

Summary
Molecules capable of directing changes to nucleic acid sequences are powerful tools for molecular biology and promising candidates for the therapeutic correction of disease-causing mutations. However, unwanted reactions at off target sites complicate their use. Here we report selective combinations of mutant editing enzyme and directing oligonucleotide. Mutations in human ADAR2 (adenosine deaminase acting on RNA 2) that introduce aromatic amino acids at position 488 reduce background RNA editing. This residue is juxtaposed to the nucleobase that pairs with the editing site adenine, suggesting a steric clash for the bulky mutants. Replacing this nucleobase with a hydrogen atom removes the clash and restores editing activity. A crystal structure of the E488Y mutant bound to abasic site-containing RNA shows the accommodation of the tyrosine side chain. Finally, we demonstrate directed RNA editing in vitro and in human cells using mutant ADAR2 proteins and modified guide RNAs with reduced off target activity.
Keywords: Site-directed RNA Editing, Bump-hole, ADAR, off-target sites, epitranscriptome
Graphical Abstract

eTOC blurb
Systems developed for genome and transcriptome editing have unwanted off-target reactions. Monteleone et al. used a bump-hole strategy to develop highly selective combinations of mutant ADARs and directing oligonucleotide. SDRE is shown in vitro and in human cells using mutant ADAR2 proteins and guide RNAs with reduced off-target activity.
Introduction
A variety of systems have been developed for directing reactions that change nucleic acid sequence, either in DNA (genome editing) or in RNA (transcriptome editing) (Montiel-Gonzalez et al., 2013; Montiel-Gonzalez et al., 2016; Nunez et al., 2016; Ran et al., 2013; Hsu et al., 2014; Cox et al., 2017; Gaudelli et al., 2017; Stafforst and Schneider, 2012). CRISPR-Cas-mediated selective cleavage of duplex DNA coupled with homology-directed repair with appropriately designed donor DNA fragments has become a popular approach for introducing specific sequence changes in genomes (Nunez et al., 2016; Ran et al., 2013; Hsu et al., 2014). Nucleotide-specific editors have been reported recently that use cleavage inactive Cas mutants fused to either cytidine or adenosine deaminase domains (Gaudelli et al., 2017; Komor et al., 2016). In complex with the appropriate single guide RNAs (sgRNAs), these proteins are capable of directing specific single nucleotide changes (C to T or A to G) in the genomes of bacteria, mammalian cells and mice (Gaudelli et al., 2017; Gao et al., 2018; Kim et al., 2017; Komor et al., 2016). Methods for directing the deamination of specific adenosines in RNA have also been described including a recent report of Cas13 fusion proteins capable of RNA editing for Programmable A to I replacement (REPAIR) (Cox et al., 2017; Hanswillemenke et al., 2015; Montiel-Gonzalez et al., 2016). This and other systems designed to direct RNA editing reactions use deaminase domains from the ADAR enzymes (Hanswillemenke et al., 2015; Montiel-Gonzalez et al., 2013; Montiel-Gonzalez et al., 2016; Cox et al., 2017; Stafforst and Schneider, 2012). ADARs (adenosine deaminases that act on RNA) are known to convert A to inosine (I) in duplex RNA (Bass, 2002; Bass and Weintraub, 1988; Goodman et al., 2012). Since I base pairs with C, it functions like G in cellular processes such as splicing, translation and reverse transcription (Figure 1A) (Bass, 2002; Nishikura, 2010). Among its many consequences on RNA function, ADAR-mediated A to I editing can alter miRNA recognition sites, redirect splicing and change the meaning of specific codons (Wang et al., 2013; Rueter et al., 1999; Yeo et al., 2010). The ability of ADARs to convert A to I has spurred efforts to develop new proteins capable of directing ADAR-catalyzed deamination to specific adenosines present in mRNAs because of disease-associated G to A mutations in the genome (Cox et al., 2017; Montiel-Gonzalez et al., 2013; Vallecillo-Viejo et al., 2018). However, in each of the reported directed RNA editing systems, off target activity is observed (Cox et al., 2017; Montiel-Gonzalez et al., 2013; Montiel-Gonzalez et al., 2016; Vallecillo-Viejo et al., 2018; Wettengel et al., 2017; Vogel et al., 2018). This is because ADAR catalytic domains are used for their deaminase activity and these domains can react with RNA substrates in the absence of a targeting domain (Zheng et al., 2017; Montiel-Gonzalez et al., 2013; Eifler et al., 2013; Hanswillemenke et al., 2015; Wang and Beal, 2016; Matthews et al., 2016; Phelps et al., 2015). Here we describe efforts to reshape an ADAR-RNA interface such that high levels of A to I editing activity are only observed at specific target sites thus reducing unwanted off target editing.
Figure 1.
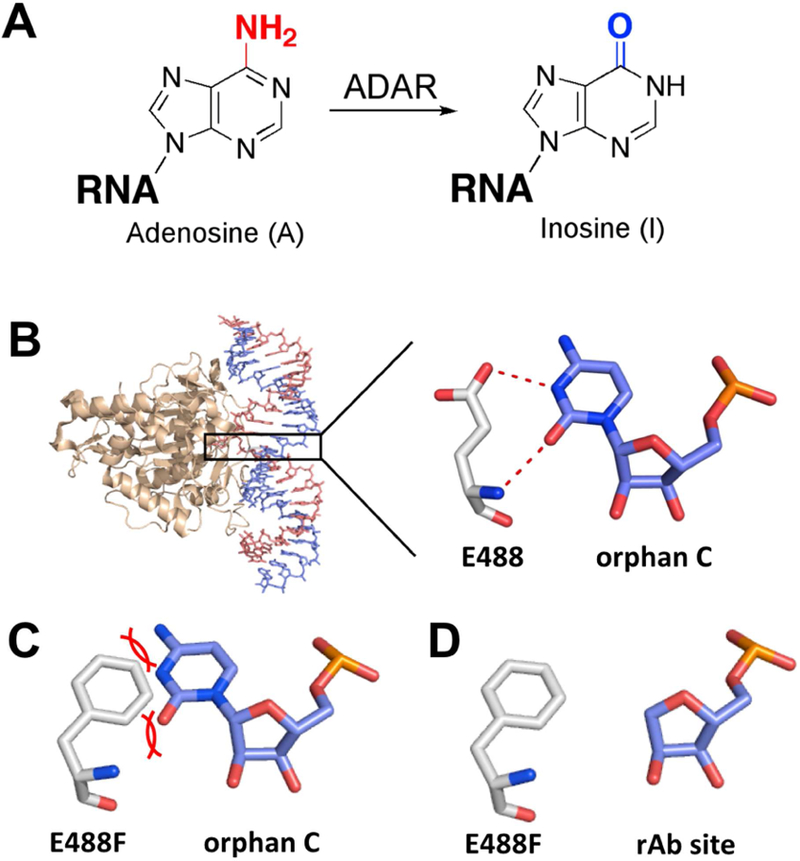
Bump-hole approach for orthogonal site-directed RNA editing with ADAR. (A) ADARs catalyze the hydrolytic deamination of adenosine to form the non-canonical nucleoside inosine. (B) Crystal structure of human ADAR2 deaminase domain bound to dsRNA substrate. ADAR flipping loop contains intercalating E488 residue, which hydrogen bonds with orphan base cytidine. (C) Mutation of 488 residue to phenylalanine (E488F) suggests steric clash (“bump”) with orphan base cytidine. (D) Incorporation of a reduced abasic site (rAb) relieves steric clash caused by E488F and acts as the “hole.”
Results
A bump-hole approach to an orthogonal A to I editing system
Our recent structural studies of the ADAR2 deaminase domain bound to RNA revealed a loop on the protein surface involved in the base flipping step of the deamination reaction (i.e. flipping loop, S486-T490) (Matthews et al., 2016). The side chain of E488 inserts into the space vacated when the reacting nucleotide occupies the deaminase active site, making direct contact to the “orphan base”; a cytosine or uracil in substrate RNAs bearing either A-C or A-U pairs at the site of reaction (Figure 1B). Interestingly, in a screen for active and inactive mutants of human ADAR2, Kuttan and Bass identified E488F and E488Y in the pool of inactive clones (Kuttan and Bass, 2012). The low activity of these mutants with typical RNA substrates is explained by the steric clash that would occur between the side chains of these large, aromatic amino acids and the orphan base (Figure 1C). However, we reasoned these mutants might be active if additional space were created in the complex to accommodate their large side chains. Furthermore, if the clash could be relieved, the ability of the aromatic side chains to engage in π stacking interactions in the RNA duplex might be advantageous. To test this idea, we overexpressed and purified the E488F and E488Y mutant ADAR2 deaminase domains. In addition, we prepared a duplex RNA substrate bearing an A-C mismatch at the target site (i.e. the optimal pairing interaction for a wild type ADAR reaction) and a substrate in which the target adenosine was paired with a reduced abasic (rAb) site (Figure 1D) (Figure 2A) (Lehmann and Bass, 2000; Wong et al., 2001). The duplex sequence used here is derived from that found near a known editing site in the human glioma factor 1 mRNA and was used in our earlier structural studies (Matthews et al., 2016). The abasic site, lacking a nucleobase, provides the “hole” to accommodate the “bump” of the E488F and E488Y mutant proteins (Alaimo et al., 2001; Belshaw et al., 1995). We evaluated the effects of these structural changes by measuring deamination rate constants under single turnover conditions (Zheng et al., 2017; Phelps et al., 2015). The combination of the wild type ADAR2 deaminase domain and an RNA substrate with an A-C mismatch led to a deamination kobs = 0.7 ± 0.2 min−1, whereas this protein deaminated the substrate with an A-rAb combination nearly 100-fold more slowly (kobs = 8.9 × 10−3 ± 0.4 × 10−3 min−1) (Figure 2B) (Table 1). This result clearly demonstrates the importance of the E488-orphan base interaction for the reaction of the wild type enzyme. Importantly, the E488F mutant shows the opposite reactivity preference with the A-rAb substrate converted to product with a kobs = 0.09 ± 0.02 min−1 and the A-C substrate reacting 71-fold more slowly at a kobs = 1.4 × 10−3 ± 0.3 × 10−3 min−1 (Figure 2C) (Table 1). The E488Y mutant also preferentially reacts with the A-rAb substrate (kobs > 3 min−1) in comparison to the A-C substrate RNA (kobs = 0.17 ± 0.04 min−1) (Figure 2D) (Table 1). Interestingly, the E488Y mutant was substantially more reactive than the E488F mutant on both RNA substrates. The E488Y mutant also had at least a five-fold higher rate of editing on the A-rAb substrate compared to the wild type protein on the A-C mismatch in an optimal flanking sequence for ADARs.
Figure 2.
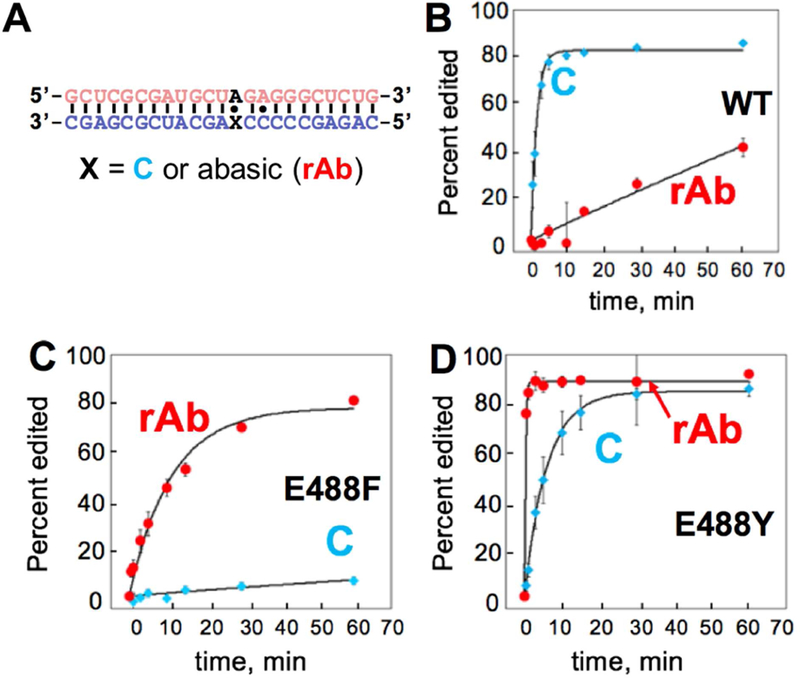
ADAR deamination kinetics. (A) Sequence of deamination substrate. Target site is bolded A in black. X is orphan base of target site either containing cytidine or reduced abasic site to form cytidine substrate or reduced abasic substrate, respectively. (B-D) Comparison of deamination product versus time with 300 nM hADAR2-D WT, hADAR2-D E488F and hADAR2-D E488Y, respectively. Deamination kinetics shown in red indicates reduced abasic substrate (rAb) and shown in blue indicates cytidine substrate (C). Error bars, s.d. (n = 3 technical replicates).
Table 1.
Kinetic parameters for the deamination of cytidine substrate and reduced abasic substrate with hADAR2 deaminase domains of WT, E488F and E488Y.
| Enzyme | Orphan | Rate constant, min−1 | Selectivity for abasic sitea |
|---|---|---|---|
| ADAR2-D WT | C | 0.7 ± 0.2 | 0.01 |
| ADAR2-D WT | rAb | 8.9 × 10−3 ± 0.4 × 10−3 | - |
| ADAR2-D E488F | C | 1.4 × 10−3 ± 0.3 × 10−3 | 71 |
| ADAR2-D E488F | rAb | 0.09 ± 0.02 | - |
| ADAR2-D E488Y | C | 0.17 ± 0.04 | > 18 |
| ADAR2-D E488Y | rAb | > 3 | - |
Selectivity reported is ratio kobs for rAb orphan / kobs for C orphan.
High-resolution structure of an engineered protein-RNA interface.
The high editing rate for the E488Y/A-rAb combination prompted us to characterize this interaction further by x-ray crystallography. For this purpose, we generated a duplex RNA with the nucleoside analog 8-azanebularine in place of the reactive adenosine paired with an rAb site (Figure 3A). We had previously shown that the 8-azanebularine modification allows one to trap the ADAR2-RNA complex in the base-flipped conformation for crystallography (Matthews et al., 2016). Crystals of hADAR2-D E488Y/A-rAb combination were grown at room temperature by the sitting drop vapor diffusion method and took several days to form. X-ray diffraction data were collected to 2.55 Å resolution. The structure was s olved by molecular replacement using the previously determined structure of ADAR2 deaminase domain complexed with Gli1 RNA (PDBID: 5ED2) (Matthews et al., 2016), and refined to a final Rfactor/Rfree of 19.0 and 25.5% respectively. (Table S1).
Figure 3.

Crystal structure of hADAR2-D E488Y bound to reduced abasic containing RNA substrate. (A) Duplex RNA for crystallization. Gli1 duplex has sequence surrounding the human Gli1-mRNA editing site. Adenosine analog 8-azanebularine (N) is across from reduced abasic site (rAb). N allows for trapping of protein-RNA complex in base flipped conformation. (B) Intercalation of Y488 from the minor groove side of RNA duplex in base-flipped conformation. Potential hydrogen bonding at N4 amine of C10’. (C) Orphan base, reduced abasic site (rAb), maintains RNA backbone found in other ADAR-RNA structures. Side chain of R510 ion-pairs with the 3’-phosphodiester of orphaned base, rAb11’. See also Table S1 and Figure S2.
As seen in the previous structures, the side chain of residue 488 in the base flipping loop intercalates into the RNA substrate (Matthews et al., 2016). Here, the tyrosine side chain fills the void created by the flipped-out base and the abasic site and stacks between the editing site flanking base pairs. However, while the electron density clearly defined the benzyl-ring of Y488, no density was observed for the hydroxyl group (Figure S2A). The presence of the E488Y mutant in the crystals was confirmed by mass spectrometry (Figure S1). Therefore, we reasoned that the mutant base-flipping loop approaching a “hole” containing substrate would likely adopt multiple conformations. Accordingly, Y488 was modeled in two conformations where the benzyl rings occupy the well-defined electron density, but the hydroxyl groups point to different positions, thus averaging out the density (Figure S2A–B). In one conformation, the Y488 side chain adopts a more “outward” position relative to the central axis of the double helix whereas in the other conformation, Y488 is more “inward”. In the “outward” conformation of Y488, the Y488 hydroxyl group is in close proximity (2.7 Å) to the cytosine N4 amine of the G:C pair containing the 3’ G, suggesting potential for hydrogen bonding although the geometry is not ideal (Figure 3B). Also, since the residue 488 may be responsible for displacing the modified base, a more “outward” position could be more effective in preventing the premature return of the flipped-out base before deamination occurs. However, the side-chain rotamer for the “outward” position is extremely uncommon, according to the MolProbity rotamer probability distribution (Lovell et al., 2000). In the case of F488, the “outward” conformation would be less stable since there is no potential for hydrogen bonding to stabilize it. Thus, the likelihood of F488 adopting this conformation is lower, which would increase the chances of the flipped-out base returning prematurely to the double-helix. This is one possible explanation for why the E488Y mutant may be more active than E488F.
The plane of the tyrosine phenol is parallel to that of the flanking U-A pair containing the U on the 5’ side of the editing site and, thus, p stacking of the tyrosine appears best with this base pair. The plane of the G-C pair containing the 3’ G is at an angle resulting in poor π overlap with the Y488 side chain (Figure S2B). In addition, overlay with previous structures shows the protein backbone of the base flipping loop bearing E488Y shifts (~ 1.5 Å) to allow the tyrosin e side chain to more effectively fill the void created by the abasic site (Figure S2C). This clearly indicates flexibility in this loop to adjust positioning of the intercalating side chain in the presence of the reduced abasic site at the orphan base position. The reduced abasic site maintains the RNA backbone conformation found in previous structures and is stabilized by contact to the side chain of R510 (Figure 3C) (Matthews et al., 2016).
Duplex RNA substrate with two good target sequences provides an in vitro test for selectivity
The preferential reactivity of the bulky ADAR2 E488X mutants suggested ADAR reaction selectivity could be controlled for substrates with multiple reactive adenosines by the appropriate positioning of the rAb site in a guide strand. To test this idea, we generated a 152 nt RNA with two adenosines with similar flanking sequences separated by 18 nt. This RNA was hybridized to four different 46 nt guide strands with each target adenosine (site 1 or site 2) paired with either C or rAb (Figure 4A). With both adenosines paired with C, the ADAR2 deaminase domain deaminated site 1 and site 2 adenosines to a yield of 36 ± 3% and 37 ± 2 %, respectively (Figure 4C). Conversely, with both adenosines paired with C, the ADAR2 deaminase domain E488Y mutant deaminated the site 2 adenosine to a yield of 10 ± 2%, while the site 1 adenosine was not deaminated by this enzyme under these conditions (Figure 4B). However, when rAb was paired with the site 1 adenosine, the editing yield for the E488Y mutant rose from undetectable to 42 ± 4% while the site 2 editing yield remained largely unchanged at 11 ± 2% under these conditions. If, on the other hand, the rAb was paired with the site 2 adenosine instead of site 1, the editing yield at this site rose to 61 ± 9%. When both site 1 and site 2 adenosines were paired with rAb, the editing yields were 24 ± 5% and 55 ± 8%, respectively (Figure 4B). These results clearly show that the reaction of the E488Y ADAR2 deaminase domain mutant can be directed to different positions in a target RNA by pairing the desired site with the rAb nucleoside analog. We have also shown that the ADAR2 deaminase domain E488F and E488W mutants show similar selectivity and dependence on rAb positioning in the guide strand for the 152 nt RNA target (Figure S3) (Table S2). At this time, reasons for differences in reactivity of site 1 and site 2 when both are paired with either C or rAb are not known, but may arise from differences in the RNA substrate at the contact site for the 5’ binding loop of the deaminase domain (Matthews et al., 2016; Wang and Beal, 2016; Wang et al., 2018).
Figure 4.
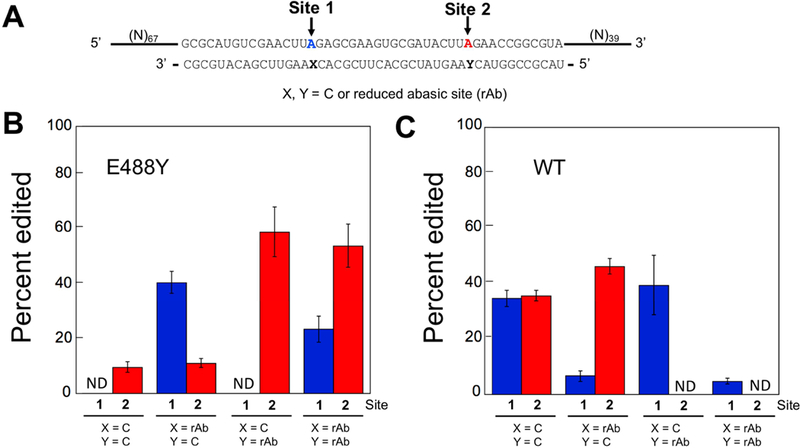
Selective editing on 152 nt RNA containing target site 1 and site 2 in optimal ADAR flanking sequence. (A) Partial sequence for four different substrates containing different combinations of cytidine or reduced abasic site at orphan base of target were tested with (B) 150 nM hADAR2-D E488Y and (C) 150 nM hADAR2-D for 15 min. Bars shown in blue and red represents editing at site 1 and site 2, respectively. X and Y are orphan base position of site 1 and site 2, respectively. Deamination of hADAR2-D E488W, hADAR2-D E488F and hADAR2-D can be found in Figure S3 and Table S2. ND = no detected editing. Error bars, s.d. (n = 3 technical replicates).
Selective oligonucleotide-directed editing in human cells with ADAR2 E488F, Y and W mutants
The studies described above suggested the combination of bulky mutations in human ADAR2 at position 488 and a guide strand bearing a rAb site opposite the targeted adenosine could be highly selective in directed RNA editing. To determine if the approach could work in living cells, we synthesized a guide oligonucleotide to direct editing by full length ADAR2 (with its two double stranded RNA binding domains (dsRBDs)) to an overexpressed target site representing a region of the 3’-UTR of β-actin mRNA (Figure S4) (Vogel et al., 2014; Vogel and Stafforst, 2014; Schneider et al., 2014). The guide RNA was designed to form a duplex containing an adenosine within a 5’-UAG-3’ sequence, the optimal flanking sequence for a human ADAR2 target (Figure 5A) (Li et al., 2009; Eifler et al., 2013; Matthews et al., 2016; Eggington et al., 2011). Opposite the targeted adenosine, the guide strand had either a cytidine or a rAb site. The guide RNA was additionally stabilized with 2’-O-methyl and phosphorothioate modifications, as previously described for directed editing in cells with chemically synthesized guide strands (Vogel et al., 2014; Schneider et al., 2014; Woolf et al., 1995). Expression of wild type human ADAR2 in HEK293T cells transfected with the guide designed to form an A-C mismatch led to 87 ± 8% editing at the target site (Figure 5B). Two other adenosines near the target site (off target 1 and off target 2, Figure 5A) were also edited under these conditions (55 ± 2% and 12 ± 2%, respectively). Expression of ADAR and transfection of the guide were both required to observe editing on this RNA (Figure S5A-D). Importantly, the E488F, E488Y and E488W mutants of ADAR2 induced high levels of target site editing (57 ± 4%, 68 ± 2% and 63 ± 2%, respectively) in the presence of the rAb guide with no editing detected at the two off target sites (Figure 5C–E, Table S3). This was not simply a result of lowering the overall ADAR activity since substantially lower levels of editing are observed with these mutants and the cytidine-containing guide (E488F, E488Y and E488W having editing levels of 21 ± 1 %, 31 ± 5% and 13 ± 2%, respectively) (Figures S6–S7, Table S4).
Figure 5.

Site-directed editing on an overexpressed target site representing a region of the 3’-UTR of β-actin RNA in HEK293T cells. (A) Partial sequence of target substrate and sequence of guide used. Bolded in green is target adenosine. Bolded in blue and grey are off-targets 1 and 2, respectively. All guide nucleotides are 2’-O-methyl modified except those bolded black which are unmodified ribonucleotides. Underlining indicates sites of phosphorothioate modification. X = cytidine (C) or reduced abasic site (rAb). (B) Percent editing at target and off-targets on overexpressed β-actin mRNA substrate and cytidine guide RNA with overexpression of hADAR2. (C-E) Percent editing at target and no detected editing (ND) at off-targets on overexpressed β-actin mRNA substrate and reduced abasic guide RNA with overexpression of hADAR2 E488F, hADAR2 E488Y and hADAR2 E488W, respectively. ND = no detected editing. Error bars, s.d. (n = 3 biological replicates). See also Figure S4, Figure S5 and Table S3.
We next tested the ability of the E488X mutant ADARs and modified guide RNAs to direct editing to specific adenosines present on endogenous mRNAs in human cells (Figure 6, Table S5). The transcripts chosen (RAB7A and β-actin) both contain adenosines in the 5’-UAG-3’ sequence context in their 3’-UTRs and both have been previously targeted for directed RNA editing (Wettengel et al., 2017). As discussed above, guide RNAs were designed to have a cytidine or a reduced abasic site across from the targeted adenosine and were further stabilized by 2’-O-methyl and phosphorothioate modifications. Under the conditions described above for targeting the overexpressed β-actin 3’ UTR fragment, no editing was observed on the endogenous β-actin mRNA. However, after optimization of the transfection of the ADAR encoding plasmid and guide RNA (see Method Details for conditions) targeted editing was observed on both the endogenous β-actin and RAB7A transcripts in HEK293T cells. Wild type ADAR2 in the presence of the RAB7A cytidine guide RNA led to an editing level of 41 ± 9 %, whereas overexpression of the E488F, E488Y and E488W ADAR2 mutants in the presence of the rAb guide RNA led to editing levels of 38 ± 3 %, 55 ± 5 % and 43 ± 1 %, respectively (Figure 6B). Thus, for this endogenous target, the ADAR2 E488X mutant / modified guide combinations led to on-target editing yields equivalent to (E488F and E488W) or superior to (E488Y) the wild type enzyme. In contrast, wild type ADAR2 edited the endogenous β-actin target in the presence of cytidine guide RNA to a level of 52 ± 2%, whereas the E488F, E488Y and E488W mutants with the reduced abasic guide RNA induced editing levels to an approximately two-fold lower level (26 ± 3%, 30 ± 1% and 25 ± 1 %, respectively) (Figure 6A). Since expression of the E488Y ADAR2 mutant in the presence of the RAB7A rAb guide led to the highest efficiency of on target editing, we evaluated off target editing under these conditions. Importantly, we found that expression of the E488Y mutant induced less efficient off-target editing in comparison to the wild type enzyme for several previously reported endogenous substrate RNAs for human ADAR2 (Figure 6C, Table S5). The sites analyzed included one that is edited by endogenous ADARs in HEK293T cells (Gli1) and five sites that are only edited in these cells with overexpressed ADAR (FLNA, TMEM63B, CYFIP2 and COG3). No editing was observed for FLNA and TMEM63B when E488Y was overexpressed while overexpression of the wild type ADAR2 clearly induced editing at these sites (FLNA 26 ± 1% and TMEM63B 37 ± 2%). For the Gli1 site, overexpression of wild type ADAR2 increased editing approximately 4-fold from 9 ± 6% to 38 ± 4%, whereas E488Y led to a 2-fold increase in editing to 17 ± 2%. Overexpression of wildtype ADAR2 increased editing on the CYFIP2 site from not detectable to 55 ± 4%, whereas E488Y caused only 18 ± 8%, editing at this site. Finally, at the COG3 sites, wild type ADAR2 increased editing from not detectable at both sites to 17 ± 1 % and 52 ± 3 %, while E488Y mutant had editing levels of 7.6 ± 0.4 % and 18 ± 4 %, respectively. Thus, for all six sites analyzed, a clear reduction in off target editing is observed for the E488Y mutant.
Figure 6.
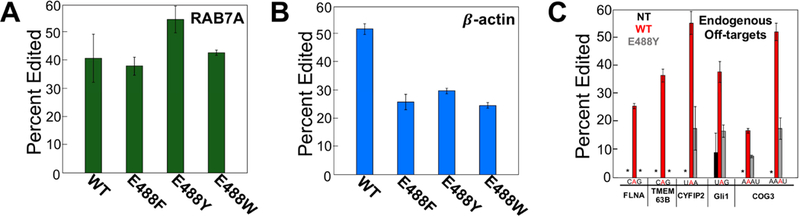
Directed editing on endogenous targets and observed endogenous off-targets. (A) Directed editing on endogenous 3’-UTR of RAB7A with overexpression of hADAR2 and cytidine guide RNA or overexpression of bulky mutants hADAR2 E488X (X = F, Y, W) and reduced abasic guide RNA. (B) Directed editing on endogenous 3’-UTR of β-actin with overexpression of hADAR2 and cytidine guide RNA or overexpression of bulky mutants hADAR2 E488X (X = F, Y, W) and reduced abasic guide RNA. (C) Percent editing on various endogenous off-target transcripts: FLNA, TMEM63B, CYFIP2, Gli1 and COG3. Bolded in red is off-target site with 5’ and 3’ flanking sequence. Bar shown in black is percent editing of off-target with no transfection. Bars shown in red are percent editing of off-targets when directing editing at endogenous RAB7A with overexpression of wildtype hADAR2 and cytidine guide RNA. Bars shown in grey are percent editing of off-targets when directing editing at endogenous RAB7A with overexpression of hADAR2 E488Y and reduced abasic guide RNA. Percent editing values can be found in Table S5. An asterisk (*) indicates no detected editing. Error bars, s.d. (n = 3 biological replicates).
Discussion
Redirecting the ADAR reaction to adenosines present in RNA as a result of disease-associated G to A mutations is a promising approach for the development of novel therapeutics (Reenan, 2014; Sinnamon et al., 2017; Heep et al., 2017; Wettengel et al., 2017). This is conceptually similar to base-specific gene editing using deaminase-Cas9 fusion proteins but with RNA, not DNA, as the target (Komor et al., 2016; Hsu et al., 2014). Directed RNA editing provides an important alternative to Cas-mediated genome editing because the RNA editing effect is transient in nature and, thus, reversible. Editing the transcriptome is inherently less risky than directing permanent sequence changes to the genome. In addition, RNA editing is possible in post-mitotic cells. For example, directing RNA editing to “repair” specific disease associated G to A mutations in the MeCP2 mRNA present in post-mitotic neurons has therapeutic potential for Rett Syndrome (Sinnamon et al., 2017). However, the intrinsic reactivity of ADAR deaminase domains, particularly hyperactive variants that have been used in some cases, can lead to off target RNA editing (Cox et al., 2017; Montiel-Gonzalez et al., 2013; Montiel-Gonzalez et al., 2016; Wettengel et al., 2017; Vogel et al., 2018). An important challenge is to identify methods that reduce off target editing activity while maintaining (or enhancing) the desired on-target, directed editing. Rosenthal has addressed this issue by targeting ADAR fusion proteins to the nucleus where off-target editing is less efficient (Vallecillo-Viejo et al., 2018). Zhang described the use of mutant ADAR catalytic domains with lower overall deaminase activity (Cox et al., 2017). Using the ADAR2 deaminase domain mutant (E488Q/T375G) fused to dCas13b, Zhang observed a decrease in off-target editing, however on-target editing was also compromised (Cox et al., 2017). Stafforst has used modified guide RNAs to block editing within the duplex containing the guide and genomic integration of the deaminase to reduce off targeting due to overexpression (Vogel et al., 2014; Vogel et al., 2018). Here we described a structure-guided approach that combines the idea of reducing ADAR activity by mutagenesis and the use of chemically-modified guide strands to re-shape the ADAR-RNA interface such that only a target adenosine opposite a nucleoside analog (e.g. a rAb site) in the guide is efficiently edited. This is an example of the bump-hole approach for controlling ligand-receptor interactions (Alaimo et al., 2001; Belshaw et al., 1995).
The abasic site was a simple starting point for modification at the orphan base position, however other modifications have potential to lead to an even more selective A to I editing systems. The high-resolution crystal structure presented here provides additional insight into other potential modifications that could be incorporated into the guide strand to facilitate editing with ADAR mutants. For instance, since the most efficient stacking of the Y488 side chain appears to occur with the U-A pair containing the 5’ U (Figure 3B), adenosine analogs with altered p-stacking properties could be beneficial in the guide strand at this position. In addition, using our structure as a starting point, one could carry out computational screening of ribose analogs to identify derivatives uniquely suited for accommodating various E488X mutants (Suter et al., 2016; Onizuka et al., 2013). Furthermore, only the 488 position was mutated here but one can easily imagine extending this concept to other residues whose side chains approach the guide strand. Combinations of specific mutations and compensatory guide strand modifications would be expected to increase further overall editing selectivity.
Other directed RNA editing approaches use artificial targeting domains fused to ADAR deaminase domains (Hanswillemenke et al., 2015; Montiel-Gonzalez et al., 2013; Montiel-Gonzalez et al., 2016; Sinnamon et al., 2017). For the directed editing in human cells described here, our editing enzymes have human ADAR2’s native targeting domain (i.e. the natural N-terminal domain containing two dsRBDs). We believe this is a demanding test of the effects of our bump-hole approach, since all natural ADAR2 sites are potential off-target sites. Interestingly, the guide RNA used to direct editing to the overexpressed β-actin 3’ UTR fragment induced wild type ADAR2 to deaminate three adenosines within a 26 nt segment of the target, including one (off target 1) that is outside the predicted binding site for the guide (Figure 5). Editing efficiency was reduced with the ADAR2 E488X mutants at both the off target sites, even though off target site 1 adenosine is not predicted to pair with a guide RNA nucleotide. While the basis for the reduced activity for this site cannot be firmly established at this time, off target site 1 may exist in duplex structure native to the overexpressed β-actin 3’ UTR fragment (Vogel et al., 2018).
We have shown that the bump-hole combinations described here can be used to direct efficient RNA editing to two different endogenous transcripts (RAB7A and β-actin) (Figure 6A, 6B). Directed editing of the RAB7A target with the bulky ADAR2 mutant / rAb guide combination occurred at levels equal to, or higher than, the wild type ADAR2 / cytidine guide combination (Figure 6A). For the β-actin target, wild type ADAR2 / cytidine guide was 1.7-fold more efficient than the best bulky mutant (E488Y) / rAb guide combination (Figure 6B). A variety of factors could be responsible for transcript-specific differences in targeting efficiencies, including differences in transcript expression levels, target accessibility and differences in the metabolic stabilities of the different guide RNAs used. Additional studies are necessary to determine the relative importance of these different parameters in controlling on-target editing yield. Nevertheless, these results show that the bulky ADAR mutant / rAb guide combinations can lead to directed editing yields comparable (within 2-fold) to the wild type ADAR2 / cytidine guide for endogenous transcripts.
Importantly, for each of the six ADAR2 sites analyzed for off target editing, the E488Y mutant induced lower editing levels than the wild type enzyme (Figure 6C). The full extent of off target editing with the bulky ADAR2 mutants described here will require transcriptome sequencing under the directed editing conditions and a comparison to wild type ADAR2 (Vogel et al., 2018). The results reported here show that, under the conditions of a directed editing scenario where efficient editing is observed with an endogenous target (e.g. RAB7A editing = 55%), off target editing is reduced through the use of a bump-hole combination. Off target activity is not completely eliminated, since some editing is observed with the E488Y mutant on the CYFIP2, Gli1 and COG3 off target sites. However, further optimization is likely possible with additional mutagenesis to disrupt the ADAR-RNA interface and new compensating guide RNA modifications. Such efforts are currently underway in our laboratory.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Beal (pabeal@ucdavis.edu).
Experimental Model and Subject Details
HEK293T cells (sex- female) were purchased from ATCC and used in this study. HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), 10 % fetal bovine serum, and 1 % anti-anti at 37 °C, 5 % CO2. HEK293T cells were used at less than 15 passages. Saccharomyces cerevisiae BCY123 was used in this study. S. cerevisiae BCY123 were grown in media without uracil (6.7 g/liter yeast nitrogen base containing (NH4)2SO4 and lacking amino acids, 10 g/liter succinic acid, 6 g/liter NaOH, 1.92 g/liter yeast synthetic dropout media without uracil, 22 mg/liter adenine hemisulfate) with 2 % dextrose (Macbeth and Bass, 2007).
Method Details
Purification of oligonucleotides
Unless otherwise noted oligonucleotides were purchased from either Dharmacon or Integrated DNA Technologies. Oligonucleotides were purified by denaturing polyacrylamide gel electrophoresis (PAGE) and visualized using UV shadowing. Bands were excised from gel and gel slices were crushed and soaked overnight at 4 °C in 500 mM NH4OAc and 100 mM EDTA. Polyacrylamide fragments were removed using a 0.2-µm filter. Oligonucleotides were ethanol precipitated and lyophilized to dryness. Oligonucleotides were then resuspended in nuclease free water and stored at −20 °C. All oligonucleotide sequences can be found in Table S6.
Protein overexpression and purification
Mutagenesis of hADAR2-D and full length hADAR2 was performed using QuickChange XL Site-Directed Mutagenesis (Agilent) and transformed into XL10-Gold Ultracompetent cells (Agilent). Primers for mutagenesis were purified as described above. hADAR2-D wt and hADAR2-D E488X (X= F, Y, W) were expressed and purified as previously described (Macbeth and Bass, 2007; Matthews et al., 2016). In brief, S. cerevisiae BCY123 cells were transformed with a pSc-ADAR construct encoding hADAR2-D WT or hADAR2-D E488X (X = F, Y, W). Cells were streaked on yeast minimal medium minus uracil (Cm-ura) plates. A single colony was used to inoculate a 15 mL Cm-ura starter culture, which was shaken at 300 r.p.m. at 30 °C ov ernight. The starter culture was used to inoculate 1.5 L yeast growth medium. After 24 h, cells were induced with 165 mL of sterile 30% galactose, and protein was expressed for 5 h. Cells were collected by centrifugation and stored at −80 °C. Cells were lys ed in 20 mM Tris-HCl, pH 8.0, 5 % glycerol, 1 mM BME, 750 mM NaCl, 35 mM imidazole, 0.01 % NP-40 supplemented with cOmplete EDTA-free protease inhibitor (Sigma Aldrich). Cell lysate was clarified by centrifugation (19,000 rpm, 50 min). Lysate was passed over a 5 mL Ni-NTA column, which was then washed with 50 mL of each wash buffer: wash I buffer (20 mM Tris-HCl, pH 8.0, 5 % glycerol, 1 mM BME, 1 M NaCl, 35 mM imidazole), wash II buffer (20 mM Tris-HCl, pH 8.0, 5 % glycerol, 1 mM BME, 500 mM NaCl, 35 mM imidazole) and wash III buffer (20 mM Tris-HCl, pH 8.0, 5 % glycerol, 1 mM BME, 100 mM NaCl, 35 mM imidazole). Protein was eluted with 20 mM Tris-HCl, pH 8.0, 5 % glycerol, 1 mM BME, 400 mM imidazole and 100 mM NaCl. Fractions containing protein were dialyzed against 20 mM Tris-HCl, pH 8.0, 20 % glycerol, 1 mM BME and 100 mM NaCl. Protein concentration was determined through BSA standards visualized by SYPRO Orange (ThermoFisher Scientific) staining on SDS-polyacrylamide gels. Purified protein was stored at −70 °C in 20 mM Tris-HCl pH 8.0, 100 mM N aCl, 20 % glycerol and 1 mM 2-mercaptoethanol.
Crystallography
The 8-azanebularine (8AN) phosphoramidite was synthesized as previously described and incorporated into RNA as mentioned prior (Haudenschild et al., 2004; Pokharel et al., 2009). RNA for crystallography was purified as described previously (Matthews et al., 2016). In brief, RNA for crystallography was purified by denaturing PAGE and visualized with UV shadowing. Bands were excised from gel and gel slices were crushed and soaked overnight at 4 °C in 500 mM NH 4OAc and 100 mM EDTA. Polyacrylamide fragments were removed using a 0.2-µm filter and was followed up by desalting on a C18 Sep-Pak column. RNA solutions were lyophilized to dryness, resuspended in nuclease-free water and quantified by absorbance at 260 nm. Oligonucleotide mass was confirmed by electrospray ionization mass spectrometry. Duplex RNA was hybridized in a 1:1 ratio by heating to 95°C for 5 min and slow cooling to 25 °C. Protein, hADAR2-D E488Y, was expressed, p urified and quantified as described above with the following exceptions (Matthews et al., 2016; Macbeth and Bass, 2007). After elution from first Ni-NTA column, fractions containing hADAR2-D E488Y were pooled and purified on a 2-mL GE Healthcare Lifesciences Hi-Trap Heparin HP column in the absence of BME. The His10 fusion protein was cleaved with an optimized ratio of 1 mg of TEV protease per 1 mg of protein. Cleavage was carried out for 1–2 h before the product was passed over another Ni–NTA column at 0.5 mL/min. The flow through and wash were collected; dialyzed against 20 mM Tris, pH 8.0, 200 mM NaCl, 5% glycerol, and 1 mM BME; and concentrated to just under 1 mL for gel filtration on a GE Healthcare HiLoad 16/600 Superdex 200 PG column. Fractions containing hADAR2-D E488Y were pooled and concentrated to 5–7 mg/mL for crystallography trials (Matthews et al., 2016). Crystals of hADAR2-D E488Y+Gli1_reduced abasic RNA complex were grown at room temperature by the sitting drop vapor diffusion method. A solution of 0.5 µL volume containing 4.5 mg/mL protein and 100 µM of Gli1_reduced abasic RNA (1:1 hADAR2-D:RNA molar ratio) was mixed with 0.5 µL of 0.1 M MES:NaOH pH 6.5 and 14% PEG 20,000. Crystals took a few days to grow at 21°C. Crystals were flashed-co oled in liquid nitrogen using 30% glycerol as a cryo-protectant. X-ray diffraction data were collected at 100K on beamline 12–2 at the Stanford Synchrotron Radiation Lightsource. Diffraction data were processed with XDS and scaled with XSCALE with an Rmerge = 6.3% to 2.55 Å resolution (Kabsch, 2010). Structure was solved by molecular replacement using the previously determined ADAR2-Gli1 RNA complex X-ray structure (PDBID: 5ED2) and the model was refined with Refmac5 (CCP4) and built using COOT (Murshudov et al., 2011; Emsley and Cowtan, 2004). Data processing and refinement statistics can be found in Table S2. Atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDBID: 6D06).
Mass spectrometry analysis of proteins present in ADAR-RNA crystals
Crystals of hADAR2-D E488Y+Gli1_reduced abasic RNA complex were pooled and carefully washed in a solution of diluted mother liquor. Sample was added to 10 µL of 4 % SDS and heated at 95 °C for 5 min. Sample was run down SDS-PAGE gel and visualized with Coomassie blue (Bio-Rad). The gel was excised and diced into 1 mm cubes using a sterile blade. Briefly, proteins were reduced and alkylated according to previously described procedures and digested with sequencing grade tryspin per manufacturer’s recommendations (Promega) (Shevchenko et al., 1996). Peptides were dried down in a vacuum concentrator after digestion, then resolubilized in 2 % acetonitrile/ 0.1 % trifluoroacetic acid. Digested peptides were analyzed by LC-MS/MS on a Thermo Scientific Q Exactive Plus Orbitrap Mass spectrometer in conjunction with Proxeon Easy-nLC II HPLC (ThermoScientific) and Proxeon nanospray source. The digested peptides were loaded on a 100 µm × 25 mm Magic C18 100 Å 5 U reverse phase trap where they were desalted online before being separated using a 75 µm × 150 mm Magic C18 200 Å 3 U reverse phase column. Peptides were eluted using a 60 min gradient with a flow rate of 300 nL/min. An MS survey scan was obtained for the m/z range 350–1600, MS/MS spectra were acquired using a top 15 method, where the top 15 ions in the MS spectra were subjected to HCD (High Energy Collisional Dissociation). An isolation mass window of 1.6 m/z was for the precursor ion selection, and normalized collision energy of 27 % was used for fragmentation. A 15 s duration was used for the dynamic exclusion.
Tandem mass spectra were extracted and charge state deconvoluted by Proteome Discoverer (ThermoScientific). All MS/MS samples were analyzed using X! Tandem (thegpm.org; version Alanine (2017. 2. 1.4)). X! Tandem was set up to search Uniprot Saccharomyces cerevisiae database (January 2018, 6079 entries), the cRAP database of common laboratory contaminants (thegpm.org/crap; 114 entries), the ADAR2 catalytic domain sequence plus an equal number of reverse protein sequences assuming the digestion enzyme trypsin. X! Tandem was searched with a fragment ion mass tolerance of 20 PPM and a parent ion tolerance of 20 PPM. Iodoacetamide derivative of cysteine was specified in X! Tandem as a fixed modification. Deamidation of asparagine and glutamine, oxidation of methionine and tryptophan, sulphone of methionine, tryptophan oxidation to formylkynurenin of tryptophan and acetylation of the N-terminus were specified in X! Tandem as variable modifications.
Criteria for Protein Identification: Version Scaffold_4.8.4 (Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they exceeded specific database search engine thresholds. X! Tandem identifications required at least –Log (Expect Scores) scores of greater than 2 .0 with a mass accuracy of 5 ppm. Protein identifications were accepted if they contained at least 2 identified peptides. Peptides pertinent to the sequence analysis were visually inspected for validation (Figure S1).
Deamination kinetics for hADAR2-D E488F and hADAR2-D E488Y
Generation of internally 32P-labeled strand: The 3’ 11 nucleotide oligonucleotide of the top (edited) strand was radiolabeled with [γ−32P] ATP (PerkinElmer Life Sciences) at the 5’ end with T4 polynucleotide kinase as previously described (Phelps et al., 2014). About 30 pmol of labeled 3’ top strand was dissolved with 40 pmol of 5’ 12 nucleotide top strand, 30 pmol of DNA splint, 0.5 µL of RNasin (1.6 U/µL), 2 µL of T4 DNA ligase 10x buffer (NEB) and 5 µL of nuclease free water (Zheng et al., 2017). The solution was heated at 65 °C for 5 min and slowly cooled to room temperature. After the addition of 1.5 µL of RNasin (1.6 U/µL), 5 µL of 4 mM ATP and 1 µL of T4 DNA ligase (NEB, 400 U/µL), reaction incubated at 30 °C for 2 h. Splint- ligated product was purified as described above. Upon hybridization internally labeled top strand and corresponding bottom strand (X = C or rAb) were heated to 95 °C f or 5 min and then slowly cooled to room temperature in 10 mM Tris-HCl, 0.1 mM EDTA pH 7.5 and 100 mM NaCl.
Deamination reactions had a final volume of 10 µL with concentrations of 10 nM RNA and 300 nM hADAR2-D wt, hADAR2-D E488F or hADAR2-D E488Y. The final reaction solution contained 16 mM Tris-HCl, pH 7.4, 3.3 % glycerol, 1.6 mM EDTA, 0.003% NP-40, 60 mM KCl, 7.1 mM NaCl, 0.5 mM DTT, 1.6 U/µL RNasin and 1 µg/mL yeast tRNA. Reactions were quenched by adding 190 µL of 95 °C n uclease free water followed by vortex of the solution and incubating at 95 °C for 5 min. Deaminated sample was purified by phenol-chloroform extraction and ethanol precipitation. The deaminated sample was lyophilized to dryness and resuspended in 50 µL of 1x TE solution followed by digestion with nuclease P1 (Sigma Aldrich). The subsequent 5’-mononucleotides were resolved by thin-layer chromatography (TLC, Macherey-Nagel). The TLC plate was visualized by exposure to a storage phosphor imaging plate (Molecular Dynamics) on a Typhoon phosphoimager (Molecular Dynamics) (O’Connell and Keller, 1994). Radioactive spots were then quantified by volume integration using ImageQuant (Molecular Dynamics) where the data were fitted in KaleidaGraph to the equation: [P]t = ∝[1-ekobst], where [P]t is percent edited at time t, ∝ is the fitted end point, and kobs is the observed rate constant. Each experiment was carried out in triplicate where the observed rate stated is the average of the replicates ± standard deviation.
Selective Editing on 152 nt Multiple Target Substrate
Using extended primers containing BamHI and HindIII restriction sites, 74 nt multiple target substrate was PCR amplified using Phusion Hot Start DNA Polymerase (ThermoScientific). PCR product was purified by agarose gel and Qiagen Gel Extraction kit. Sample was then resuspended in nuclease free water and double digested at BamHI and HindIII restriction sites (NEB). Double digested product was inserted into T7-promoter containing vector by standard cloning procedures. Plasmid containing 152 nt target substrate was single digested by BamHI and 152 nt RNA was transcribed from this DNA template with MEGAscript® T7 Kit (ThermoFisher). Transcribed product was purified as described above.
RNA bottom strands containing orphan nucleotide for site 1 (3 nmols) were first phosphorylated by T4 PNK (NEB, 10,000 U/mL) in 10x T4 PNK buffer and 100 mM ATP. After denaturation of PNK, RNA bottom strand containing orphan nucleotide for site 2 (3 nmols), DNA splint (3 nmols) and RNasin (1.6 U/µL) were dissolved in solution with phosphorylated RNA bottom strand containing site 1. Solution was heated to 95 °C for 5 min and then slowly cooled to room temperature. Additional RNasin (1.6 U/µL) was added to the cooled solution followed by T4 DNA ligase (400 U/µL). The reaction was incubated at 30 °C for 2 h. Ligated sample was then phenol-chloroform extracted, ethanol precipitated, lyophilized to dryness, and resuspended in nuclease-free water. Samples were then DNase treated with RQ1 RNase-free DNase (Promega). DNase treated splint ligated product was purified as described above. Upon hybridization, 152 nt transcribed top strand and corresponding ligated bottom strand (varying X and Y containing C or rAb) were heated to 95 °C for 5 min and then slowly cooled to room temperature in 10 mM Tris-HCl, 0.1 mM EDTA pH 7.5 and 100 mM NaCl.
Deamination reactions had a final volume of 10 µL with concentrations of 10 nM RNA and 1.2 µM hADAR2-D E488F, 150 nM hADAR2-D E488Y, or 300 nM hADAR2-D E488W. The final reaction solution contained 16 mM Tris-HCl, pH 7.4, 3.3 % glycerol, 1.6 mM EDTA, 0.003% NP-40, 60 mM KCl, 7.1 mM NaCl, 0.5 mM DTT, 1.6 U/µL RNasin and 1 µg/mL yeast tRNA. Reaction was quenched as described above. Reaction was quenched after 30 min, 15 min, and 30 min for hADAR2-D E488F, hADAR2-D E488Y, and hADAR2-D E488W, respectively. RT-PCR of deaminated samples was performed using Access RT-PCR kit (Promega) for 24 cycles. PCR product was purified using Zygmo DNA Clean and Concentrator kit. Purified samples were submitted for Sanger Sequencing and sequence traces were analyzed by 4Peaks (Nucleobytes) to quantify percent editing.
Directed editing on overexpressed β-actin in HEK293T cells and analysis
Full length hADAR2 wt and HA tag were subcloned into pcDNA3.1 vector using Gibson Assembly Cloning Kit (NEB). QuickChange XL Site-Directed Mutagenesis (Agilent) was used to incorporate E488X (X= F, Y, W) mutation in pcDNA3.1 vector containing hADAR2 sequence. Plasmid containing β-actin target sequence was ordered from ThermoFisher. β-actin target region was amplified by PCR using primers containing non-native sequence at 5’ ends of forward and reverse primers (Figure S2). β-actin target was then subcloned into pcDNA3.1 vector using Gibson Assembly Cloning Kit (NEB). HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), 10 % fetal bovine serum, and 1 % anti-anti at 37 °C, 5 % CO2. Once cultivated cells reached 70–90 % confluency, 1.5 × 105 cells were seeded into 24 well plates. Cells were transfected 24 h later using Lipofectamine 2000 (ThermoFisher Scientific). Transfection of plasmids and guide RNA was as followed: 500 ng ADAR plasmid, 500 ng β-actin plasmid, and 50 nM chemically synthesized guide RNA. After incubation of transfection reagent, specified plasmids and guide RNAs in Opti-MEM Reduced Serum Media (ThermoFisher Scientific), solution was added to designated well and incubated at 37 °C, 5 % CO2 for 24 h. Total RNA was then collected using RNAqueous Total RNA Isolation Kit (ThermoFisher Scientific) and DNase treated with RQ1 RNase-free DNase (Promega). Nested RT-PCR was performed using Access RT-PCR kit (Promega) for 15 cycles and then followed by Phusion Hot Start DNA Polymerase (ThermoScientific) for the second PCR of 25 cycles. PCR product was purified by agarose gel and Qiagen Gel Extraction kit. Product was submitted for Sanger Sequencing and sequence traces were analyzed by 4Peaks (Nucleobytes) to quantify percent editing.
Directed editing on RAB7A and endogenous β-actin in HEK293T cells and analysis
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), 10 % fetal bovine serum, and 1 % anti-anti at 37 °C, 5 % CO2. Once cultivated cells reached 70–90 % confluency, 6.4 × 103 cells were seeded into 96 well plates. Cells were transfected 24 h later using Lipofectamine 2000 (ThermoFisher Scientific). Transfection of plasmids and guide RNA was as followed: 500 ng ADAR plasmid and 50 nM chemically synthesized guide RNA. After incubation of transfection reagent, specified plasmids and guide RNAs in Opti-MEM Reduced Serum Media (ThermoFisher Scientific), solution was added to designated well and incubated at 37 °C, 5 % CO2 for 48 h. Total RNA was then collected using RNAqueous Total RNA Isolation Kit (ThermoFisher Scientific) and DNase treated with RQ1 RNase-free DNase (Promega). Nested RT-PCR was performed using Access RT-PCR kit (Promega) for 20 cycles and then followed by Phusion Hot Start DNA Polymerase (ThermoScientific) for the second PCR of 30 cycles. PCR product was purified by agarose gel and Qiagen Gel Extraction kit. Product was submitted for Sanger Sequencing and sequence traces were analyzed by 4Peaks (Nucleobytes) to quantify percent editing.
Detection of full length ADAR2 proteins in transfected HEK293T cells
Transfection for Western blotting was performed as described above. Cells were lysed with 300 µL of lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 % (v/v) NP-40 supplemented with Halt protease inhibitor cocktail) (ThermoFisher) by shaking at 4 °C for 30 min. Samples were resolved on an SDS-PAGE gel alongside PageRuler Prestained Plus Protein Ladder (ThermoFisher Scientific). Western blotting was carried out using primary antibody HA Tag Monoclonal Antibody (2–2.2.14) (ThermoFisher) at 1:10,000 dilution and anti-mouse IgG with alkaline phosphatase-conjugated secondary antibody (Santa Cruz Biotechnology) at 1:2,000 dilution. The ADAR proteins were detected using ECF substrate (GE Healthcare) on a Typhoon Trio Variable Mode Imager (GE Healthcare).
Quantification and Statistical Analysis
Statistical details can be found in corresponding figure legends. Bar graphs are plotted as means ± SD. Error bars represent standard deviation of n = 3 replicates. Quantification of Western blots was carried out using ImageQuant. Sanger Sequencing and sequence traces were analyzed by 4Peaks (Nucleobytes) to quantify percent editing.
Supplementary Material
Highlights.
Bump-hole strategy developed for selective site-directed RNA editing (SDRE) system
SDRE in vitro and in human cells with bulky mutant ADAR2 proteins and guide RNAs
Directed editing on endogenous targets with reduced off-target activity
Crystal structure of ADAR2-D E488Y with RNA duplex containing reduced abasic site
Significance.
Genome and transcriptome editing tools have the potential to revolutionize molecular biology and medicine. However, it is essential that such tools be highly precise as unexpected changes in nucleic acid sequences could be lethal. Here we show the classic bump-hole approach from chemical biology can be applied to controlling the selectivity of directed RNA editing by adenosine deamination. In principle, this approach could be applied to each of the published directed RNA editing platforms that use ADAR-derived adenosine deaminase domains and guide RNAs. Furthermore, the bump-hole approach described here for optimizing targeted nucleic acid editing should prove useful for other systems where use of native components leads to unacceptable levels of off targeting and high resolution structures of the protein-nucleic acid complexes exist.
Acknowledgments
The authors acknowledge funding from the US National Institutes of Health (NIH) grant R01GM061115 (P.A.B). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02–76SF00515. The authors acknowledge the technical assistance of Michelle Salemi at the University of California Davis Proteomics Core. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data and Software Availability
The atomic coordinates and structure factors for hADAR2-D E488Y:hGli1_abasic-8AN reported in this paper, have been deposited in the Protein Data Bank (www.rcsb.org) under the accession code 6D06.
Declaration of Interest
The authors declare competing financial interests: L.R.M, A.J.F and P.A.B have filed a patent application on this work. P.A.B is a consultant for Beam Therapeutics, Cambridge, MA, USA and ProQR Therapeutics, Leiden, Netherlands.
References
- Alaimo PJ, Shogren-Knaak MA and Shokat KM (2001). Chemical genetic approaches for the elucidation of signaling pathways. Curr Opin Chem Biol 5, 360–367. [DOI] [PubMed] [Google Scholar]
- Bass BL (2002). RNA editing by adenosine deaminases that act on RNA. Annu. Rev. of Biochem 71, 817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL and Weintraub H (1988). An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55, 1089–1098. [DOI] [PubMed] [Google Scholar]
- Belshaw PJ, Schoepfer JG, Liu KQ, Morrison KL and Schreiber SL (1995). Rational Design of Orthogonal Receptor–Ligand Combinations. Angew. Chem 34, 2129–2132. [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J and Zhang F (2017). RNA editing with CRISPR-Cas13. Science 358, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Greene T and Bass BL (2011). Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun 2, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eifler T, Pokharel S and Beal PA (2013). RNA-Seq analysis identifies a novel set of editing substrates for human ADAR2 present in Saccharomyces cerevisiae. Biochemistry 52, 7857–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Gao X, Tao Y, Lamas V, Huang M, Yeh WH, Pan B, Hu YJ, Hu JH, Thompson DB, Shu Y, Li Y, Wang H, Yang S, Xu Q, Polley DB, Liberman MC, Kong WJ, Holt JR, Chen ZY and Liu DR (2018). Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI and Liu DR (2017). Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RA, Macbeth MR and Beal PA (2012). ADAR proteins: structure and catalytic mechanism. Curr. Top. Microbiol 353, 1–33. [DOI] [PubMed] [Google Scholar]
- Hanswillemenke A, Kuzdere T, Vogel P, Jekely G and Stafforst T (2015). Site-Directed RNA Editing in Vivo Can Be Triggered by the Light-Driven Assembly of an Artificial Riboprotein. J. Am. Chem. Soc 137, 15875–15881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudenschild BL, Maydanovych O, Veliz EA, Macbeth MR, Bass BL and Beal PA (2004). A transition state analogue for an RNA-editing reaction. J. Am. Chem. Soc 126, 11213–11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heep M, Mach P, Reautschnig P, Wettengel J and Stafforst T (2017). Applying Human ADAR1p110 and ADAR1p150 for Site-Directed RNA Editing-G/C Substitution Stabilizes GuideRNAs against Editing. Genes (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES and Zhang F (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Ryu SM, Kim ST, Baek G, Kim D, Lim K, Chung E, Kim S and Kim JS (2017). Highly efficient RNA-guided base editing in mouse embryos. Nat Biotechnol 35, 435–437. [DOI] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA and Liu DR (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttan A and Bass BL (2012). Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. U. S. A 109, E3295–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann KA and Bass BL (2000). Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39, 12875–12884. [DOI] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y and Church GM (2009). Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324, 1210–1213. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Word JM, Richardson JS and Richardson DC (2000). The penultimate rotamer library. Proteins 40, 389–408. [PubMed] [Google Scholar]
- Macbeth MR and Bass BL (2007). Large-scale overexpression and purification of ADARs from Saccharomyces cerevisiae for biophysical and biochemical studies. Methods Enzymol 424, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, Scott AI, Havel J, Fisher AJ and Beal PA (2016). Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol 23, 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-Gonzalez MF, Vallecillo-Viejo I, Yudowski GA and Rosenthal JJ (2013). Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc. Natl. Acad. Sci. U. S. A 110, 18285–18290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-Gonzalez MF, Vallecillo-Viejo IC and Rosenthal JJ (2016). An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res 44, e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F and Vagin AA (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K (2010). Functions and regulation of RNA editing by ADAR deaminases. Annual Review of Biochemistry 79, 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez JK, Harrington LB and Doudna JA (2016). Chemical and Biophysical Modulation of Cas9 for Tunable Genome Engineering. ACS Chem. Biol 11, 681–688. [DOI] [PubMed] [Google Scholar]
- O’connell MA and Keller W (1994). Purification and properties of double-stranded RNA-specific adenosine deaminase from calf thymus. Proc. Natl. Acad. Sci. U. S. A 91, 10596–10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onizuka K, Harrison JG, Ball-Jones AA, Ibarra-Soza JM, Zheng Y, Ly D, Lam W, Mac S, Tantillo DJ and Beal PA (2013). Short interfering RNA guide strand modifiers from computational screening. J. Am. Chem. Soc 135, 17069–17077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps KJ, Ibarra-Soza JM, Tran K, Fisher AJ and Beal PA (2014). Click modification of RNA at adenosine: structure and reactivity of 7-ethynyl- and 7-triazolyl-8-aza-7-deazaadenosine in RNA. ACS Chem. Biol 9, 1780–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps KJ, Tran K, Eifler T, Erickson AI, Fisher AJ and Beal PA (2015). Recognition of duplex RNA by the deaminase domain of the RNA editing enzyme ADAR2. Nucleic Acids Res 43, 1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokharel S, Jayalath P, Maydanovych O, Goodman RA, Wang SC, Tantillo DJ and Beal PA (2009). Matching active site and substrate structures for an RNA editing reaction. J. Am. Chem. Soc 131, 11882–11891. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reenan R (2014). Correcting mutations by RNA repair. N. Engl. J. Med 370, 172–174. [DOI] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR and Emeson RB (1999). Regulation of alternative splicing by RNA editing. Nature 399, 75–80. [DOI] [PubMed] [Google Scholar]
- Schneider MF, Wettengel J, Hoffmann PC and Stafforst T (2014). Optimal guideRNAs for re-directing deaminase activity of hADAR1 and hADAR2 in trans. Nucleic Acids Res 42, e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O and Mann M (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Sinnamon JR, Kim SY, Corson GM, Song Z, Nakai H, Adelman JP and Mandel G (2017). Site-directed RNA repair of endogenous Mecp2 RNA in neurons. Proc. Natl. Acad. Sci. U. S. A 114, 9395–9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafforst T and Schneider MF (2012). An RNA-deaminase conjugate selectively repairs point mutations. Angew. Chem. Int. Ed. Engl 51, 11166–11169. [DOI] [PubMed] [Google Scholar]
- Suter SR, Sheu-Gruttadauria J, Schirle NT, Valenzuela R, Ball-Jones AA, Onizuka K, Macrae IJ and Beal PA (2016). Structure-Guided Control of siRNA Off-Target Effects. J. Am. Chem. Soc 138, 8667–8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallecillo-Viejo IC, Liscovitch-Brauer N, Montiel-Gonzalez MF, Eisenberg E and Rosenthal JJC (2018). Abundant off-target edits from site-directed RNA editing can be reduced by nuclear localization of the editing enzyme. RNA Biol 15, 104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel P, Moschref M, Li Q, Merkle T, Selvasaravanan KD, Li JB and Stafforst T (2018). Efficient and precise editing of endogenous transcripts with SNAP-tagged ADARs. Nat. Methods 15, 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel P, Schneider MF, Wettengel J and Stafforst T (2014). Improving site-directed RNA editing in vitro and in cell culture by chemical modification of the guideRNA. Angew. Chem. Int. Ed. Engl 53, 6267–6271. [DOI] [PubMed] [Google Scholar]
- Vogel P and Stafforst T (2014). Site-directed RNA editing with antagomir deaminases--a tool to study protein and RNA function. ChemMedChem 9, 2021–2025. [DOI] [PubMed] [Google Scholar]
- Wang Q, Hui H, Guo Z, Zhang W, Hu Y, He T, Tai Y, Peng P and Wang L (2013). ADAR1 regulates ARHGAP26 gene expression through RNA editing by disrupting miR-30b-3p and miR-573 binding. RNA 19, 1525–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y and Beal PA (2016). Probing RNA recognition by human ADAR2 using a high-throughput mutagenesis method. Nucleic Acids Res 44, 9872–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Park S and Beal PA (2018). Selective Recognition of RNA Substrates by ADAR Deaminase Domains. Biochemistry 57, 1640–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettengel J, Reautschnig P, Geisler S, Kahle PJ and Stafforst T (2017). Harnessing human ADAR2 for RNA repair - Recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res 45, 2797–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SK, Sato S and Lazinski DW (2001). Substrate recognition by ADAR1 and ADAR2. RNA 7, 846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf TM, Chase JM and Stinchcomb DT (1995). Toward the therapeutic editing of mutated RNA sequences. Proc. Natl. Acad. Sci. U. S. A 92, 8298–8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo J, Goodman RA, Schirle NT, David SS and Beal PA (2010). RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc. Natl. Acad. Sci. U. S. A 107, 20715–20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Lorenzo C and Beal PA (2017). DNA editing in DNA/RNA hybrids by adenosine deaminases that act on RNA. Nucleic Acids Res 45, 3369–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.