

Abstract
The major barrier to eradicating human immunodeficiency virus-1 (HIV) infection is the generation and extended survival of HIV reservoirs. Thus, in order to eradicate HIV infection, it is essential to detect, quantify, and characterize circulating and tissue-associated viral reservoirs in infected individuals. Currently, PCR-based technologies and Quantitative Viral Outgrowth Assays (Q-VOA) are the gold standards to detect viral reservoirs. However, these methods are limited to detecting circulating viral reservoirs, and it has been shown that they misrepresent the size of the reservoirs, largely because they detect only one component of the HIV life cycle and are unable to detect viral reservoirs in tissues. Here, we described the use of multiple detection systems to identify integrated HIV DNA or viral mRNA and several HIV proteins in circulating and tissue reservoirs using improved staining and microscopy techniques. We believe that this imaging-based approach for detecting HIV reservoirs will lead to breakthroughs necessary to eradicate these reservoirs.
Keywords: AIDS, HIV, T cells, macrophages, transmission, QVOA
Introduction
HIV/AIDS represents a major public health concern worldwide with an estimated 36 million infected individuals worldwide and 1.2 million infected in the US. The introduction of anti-retroviral drugs has increased the lifespan of HIV-infected individuals but has not led to a cure. Our inability to eradicate HIV has been linked to the capacity of the HIV to persist in the body as viral reservoirs (Richman, Margolis et al. 2009, Siliciano and Greene 2011, Kimata, Rice et al. 2016, Wong and Yukl 2016). However, the size and localization of these viral reservoirs and the mechanisms of viral silencing and reactivation are still poorly understood (Cary, Fujinaga et al. 2016, Churchill, Deeks et al. 2016, Cihlar and Fordyce 2016, Wong and Yukl 2016).
A critical barrier for HIV eradication is that most available techniques to detect viral reservoirs (i.e. viral DNA and mRNA PCR, episomal DNA, and cell activation/ amplification techniques including Q-VOA [Quantitative Viral Outgrowth Assays] or TILDA [Tat/rev Induced Limiting Dilution Assay]) have significant limitations in terms of accuracy, precision, sensitivity, cost, timing, and requirement for large blood volumes from patients (Eriksson, Graf et al. 2013, Graf and O’Doherty 2013, Spina, Anderson et al. 2013, Strain and Richman 2013, Deere, Kauffman et al. 2014). In addition, most of these techniques can detect circulating viral reservoirs and a small pool of HIV-infected cells only upon reactivation (Costiniuk and Jenabian 2015, Banga, Procopio et al. 2016). However, large populations of latently HIV-infected cells, mostly present in tissues cannot be quantified, and it is likely that these tissue reservoirs generate a constant efflux of HIV-infected cells into the circulation. Furthermore, all the current techniques are based on detecting only one component of the viral life cycle (e.g. viral DNA, mRNA or viral proteins) resulting in false positive results to detect viral reservoirs (Hilldorfer, Cillo et al. 2012, Eriksson, Graf et al. 2013, Rouzioux and Richman 2013, Spina, Anderson et al. 2013, Deere, Kauffman et al. 2014). Finally, most infected cells contain replication- incompetent HIV, therefore, detecting a single viral component can lead to misinterpretation of the data and incorrect estimation of replication-competent viruses within the reservoir (Blankson, Persaud et al. 1999, Shen and Siliciano 2008, Eriksson, Graf et al. 2013, Churchill, Deeks et al. 2016).
Here, we describe a protocol for simultaneous detection of integrated HIV DNA, viral mRNA and HIV/host proteins using microscopy and imaging analysis. The described methods are based on cutting-edge staining and microscopy techniques that work with unprecedentedly high resolution, specificity, and sensitivity, to identify low amounts of integrated HIV DNA, HIV mRNA, several HIV proteins, and host cell markers in latently infected cells in vivo and in vitro (Rella, Ruel et al. 2014, Eugenin and Berman 2016). It is possible to detect both circulating and tissue-associated viral reservoirs in individuals with undetectable HIV replication using these techniques. Our platform can evaluate the expression of several viral components (e.g. viral integrated DNA or viral mRNA and several viral proteins) and cellular markers in a single test, significantly reducing potential ambiguities in interpreting the results. These innovations will guide efforts to map HIV reservoirs within the infected population.
Materials
REAGENTS
PNA, BNA, Stellaris, or RNAscope probes
PNA ISH detection kit (Dako, K5201)
0.5 M EDTA, pH 8.0 (Gibco, 15575-038)
Fish gelatin from cold water 45% (Sigma, G7765)
Albumin from bovine serum fraction V (Fisher, BP1605)
Horse serum (Sigma, H1138)
HIV-p24 antibody (National Institute of Health AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, Germantown, MD or Genetex, Catalog Number, GTX40278, Irvine, CA).
Rabbit anti-human CD3 (Abcam, ab5690)
Rat anti-human CD4 (Abcam, ab19641)
Rabbit anti-human Iba-1 (Wako, 019-19941)
Hydrophobic barrier pen (ACD bio, catalog number 310018)
Positive control probe (ACD bio, catalog number 313901)
Negative control probe (ACD bio, catalog number 310043)
EQUIPMENT
HybEZ hybridization system (HybEZ, 110V AC, Cat No. 310010)
HybEZ humidity control tray with lid (HybEZ, Cat. Number 310012)
Incubators (Lab Companion SI 600)
Confocal or epifluorescence microscope (Nikon A1, Japan or Leica SP8, Germany)
Imaging working stations (NIS elements, Nikon, Tokyo, Japan)
REAGENT SETUP BEFORE START THE MAIN PROTOCOL
Dissolving DNA and mRNA probes (testing, negative and positive control probes): Probes are re-suspended in formamide at a concentration of 100 μM.
Blocking solution: 1 ml 0.5 M EDTA, pH 8.0 (Gibco catalog # 5575-038), 100 μl fish gelatin from cold water 45% (Sigma #G7765), 0.1 g. albumin from bovine serum-immunoglobulin free (Sigma, A2058), 100 μl horse serum (Sigma, H1138), 5% human serum (Corning, CA) and 9 ml double distillated H20.
TBS buffer (Tris-Buffered Saline, pH 7.4): mix 50 mM Tris and 150 mM NaCl. Prepare fresh. Also comes in the PNA ISH kit (Dako).
Citrate buffer for antigen retrieval: 10 mM sodium citrate, 0.05% Tween 20, pH 6.0. Prepare fresh every time and do not store.
BASIC PROTOCOL
The protocol described below correspond to the sample preparation, HIV DNA, HIV mRNA, viral proteins, and cellular markers staining in this order.
Sample fixation
Tissues (humanized mice, monkeys, or human) and cells can be fixed in multiple fixatives including formalin, aldehydes, glutaraldehyde, ethanol, or acetone. The staining can be performed in tissues from paraffin blocks, cryostat or vibrotome tissue slices. Non-adherent cells can be pelleted or smeared, while tissue sectioning thickness must be chosen by the user. We recommend using 10-300μM sections to preserve signal-to-noise ratio derived from sample thickness, and also to provide a sufficient number of cells to be statistically representative of the reservoir population.
Positive and Negative Controls
For negative controls use scrambled probes+ control isotype antibodies+ DAPI (to detect unspecific binding of probes and antibodies)
Unstained tissue sections (to detect baseline auto-fluorescence)
Positive and negative controls. ACH-2 or OM-10 cells (NIH repository, Germantown, MD). These cell lines are a human lymphoid cell with only one integrated copy of HIV-1 DNA. As negative controls use uninfected tissues or uninfected PBMCs or HL-60 that correspond to the uninfected counterpart of ACH-2 and OM-10 cells, respectively.
Sample preparation
Rehydration Stage (Apply to all the potential samples described above)
**CRITICAL – All steps are to be performed at room temperature and all reagents should be prepared fresh. In case of using paraffin sections, remove extra wax and proceed to de-paraffinization.
1. Immerse slides containing tissues or cells into separate xylene solutions, 5 minutes each.
2. Immerse slides into two separate solutions containing 99% ethanol dissolved in distilled water (dH20), 3 minutes each.
3. Immerse slides into two separate solutions containing 96% ethanol dissolved in dH20, 3 minutes each.
4. Repeat the process described above with 90, 80, 70 and 50% ethanol.5.
5. Immerse slides into dH20 for at least 3 minutes.
6. Encircle the specimen with DAKO PAP Pen to reduce the reagent volume to cover the specimens.
7. Immerse slides into a solution of dH20 for 3 minutes
Pre-treatment
All steps are to be performed at room temperature unless otherwise stated.
8. Place slides in a humidity chamber to avoid evaporation of the reagents.
9. Dilute the needed amount of proteinase K. Tissue sections require 1:10 dilution of proteinase K diluted in TBS and incubated for 20-30 min. Cells in suspension or culture monolayers require 1:2000 dilution of proteinase K in TBS with 10 min incubation (This step may need further optimization depending the size and thickness of the sample).
10. Immerse slides into dH2O for 3 minutes
11. Immerse slides in 96% ethanol for ~ 10-30 seconds.
12. To inactivate Proteinase K activity, immerse the sections/cells into an ethanol series - 70%, 85%, 95% each for 3 minutes
13. Air dry the slides for ~ 5 minutes
14. Treat the samples, if necessary, to eliminate auto-fluorescence. Refer to “elimination of auto-fluorescence” in sections “critical parameters and troubleshooting” for suggestions.
DNA probe Hybridization
** Critical – All steps should be performed at the suggested temperatures. The use of different equipment may be required further optimization. It is essential to determine the best timing of incubation and temperatures for HIV DNA or RNA probe hybridization to the target. These variables depend on the tissue used and the size and thickness of the section. It is essential to use DNAse 1 or RNAase as negative controls to ensure proper binding of the probe. In addition, it is necessary to use internal positive controls to ensure proper probe binding. In our case, we used Alu repeat probes because every cell has multiple copies of Alu repeats.
15. Critical for cells only – Place slides in a preheated incubator adjusted to 80°C, then place slides in the dark for 30 minutes at room temperature. This step is important for cells only to enable a better penetration of the treatment.
16. Dilute probe stocks to 10 μM in TBS and cover the tissue/cells with the solution. Due to the low numbers of cells containing HIV genome, mRNA or protein in samples obtained from suppressed individuals, we recommend conjugating the HIV probes to biotin and/or tyramine to further amplify the signal.
Our system involves the detection of at least two HIV components as well as 2-3 cell markers. We used peptide-nucleic acid (PNAbio, Thousand Oaks, CA) probes for HIV DNA detection, Bridge Nucleic acid (BNA, Biosynthesis, Lewisville, TX), stellaris probes (Biosearch Technologies, Petaluma, CA) or RNA scope (RNAscope, Newark, CA) for HIV RNA detection and purified antibodies to detect HIV proteins or host markers. The combination of probes and their characteristic of chemical stability, Tm and hybridization affinity (http://www.biosyn.com/tew/bna-pna-lna-dna-comparison.aspx) enable to detect DNA and mRNA in the same reaction using sequential incubations. Depending on the combination used, the mix of fluorophores can change, but the protocol is consistent (see Table 1).
Table 1.
Probes information and conditions
| Probe | Probe sequence | Target | Hybridization temperature (°C) | Probe concentration | Conjugation | Additional immune detection |
|---|---|---|---|---|---|---|
| DNA-LTR (PNA) | CAT ATA AGC AGC TGC TTT TTG CCT GTA CTG GGT | HIV-LTR | 42° | 10μM | 5′-Biotin or direct conjugation to any color | Yes |
| DNA-Nef (PNA) | GCA GCT TCC TCA TTG ATG G | HIV-Nef | 42° | 10μM | 5′-Biotin or direct conjugation to any color | Yes |
| DNA-Alu (PNA) | GCC TCC CAA AGT GCT GGG ATT ACA G | Alu repeats | 42° | 10μM | 5′-Biotin or direct conjugation to any color | Yes |
| RNA-Gag- Pol (BNA) | Advanced Cell Diagnotics Catalog # 317691 | Gag- Pol mRNA | 40° | 1μM | 5′-Biotin or direct conjugation to any color | Yes |
| RNA-Stellaris probes | Biosearch technologies | Nef mRNA | 40° | See commercial information | See commercial information | Limited, single molecule detection, but analysis by confocal is limited |
17. Place slides in a pre-warmed humidity chamber at 42°C.
18. Incubate slides at 42°C for 30 minutes, then at 55°C for 1 hour.
DNA probe stringent wash
**Critical – make sure to preheat stringent wash prior to this step.
19. Preheat stringent wash working solution (diluted 1:60 in TBS) to 55°C in a water-bath.
20. Place slides in stringent wash solution for 25 minutes in an orbital shaker at 55°C
21. Place samples in a series of ethanol 70%, 85%, and 95% for 5 minutes each.
22. Equilibrate the slides to room temperature by a brief immersion in TBS for 10-30 seconds (avoid drying the samples from this point).
DNA Probe detection
Note: Before starting this section, review the selection of the dyes and amplification methods used to avoid any cross-reaction with antibody labelings such as biotin or tyramine systems (see details in (Eugenin 2014, Eugenin and Berman 2016, Rao, Eugenin et al. 2016)). For example, few copies of HIV integrated DNA are found in host cells in HIV suppressed individuals, therefore, a strong and stable fluorophore needs to be used. We suggest using far-red dyes such as Cy5 or Alexa647 or 700 for the HIV or Alu probes due to the low auto-fluorescence into these channels.
23. Place slides in a humidity chamber to avoid evaporation.
24. Add Texas Red-streptavidin (Molecular Probes, catalog # 521374, 1 μl into 700 μl of pure ethanol) or any other dye conjugated to a fluorophore directly to sample. Be sure to select a fluorophore that does not interfere with the Cy5 Alu repeat probes or your subsequent antibody labeling.
25. Incubate for 30 minutes to 2 hours at room temperature.
26. Wash 3 times with dH20.
27. To perform HIV mRNA after the HIV DNA staining follows the protocol described by the company for RNAscope 2.5 HD detection reagent. https://acdbio.com/search/site/%252A2.5%20hd%20detection%20reagent%252A/cms/product.
For RNA detection, RNAase-free conditions are necessary during the entire protocol. This protocol enables to detect low amounts of HIV mRNA that can be visualized in the red channel. We do not recommend to use a colorimetric determination because sometimes false positive is detected even in uninfected samples may be due to the accumulation of different pigments and artifacts. Instead, we recommend confirming positive staining using fluorescence microscopy and spectral detection to eliminate the probability of false positives (wavelengths 568-580 nm).
28. Develop the colorimetric method as described on the company website. The development of the mRNA viral signal needs to be low, do not overdevelop. Stop the reaction as soon the red color is observed.
29. Wash 3 times with dH20.
30. If the HIV DNA and HIV mRNA are the final stainings, mount the slides with Prolong Diamond Antifade Mount medium containing DAPI (Thermo-Fisher Scientific, Catalog Number, P36971).
This point only applies if immunofluorescence for protein host and viral detection is not performed. If immunofluorescence for host and viral proteins is performed processed with the next steps described below.
Blocking unspecific binding sites (the following steps are necessary for immunofluorescence for detection of host and viral proteins)
Note: Endogenous biotin, antigen retrieval, elimination of auto-fluorescence, and blocking unspecific binding sites is strongly recommended if immunofluorescence will be done (use of antibodies after the hybridization steps). For example, upon inflammation, cancer, and other damaging conditions, several tissues express or accumulate significant amounts of endogenous biotin (Bussolati, Gugliotta et al. 1997, Lu, Kashima et al. 2000, Fahmy, Woo et al. 2014).
31. Since our detection and signal amplification systems use biotin-related systems, it is necessary to eliminate the presence of endogenous biotin in cells or tissues using any biotin-blocking system (for example, DAKO catalog number X0590 or similar product).
32. Antigen retrieval: Incubate the slices in antigen retrieval solution (Advanced Cell Diagnostics, 30 minutes or homemade as described above) or in citrate buffer for 40 minutes at 95 °C, and then allow to cool down for 20 minutes.
33. Perform elimination of auto-fluorescence if required (see notes in critical “Parameters and Troubleshooting”).
34. To block unspecific binding sites (mainly due to the presence of Fc receptors on immune cells) a blocking solution is used. Incubate the samples with blocking solution at least 1-4 hours or overnight. Furthermore, since latently infected cells and activated immune cells express Fc receptors, we recommend to including a high concentration of human serum (2-3 %) in the blocking solution to compete for these binding sites (Beckton Dickinson and Milteny Biotech offer similar products for elimination of Fc related background). Pause point: Slides can be stored at 4°C for 24 to 72 hours (Note: caution, do not to let the blocking solution to dry out).
Immuno-staining
35. Add the proper diluted primary antibodies such as viral and cell markers to the samples, including, but not limited to HIV-p24, CD3, CD4, macrophage and dendritic cell markers. Cover the entire surface of the sample with the solution containing the diluted antibodies. The number of antigens used for simultaneous staining is limited by the microscope detection capabilities. Pause point: Slides can be stored at 4°C for 2-7 days.
36. Wash at least 3 times with TBS every 5 minutes to eliminate the unbound antibodies.
37. Add the secondary antibodies at the right dilution and incubate for at least 2-3 hours.
38. Wash the samples at least 3 washes with PBS or TBS every 5 minutes to eliminate the unbound antibodies.
39. Mount the slides with Prolong Diamond Antifade Mount medium containing DAPI.
** Critical point – Slides should be kept at 4°C and in the dark to minimized decay of the fluorescence.
40. Use epifluorescence or confocal microscopy to observe the samples (see details below).
Microscopy and data interpretation
Note: To identify the probes and antibody labeled signals we used a Nikon A1 or SP8 confocal microscope with spectral detection or a similar equipment to further narrow the wavelengths. However, an epifluorescence microscope also can be used.
** Important: due to the low numbers of HIV infected cells and the low signal generated by viral reservoirs, it is essential to perform an accurate analysis of the samples by comparing them to all the controls suggested (negative and positive controls).
-
41. Visualization and Analysis by microscopy. Identify areas with positive staining for HIV DNA or HIV proteins.
The microscope automatization enables the identification of areas positive for the different staining.
42. Examine the co-localization of these components with DAPI and Alu repeats or Nef DNA signals. Observation of cytoplasmic markers for HIV mRNA and viral proteins may occur after confidence in the HIV DNA probe and Alu repeat probe colocalized with the nucleus. HIV-DNA staining must co-localize with DAPI and Alu repeats. For example, we expect that Alu repeats will co-localize or be close to HIV Nef DNA and both will co-localize with DAPI. With HIV-RNA, expect most of the staining will not co-localize with Alu repeats or DAPI and will be present mainly in the cytoplasm.
43. Use software packages like NIS elements (Nikon, Japan) and other proprietary software to quantify the data. These automatized system allow the detection of small signals in large pieces of tissues or significant cell numbers.
44. Numbers of positive cells, cell populations with HIV components, as well as subcellular localization, are some potential quantifications.
TIME CONSIDERATIONS
Steps 1- 7, Rehydration stage, about 30 minutes
Steps 8-14, Pretreatment, about 40 minutes
Steps 15-19, DNA Hybridization, about 1 ½ hours
Steps 20-23, Wash, about 30 minutes
Steps 24-31, DNA probe detection, 30 minutes to 2 hours
Steps 32-35, Block and Immunofluorescence, 1 h to 2 days
Steps 36-41, Immunostaining, 4 hours to 2 days
Steps 42-45, Microscopy and data interpretation, 1-2 days
Critical Parameters and Troubleshooting
Step: 1-31 – Probes not labeling the correct location or lack of staining
Reason 1: Probe quality is extremely important as well as the solutions used. Any contaminated solution could ruin the staining. Use new solutions.
Reason 2: Proteinase K treatment is used to digest fixed proteins and increase probe access to DNA. However, over-digestion may lead to tissue damage and reduced signal intensity. Shorter times also can results in under digestion and low probe penetration into tissues and cells.
Reason 3: Probe concentration – higher probe concentrations may lead to the increased background.
Reason 4: Hybridization temperature – needs to be optimized for each probe. Here, we described the Alu and HIV Nef probes. However, probes to other genes such as LTR, Vif, Vpr, and tat also can be used with similar degree of success (see table 1 for details).
Reason 5: Tissue fixation– tissues have been stored for a long time or were not fixed properly
Steps 32-45: Elimination of Auto-fluorescence: As described above, check fixation and setting on the microscope.
Reason 1: There are several forms of auto-fluorescence that can be reduced or eliminated before staining with probes or antibodies to increase the sensitivity of the system. Natural auto-fluorescence is due to flavins, porphyrins, and chlorophyll (mostly in plants). The main problem with these endogenous fluorescent compounds present in cells and tissues is that, during sectioning and solvent treatments, they become redistributed, resulting in background fluorescence. However, several alternatives or pre-treatments can be used to partially eliminate this problem. For instance, samples can be treated with Sudan Back (0.3% in 70% ethanol) stirred in the dark for 2 hours to reduce or eliminate auto-fluorescence produced by lipofuscins without the necessity for further adjustment. Lipofuscins are fine yellow-brown pigment granules composed of lipid-containing residues of lysosomal digestion and auto-fluoresce in several channels.
Another source of auto-fluorescence is related to elastin and collagen, which are mainly present in blood vessels. Elastin contains several potential fluorophores that upon fixation or cross-linking of tricarboxylic amino acid with pyridinium rings become fluorescent (Deyl, Macek et al. 1980, Deyl, Horakova et al. 1981). Normally, detection of these products is minimal in small vessels, but in large vessels, it is a significant problem. Incubation of these tissues in 0.5% pontamine sky blue and 6.6′-[(3, 3′-dimethoxy[1,1′-biphenyl]-4,4′-diyl)bis(azo)]bis[4-aminuteso-5-hydroxy-1,3-naphthalenedisulfonic acid], tetra-sodium salt dissolved in 50 mM Tris buffer, pH 7.5, before mounting the samples reduces most of the auto-fluorescence. However, the use of both compounds requires calibration, because pontamine sky blue fluoresces in the red channel. Thus, if these compounds are used, it is optimal to avoid using fluorophores in the red channel.
In most samples, cells and tissues are fixed with aldehydes such as glutaraldehyde, formaldehyde, and formalin. These aldehydes react with amines and proteins to generate fluorescent products, especially in samples incubated in fixatives for a long time. To reduce this kind of auto-fluorescence, incubate the tissues or cells in a solution of fresh sodium borohydride (1 mg/ml dissolved in PBS, pH 7.5, and prepared on ice) for 15 minutes. After this process, wash the samples in PBS 3 times and discard the leftover sodium borohydride (do not store or re-use). However, all these types of auto-fluorescence are diffuse and are extremely different than the specific fluorescence produced by the HIV DNA and HIV mRNA probes.
Reason 2: Proteinase K – the use of proteinase K is essential to allow access to the probes and antibodies to the selected tissues or cells; however, over-incubation results in the total dissolution of the samples.
Reason 3: Finally, the use of alternative probes – an alternative probe for tissue and cell staining is TelC-Cy3 for pan centromere. This corresponds to a widely used probe to detect chromosomes and nuclear DNA. This probe is mostly used to detect human epidermal growth factor receptor-related -2 (HER2) detection for several types of cancers.
Limitations of Protocol
- Viral mutations: Mutations or deletions in the areas of the viral DNA analyzed reduce or eliminate binding of the probes. In this case sequencing of the virus is required to adjust the probes or change the annealing temperature.
- Degradation of RNA/DNA: As discussed above, this issue is critical. Depending on the fixative used to initially prepare the sample after collection, most host and viral RNA and DNA sequences only last for a couple of months or several years.
- Cell numbers: It is essential to quantify millions of cells to identify the viral reservoirs that are in low abundance. Thus, samples with few cells may not accurately reflect the identity and abundance of viral reservoirs. Most protocols involve the use of thicker tissue sections, 10 to 300 μm to collect enough data and cell numbers.
ANTICIPATED RESULTS
- Detection of HIV-DNA in one cell among millions of uninfected cells. We are able to quantify greater numbers of cells as well as maintain the 3D structure of the tissue by using thicker sections (10 to 300 μm, in contrast to 5-7 μm tissue sections used in most studies). The described techniques allow the detection of a single copy of integrated HIV-DNA in one HIV infected cell among millions of uninfected cells. These techniques are based on those used in cytogenetic clinical analysis aimed at detecting specific chromosome sequences or mRNAs (Fox, Kotler et al. 1989, Webb 2000). The efficiency of analysis depends on microscope speed and available software.
For HIV DNA and HIV protein detection: We used several types of human tissues, including biopsies and necropsies from vaginal, lymph nodes, spleen, urethra, and brains, obtained from uninfected and HIV infected individuals who, according to clinical assays, exhibited undetectable viral replication for ≥5 years. We were able to detect HIV integrated DNA (HIV Nef and LTR DNA) in all cases analyzed (see Table 1). All individual samples analyzed have a threshold of 20 copies/ml as determined by COBAS Roche Amplicor v 1.5, a widely used commercial method to detect HIV mRNA in fluids. No signal was detected in uninfected tissues. As our system is combined with detection of not only HIV-DNA but also HIV proteins and cellular markers, we are able to detect whether viral replication is occurring and which cell type is producing the virus (see Figure 1 and Table 2). Six examples obtained from PBMCs isolated from 6 different individuals without detectable HIV replication are shown (Fig. 1). Arrows indicate cells with integrated HIV DNA and arrowheads indicate cells expressing HIV-p24. Depending of the patient analyzed, 1.2×107 to 1.6×109 cells were required to identify cells containing integrated HIV DNA and/or producing HIV-p24 protein. Thus, the pictures in Fig. 1 correspond to a representative picture of thousands of field analyzed that were negative for HIV integrated DNA. For patient 2, no cells were detected despite the analysis of 1.2×107 cells.
Figure 1.
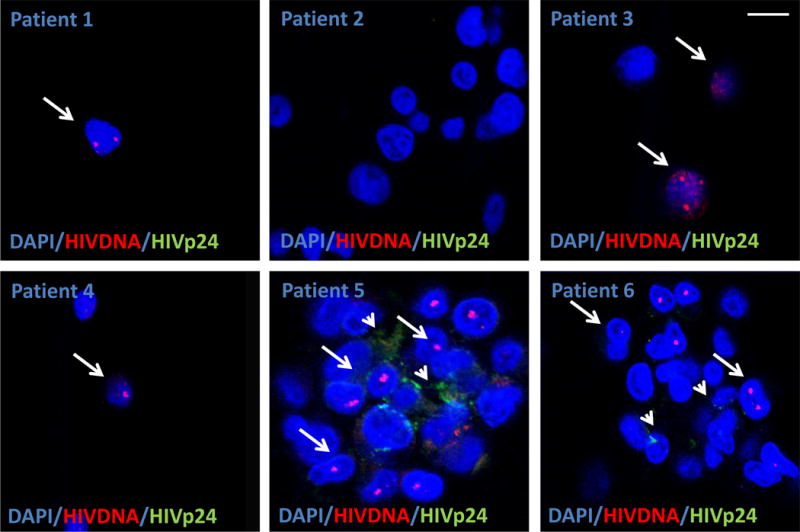
Detection of HIV Nef DNA and HIV-p24 protein in PBMCs smears obtained from HIV infected individuals with no viral replication detected by at least 5 years. Six representative images of areas with HIV Nef DNA from six different individuals with undetectable replication. Using the technique described, we are able to detect a single copy of HIV integrated DNA (arrows) in non- replicating CD4+ T lymphocytes as determined by ELISA and PCR (>20 copies/ml). To demonstrate that these sequences of integrated DNA are productive, we stained for HIV-p24 (arrowheads). The merged picture shows the colocalization of nuclei (DAPI, blue staining to quantify a total number of cells), HIV DNA (Red nuclear staining), and HIV protein (HIV-p24, green staining). Note that not all cells with inserted DNA produce HIV-p24 protein. Note that in patient 2, we are unable to detect any HIV Nef DNA despite that we analyzed 1.2×107 cells. No positive signal was detected in uninfected individuals with HIV-DNA or HIV-p24 protein. Each picture corresponds just to the positive cells in thousands of negative fields. Most of the circulating cells with HIV Nef DNA were CD4 positive cells. Arrows denote cells with integrated HIV Nef DNA and the arrowhead indicates placed where HIV-p24 is accumulated.
Table 2.
Samples analyzed
| Samples | Peripheral HIV replication | ART status | HIV DNA | HIV RNA | HIV-p24 | Cell type positive for HIV |
|---|---|---|---|---|---|---|
| Blood | n=12, control(/br)n=15, HIV (4 no detected replication and 4 with detected replication) | Yes | (++) | (+) | (+) | CD3, CD4, and a small population of monocytes |
| Vaginal tissue | n=5, control(/br)n=3, HIV | Yes | (++) | (+) | (+) | CD3, CD4, and resident macrophages |
| Lymph nodes | n= 10, control(/br)n= 22, HIV | Yes | (++) | (+) | (+) | CD3, CD4, and a population of macrophages/dendritic cells |
| Brain(/br)HIV-p24 | n= 19, control(/br)n= 25, HIV | Yes | (+) | (+/−) | (+/−) | Macrophage/microglia and a small population of astrocytes |
Figure 2 shows staining for human primary macrophages after 24 hours of infection with HIVADA, which corresponds to ~1 cycle of infection. HIV DNA Nef staining was not detected in uninfected cells (Fig. 2, uninfected), but the significant insertion of HIV-DNA Nef was detected in cultures of macrophages infected with HIV only after 24 h post-infection (Fig. 2, HIV). We detected perfect colocalization between DAPI, Nef DNA probe, and Alu repeats (see Fig. 2, merge). Figure 3 shows the staining for HIV DNA Nef and the macrophage marker, Iba-1, Alu repeats and DAPI to identify HIV infected macrophages in vaginal tissue obtained from an individual with effective HIV replication suppression by years (Fig. 3). A summary of the data, as well as several examples, are presented in Table 2.
Figure 2.
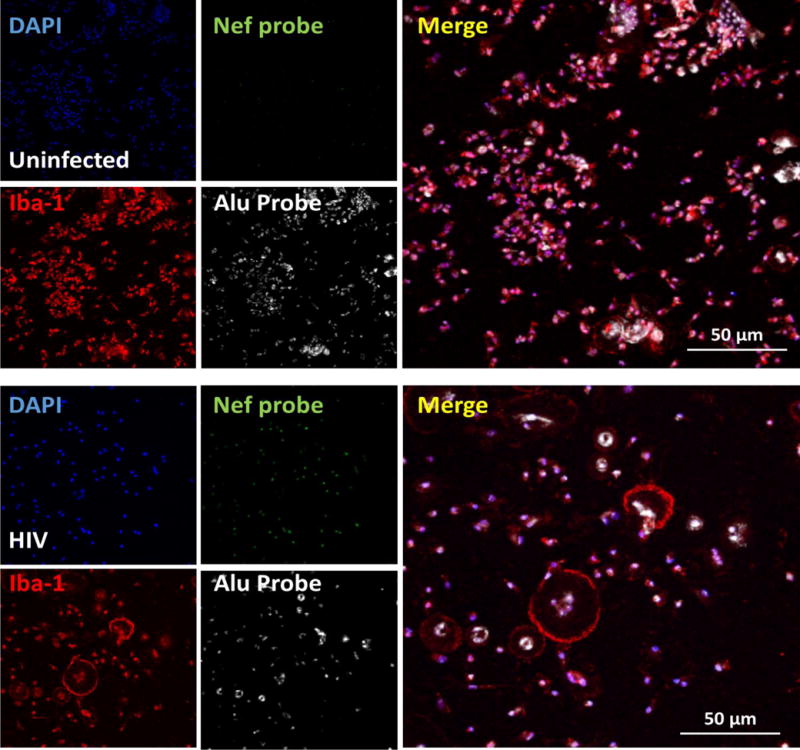
Detection of HIV Nef DNA, Alu repeats (host DNA), nucleus (DNA) and a macrophage marker (Iba-1) in primary cultures of human macrophages after 2 days post-HIV infection. Using the described technique, we are able to detect the first and maybe the second cycle of HIV replication in these primary cells. Similar results were found using viral reservoirs, latently infected T cells and macrophages. The pictures show the staining of human primary cultures of macrophages for nuclei (DAPI, blue staining to quantify a total number of cells), HIV Nef DNA (green nuclear staining), and a macrophage marker (red staining, Iba-1) in uninfected and HIV infected conditions. Note the perfect colocalization of the Alu repeat and Nef probe as well as DAPI in the HIV infected cultures. This point is essential to demonstrate insertion of HIV DNA into the host DNA.
Figure 3.
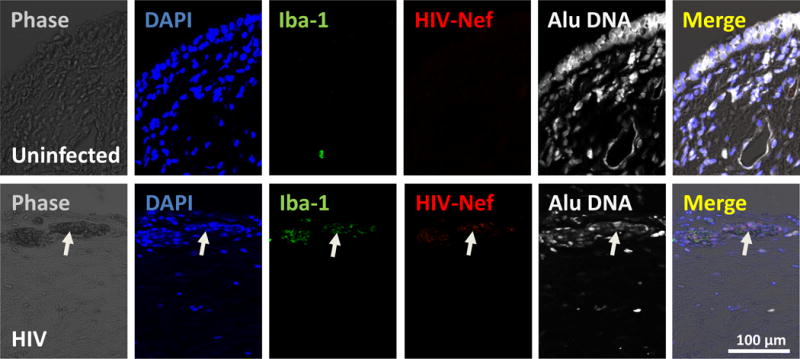
Detection of HIV Nef DNA in human vaginal tissue obtained from HIV infected individuals in ART for at least 15 years. Using the described techniques, we are able to detect few HIV infected cells in clinically suppressed individuals. Staining corresponds to nuclear (DAPI, blue staining), a macrophage marker (Iba-1, green staining), HIV Nef DNA (HIV Nef DNA, red staining), and Alu repeats (Alu sequences, white staining). Merge of all colors and phase contrast is also shown. Arrow represents a single positive area in a total area of 0.5 mm2 of the biopsy. Clearly, the staining shows a population of HIV infected macrophages and T cells that are positive for the virus. Thus, this technique allows the identification and quantification of HIV reservoirs in multiple tissues even in patient samples without reactivation.
Together, our data indicate that examining only circulating CD4+ T lymphocytes for viral components neglects HIV reservoirs in tissues, thus underestimating the extent and distribution of HIV reservoirs. The ability to detect viral DNA, mRNA, and protein in the same cell circumvents current challenges in the field of viral reservoirs, such as detection of “abortive” viral DNA sequences that do not produce mRNA or proteins. Furthermore, some viral reservoirs do not express mRNA or proteins unless they have been reactivated.
Summary of results obtained with different systems and sample types
Integrated HIV DNA
We analyzed purified PBMCs, brain sections, and lymph nodes obtained from HIV infected individuals undergoing effective antiretroviral treatment (ART) with no detectable viral replication. In fresh PBMCs, we detected HIV DNA positive cells among 106-107 uninfected cells (n=15, HIV infected samples, and 12 uninfected samples). In brain tissue sections, using 3D reconstruction and deconvolution, we observed 5.1±3.2 and 11.98±8.77 % of astrocytes and microglia/macrophages, respectively, were positive for HIV integrated DNA (n=25). In lymph nodes, we found that only 0.0005 ±0.00002 % (XXXX) of the CD4 T cells and a small population of dendritic cells and macrophages had integrated DNA (n=22, HIV infected samples, and 10 uninfected samples).
HIV mRNA
We found that most circulating HIV DNA positive cells were negative for gag mRNA. If we combine all the data of all patients analyzed, only 15.56 ±11.23 % cells with HIV DNA also contain HIV-gag mRNA in un-activated PBMCs obtained from HIV infected individuals with undetectable replication. In contrast, in tissues obtained from individuals with low to undetectable HIV replication (20 copies/ml), 6.48 ± 2.65 % of the HIV DNA positive cells are also positive for gag mRNA.
HIV protein
We detected several HIV proteins, including HIV-p24, Nef, RT, integrase, and Vif, in up to 0.78 ± 0.63 % of cells also positive for HIV DNA in the absence of reactivation. In addition, we detected several positive cells for HIV-p24 and integrase with no integrated HIV-DNA, suggesting that some cells in circulation can take up viral proteins without viral integration. We hypothesize that these HIV-p24 and integrase positive cells had taken up these proteins from dead cells, exosomes or proteins released by nearby tissue viral reservoirs. Our immuno-histological methods can be used to can detect several viral markers (HIV-DNA, viral mRNA, and several HIV proteins) in various populations of cells, such as those positive for CD4, Iba-1 (a macrophage marker), DC-SIGN (a dendritic cell marker), and GFAP (an astrocyte marker), depending on the tissue analyzed.
In summary, some key features of our detection system are: 1) High sensitivity, with identification of one copy of integrated HIV DNA within one cell among millions (106-109) of negative cells, 1-3 viral mRNAs and 3-5 complexes of viral proteins per cell; 2) High accuracy, with no signal detection in uninfected samples, auto-fluorescence or in samples infected with other viruses; 3) High reproducibility; 4) No requirement for cell activation or HIV amplification; 5) Simultaneous use of multiple viral markers, such as viral DNA, viral mRNA and viral protein in the same sample in addition to cellular and molecular host markers; 6) Cost effectiveness; 7) Time effectiveness: results can be obtained in 1 to 2 days; 8) Compatibility with a variety of samples types, including circulating cells as well as tissues; 9) Flexibility: different mutated DNA sequences can be assayed by altering probe sequences; 10) Low cell numbers/blood volumes required for testing; 11) Adaptability to high throughput assays using robotic improvements and algorithms for automated detection;12) Combinability with several cellular/inflammatory/viral markers; 13) Easy scalability for clinical analysis; 14) Isolation of viral reservoirs by using laser capture microscopy using the coordinates identified by confocal imaging. Our innovation in detecting viral reservoirs in blood and tissues will expand research into tissue-associated viral reservoirs and provide novel insights to help eradicate these reservoirs in HIV infected individuals.
Significance Statement.
The major barrier to eradicate HIV infection is the generation and extended survival of HIV reservoirs in several cell types and tissues. An additional complication is the poor detection and quantification of these reservoirs by using PCR and cell culture amplification-based techniques, which are controversial and do not represent the size of the circulating reservoirs. Our protocol is designed to detect, quantify and localize viral reservoirs using new and improved imaging and staining technologies to simultaneously detect viral DNA, viral mRNA, and viral proteins in vivo and in vitro.
Acknowledgments
We would like to thank National NeuroAIDS Tissue Consortium (NNTC) and CNS HIV Anti-Retroviral Therapy Effects Research (CHARTER) for providing all samples. This work was funded by The National Institute of Mental Health grant, MH096625, the National Institute of Neurological Disorders and Stroke, NS105584, and UTMB internal funding (to E.A.E).
Footnotes
AUTHOR CONTRIBUTIONS. L.P., N.R., P.C., M.B., S.M., and EAE performed the experiments. C.S., S.M., C.V., M.B., S.M., and EAE established material collection, preparation and microscope settings.
COMPETING FINANCIAL INTERESTS. The authors declare no competing financial interests.
References
- Banga R, Procopio FA, Perreau M. Current approaches to assess HIV-1 persistence. Curr Opin HIV AIDS. 2016;11(4):424–431. doi: 10.1097/COH.0000000000000282. [DOI] [PubMed] [Google Scholar]
- Blankson J, Persaud D, Siliciano RF. Latent reservoirs for HIV-1. Curr Opin Infect Dis. 1999;12(1):5–11. doi: 10.1097/00001432-199902000-00002. [DOI] [PubMed] [Google Scholar]
- Bussolati G, Gugliotta P, Volante M, Pace M, Papotti M. Retrieved endogenous biotin: a novel marker and a potential pitfall in diagnostic immunohistochemistry. Histopathology. 1997;31(5):400–407. doi: 10.1046/j.1365-2559.1997.3020895.x. [DOI] [PubMed] [Google Scholar]
- Cary DC, Fujinaga K, Peterlin BM. Molecular mechanisms of HIV latency. J Clin Invest. 2016;126(2):448–454. doi: 10.1172/JCI80565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, Swanstrom R. HIV reservoirs: what, where and how to target them. Nat Rev Microbiol. 2016;14(1):55–60. doi: 10.1038/nrmicro.2015.5. [DOI] [PubMed] [Google Scholar]
- Cihlar T, Fordyce M. Current status and prospects of HIV treatment. Curr Opin Virol. 2016;18:50–56. doi: 10.1016/j.coviro.2016.03.004. [DOI] [PubMed] [Google Scholar]
- Costiniuk CT, Jenabian MA. HIV reservoir dynamics in the face of highly active antiretroviral therapy. AIDS Patient Care STDS. 2015;29(2):55–68. doi: 10.1089/apc.2014.0173. [DOI] [PubMed] [Google Scholar]
- Deere JD, Kauffman RC, Cannavo E, Higgins J, Villalobos A, Adamson L, Schinazi RF, Luciw PA, North TW. Analysis of Multiply Spliced Transcripts in Lymphoid Tissue Reservoirs of Rhesus Macaques Infected with RT-SHIV during HAART. PLoS One. 2014;9(2):e87914. doi: 10.1371/journal.pone.0087914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyl Z, Horakova M, Vancikova O. Changes in pyridinoline content of elastin during ontogeny. Mech Ageing Dev. 1981;17(4):321–325. doi: 10.1016/0047-6374(81)90050-6. [DOI] [PubMed] [Google Scholar]
- Deyl Z, Macek K, Adam M, Vancikova O. Studies on the chemical nature of elastin fluorescence. Biochim Biophys Acta. 1980;625(2):248–254. doi: 10.1016/0005-2795(80)90288-3. [DOI] [PubMed] [Google Scholar]
- Eriksson S, Graf EH, Dahl V, Strain MC, Yukl SA, Lysenko ES, Bosch RJ, Lai J, Chioma S, Emad F, Abdel-Mohsen M, Hoh R, Hecht F, Hunt P, Somsouk M, Wong J, Johnston R, Siliciano RF, Richman DD, O’Doherty U, Palmer S, Deeks SG, Siliciano JD. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 2013;9(2):e1003174. doi: 10.1371/journal.ppat.1003174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Berman JW. Improved Methods to Detect Low Levels of HIV Using Antibody-Based Technologies. Methods Mol Biol. 2016;1354:265–279. doi: 10.1007/978-1-4939-3046-3_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahmy N, Woo M, Alameldin M, Lee JK, MacDonald K, Goneau LW, Cadieux P, Burton J, Pautler S. Endogenous biotin expression in renal and testicular tumours and literature review. Can Urol Assoc J. 2014;8(7-8):268–272. doi: 10.5489/cuaj.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CH, Kotler D, Tierney A, Wilson CS, Fauci AS. Detection of HIV-1 RNA in the lamina propria of patients with AIDS and gastrointestinal disease. J Infect Dis. 1989;159(3):467–471. doi: 10.1093/infdis/159.3.467. [DOI] [PubMed] [Google Scholar]
- Graf EH, O’Doherty U. Quantitation of integrated proviral DNA in viral reservoirs. Curr Opin HIV AIDS. 2013;8(2):100–105. doi: 10.1097/COH.0b013e32835d8132. [DOI] [PubMed] [Google Scholar]
- Hilldorfer BB, Cillo AR, Besson GJ, Bedison MA, Mellors JW. New tools for quantifying HIV-1 reservoirs: plasma RNA single copy assays and beyond. Curr HIV/AIDS Rep. 2012;9(1):91–100. doi: 10.1007/s11904-011-0104-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata JT, Rice AP, Wang J. Challenges and strategies for the eradication of the HIV reservoir. Curr Opin Immunol. 2016;42:65–70. doi: 10.1016/j.coi.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CS, Kashima K, Daa T, Yokoyama S, Yanagisawa S, Nakayama I. Immunohistochemical study of the distribution of endogenous biotin and biotin-binding enzymes in ductal structures of salivary gland tumours. J Oral Pathol Med. 2000;29(9):445–451. doi: 10.1034/j.1600-0714.2000.290905.x. [DOI] [PubMed] [Google Scholar]
- Rao VR, Eugenin EA, Prasad VR. Evaluating the Role of Viral Proteins in HIV-Mediated Neurotoxicity Using Primary Human Neuronal Cultures. Methods Mol Biol. 2016;1354:367–376. doi: 10.1007/978-1-4939-3046-3_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rella CE, Ruel N, Eugenin EA. Development of imaging techniques to study the pathogenesis of biosafety level 2/3 infectious agents. Pathog Dis. 2014 doi: 10.1111/2049-632X.12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. The challenge of finding a cure for HIV infection. Science. 2009;323(5919):1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- Rouzioux C, Richman D. How to best measure HIV reservoirs? Curr Opin HIV AIDS. 2013;8(3):170–175. doi: 10.1097/COH.0b013e32835fc619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Siliciano RF. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J Allergy Clin Immunol. 2008;122(1):22–28. doi: 10.1016/j.jaci.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano RF, Greene WC. HIV latency. Cold Spring Harb Perspect Med. 2011;1(1):a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, Mau M, Ruelas D, Saleh S, Shirakawa K, Siliciano RF, Singhania A, Soto PC, Terry VH, Verdin E, Woelk C, Wooden S, Xing S, Planelles V. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013;9(12):e1003834. doi: 10.1371/journal.ppat.1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strain MC, Richman DD. New assays for monitoring residual HIV burden in effectively treated individuals. Curr Opin HIV AIDS. 2013;8(2):106–110. doi: 10.1097/COH.0b013e32835d811b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb GC. Radioactive in situ hybridization to animal chromosomes. Methods Mol Biol. 2000;123:29–50. doi: 10.1385/1-59259-677-0:29. [DOI] [PubMed] [Google Scholar]
- Wong JK, Yukl SA. Tissue reservoirs of HIV. Curr Opin HIV AIDS. 2016;11(4):362–370. doi: 10.1097/COH.0000000000000293. [DOI] [PMC free article] [PubMed] [Google Scholar]