicrob Agents Chemother 49, 4957–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson AJ, Pomerantz WC, Neilsen KJ, Gellman SH, and Palecek SP (2009). Effect of Sequence and Structural Properties on 14-Helical beta-Peptide Activity against Candida albicans Planktonic Cells and Biofilms. ACS Chem Biol 4, 567–579. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Chikushi A, Tougu S, Imura Y, Nishida M, Yano Y, and Matsuzaki K (2004). Membrane translocation mechanism of the antimicrobial peptide buforin 2. Biochemistry 43, 15610–15616. [DOI] [PubMed] [Google Scholar]
- Kondejewski LH, Jelokhani-Niaraki M, Farmer SW, Lix B, Kay CM, Sykes BD, Hancock REW, and Hodges RS (1999). Dissociation of antimicrobial and hemolytic activities in cyclic peptide diastereomers by systematic alterations in amphipathicity. J Biol Chem 274, 13181–13192. [DOI] [PubMed] [Google Scholar]
- Krauthauser S, Christianson LA, Powell DR, and Gellman SH (1997). Antiparallel sheet formation in beta-peptide foldamers: Effects of beta-amino acid substitution on conformational preference. J Am Chem Soc 119, 11719–11720. [Google Scholar]
- Lai YP, and Gallo RL (2009). AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends in Immunology 30, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis T, He Y, and Prieto L (2013). Membrane Interactions and Pore Formation by the Antimicrobial Peptide Protegrin. Biophys J 104, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Sun Y, Qian S, and Huang HW (2011). Transmembrane Pores Formed by Human Antimicrobial Peptide LL-37. Biophys J 100, 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MR, Raman N, Gellman SH, Lynn DM, and Palecek SP (2014). Hydrophobicity and Helicity Regulate the Antifungal Activity of 14-Helical beta-Peptides. ACS Chem Biol 9, 1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrer RI, Barton A, Daher KA, Harwig SSL, Ganz T, and Selsted ME (1989). Interaction of human defensins with Escherichia coli - mechanisms of bactericidal activity. J Clin Invest 84, 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienkamp K, Madkour AE, and Tew GN (2013). Antibacterial Peptidomimetics: Polymeric Synthetic Mimics of Antimicrobial Peptides In Polymer Composites - Polyolefin Fractionation - Polymeric Peptidomimetics - Collagens, Abe A, Kausch HH, Moller M, and Pasch H, eds. (Berlin: Springer-Verlag Berlin; ), pp. 141–172. [Google Scholar]
- Magzoub M, Oglecka K, Pramanik A, Eriksson LEG, and Graslund A (2005). Membrane perturbation effects of peptides derived from the N-termini of unprocessed prion proteins. Biochim Biophys Acta-Biomembr 1716, 126–136. [DOI] [PubMed] [Google Scholar]
- Mahlapuu M, Hakansson J, Ringstad L, and Bjorn C (2016). Antimicrobial Peptides : An Emerging Category of Therapeutic Agents. Front Cell Infect Microbiol 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason AJ, Marquette A, and Bechinger B (2007). Zwitterionic phospholipids and sterols modulate antimicrobial peptide-induced membrane destabilization. Biophys J 93, 4289–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K, Murase O, and Miyajima K (1995). Kinetics of pore formation by an antimicrobial peptide, Magainin-2, in phospholipid bilayers. Biochemistry 34, 12553–12559. [DOI] [PubMed] [Google Scholar]
- Michaelson DM, Horwitz AF, and Klein MP (1974). Head group modulation of membrane fluidity in sonicated phospholipid dispersions. Biochemistry 13, 2605–2612. [DOI] [PubMed] [Google Scholar]
- Nishikata M, Kanehira T, Oh H, Tani H, Tazaki M, and Kuboki Y (1991). Salivary histatin as an inhibitor of a protease produced by the oral bacterium Bacteroides gingivalis. Biochem Biophys Res Commun 174, 625–630. [DOI] [PubMed] [Google Scholar]
- Nizet V (2006). Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr Issues Mol Biol 8, 11–26. [PubMed] [Google Scholar]
- Otvos L, O I, Rogers ME, Consolvo PJ, Condie BA, Lovas S, Bulet P, and Blaszczyk-Thurin M (2000). Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 39, 14150–14159. [DOI] [PubMed] [Google Scholar]
- Park CB, Kim HS, and Kim SC (1998). Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun 244, 253–257. [DOI] [PubMed] [Google Scholar]
- Patrzykat A, Friedrich CL, Zhang LJ, Mendoza V, and Hancock REW (2002). Sublethal concentrations of pleurocidin-derived antimicrobial peptides inhibit macromolecular synthesis in Escherichia coli. Antimicrob Agents Chemother 46, 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschel A, and Sahl HG (2006). The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat Rev Microbiol 4, 529–536. [DOI] [PubMed] [Google Scholar]
- Porter EA, Wang XF, Lee HS, Weisblum B, and Gellman SH (2000). Antibiotics - Non-haemolytic beta-amino-acid oligomers. Nature 404, 565–565. [DOI] [PubMed] [Google Scholar]
- Pouny Y, Rapaport D, Mor A, Nicolas P, and Shai Y (1992). Interaction of antimicrobial dermaseptin and its fluorescently labeled analogs with phospholipid membranes. Biochemistry 31, 12416–12423. [DOI] [PubMed] [Google Scholar]
- Pouny Y, and Shai Y (1992). Interaction of D-amino acid incorporated analogs of paradoxin with membranes. Biochemistry 31, 9482–9490. [DOI] [PubMed] [Google Scholar]
- Pukala TL, Brinkworth CS, Carver JA, and Bowie JH (2004). Investigating the importance of the flexible hinge in caerin 1.1: Solution structures and activity of two synthetically modified caerin peptides. Biochemistry 43, 937–944. [DOI] [PubMed] [Google Scholar]
- Puri S, and Edgerton M (2014). How Does It Kill?: Understanding the Candidacidal Mechanism of Salivary Histatin 5. Eukaryot Cell 13, 958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raguse TL, Porter EA, Weisblum B, and Gellman SH (2002). Structure-activity studies of 14-helical antimicrobial beta-peptides: Probing the relationship between conformational stability and antimicrobial potency. J Am Chem Soc 124, 12774–12785. [DOI] [PubMed] [Google Scholar]
- Raman N, Lee M-R, Lynn DM, and Palecek SP (2015). Antifungal activity of 14-helical -peptides against planktonic cells and biofilms of Candida Species. Pharmaceuticals 8, 483–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport D, and Shai Y (1991). Interaction of fluorescently labeled pardaxin and its analogs with lipid bilayers. J Biol Chem 266, 23769–23775. [PubMed] [Google Scholar]
- Schmidtchen A, Frick IM, Andersson E, Tapper H, and Bjorck L (2002). Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol 46, 157–168. [DOI] [PubMed] [Google Scholar]
- Seebach D, Gademann K, Schreiber JV, Matthews JL, Hintermann T, Jaun B, Oberer L, Hommel U, and Widmer H (1997). ‘Mixed’ beta-peptides: A unique helical secondary structure in solution. Helv Chim Acta 80, 2033–2038. [Google Scholar]
- Seebach D, and Matthews JL (1997). beta-peptides: a surprise at every turn. Chem Commun, 2015–2022.
- Shai Y (1999). Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta-Biomembr 1462, 55–70. [DOI] [PubMed] [Google Scholar]
- Smith JJ, Travis SM, Greenberg EP, and Welsh MJ (1996). Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 85, 229–236. [DOI] [PubMed] [Google Scholar]
- Takeshima K, Chikushi A, Lee KK, Yonehara S, and Matsuzaki K (2003). Translocation of analogues of the antimicrobial peptides magainin and buforin across human cell membranes. J Biol Chem 278, 1310–1315. [DOI] [PubMed] [Google Scholar]
- Tamba Y, and Yamazaki M (2005). Single giant unilamellar vesicle method reveals effect of antimicrobial peptide magainin 2 on membrane permeability. Biochemistry 44, 15823–15833. [DOI] [PubMed] [Google Scholar]
- Walther A, and Wendland J (2004). Polarized hyphal growth in Candida albicans requires the Wiskott-Aldrich syndrome protein homolog Wal1p. Eukaryot Cell 3, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warschawski DE, Arnold AA, Beaugrand M, Gravel A, Chartrand E, and Marcotte I (2011). Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim Biophys Acta-Biomembr 1808, 1957–1974. [DOI] [PubMed] [Google Scholar]
- Wieprecht T, Dathe M, Beyermann M, Krause E, Maloy WL, MacDonald DL, and Bienert M (1997). Peptide hydrophobicity controls the activity and selectivity of magainin 2 amide in interaction with membranes. Biochemistry 36, 6124–6132. [DOI] [PubMed] [Google Scholar]
- Wieprecht T, Dathe M, Schumann M, Krause E, Beyermann M, and Bienert M (1996). Conformational and functional study of magainin 2 in model membrane environments using the new approach of systematic double-D-amino acid replacement. Biochemistry 35, 10844–10853. [DOI] [PubMed] [Google Scholar]
- Wilson KJ, Honegger A, Stotzel RP, and Hughes GJ (1981). The Behavior of Peptides on Reverse-Phase Supports During High-Pressure Liquid-Chromatograpy. Biochem J 199, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Hong T, Waring A, Lehrer RI, and Hong M (2002). Solid-State NMR Investigations of Peptide-Lipid Interaction and Orientation of a -Sheet Antimicrobial Peptide, Protegrin. Biochemistry 41, 9852–9862. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Huster D, Waring A, Lehrer RI, Kearney W, Tack BF, and Hong M (2001). Orientation and dynamics of an antimicrobial peptide in the lipid bilayer by solid-state NMR spectroscopy. Biophys J 81, 2203–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Harroun TA, Heller WT, Weiss TM, and Huang HW (1998). Neutron off-plane scattering of aligned membranes. I. Method of measurement. Biophys J 75, 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Harroun TA, Weiss TM, Ding L, and Huang HW (2001). Barrel-stave model or toroidal model? A case study on melittin pores. Biophys J 81, 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman MR, and Yount NY (2003). Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 55, 27–55. [DOI] [PubMed] [Google Scholar]
- Yonezawa A, Kuwahara J, Fujii N, and Sugiura Y (1992). Binding of Tachyplesin-I to DNA revealed by footprinting analysis - significant contribution of secondary structure to DNA binding and implication for biological action. Biochemistry 31, 2998–3004. [DOI] [PubMed] [Google Scholar]
- Zasloff M (2002). Antimicrobial peptides of multicellular organisms. Nature 415, 389–395. [DOI] [PubMed] [Google Scholar]
- Zhou L, Narsimhan G, Wu XY, and Du FP (2014). Pore formation in 1,2-dimyristoyl-sn-glycero-3-phosphocholine/cholesterol mixed bilayers by low concentrations of antimicrobial peptide melittin. Colloid Surf B-Biointerfaces 123, 419–428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CLSM and SR-SIM Images were processed using Zeiss ZEN software (https://www.zeiss.com/).